Introduction
Cancer is the second most common cause of death
worldwide, preceded only by heart diseases (1). Breast cancer is one of the most common
in women, with high morbidity. Generally, treatment occurs via
surgery, chemotherapy, radiotherapy and immunotherapy. However,
despite the various therapeutic strategies that can be adopted, the
mortality rate and the side effects of the treatment remain a
challenge (2,3). Therefore, the search for new
alternative treatments with less side effects is under
investigation.
In such context, there is great interest in the
pharmaceutical industry to target pathways that regulate cell
proliferation and apoptosis in cancer. These pathways are initiated
from various cell surface receptors, and may converge on the MAPK
cascade, a module consisting of MAP kinase kinase (MEK) and MAPK
(4–6). The MAPKs are serine/threonine protein
kinases that participate in different intracellular processes such
as proliferation, differentiation, cellular stress responses, and
apoptosis (7,8). ERK1/2, JNK1/2/3 and p38α/β/γ/δ
constitute the main mammalian MAPKs studied in cancer area. ERK
pathway is activated by mitogen factors, and this pathway is one of
the most mutated in cancer often leading to an increase of cell
proliferation and generally a decrease of apoptosis (9). On the other hand, p38 and JNK pathways
are activated by stress factors, but their roles in cancer remain
unclear, depending on cancer stage, cell type or MAPK isoforms
(10–13).
Another pathway that called attention and has also
been described as crucial in cancer progression is the NF-κB
pathway (14,15). The transcription factor NF-κB
controls many intracellular signals including cell cycle (e.g.
cyclin D1), suppression of apoptosis (e.g. Bcl-2 and Bcl-xL) and
inflammation (e.g. IL-6) (16). In
response to a wide variety of stimuli, NF-κB becomes active via
canonical or non-canonical pathways in cancer. Generally, in the
canonical pathway NF-κB activation is preceded by the
phosphorylation of IKK-αβ and IκBα (17,18).
Nowadays, the major interest is to identify relevant
molecular targets that may offer a specific therapeutic alternative
treatment to cancer. In this context, MAPKs and NF-κB pathways
represent an interesting signaling network for investigation. In
the present study, we used a peptide (named LyeTx II) derived from
the venom of the spider Lycosa erythrognatha, that induced
an exacerbated proliferation of MDA-MB-231 breast cancer cells, to
evaluate the importance of the MAPKs and NF-κB pathways in
proliferation and migration of this breast cancer cell line
considered as aggressive. According to our data, the proliferative
and migratory effect induced by the peptide occurred mainly through
upregulation of p38 pathway activation, without involvement of ERKs
and JNKs. This study provides new insights into the role of p38 in
aggressive breast tumor indicating that p38 may be a suitable
target in this cancer type.
Materials and methods
Reagents
DMEM and fetal bovine serum (FBS) (Gibco) were from
Thermo Fisher Scientific (Waltham, MA, USA). Thiazolyl Blue
Tetrazolium Blue (MTT) and RIPA buffer were purchased from
Sigma-Aldrich (St. Louis, MO, USA). p38 inhibitor SB203580 were
obtained from InvivoGen (San Diego, CA, USA). Protease cocktail
inhibitor and phosphatase cocktail inhibitor were acquired from
Roche (Mannheim, Germany). Monoclonal antibodies against
phospho-p44/42 (ERK1/2), phospho-p38 MAPK, phospho-JNK MAPK,
β-actin and peroxidase-conjugated goat anti-mouse IgG and goat
anti-rabbit IgG were obtained from Cell Signaling Technology
(Danvers, MA, USA). Apoptosis, DNA Damage and Cell Proliferation
Kit were obtained from BD Biosciences (Franklin Lakes, NJ, USA).
Immobilon-P membranes and Luminat were acquired through Merck
Millipore (Darmstadt, Germany).
Peptide information, synthesis and
purification
LyeTx II is a 19 amino acid peptide, which includes
four lysine residues conferring positive charge to it. LyeTx II
synthesis and purification was performed at Professor J.M.
Resende's Laboratory, from the Chemistry Department of UFMG,
Brazil, according to methodology described by Santos et al
(19). The purity of LyeTx II was
checked by mass spectometry.
Cell culture conditions
The breast tumor cell lines MDA-MB-231 and MCF-7
were gifts from the Laboratory of Prof. A.M. Goes. MACL-1 and
MGSO-3 cell lines were derived from primary tumor samples in the
same laboratory (20). Cells were
grown at 37°C in a humidified atmosphere of 5% CO2 in
DMEM supplemented with 10% heat inactivated FBS. For starvation
conditions, cells were incubated with serum-free medium for 2 h
before the western blot assay.
MTT assay
Breast cancer cells (104/well) were
plated in 24-well plates in DMEM with 10% serum. Cells were
incubated for a further 96 h in the following conditions: medium
alone, LyeTx II (from 0.1 to 100 nM), SB203580 (10 µM). After 96 h,
the medium was removed and cells were incubated with 100 µl/well of
MTT at 5 mg/ml for 2 h, at 37°C. After removing the medium, 200 µl
of DMSO was added to dissolve the formazan crystals and the
absorbance was measured in a microplate reader at a wavelength of
595 nm. A value of 100% was assigned to untreated control cultures.
Results were derived from at least three independent sets of
triplicate experiments.
Western blotting
Cells seeded in 6-well plates (106
cells/well) were serum-starved for 2 h. Then, cells were treated or
not with LyeTx II (100 nM) in DMEM containing 10% FBS for the
indicated time-points. 5z-7-oxozeaenol was added 30 min before the
addition of LyeTx II. Cells were harvested and lysed in RIPA buffer
supplemented with phosphatase inhibitor and protease inhibitor
cocktail according to the manufacturer's instructions. Protein
lysates were separated by polyacrylamide gel electrophoresis on 12%
gels, and electrotransferred to Immobilon-P membranes. Membranes
were incubated with primary antibodies. After incubation with
peroxidase-conjugated secondary antibody, protein expression was
detected using Luminat reagent.
Measurement of cell proliferation by
BrdU incorporation
This assay was realized using the Apoptosis, DNA
Damage, and Cell Proliferation kit from BD Biosciences in accord
with the manufacturer's instructions. Briefly, cells were treated
or not with LyeTx II (100 nM) in DMEM containing 10% FBS and
incubated with BrdU (10 µM) for 8 h. When used, SB203580 was added
30 min before the addition of LyeTx II. After labeling, cells were
fixed and permeabilized. After several washing, cells were treated
with DNase (30 µg/106 cell) for 1 h. Following this
treatment, cells were simultaneously stained with PerCP-Cy™5.5
anti-BrdU and Alexa-647 anti-γH2AX for 20 min at room temperature.
After washing, cells were suspended in staining buffer and analyzed
by flow cytometry. The data were collected by the cell analyzing
LSRFortessa (BD Biosciences - Immunocytometry Systems) using BD
FACSDiva™ software (BD Biosciences) and analyzed with FlowJo (Tree
Star) software.
Cell counting
Cells were seeded in a density of 1×104
cells/well in 24-well plates. Cells were treated with LyeTx II (100
nM) in DMEM containing 10% FBS and incubated for 24, 48, 72 and 96
h. In each time, cells were harvested, stained with Tripan Blue
(Gibco) and then counted in Neubauer plate. Number of cells = media
of three counting ×104/ml.
