Introduction
Radiotherapy is one of the main treatments used to
deal with malignancy. More than 50% of patients with malignant
tumors receive radiotherapy during their treatment (1). Radiation-induced DNA damage is one of
main mechanisms underlying the cell killing effect of radiotherapy.
Compared with the direct killing effect of irradiation, the
indirect killing effect that results from reactive oxygen species
(ROS) plays a pivotal role in DNA damage (2). When high-energy rays act on tumor
cells, a booming growth of ROS levels occurs in the cytoplasm, then
combines with DNA bases and sugar backbones to cause DNA chain
fractures. These fractures, if not repaired in time, trigger the
process of apoptosis of the cells and ultimately cell death
(3).
Statin is a type of 3-hydroxy-3-methyl-glutaryl-CoA
(HMGCoA) reductase inhibitor which has been used to lower plasma
cholesterol in clinical practice (4). Recently, studies have discovered that
statins have the potential to increase the radiation-mediated
killing of malignant cells. Park et al reviewed 6 studies
and concluded that statins have a potential benefit to prostate
cancer patients who receive radiotherapy (5). He et al found that atorvastatin
(ATO) may increase the radiation-mediated killing of PC-3 cells
(6). Fritz et al found that
lovastatin increased the cell killing effect of irradiation in a
variety of tumors (7). Moreover,
several studies have also reported that statins can influence the
intracellular ROS level in tumor cells. Mihăilă found that
simvastatin can increase the ROS level in lymphoblasts (8), but Song et al reported that
lovastatin may inhibit the ROS generation in B-cell lymphoma
(9). However, the effects of
statins on radiation-induced ROS levels in malignant cells have
rarely been investigated.
ATO is a third generation statin. Studies have
reported that ATO can influence ROS generation in vascular cells
(10). The PC-3 cell line is a
typical castrate-resistant prostate cancer (CRPC) cell line which
has poor radiosensitivity and strongly resists the cell killing
effect of irradiation (11). In the
present study, we hypothesized that ATO may also interfere with the
ROS generation in PC-3 cells, thereby influencing the
radiation-induced ROS levels and the cell killing effect. Thus, the
clonogenic ability, apoptosis rate, intracellular ROS level and
levels of regulatory proteins, such as NOX2, NOX4 and SOD1, were
evaluated.
Materials and methods
Reagents
ATO was purchased from Cayman Chemical Co. (Ann
Arbor, MI, USA). Tempol was purchased from Enzo Life Sciences, Inc.
(Farmingdale, NY, USA) and an apoptosis detection kit was purchased
from BD Biosciences (Franklin Lakes, NJ, USA). An ROS assay kit and
a SOD assay kit were purchased from Beyotime Institute of
Biotechnology (Shanghai, China). Hoechst 33342 staining solution
was purchased from Bridgen Company (Beijing, China). A rabbit
monoclonal anti-NOX2 antibody and a rabbit monoclonal anti-SOD1
antibody were purchased from Biosynthesis Biotechnology Corp.
(Beijing, China) and a rabbit monoclonal anti-NOX4 antibody was
obtained from Abcam (Cambridge, UK).
Cell culture
The PC-3 cell line was purchased from the American
Type Culture Collection (ATCC, Manassas, VA, USA) and the cells
were maintained in RPMI-1640 medium that was supplemented with 10%
(v/v) fetal bovine serum (FBS) and cultured at 37°C in an incubator
with 5% CO2 and a humid atmosphere.
Experimental schedule
To evaluate the radiosensitization potential of ATO
on PC-3 cells, clonogenic and apoptosis assays were first
conducted. Next, we transiently transfected NOX2 and
NOX4 genes into the PC-3 cells so that by increasing the
expression level of NOXs in the cells and with the addition of
tempol, an SOD mimetic, into the cell culture medium, the ROS
scavenging ability of the PC-3 cells was increased. In the
following experiments, the cells were divided into four groups:
vehicle, ATO alone, ATO plus tempol and ATO plus transfected
NOXs. These grouped cells were applied to an ROS detection
assay, an immunoblotting assay and SOD activity detection. Finally,
we explored the change in the survival fraction of the PC-3 cells
with the clonogenic assay by combining ATO with tempol or by the
transfected NOXs. All the cells from the study groups were
treated with 10 µM ATO for 24 h and the cells with the addition of
tempol were treated with 3 mM tempol for another 10 h. Dimethyl
sulfoxide (DMSO) was applied to the vehicle as a negative
control.
Clonogenic assay
Cells were seeded into seven 6-well plates at
different quantities and cultured at 37°C in an incubator for
>24 h. After being pretreated with the vehicle, ATO, tempol or
transfected NOXs, the medium was renewed and the cells were
irradiated with different doses (0, 0.5, 1, 2, 4, 6 and 8 Gy) by a
6 MV X-ray linear accelerator (4 Gy/min at room temperature).
Subsequently, the cells were cultured at 37°C in an incubator for
14 days. When the clusters were >50 cells, colonies were fixed
with methanol and stained using methylene blue for cell
counting.
A cell survival curve was plotted by GraphPad Prism
6 software according to survival fractions and was calculated as
the clonogenic efficiency of the irradiated cells divided by that
of the unirradiated cells. The clonogenic efficiency was calculated
as the percentage of the clones of the treated cells divided by
that of the control. To calculate the radiosensitivity parameters,
including D0, Dq, N, SF2 and SERs
(12), a cell survival curve was
fitted by the multi-target single hit equation: SF = 1 - (1 -
e−D/D0)N (13).
Apoptosis assay
After ATO treatment, the cells from the study group
received a 1 Gy dose of irradiation using an Elekta linear
accelerator (4 Gy/min at room temperature). Six hours later, all
the cells were collected into a 15 ml centrifuge tube, digested
with 0.25% trypsin (without 0.05% EDTA) and rinsed thrice with
ice-cold phosphate-buffered saline (PBS). Subsequently, the cells
were resuspended with 1X Annexin-binding buffer, and then
1×105 cells were transferred into a 1.5 ml Eppendorf
tube and 5 µl Annexin-V FITC and 5 µl propidium iodide were added.
After 15 min of lucifugal incubation at room temperature, cell
apoptosis was detected by flow cytometry. The apoptosis rate was
calculated as the early apoptosis rate plus the late apoptosis
rate.