Transwell assay
The 24-well Boyden chamber with 8 µm pore size
polycarbonate membrane Millipore (Millicell Hanging Cell Culture
Inserts, Millipore, EUA) was used to analyze cell motility. For
invasion assay, the membrane was pre-coated for 1 h with 300 µl of
free serum DMEM. Cells (106)were seeded on the upper
chamber with serum-free medium with or without LyeTx II (100 nM).
When used, SB203580 (10 µM) was added 30 min before LyeTx II
addition also in the upper chamber. Medium 700 µl (or until the
volume reaches the upper chamber membrane) with 10% serum was added
to the lower chamber as a chemoattractant. After 24 h of
incubation, the non-motile cells at the top of the membranes were
removed with cotton swabs, then the membranes were fixed and
stained with 0.5% crystal violet (Sigma). Five visual fields of
×200 magnification of each membrane were randomly selected and
counted.
Results
Peptide LyeTx II enhances MDA-MB-231
cell population growth
Without initial information about LyeTx II activity,
we predicted that this peptide, derived from the venom of the
Brazilian spider Lycosa erythrognatha, could interact with
the anionic membrane of the cancer cells due to its amino acid
composition (four lysine residues conferring positive charge) and
its secondary structure (α-helix). Therefore, we have evaluated the
effect of the peptide on four different breast cancer cell lines
(MDA-MB-231, MCF-7, MACL-1 and MGSO-3). Cells were incubated with
different peptide concentrations varying from 0.1 to 100 nM and an
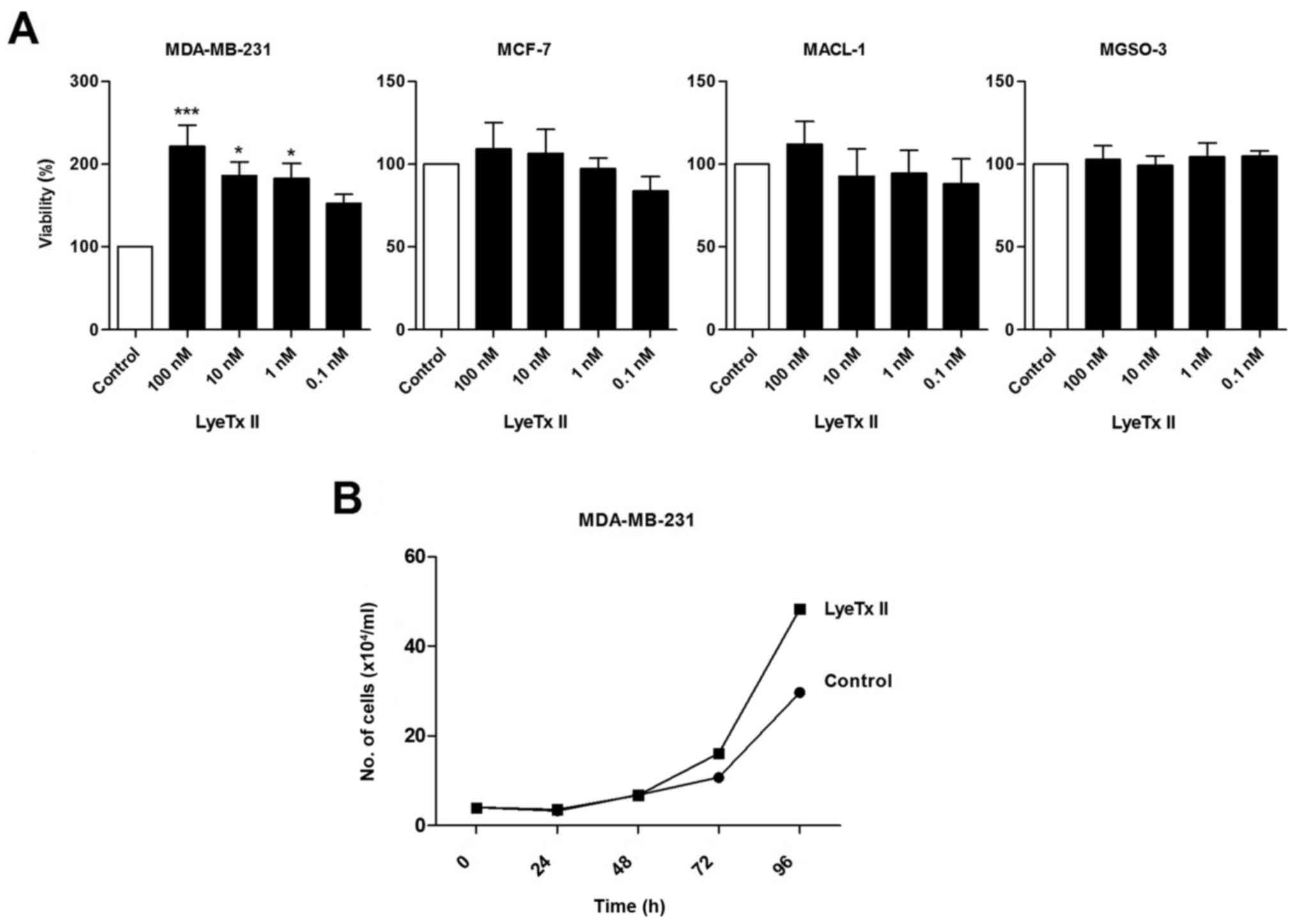
MTT assay was performed to evaluate cell viability (Fig. 1A). Of note, LyeTx II induced an
increase of MDA-MB-231 cell viability after 4 days and this effect
was more marked at 100 nM. No effect induced by the peptide was
observed on the other breast cancer cell lines indicating that only
the most aggressive cell line was responsive to LyeTx II. As MTT
assay measures viable cell metabolism and not specifically cell
proliferation, we confirmed our data by cell counting. According to
Fig. 1B, an increased cell number
was noted after 72 and 96 h of treatment with LyeTx II when
compared to the control group.
Peptide LyeTx II enhances MDA-MB-231
cell proliferation
We sought to define whether the increased cell
number observed in the presence of LyeTx II after 96 h was due to a
direct activation of the proliferative machinery or liberation of
secondary compounds. For this purpose, a BrdU assay was performed,
that allowed us to correlate the proliferative effect induced by
the peptide and the incorporation of BrdU that is a synthetic
nucleoside analog of thymidine. Firstly, we pregated cells
according to size (SSC) and granulometry (FSC). To select the
population in division, H2AX in its phosphorylated form (γH2AX) has
been used as a marker of the DNA doubled stranded breaks. The
detection of the incorporation of BrdU and the presence of γH2AX
has been achieved by using antibody stained with PerCP-Cy5.5 and
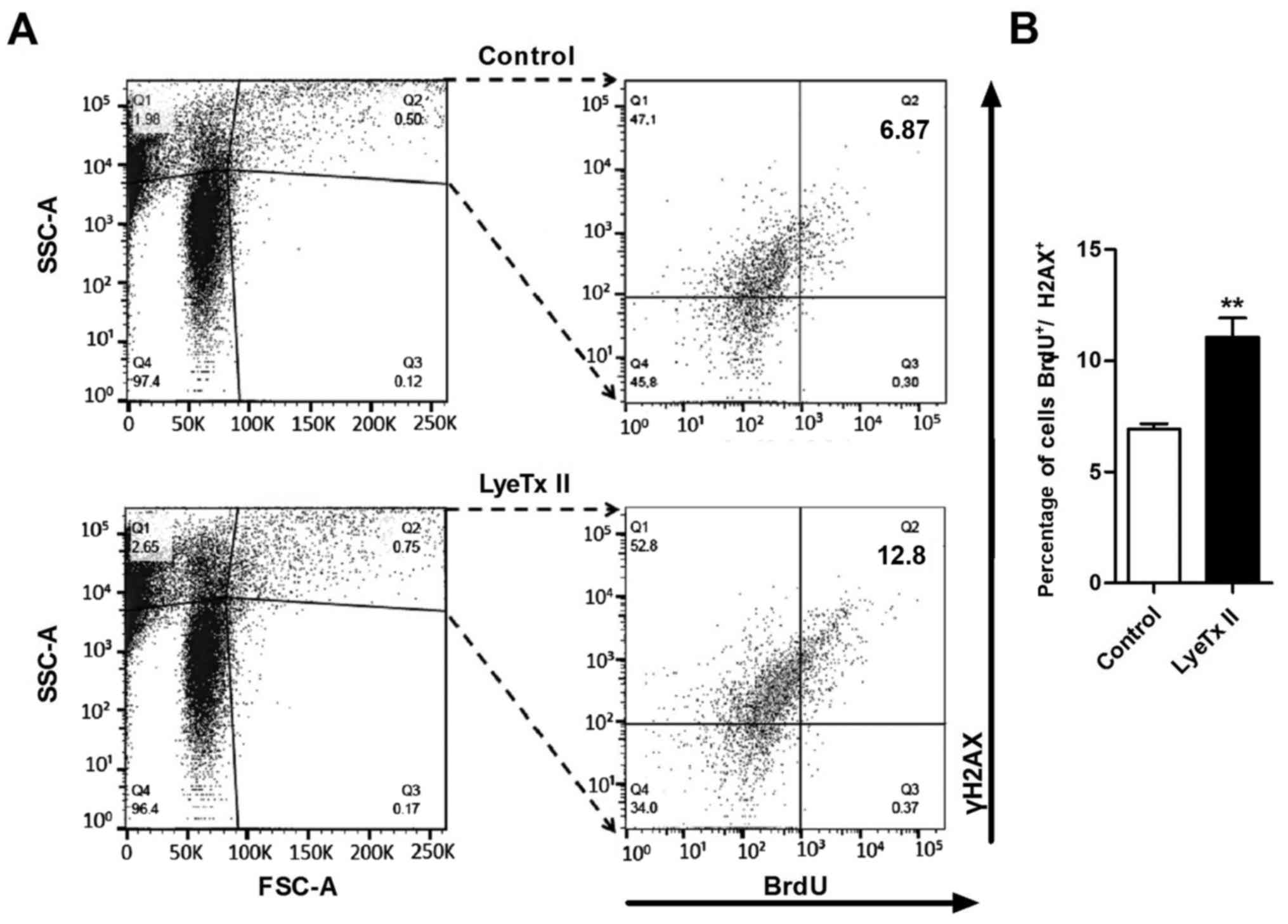
Alexa-647, respectively. As shown in the Fig. 2A, by double immunodetection, after 8
h of incubation with LyeTx II and BrdU, augmentation of the
BrdU+/γH2AX+ population was noted when
compared with the control cell population. The
BrdU+/γH2AX+ population increased from 6.9 to
11.0% (Fig. 2B). Taken together,
these data suggest that LyeTx II is able to interfere directly on
the proliferative machinery of MDA-MB-231 cells. These findings led
us to investigate the intracellular signaling pathways involved in
the increased proliferation induced by LyeTx II.
p38 and NF-κB pathway activation are
upregulated in the presence of LyeTx II
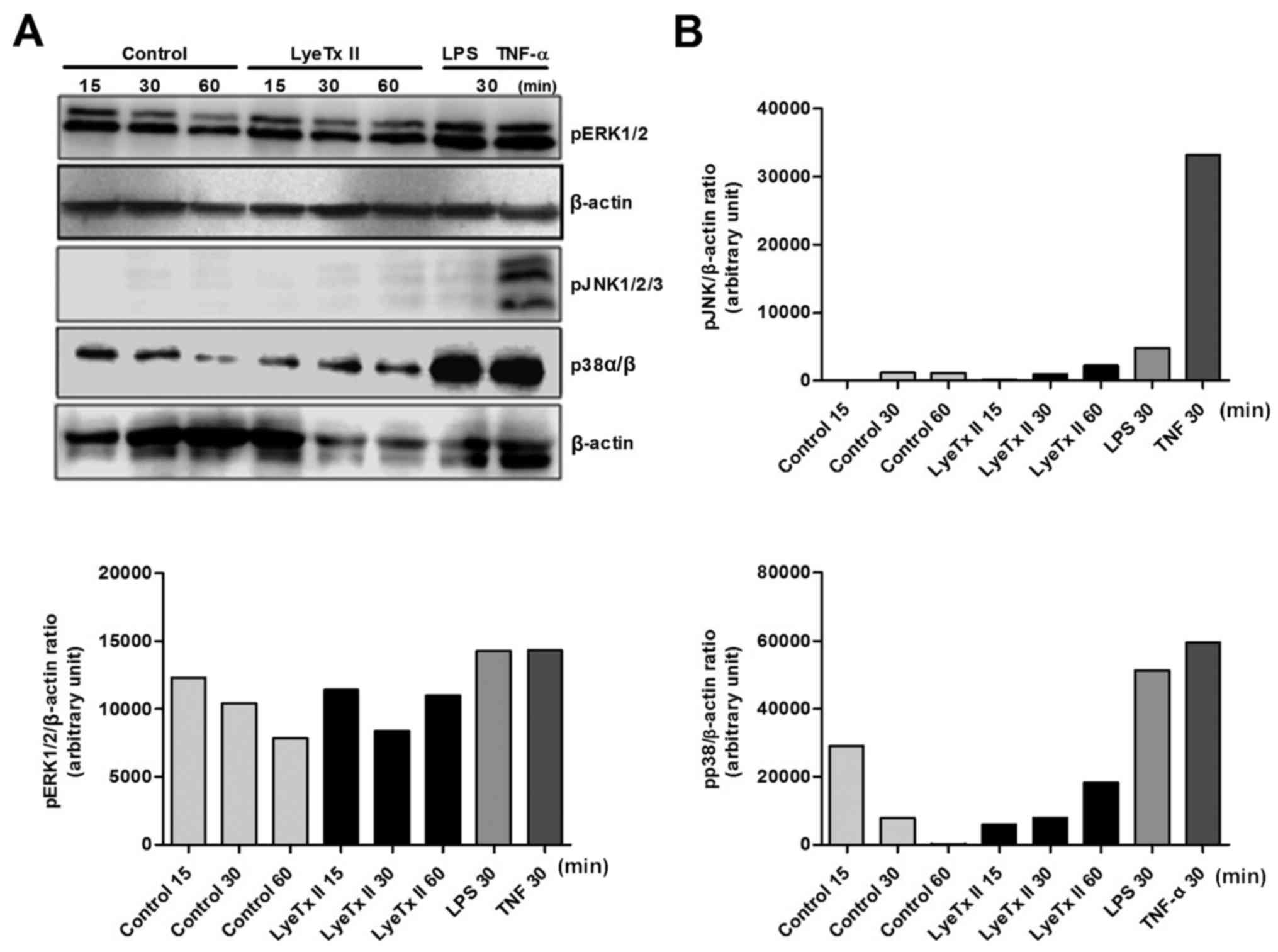
Considering that MAPKs are frequently involved in
the balance between cell proliferation and cell death, we sought to
evaluate the phosphorylation state of the three main MAPKs (ERKs,
JNKs and p38s) in the presence of LyeTx II (Fig. 3A). LPS (1 µg/ml) and TNF-α (20
pg/ml) were used as positive control for the MAPK phosphorylation.