RNA extraction and DNA
amplification
Total RNA was extracted by TRIzol method according
to the manufacturers instructions (Invitrogen, Carlsbad, CA, USA),
c-DNA was synthesized by Reverse transcription PCR kit (Tiangen
Biotech Co., Ltd. (Beijing, China). NOX2 and NOX4
cDNA were then amplified by a PCR kit (Sangon, Biotech Co., Ltd.,
Shanghai, China). The NOX2 primer sequences (sense primer,
5′-TCACTGGAGTTGTCATCACGCTGT-3 and antisense primer,
5′-AAGGGCCCATCAACCGCTATCTTA-3) and the NOX4 primer sequences
(sense primer, 5′-AGCAGAGCCTCAGCATCTGTTCTT-3 and antisense primer,
5′-AGCTTGGAATCTGGGCTCTTCCAT-3) are referred to in published data
(14). Briefly, the PCR reaction
system was composed of diluted NOXs cDNA, 0.1 mM dNTP, 0.2
U/µl Taq polymerase, 0.4 pmol/µl of each primer, PCR buffer
and deionized water. The procedure steps were as follows: the first
step was at 95°C for 5 min, then the second step to the fourth step
were run for 30 cycles at 95°C for 30 sec, 55°C for 30 sec and 72°C
for 30 sec and the fifth step was conducted at 72°C for 7 min.
Recombinant plasmid construction
The amplified DNA was transfected into DH5α
competent cells (GenStar, Biosolutions Co., Ltd., Beijing, China)
and the plasmid was extracted using a plasmid extraction kit (Life
Technologies, Grand Island, NY, USA). The plasmid and pcDNA3.1
vector (Invitrogen) were digested by the restriction enzymes
BamHI and HindIII. Next, NOX genes and
pcDNA3.1 DNA were purified by agarose gel electrophoresis and
extracted by DNA gel extraction kit (ATOM, China) and then the
NOX genes and the vector were linked by T4 DNA ligase.
Finally, the recombinant vector was transfected into DH5α cells
again and plasmid extraction was conducted and identified by enzyme
digestion, electrophoresis and sequencing.
Transient transfection
Before transfection, the PC-3 cells were seeded into
6-well plates at 2×105 cells/well and cultured in a 37°C
incubator for 48 h. DNA and Lipo3000 transfection reagent
(Invitrogen) were diluted using non-serum RPMI-1640 medium, mixed
and maintained at room temperature for 15 min. Then, the cells were
incubated with Lipo3000-DNA complexes in non-serum RPMI-1640
medium, cultured in a 37°C incubator for 6 h. Next, the old media
was replaced with fresh RPMI-1640 (with 10% FBS) and cultured at
37°C for 24 h. Finally, the transfected cells were screened using
G418 antibiotic (Amresco, LLC, Solon, OH, USA) in fresh RPMI-1640
for 14 days, and the clones which were enlarged in the culture
medium were selected.
ROS detection assay
PC-3 cells for flow cytometry assay were seeded into
6-well plates. Before probe treatment, the cells were collected
into 15 ml tubes digested with 0.25% trypsin (with 0.05% EDTA)
digestion. The cells for laser scanning confocal microscopy assay
were seeded into a confocal dish. During ROS detection, part of the
cells were suspended in a positive control that was diluted with
non-serum RPMI-1640 medium in 1:1,000 titer and cultured in a 37°C
incubator. Fifteen minutes later, all the cells were rinsed thrice
with non-serum RPMI-1640 medium, then suspended in DCFH-DA solution
that was diluted with non-serum RPMI-1640 medium in 1:1,000 titer.
The cells were cultured in a 37°C incubator for 20 min and then
immediately detected by flow cytometry or observed under a laser
scanning confocal microscope.
For the irradiation study, the cells received a 1 Gy
dose of irradiation using an Elekta linear accelerator after ATO
alone or combined treatment and then the cells were cultured in a
37°C incubator for 2 h before following the same steps as
aforementioned.
Immunoblotting assay
The cells were rinsed twice with ice-cold PBS, and
lysed with lysis buffer, which was supplemented with a protease
inhibitor cocktail. Cell lysates were collected using a scraper and
transferred into an Ependorf tube for high speed centrifugation
(4°C, 12,000 rpm, 20 min), The liquid supernatant was divided into
segments of protein samples and the concentrations were determined.
The protein samples were diluted with 5X loading buffer and
denaturated at 100°C for 5 min.
For immunoblotting, an equal amount of total
proteins from each group was separated by SDS-polyacrylamide gel
electrophoresis and transferred onto PVDF membranes. Next, the
antigens on the membranes were blocked with 5% dried skimmed
milk/1X TBST for 1 h, and incubated overnight with corresponding
primary antibodies against NOX2, NOX4, SOD1 and GAPDH,
respectively. Subsequently, the membranes were washed thrice with
1X TBST and incubated with horseradish peroxidase-coupled secondary
antibodies for 1 h. After washing thrice again with 1X TBST, blots
on the membranes were finally detected by ECL western blotting
detection reagent.
SOD activity detection
Total proteins of the PC-3 cells were extracted as
aforementioned. When the protein concentration was determined, the
assay was carried out according to the instructions from the total
SOD assay kit with WST-8. Briefly, an equal amount of proteins was
adjusted by SOD detection buffer and transferred into a 96-well
plate. Then WST-8/enzyme operating solution and reaction activating
solution were added. The reaction system was incubated for 30 min
at 37°C and then the absorption was detected with photometry using
a microplate reader at 459 nm.
Statistical analysis
Data are expressed as the mean ± SEM. Statistic
software SPSS 13.0 and GraphPad Prism 6 were used to analyze data.
ImageJ software was applied to analyze the relative intensity of
the protein blots. A coupled t-test was used to contrast
radiosensitivity parameters and a one-way ANOVA was used to
evaluate the apoptosis rates, ROS levels, protein levels and SOD
inhibition rates. P<0.05 was defined as statistically
significant; P<0.01 was considered as a highly significant
difference.
Results
ATO decreases the survival fraction of
irradiated PC-3 cells
To evaluate the influence of ATO on the
radiosensitivity of PC-3 cells, a clonogenic assay was first
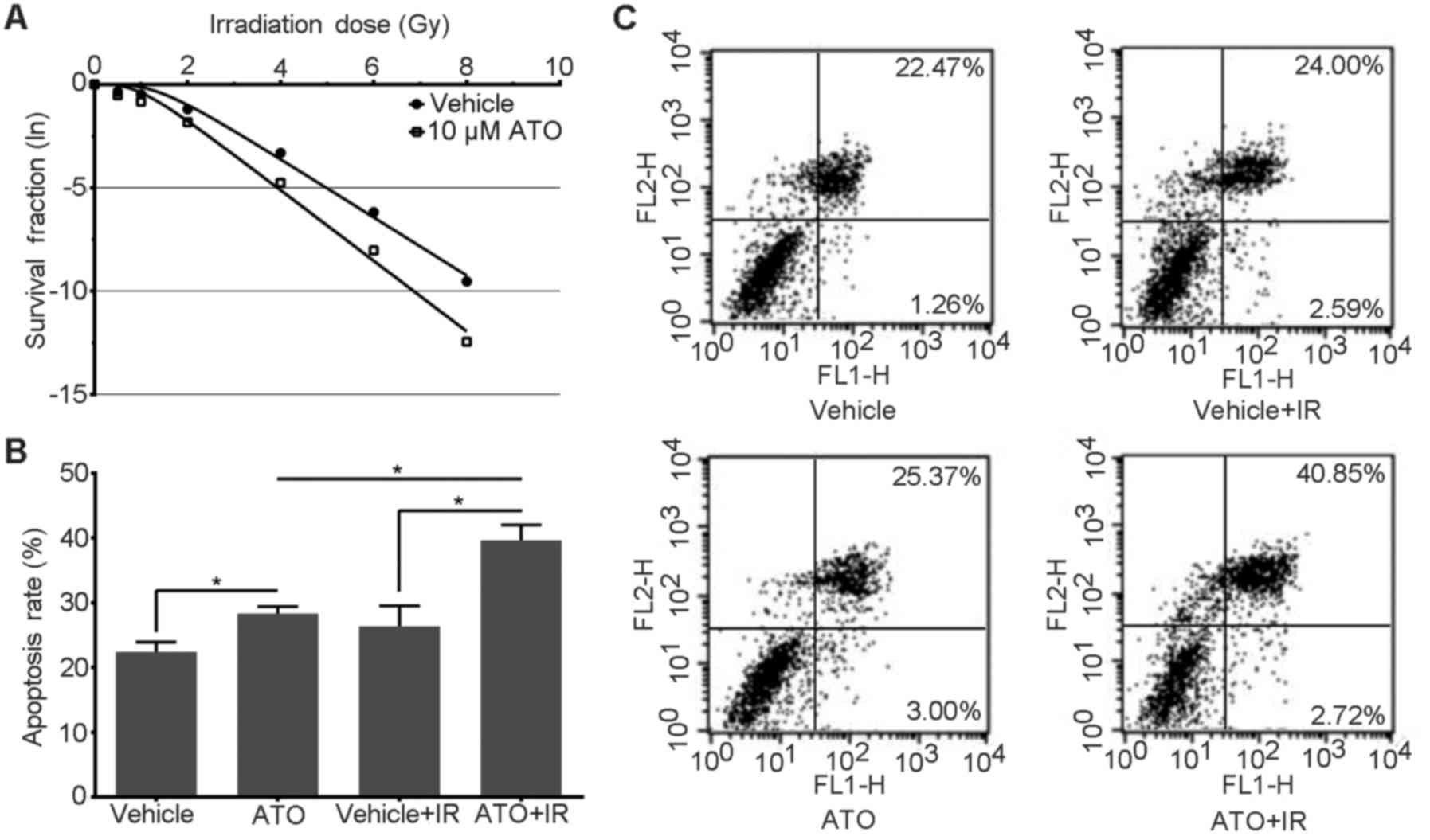
conducted and a cell survival curve was plotted. As shown in the
survival curve (Fig. 1A), the
survival fractions of the study group were less than those of the
control, especially the cells irradiated with high doses. The cell
survival curves of the two groups are separated with an ampliative
trend. This result implies that there was more cell killing under
ATO treatment.
Using GraphPad Prism 6 fitted cell survival curve
with multi-target single hit equation, the results of the
radiosensitivity parameters (12)
indicated that the mean values for D0, Dq, N
and SF2 of the ATO group were universally lower than
that of the control (0.585 vs. 0.710 Gy, 1.006 vs. 1,43 Gy, 5.582
vs. 7.495 Gy, 0.161 vs. 0.301 Gy, respectively). Moreover, the SERs
were all >1.2 in the ATO group (Table I). A coupled t-test displayed a
statistically significant difference in parameters between the two
groups (P<0.05), except for the N-value (P>0.05). This
indicated that ATO sensitized PC-3 cells to irradiation-mediated
killing.
 | Table I.Parameters of the cell survival
curves fitted by the multi-target single hit equation. |
Table I.
Parameters of the cell survival
curves fitted by the multi-target single hit equation.
| Parameters | n | Control | ATO group | t-stat | P-value |
|---|
| D0
(Gy) | 3 | 0.710±0.021 |
0.585±0.017b |
8.013 | 0.001 |
| Dq
(Gy) | 3 | 1.430±0.130 |
1.006±0.132b |
3.964 | 0.008 |
| N | 3 | 7.495±1.770 | 5.582±1.502 |
1.427 | 0.113 |
| SF2 | 3 | 0.301±0.020 |
0.161±0.008a |
8.774 | 0.013 |
|
SERD0 | 3 | 1 |
1.277±0.065b | −7.381 | 0.001 |
|
SERDq | 3 | 1 |
1.356±0.196a | −3.146 | 0.017 |
|
SERSF2 | 3 | 1 |
1.739±0.222b | −5.766 | 0.002 |
ATO increases the apoptosis rate of
irradiated PC-3 cells
Apoptosis is the main form of cell death following
irradiation (15). As the
scatterplot of the flow cytometry shows (Fig. 1C), although ATO had an inducive
effect on cell apoptosis in the unirradiated groups, in the
irradiated groups, the apoptosis rate of the study groups was
significantly increased compared with the control and late
apoptosis cells were mainly predominant. The apoptosis rate in the
histogram (Fig. 1B) also shows a
more obvious increase in the irradiated study group compared with
the control. A one-way ANOVA displayed a statistically significant
difference between the two groups (P<0.05).
In addition, the apoptosis rate of the irradiated
cells was significantly increased when compared with this rate in
the unirradiated ATO-treated group. However, there was no
statistical significance in the apoptosis rate between the
irradiated and unirradiated cells in the control group.