At first, we observed that MDA-MB-231 cells did not use the JNK
pathway, not in the presence of serum or in the presence of the
peptide. However, we verified, that MDA-MB-231 cells were able to
phosphorylate JNK1/2/3 under stress stimulation when TNF-α induced
activation of the three isoforms of JNK. This constitued a direct
evidence that LyeTx II did not require JNK activation to induce its
proliferative effect. Concerning ERK1/2 pathway, no significant
change in the kinetic phosphorylation of this MAPK was detected in
the presence of LyeTx II. The most significant impact induced by
the peptide was the modulation of p38 phosphorylation. We observed
that the phosphorylation of p38 was upregulated during stimulation
with LyeTx II, suggesting that the peptide effect might be related
to p38 pathway activation. As shown in the Fig. 3A, phosphorylation of p38 remained
elevated when compared to control group, even 60 min after peptide
stimulation.
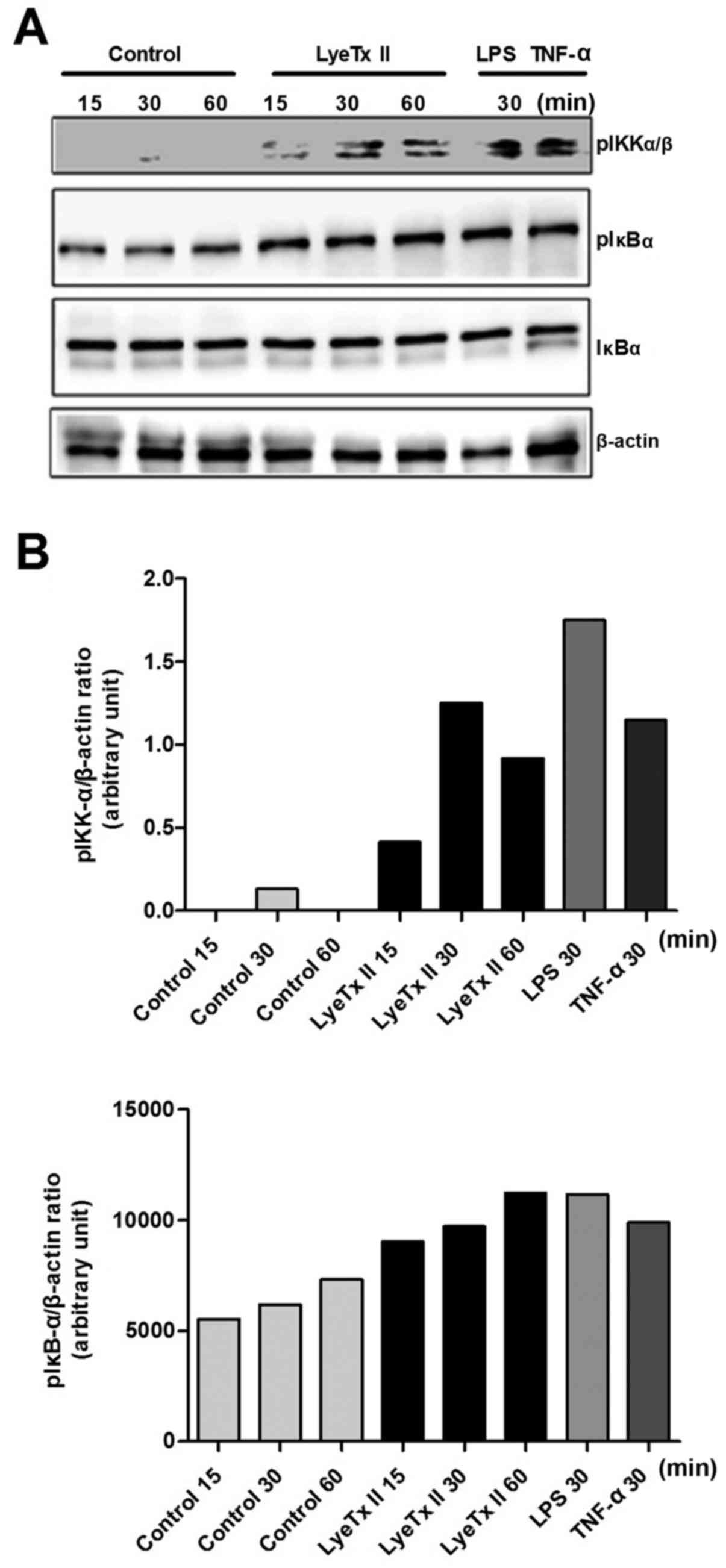
In parallel, we explored the NF-κB pathway, another
pathway well described as having an important role in cancer
development. As shown in Fig. 4A,
the peptide was able to enhance IκBα phosphorylation as well as
IKK-αβ, two of the main kinases involved in the canonical NF-κB
pathway activation. These data suggest a possible involvement of
NF-κB pathway in MDA-MB-231 cell proliferation under stimulation
with LyeTx II.
Upregulation of p38 pathway is
associated with the accelerated proliferation of MDA-MB-231 in the
presence of LyeTx II
To evaluate whether the peptide effect was dependent
on IκBα and p38 activation, we used two selective inhibitors: BAY
11–7082 for NF-κB pathway (21) and
SB203580 for p38 pathway (22). It
was not possible to define the involvement of NF-κB pathway in the
peptide activity, since IκBα inhibitor induced MDA-MB-231 cell
death in the concentration range used (2.5-20 µM), that represent
the concentrations able to inhibit IκBα activity as previously
reported (21). To correlate the
peptide effect with the p38 pathway activation, we performed an MTT
assay using SB203580 at concentration that did not affect the basal
cell proliferation in our system. As shown in Fig. 5C, SB203580 was able to abrogate the
proliferative effect induced by the peptide. To confirm the
relationship between p38 activation and the proliferative effect
induced by the peptide, we evaluated the BrdU incorporation in the
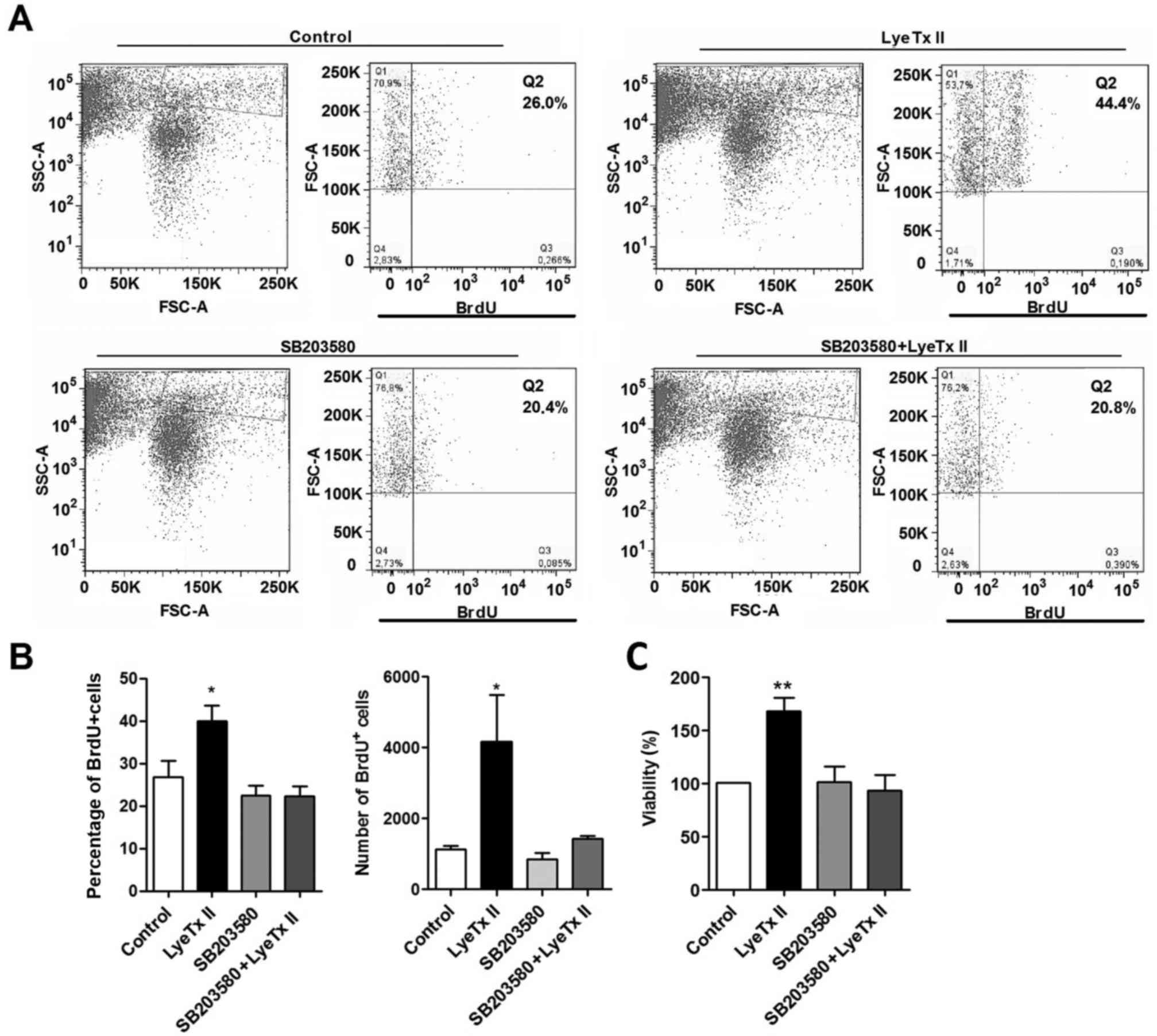
presence of LyeTx II, SB203580 and SB203580 plus LyeTx II (Fig. 5A and B). Corroborating with the MTT
assay, SB203580 was able to abolish the peptide effect maintaining
the proliferative rate at a basal level. More precisely, in the
presence of SB203580, the frequency of BrdU+ cells was
significantly reduced in LyeTx II group comparing with LyeTx II
group pretreated with SB203580. Collectively, the data confirmed
that p38 plays a role in the increase of MDA-MB-231 cell
proliferation when stimulated with LyeTx II.
p38 pathway upregulation is associated
with the increased migration of MDA-MB-231 cells in the presence of
LyeTx II
We hypothesized that accelerated proliferation and
migration events might share the same signaling pathways in
MDA-MB-231 cells. In such context, we evaluated whether the peptide
could increase MDA-MB-231 cell migration by using the transwell
assay. By placing the cells on one side of the membrane and using
FBS as a chemoattractant on the other side, migration was
determined by counting those cells that traversed the
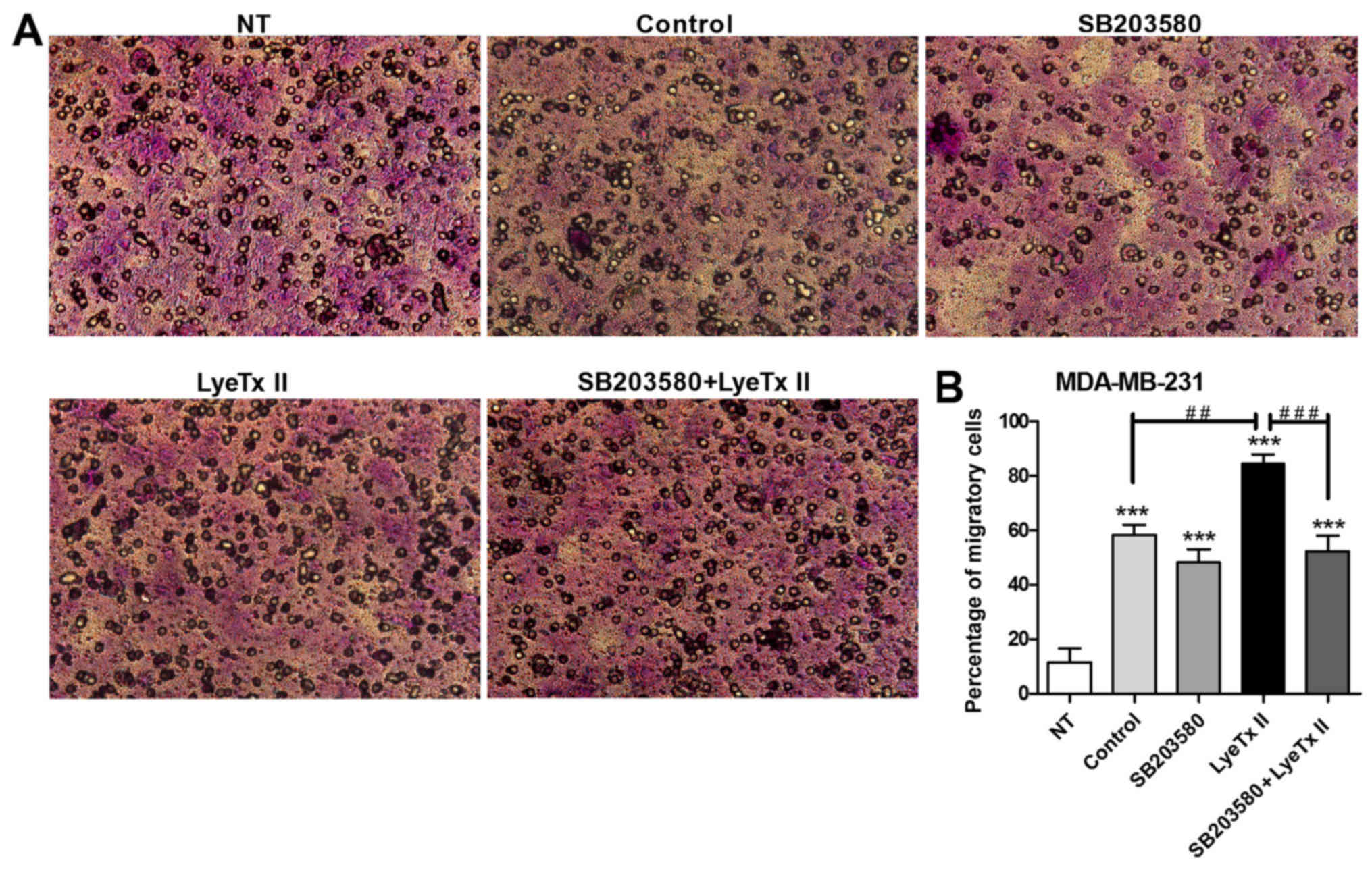
cell-permeable membrane. As shown in Fig. 6A and B, LyeTx II at 100 nM was also
capable of enhancing cell migration in a significant way when
compared to the control group. It is important to note that
MDA-MB-231 cell line is considered as invasive, that explains the
high number of cells that can pass through the membrane pores
observed in serum conditions. The involvement of p38 pathway in
MDA-MB-231 cell migration was confirmed when it was shown that
SB203580 reduced the peptide effect on cell migration.