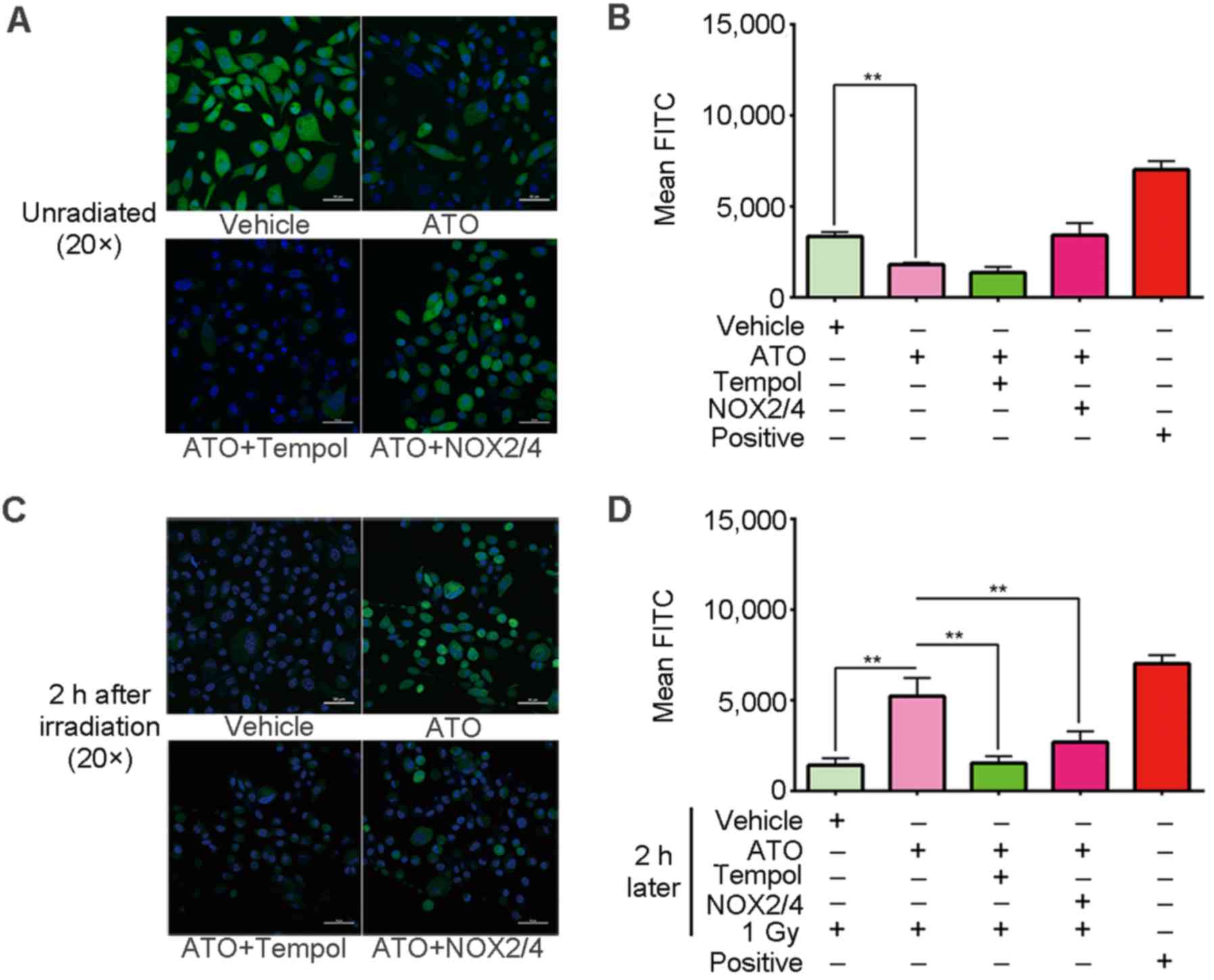
ATO decreases the endogenetic ROS
level in the PC-3 cells
To explain the radiosensitization effect of ATO, the
intracellular ROS level of PC-3 cells was then detected. ROS play
an important role in the cell killing process resulting from
irradiation. The ROS level in cells before irradiation corresponds
to the endogenetic ROS level. Our results showed that the
endogenetic ROS level of the study group was obviously decreased
with ATO treatment. As the result from laser scanning confocal
microscopy shows (Fig. 2A), the
DCF-tagged ROS fluorescence in the study group was less than that
of the control. The histogram of the flow cytometry results
(Fig. 2B) also shows that the ROS
level of the study group dropped to nearly half the level of the
negative control. A one-way ANOVA displayed a statistically
significant difference between the two groups (P<0.01). These
results imply that ATO decreased the endogenetic ROS level in the
PC-3 cells.
However, this trend was abrogated by NOX2 and
NOX4 gene transfection, and was exacerbated by combined
treatment with tempol, which is a mimetic of SOD (Fig. 2A and B). This indicates that NOXs
and SOD were responsible for the endogenetic ROS decrease.
ATO prolongs the lifespan of
radiation-induced ROS
Next, the change in ROS level after irradiation was
detected. Generally, radiation-induced ROS are only maintained
~10−9-10−7 sec in cells (16,17).
However, our results showed that compared with the ROS level of the
control that had returned to a low level, the radiation-induced ROS
of the study group was still at a high level even when the
irradiation had been terminated for 2 h. The results of the laser
scanning confocal microscopy showed that the DCF-tagged ROS
fluorescence of the study group was more intense than that of the
control (Fig. 2C). The histogram of
the flow cytometry results shows that the ROS level of the study
group was 4-fold more than that of the control (Fig. 2D). A one-way ANOVA displayed a
statistically significant difference between the two groups
(P<0.01). These results imply that ATO may maintain
radiation-induced ROS at a lingering high level.
On the other hand, the DCF-tagged ROS fluorescence
of the combined treatment groups, including ATO plus tempol or
NOX transfection, dropped to normal levels similarly to the
control (Fig. 2C and D), which
indicate NOXs and SOD were responsible for the high level of
radiation-induced ROS as well.
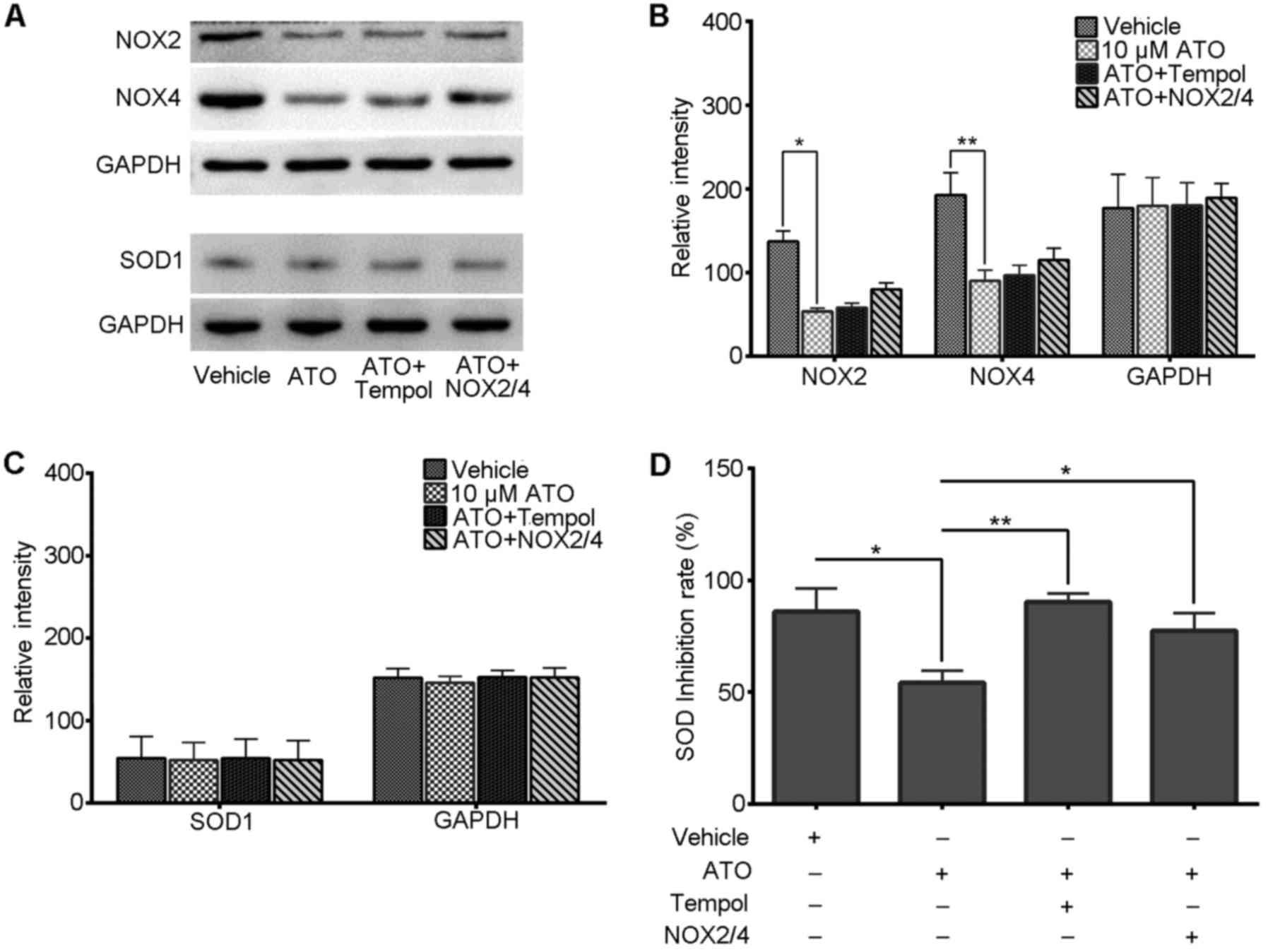
ATO reduces the level of NOXs in the
PC-3 cells
NOX2 and NOX4 are two isoforms that are involved in
prostate cancer progression and are two main sources of endogenous