p38 and IκBα phosphorylation induced
by LyeTx II is dependent on TAK1
According to our data, LyeTx II was able to
upregulate p38 and NF-κB pathway activation, so we sought to
determine possible crosstalk between these pathways. TAK1 has been
previously described as a common upstream kinase for both pathways
(23). Further, other groups have
previously described the involvement of TAK1 in the MDA-MB-231 cell
survival (24,25). Thus, we explored the impact of TAK1
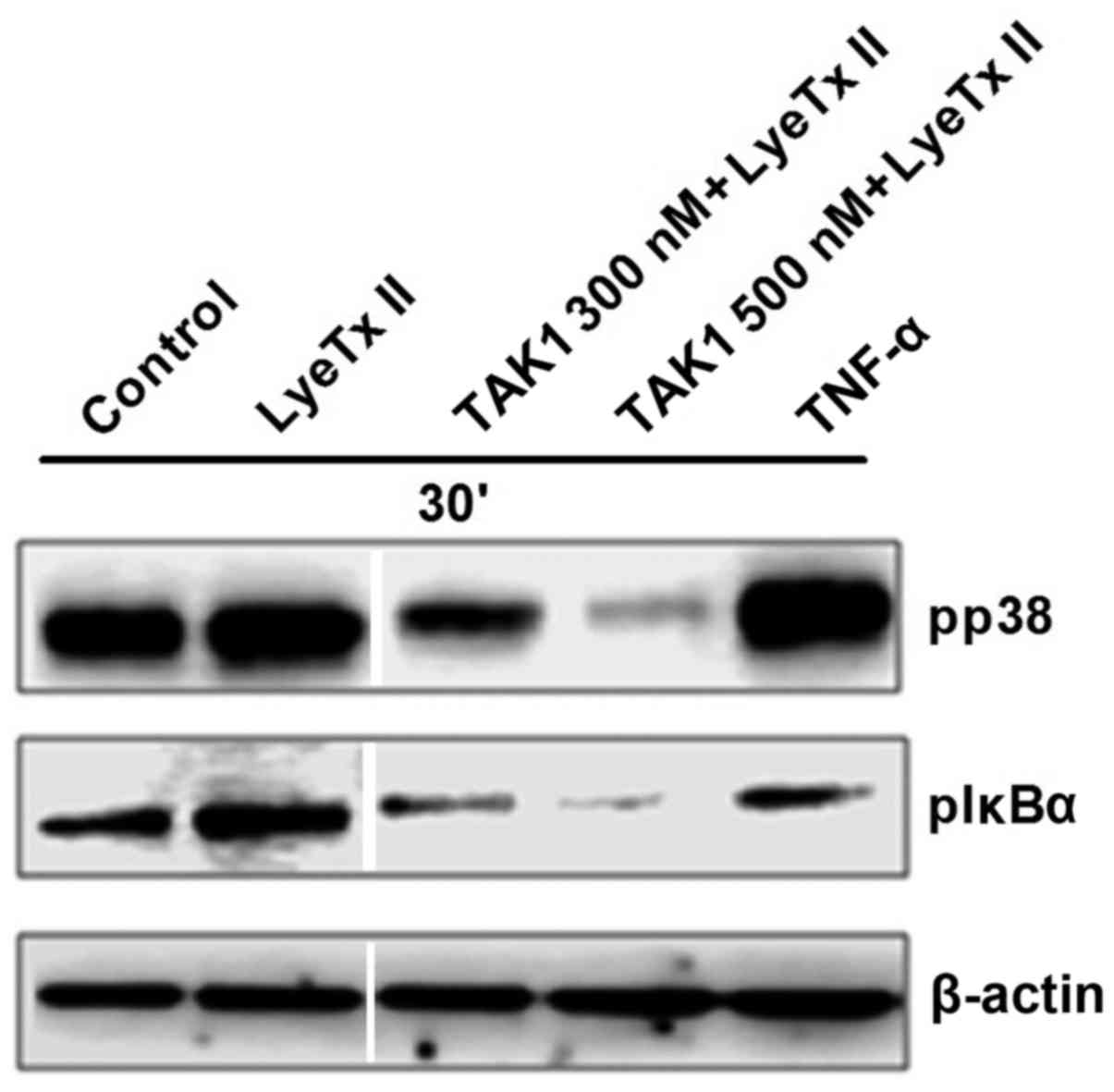
inhibition on the peptide effect. As shown in Fig. 7, when cells were stimulated with the
peptide, they were still able to phosphorylate p38 and IκBα in the
presence of TAK1 at 300 nM. However, at 500 nM, TAK1 inhibitor
reduced significantly p38 and IκBα phosphorylation in LyeTx
II-treated cells. Taken together, these data indicate that LyeTx II
upregulates p38 and NF-κB pathways in a TAK1-dependent manner.
Discussion
A peptide derived from the venom of the Brazilian
spider Lycosa erythrognatha was used to correlate activated
signaling pathways and aggressive breast cancer cell proliferation.
There are several studies describing p38 as a proliferative agent
(26,27); on the other hand, some studies have
defined p38 as an apoptosis inducer (28–30).
Herein, we clearly defined that p38 was involved in the peptide
induced-MDA-MB-231 cell gain functions such as proliferation and
migration. Further, these data show a direct role for p38 in the
cell cycle since the use of p38 inhibitor SB203580 abrogates the
peptide effect on the BrdU incorporation in the first hours. In
fact, it has been previously reported that p38 may regulate both
the G2/M as well as G1/S cell cycle checkpoints (31–33).
Different hypothesis may be advanced like the upregulation of
cyclin D1 expression in a p38-dependent manner (34) or an additional mechanism that may
contribute to augmentation of proliferation in the presence of the
peptide. As p38 has also a well-known role in inflammation
(35), we investigated a possible
role of inflammatory mediators in the peptide effect. Since no
augmentation of cytokine secretion was observed in the presence of
LyeTx II, we were able to discard the correlation between the
pro-inflammatory property of p38 and the peptide activity (data not
shown). In addition, the use of rhTNF-α at a very low concentration
(20 pg/ml) enhanced MDA-MB-231 proliferation (data not shown) and
MAPK phosphorylation (including ERK and JNK) in a more pronounced
way than the peptide. Taken together, these data support the idea
that the proliferation effect induced by LyeTx II is independent on
inflammatory processes.
It has also been reported that p38 may play an
important role in many steps of metastasis, such as
invasion/migration, in pancreatic, hepatocellular and head and neck
squamous carcinoma cell lines on the basis of the use of SB203580
and dominant-negative mutants (36). Herein we demonstrated that LyeTx II,
through upregulation of p38 phosphorylation, enhanced MDA-MB-231
cell migration, reinforcing the involvement of p38 in the
augmentation of the aggressive character of the tumor cells.
We sought to define whether the increased
proliferation was due to a combination of events involving
proliferation and/or anti-apoptotic pathways. Given the role of the
transcription factor NF-κB in prosurvival pathways (37), we analyzed the capacity of the
peptide to upregulate the NF-κB pathway. At first, it was of
interest to note that while MDA-MB-231 cells were capable of
phosphorylating IKK-αβ and IκBα in the presence of serum, MCF-7 did
not (data not shown). This is in accord with a model proposed by
Lee et al (38) where
ER− breast cancer cells, such as MDA-MB-231, use
constitutively NF-κB pathway and ER+ breast cancer
cells, such as MCF-7, do not. The peptide activity fits this model
by enhancing IKK-αβ and IκBα phosphorylation only in MDA-MB-231
cells. This suggests that LyeTx II, besides its proliferative
activity, might play a pro-survival role through upregulation of
NF-κB pathway. This might explain the significant impact of this
peptide on MDA-MB-231 cell proliferation and migration at low
concentration (approximately 100 nM). Peptides used in cancer
studies act generally at concentration superior to 1 µM (39,40).
Considering that LyeTx II modulated p38 and NF-κB
pathways activation, we hypothesized that a common molecule could
regulate both pathways in response to the peptide. TAK1 has been
reported as an upstream kinase of p38 and NF-κB (41–43),
so it appeared relevant in our model to correlate p38 and IκBα
activation with TAK1 activity. Our data suggest that the
phosphorylation of p38 and IκBα is dependent on TAK1 activation.
Noteworthy, it has been reported that p38 may exert a positive
feedback on TAK1 phosphorylation (44), which may occur in our model since a
sustained phosphorylation of p38 has been observed in presence of
LyeTx II. Such feedback mechanism may contribute to the peptide
activity.
An interesting finding in this study was the
divergence of cellular response between the non-aggressive MCF-7
and aggressive MDA-MB-231 tumor cells to LyeTx II, probably due to
the differential use of signaling pathways by cell lines.
Comparative phosphoproteosome analysis reveals differences in
levels of various phosphoproteins in these two cell lines (45). This indicates that the sole presence
or lack of receptors (ER, PR, HER2) may not be sufficient to
classify tumor cells and to predict treatment.
In conclusion, we were able to associate a
dysregulation of MAPK signaling pathways with a specific breast
tumor cell function. We identified, by using the synthetic peptide
LyeTx II, that upregulation of the p38 pathway in a TAK1-dependent
manner led to an accelerated proliferation and migration rate in
MDA-MB-231 cells. This study highlights the importance of using
compounds derived from animal venoms that may contribute to
identify new targets for breast cancer treatment. Furthermore,
these data open new perspectives for the use of p38 and TAK1
inhibitors as a targeted-treatment in cancer.
Acknowledgements
This study was funded by Fundação de Amparo à
Pesquisa do Estado de Minas Gerais (FAPEMIG) (grant no.
536-APQ-01682-13), Conselho Nacional de Desenvolvimento Científico
e Tecnológico (CNPq), CAPES (Coordenação de Aperfeiçoamento de
Pessoal de Nível Superior) and INCTTOX (Instituto Nacional de
Ciência e Tecnologia em Toxinas). H.W.H. is a Ph.D. student fellow
from CAPES. D.M.S. and H.D.G are post-doc and Ph.D. research
fellows from CNPq and CAPES, respectively. C.R. is senior
researcher from CNPq. M.E.L. is a professor and has received grants
from the cited agencies. The authors thank the Program for
Technological Development in Tools for Health-PDTIS - FIOCRUZ (Belo
Horizonte, Minas Gerais, Brazil) for use of its facilities. We are
grateful to Professor Aristóbolo Mendes (Depto. de Morfologia;
UFMG; Brazil) Professor D. Gomes (Depto. Bioquímica Imunologia;
UFMG; Brazil) for use of their laboratory facilities.
Glossary
Abbreviations
Abbreviations:
|
MAPK
|
mitogen-activated protein kinase
|
References
|
1
|
Banerji B, Pramanik SK, Pal U and Maiti
NC: Potent anticancer activity of cystine-based dipeptides and
their interaction with serum albumins. Chem Cent J. 7:912013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Misale S, Yaeger R, Hobor S, Scala E,
Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M,
Siravegna G, et al: Emergence of KRAS mutations and acquired
resistance to anti-EGFR therapy in colorectal cancer. Nature.
486:532–536. 2012.PubMed/NCBI
|
|
3
|
Diaz LA Jr, Williams RT, Wu J, Kinde I,
Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, et al:
The molecular evolution of acquired resistance to targeted EGFR
blockade in colorectal cancers. Nature. 486:4–7. 2012.
|
|
4
|
Dhillon AS, Hagan S, Rath O and Kolch W:
MAP kinase signalling pathways in cancer. Oncogene. 26:3279–3290.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen-activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Boutros T, Chevet E and Metrakos P:
Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase
regulation: Roles in cell growth, death, and cancer. Pharmacol Rev.
60:261–310. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kyriakis JM and Avruch J: Mammalian MAPK
signal transduction pathways activated by stress and inflammation:
A 10-year update. Physiol Rev. 92:689–737. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cargnello M and Roux PP: Activation and
function of the MAPKs and their substrates, the MAPK-activated
protein kinases. Microbiol Mol Biol Rev. 75:50–83. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Samatar AA and Poulikakos PI: Targeting
RAS-ERK signalling in cancer: Promises and challenges. Nat Rev Drug
Discov. 13:928–942. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Igea A and Nebreda AR: The stress kinase
p38α as a target for cancer therapy. Cancer Res. 75:3997–4002.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Koul HK, Pal M and Koul S: Role of p38 MAP
kinase signal transduction in solid tumors. Genes Cancer.