ROS generation (18–20). To understand the reason for
endogenetic ROS change under ATO treatment, the levels of these two
protein were detected by immunoblotting. Our results showed that
NOX2 and NOX4 protein blots from the study group were weaker than
those noted in the control (Fig.
3A), which signifies a low expression of NOX2 and NOX4 in the
study group. The histogram of relative intensity (Fig. 3B) also indicates that NOX2 and NOX4
expression levels were decreased and one-way ANOVA displayed a
statistically significant difference between the two groups
(P<0.05 and P<0.01). This result implies that ATO may reduce
the level of NOXs in PC-3 cells.
Furthermore, our results showed that, if we
transfected NOX2 and NOX4 genes into cells of the
study group, their expression did not increase as expected, but
partly increased and less than that of the levels in the control
(Fig. 3A and B). On the other hand,
our results also found that the addition of tempol to the study
group slightly increased NOX2 and NOX4 expression, which implies
that there is an association between the level of SOD and the
expresssion of the NOX genes (Fig. 3A
and B).
ATO reduces SOD activity in the PC-3
cells
The SOD family is able to eliminate redundant
intracellular ROS in time. Based on the aforementioned results, the
level of SOD was assessed. In our results, western blot analysis
revealed little difference in the thickness and relative intensity
of the SOD1 blots in all groups (Fig.
3A and C) and a one-way ANOVA also displayed no statistically
significant difference between the groups (P>0.05), which
suggests that the SOD1 expression underwent little change under
ATO-alone treatment or the combination of treatments.
However, on the other hand, we found that the SOD
activity decreased in the ATO-alone group. As our results showed
(Fig. 3D), the SOD inhibition rate
of the ATO-alone group decreased to nearly half of that of the
control, but was restored largely when combined with tempol and was
maintained at a relatively high level in the NOX transfection
groups. This implies that ATO can inhibit SOD activity effectively,
and this effect is related to the intracellular NOX level.
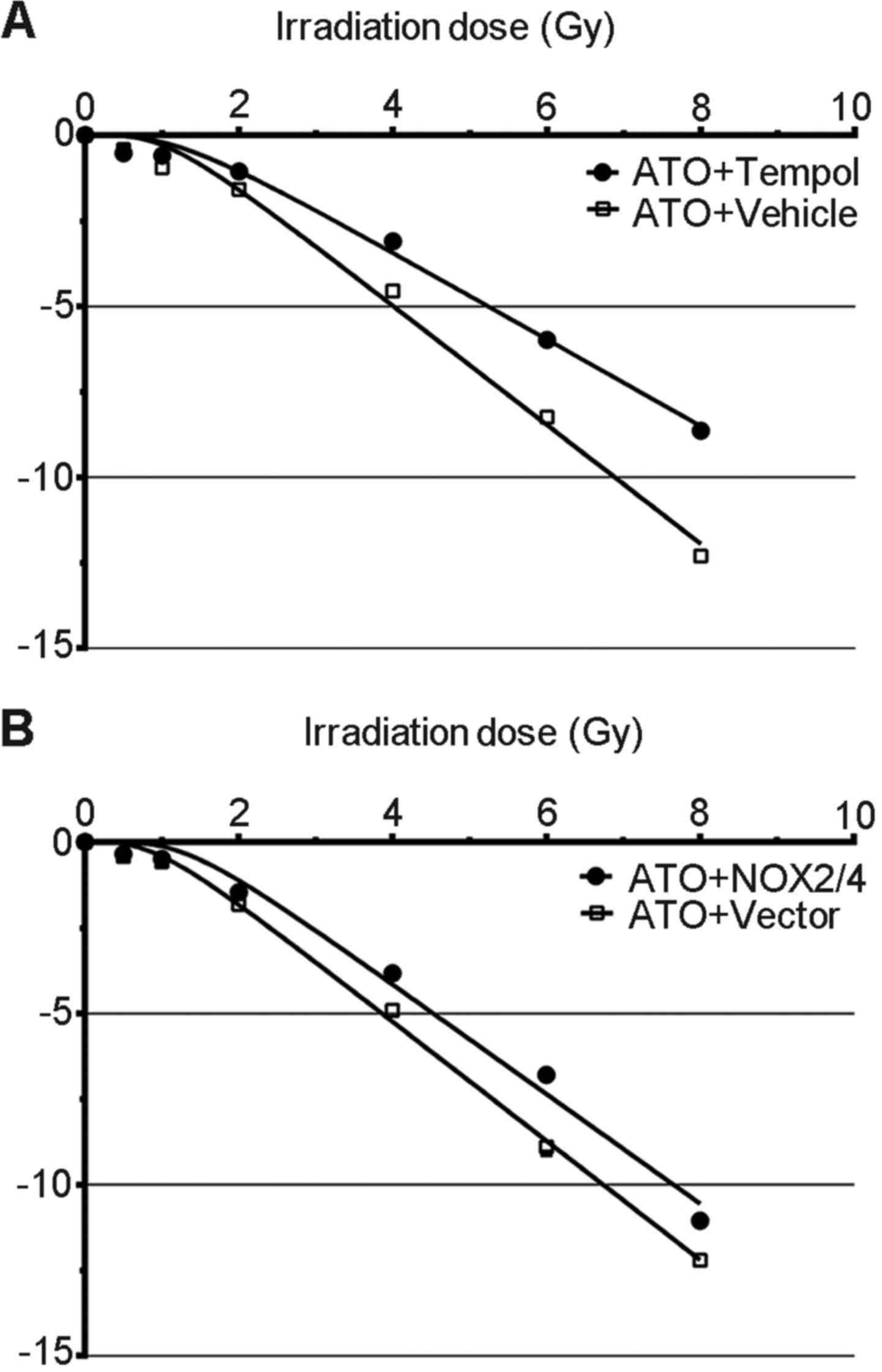
Combining ATO with tempol decreases
radiosensitivity
The aforementioned results imply that a low level of
endogenetic ROS and a high level of radiation-induced ROS are
related to the radiosensitization effect of ATO, and a low
expression of NOXs and decreased SOD activity contribute to these
changes. Next, we combined ATO with NOX transfection or
tempol to increase endogenetic ROS, or decrease radiation-induced
ROS, in the clonogenic assay.