4:342–359. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wagner EF and Nebreda AR: Signal
integration by JNK and p38 MAPK pathways in cancer development. Nat
Rev Cancer. 9:537–549. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tournier C: The 2 faces of JNK signaling
in cancer. Genes Cancer. 4:397–400. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hoesel B and Schmid JA: The complexity of
NF-κB signaling in inflammation and cancer. Mol Cancer. 12:862013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gyrd-Hansen M and Meier P: IAPs: From
caspase inhibitors to modulators of NF-kappaB, inflammation and
cancer. Nat Rev Cancer. 10:561–574. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sung B, Prasad S, Yadav VR and Aggarwal
BB: Cancer cell signaling pathways targeted by spice-derived
nutraceuticals. Nutr Cancer. 64:173–197. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Erstad DJ, Cusack JC Jr, et al: Targeting
the NF-κB pathway in cancer therapy. Surg Oncol Clin N Am.
22:705–746. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gilmore TD: Introduction to NF-kappaB:
Players, pathways, perspectives. Oncogene. 25:6680–6684. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Santos DM, Verly RM, Piló-Veloso D, de
Maria M, de Carvalho MA, Cisalpino PS, Soares BM, Diniz CG, Farias
LM, Moreira DF, et al: LyeTx I, a potent antimicrobial peptide from
the venom of the spider Lycosa erythrognatha. Amino Acids.
39:135–144. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Correa CR, Bertollo CM and Goes AM:
Establishment and characterization of MACL-1 and MGSO-3 cell lines
derived from human primary breast cancer. Oncol Res. 17:473–482.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pierce JW, Schoenleber R, Jesmok G, Best
J, Moore SA, Collins T and Gerritsen ME: Novel inhibitors of
cytokine-induced IkappaBalpha phosphorylation and endothelial cell
adhesion molecule expression show anti-inflammatory effects in
vivo. J Biol Chem. 272:21096–21103. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bain J, Plater L, Elliott M, Shpiro N,
Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR and
Cohen P: The selectivity of protein kinase inhibitors: A further
update. Biochem J. 408:297–315. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Clark K, Nanda S and Cohen P: Molecular
control of the NEMO family of ubiquitin-binding proteins. Nat Rev
Mol Cell Biol. 14:673–685. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Martin SE, Wu ZH, Gehlhaus K, Jones TL,
Zhang YW, Guha R, Miyamoto S, Pommier Y and Caplen NJ: RNAi
screening identifies TAK1 as a potential target for the enhanced
efficacy of topoisomerase inhibitors. Curr Cancer Drug Targets.
11:976–986. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Safina A, Ren MQ, Vandette E and Bakin AV:
TAK1 is required for TGF-beta 1-mediated regulation of matrix
metalloproteinase-9 and metastasis. Oncogene. 27:1198–1207. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen L, Mayer JA, Krisko TI, Speers CW,
Wang T, Hilsenbeck SG and Brown PH: Inhibition of the p38 kinase
suppresses the proliferation of human ER-negative breast cancer
cells. Cancer Res. 69:8853–8861. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Leelahavanichkul K, Amornphimoltham P,
Molinolo AA, Basile JR, Koontongkaew S and Gutkind JS: A role for
p38 MAPK in head and neck cancer cell growth and tumor-induced
angiogenesis and lymphangiogenesis. Mol Oncol. 8:105–118. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kummer JL, Rao PK and Heidenreich KA:
Apoptosis induced by withdrawal of trophic factors is mediated by
p38 mitogen-activated protein kinase. J Biol Chem. 272:20490–20494.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bulavin DV, Kovalsky O, Hollander MC,
Fornace AJ Jr, et al: Loss of oncogenic H-ras-induced cell cycle
arrest and p38 mitogen-activated protein kinase activation by
disruption of Gadd45a. Mol Cell Biol. 23:3859–3871. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
She QB, Bode AM, Ma WY, Chen NY and Dong
Z: Resveratrol-induced activation of p53 and apoptosis is mediated
by extracellular-signal-regulated protein kinases and p38 kinase.
Cancer Res. 61:1604–1610. 2001.PubMed/NCBI
|
|
31
|
Mikhailov A, Shinohara M and Rieder CL:
Topoisomerase II and histone deacetylase inhibitors delay the G2/M
transition by triggering the p38 MAPK checkpoint pathway. J Cell
Biol. 166:517–526. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bulavin DV, Amundson SA and Fornace AJ:
p38 and Chk1 kinases: Different conductors for the G(2)/M
checkpoint symphony. Curr Opin Genet Dev. 12:92–97. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Thornton TM and Rincon M: Non-classical
p38 map kinase functions: Cell cycle checkpoints and survival. Int
J Biol Sci. 5:44–51. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huth HW, Albarnaz JD, Torres AA, Bonjardim
CA and Ropert C: MEK2 controls the activation of MKK3/MKK6-p38 axis
involved in the MDA-MB-231 breast cancer cell survival: Correlation
with cyclin D1 expression. Cell Signal. 28:1283–1291. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Schieven GL: The biology of p38 kinase: A
central role in inflammation. Curr Top Med Chem. 5:921–928. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
del Barco Barrantes I and Nebreda AR:
Roles of p38 MAPKs in invasion and metastasis. Biochem Soc Trans.
40:79–84. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pal S, Bhattacharjee A, Ali A, Mandal NC,
Mandal SC and Pal M: Chronic inflammation and cancer: potential
chemoprevention through nuclear factor kappa B and p53 mutual
antagonism. J Inflamm (Lond). 11:232014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee ST, Li Z, Wu Z, Aau M, Guan P,
Karuturi RKM, Liou YC and Yu Q: Context-specific regulation of
NF-κB target gene expression by EZH2 in breast cancers. Mol Cell.
43:798–810. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Han R, Liang H, Qin ZH and Liu CY:
Crotoxin induces apoptosis and autophagy in human lung carcinoma
cells in vitro via activation of the p38MAPK signaling pathway.
Acta Pharmacol Sin. 35:1323–1332. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu Z, Zhao Y, Li J, Xu S, Liu C, Zhu Y
and Liang S: The venom of the spider Macrothele raveni induces
apoptosis in the myelogenous leukemia K562 cell line. Leuk Res.
36:1063–1066. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ninomiya-Tsuji J, Kishimoto K, Hiyama A,
Inoue J, Cao Z and Matsumoto K: The kinase TAK1 can activate the
NIK-I kappaB as well as the MAP kinase cascade in the IL-1
signalling pathway. Nature. 398:252–256. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
McDermott EP and O'Neill LA: Ras
participates in the activation of p38 MAPK by interleukin-1 by
associating with IRAK, IRAK2, TRAF6, and TAK-1. J Biol Chem.
277:7808–7815. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mihaly SR, Ninomiya-Tsuji J and Morioka S:
TAK1 control of cell death. Cell Death Differ. 21:1667–1676. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sakurai H: Targeting of TAK1 in
inflammatory disorders and cancer. Trends Pharmacol Sci.
33:522–530. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kabir MH, Suh EJ and Lee C: Comparative
phosphoproteome analysis reveals more ERK activation in MDA-MB-231
than in MCF-7. Int J Mass Spectrom. 309:1–12. 2012. View Article : Google Scholar
|