Our results indicate that combined treatment of ATO
and NOX transfection moderately increased the survival
fractions of the PC-3 cells, which had been decreased by ATO-alone
treatment (Fig. 4A). On the other
hand, the ATO and tempol combination greatly increased the survival
fractions of the PC-3 cells compared with the ATO-alone group
(Fig. 4B). These results indicate
that low NOX levels and low SOD activity contribute to the
sensitivity of PC-3 cells to irradiation-mediated killing.
Discussion
In the present study, we verified that ATO could
increase the radiosensitivity of PC-3 cells. According to the cell
survival curve, the D0, Dq, N, and
SF2 values of the study group were all lower compared
with the control. A low D0 value implies that the mean
lethal dose is lower, in other words, the dose for reducing the
survival fraction from 0.1–0.037 is lower than that of the control
(12), which means that PC-3 cells
are prone to be killed by irradiation under ATO treatment. A low
Dq and N-value imply the ability of the sublethal
damage-repair to be suppressed (12), it also means that ATO-treated cells
are liable to be killed by irradiation even when receiving low
doses. Moreover, a low SF2 value implies that the
quantity of cells that survived that received a 2 Gy irradiation is
less than that of the control (12,21).
Additionally, sensitivity enhancement ratios (SERs), such as
SERD0, SERDq and SERSF2 values
were all >1.2, which provides us with further evidence to
conclude that ATO increases the sensitivity of PC-3 cells to
radiation-induced cell killing.
Similarly, the apoptosis assay also indicated that
ATO increased the apoptosis rate of the irradiated PC-3 cells. In
our study, the cells that underwent ATO pretreatment and 1 Gy
irradiation showed a higher apoptosis rate than the other groups,
especially the cells treated with ATO alone, which implies that ATO
increases radiation-induced cell killing. Apoptosis is the main
form of cell death during irradiation (15). After exposure to irradiation, cells
undergo either a direct effect by irradiative rays or an indirect
effect by radiation-induced ROS and consequently the apoptosis
process begins in tumor cells and leads to cell death. Therefore, a
high apoptosis rate means more cell apoptosis under the same
irradiation dose, and implies a higher radiation-induced killing
effect on cells. On the other hand, a high apoptosis rate following
a low dose of irradiation in cells usually implies that increased
apoptosis would occur under a high dose of irradiation.
DNA damage is the key event following irradiation
that causes cell death. Radiation-induced DNA damage can be defined
as two types: direct damage and indirect damage. For indirect
damage, its occurrence and degree are closely related to the ROS
levels evoked by irradiation (22).
In this respect, the more ROS generated, the more indirect damage
will occur. Generally, radiation-induced ROS are only maintained
for 10−9-10−7 sec in the cell. However, our
results revealed that ATO maintained radiation-induced ROS at a
high level even when the irradiation was terminated for 2 h,
indicating that the radiation-induced ROS levels were maintained in
the cytoplasm causing persistent indirect damage to DNA.
Additionally, it also implied that the ROS scavenging system, such
as SOD, failed to eliminate ROS effectively.
In addition, we also found that ATO significantly
decreased endogenous ROS levels in the PC-3 cells. Normally, cells
may generate endogenous ROS in metabolic processes, and these play
an important role in signal transduction and ensuring cell growth
(23). ROS are the product of a
redox reaction. A redox reaction usually maintains specific
homeostasis in different types of cells (24). Normal cells maintain redox reaction
homeostasis at an appropriate level and only create a moderate
quantity of ROS. Conversely, tumor cells often display a
hypermetabolic state so that the ROS generation is maintained at a
relatively high level (24). High
ROS levels may induce high expression of ROS scavengers so as to
maintain redox homeostasis. However, enhanced ROS-elimination
ability allows tumor cells to endure more ROS damage, such as
radiation-induced ROS and possess a radioresistant phenotype
(25). Therefore, a relatively low
level of endogenous ROS means a relatively high sensitivity to
irradiation-induced damage and the ATO-reduced endogenous ROS level
of PC-3 cells would increase its susceptibility to
radiation-induced cell killing.
This effect was confirmed by adding tempol and by
NOX tranfection. Tempol is a mimetic of SOD and the addition
of tempol to culture medium enhanced the ROS-elimination ability of
the PC-3 cells. Moreover, NOX tranfection increased ROS
generation. In our results, the cells treated with ATO and tempol
did not show a high level of radiation-induced ROS as
aforementioned, but displayed a nearly normal level. This
phenomenon confirmed that low ROS-elimination ability was the main
reason for ROS accumulation. On the other hand, combining ATO with
NOX transfection not only restored the ROS level to a normal
level before irradiation, but partly weakened the high level of
radiation-induced ROS as aforestated, which proves that redundant
ROS that was generated by NOX transfection induced the
ROS-elimination ability to be enhanced through a redox homeostasis
mechanism.
As a crucial factor in redox reactions, NOXs are
essential for endogenous ROS generation. NOX1-NOX5 are five main
isoforms of the NOX family (26).
In the present study, we found that ATO reduced the expression of
NOX2 and NOX4, which were reported as main generators of endogenous
ROS in prostate cancer (18–20).
Low expression of NOX2 and NOX4 imply a reduction in endogenous
ROS, and this inference complies with the result of the endogenous
ROS detection aforementioned. Functionally, ATO has been reported
to have antioxidant capacity in vascular endothelial cells, which
is closely linked to NOXs (10).
Goettsch et al found that ATO can inhibit NOX4
overexpression in endotheliocytes (27). Moreover, Pignatelli et al
reported that ATO can directly inhibit the activation of NOX2 in
platelets (28). Therefore, these
results offer evidence to support the relationship between ATO and
NOXs in PC-3 cells. In the present study, we transfected
NOX2 and NOX4 genes into PC-3 cells and then treated
these cells with ATO. However, the expression level of NOXs did not
increase as expected, indicating that ATO decreased NOX2 and NOX4
expression.
The SOD family is the main scavenging system in
cells for ROS elimination, especially superoxide anion
(O2•−), a main form of endogenous ROS
(29). There are four SOD isoforms
distributed in nature, including SOD1-SOD4 (30). SOD1 is a main isoform which is
highly expressed in eukaryotic cytoplasm (31). In the present study, our findings
suggested that ATO had little impact on the SOD1 level, but reduced
the total SOD activity significantly. As aforementioned, redox
reaction homeostasis is crucial for cell metabolism. According to
our results, NOX reduction broke down previous redox homeostasis
and caused a decrease in ROS. Then, the ROS scavenging system, such
as SOD, was attenuated and tried to recover new homeostasis.
Therefore, even when SOD1 protein levels are stably maintained, the
total SOD activity attenuation still inhibits the reduction of ROS
and thus can be regarded as a compensation to maintain redox
homeostasis.
In contrast, a decrease in SOD activity may
attenuate the ROS-eliminating capability during irradiation.
Radiation-induced ROS is the product of radiolytic hydrolysis
during irradiation, including superoxide anion
(O2•−), hydroxy radical (OH•) and hydrogen
peroxide (H2O2) (32). SOD plays a pivotal role in
eliminating these ROS so as to resist DNA damage. However, the
decreased SOD activity fails to eliminate these ROS effectively,
leading to accumulation of irradiation-induced ROS in the cells.
The existence of irradiation-induced ROS accumulation aggravates
the indirect DNA damage and causes more cell death.
This effect was finally verified by irradiative
clonogenic assay. When we treated cells with ATO and tempol, the
cell survival fraction was markedly increased compared with that of
the cells treated with ATO alone, which proves that low SOD
activity is responsible for the radiosensitization effect of ATO.
However, when we treated cells with ATO and NOX
transfection, the cell survival fraction also increased, but only
to a level less than that of the cells treated with ATO alone,
which further confirms that redundant ROS generation enhances SOD
ability.
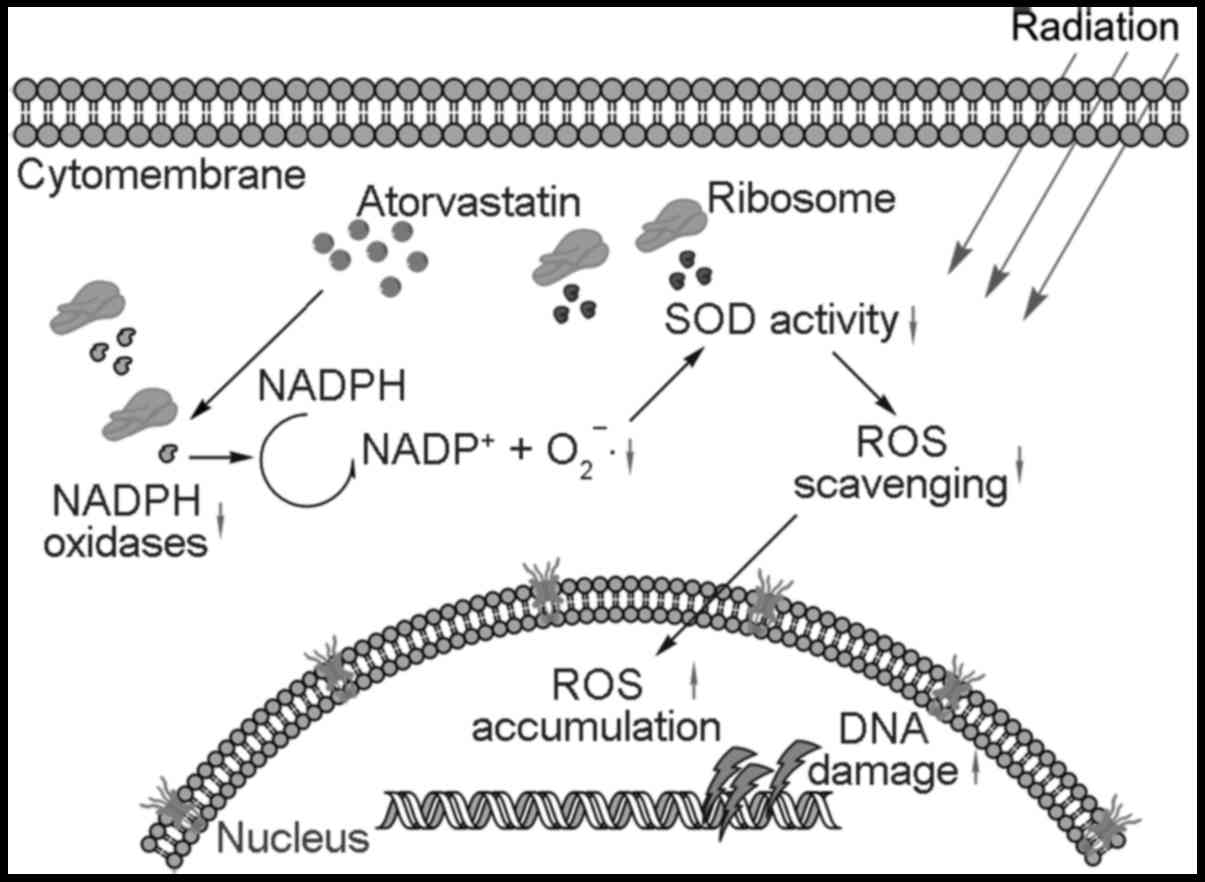
In conclusion, our data indicate that ATO is
effective in decreasing endogenous ROS levels through the reduction
in NOX2 and NOX4 expression and in prolonging the lifespan of
irradiation-induced ROS through the attenuation of SOD activity.
These effects not only increased the DNA susceptibility to
irradiation, but also increased the indirect DNA damage of
irradiation-induced ROS (Fig. 5)
and thus enhanced the cell killing effect of irradiation in PC-3
cells.
Acknowledgements
We sincerely thank Professor De-Min Zhou for
providing access to the laboratory equipment, facilities and
reagents. Professor De-Min Zhou is the director of the State Key
Laboratory of Natural and Biomimetic Drugs of Peking University
(Beijing, China). This study was supported by the Department of
Radiation Oncology, Peking University First Hospital, (Beijing,
China).
References
|
1
|
Delaney G, Jacob S, Featherstone C and
Barton M: The role of radiotherapy in cancer treatment: Estimating
optimal utilization from a review of evidence-based clinical
guidelines. Cancer. 104:1129–1137. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Seong KM, Kim CS, Jeon HY, Oh SH, Nam SY,
Yang KH, Kim JY and Jin YW: Intrinsic radiosensitivity correlated
with radiation-induced ROS and cell cycle regulation. Mol Cell
Toxicol. 6:1–7. 2010. View Article : Google Scholar
|
|
3
|
Cooke MS, Evans MD, Dizdaroglu M and Lunec
J: Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB
J. 17:1195–1214. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Goldstein JL and Brown MS: Regulation of
the mevalonate pathway. Nature. 343:425–430. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Park HS, Schoenfeld JD, Mailhot RB, Shive
M, Hartman RI, Ogembo R and Mucci LA: Statins and prostate cancer
recurrence following radical prostatectomy or radiotherapy: A
systematic review and meta-analysis. Ann Oncol. 24:1427–1434. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
He Z, Mangala LS, Theriot CA, Rohde LH, Wu
H and Zhang Y: Cell killing and radiosensitizing effects of
atorvastatin in PC3 prostate cancer cells. J Radiat Res (Tokyo).
53:225–233. 2012. View Article : Google Scholar
|
|
7
|
Fritz G, Brachetti C and Kaina B:
Lovastatin causes sensitization of HeLa cells to ionizing
radiation-induced apoptosis by the abrogation of G2 blockage. Int J
Radiat Biol. 79:601–610. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mihăilă RG: Advances in the management of
malignant hemopathies: The role of statins. Recent Pat DNA Gene
Seq. 7:57–61. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Song X, Liu BC, Lu XY, Yang LL, Zhai YJ,
Eaton AF, Thai TL, Eaton DC, Ma HP and Shen BZ: Lovastatin inhibits
human B lymphoma cell proliferation by reducing intracellular ROS
and TRPC6 expression. Biochim Biophys Acta. 1843:894–901. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wassmann S, Laufs U, Müller K, Konkol C,
Ahlbory K, Bäumer AT, Linz W, Böhm M and Nickenig G: Cellular
antioxidant effects of atorvastatin in vitro and in vivo.
Arterioscler Thromb Vasc Biol. 22:300–305. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Leith JT, Quaranto L, Padfield G,
Michelson S and Hercbergs A: Radiobiological studies of PC-3 and
DU-145 human prostate cancer cells: X-ray sensitivity in
vitro and hypoxic fractions of xenografted tumors in vivo. Int
J Radiat Oncol Biol Phys. 25:283–287. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tucker SL: Parameters of radiosensitivity.
Radiat Res. 108:226–229. 1986. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Oliver R and Shepstone B: Some practical
considerations in determining the parameters for multi-target and
multi-hit survival curves. Phys Med Biol. 9:167–175. 1964.
View Article : Google Scholar
|
|
14
|
Jones KJ, Chetram MA, Bethea DA, Bryant
LK, Odero-Marah V and Hinton CV: Cysteine (C)-X-C receptor 4
regulates NADPH oxidase-2 during oxidative stress in prostate
cancer cells. Cancer Microenviron. 6:277–288. 2013. View Article : Google Scholar :
|
|
15
|
Verheij M and Bartelink H:
Radiation-induced apoptosis. Cell Tissue Res. 301:133–142. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim GJ, Fiskum GM and Morgan WF: A role
for mitochondrial dysfunction in perpetuating radiation-induced
genomic instability. Cancer Res. 66:10377–10383. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lesser MP: Oxidative stress in marine
environments: Biochemistry and physiological ecology. Annu Rev
Physiol. 68:253–278. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kumar B, Koul S, Khandrika L, Meacham RB
and Koul HK: Oxidative stress is inherent in prostate cancer cells
and is required for aggressive phenotype. Cancer Res. 68:1777–1785.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lu JP, Hou ZF, Duivenvoorden WC, Whelan K,
Honig A and Pinthus JH: Adiponectin inhibits oxidative stress in
human prostate carcinoma cells. Prostate Cancer Prostatic Dis.
15:28–35. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lu JP, Monardo L, Bryskin I, Hou ZF,
Trachtenberg J, Wilson BC and Pinthus JH: Androgens induce
oxidative stress and radiation resistance in prostate cancer cells
though NADPH oxidase. Prostate Cancer Prostatic Dis. 13:39–46.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Deacon J, Peckham MJ and Steel GG: The
radioresponsiveness of human tumours and the initial slope of the
cell survival curve. Radiother Oncol. 2:317–323. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kryston TB, Georgiev AB, Pissis P and
Georgakilas AG: Role of oxidative stress and DNA damage in human
carcinogenesis. Mutat Res. 711:193–201. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Boonstra J and Post JA: Molecular events
associated with reactive oxygen species and cell cycle progression
in mammalian cells. Gene. 337:1–13. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang Y and Martin SG: Redox proteins and
radiotherapy. Clin Oncol (R Coll Radiol). 26:289–300. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Trachootham D, Alexandre J and Huang P:
Targeting cancer cells by ROS-mediated mechanisms: A radical
therapeutic approach? Nat Rev Drug Discov. 8:579–591. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lambeth JD: NOX enzymes and the biology of
reactive oxygen. Nat Rev Immunol. 4:181–189. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Goettsch C, Goettsch W, Muller G, Seebach
J, Schnittler HJ and Morawietz H: Nox4 overexpression activates
reactive oxygen species and p38 MAPK in human endothelial cells.
Biochem Biophys Res Commun. 380:355–360. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pignatelli P, Carnevale R, Pastori D,
Cangemi R, Napoleone L, Bartimoccia S, Nocella C, Basili S and
Violi F: Immediate antioxidant and antiplatelet effect of
atorvastatin via inhibition of Nox2. Circulation. 126:92–103. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nathan C and Cunningham-Bussel A: Beyond
oxidative stress: An immunologists guide to reactive oxygen
species. Nat Rev Immunol. 13:349–361. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Youn HD, Kim EJ, Roe JH, Hah YC and Kang
SO: A novel nickel-containing superoxide dismutase from
Streptomyces spp. Biochem J. 318:889–896. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Juarez JC, Manuia M, Burnett ME,
Betancourt O, Boivin B, Shaw DE, Tonks NK, Mazar AP and Doñate F:
Superoxide dismutase 1 (SOD1) is essential for
H2O2-mediated oxidation and inactivation of
phosphatases in growth factor signaling. Proc Natl Acad Sci USA.
105:7147–7152. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Riley PA: Free radicals in biology:
Oxidative stress and the effects of ionizing radiation. Int J
Radiat Biol. 65:27–33. 1994. View Article : Google Scholar : PubMed/NCBI
|