Introduction
Hepatocellular carcinoma (HCC) is the most common
malignancy of the liver. It is the fifth most common cancer in men
and the seventh amongst women worldwide (1,2). There
is a high incidence of HCC in China due to hepatitis B virus (HBV)
infection (3). More than 80% of HCC
tumors are inoperable with poor prognosis, and only 10–20% of HCC
patients undergo curative treatments (4,5).
Results from clinical trials show a lack of survival benefits
following HCC treatment with chemotherapeutic agents and
conventional drugs. Thus, effective and well-tolerated treatment
strategies for advanced HCC are urgently needed (6).
Sorafenib, an oral multikinase inhibitor of
BRAF, RAF1, FLT3, KIT, VEGFR and
PDGFR, has been approved for the treatment of advanced HCC.
The Sorafenib HCC Assessment Randomized Protocol trial and the
Asia-Pacific trial demonstrated that sorafenib improves the
survival of patients with advanced HCC (7,8). Since
this major development in HCC treatment, a focus has been shifted
to identify novel agents that target driver genes and key molecular
pathways in hepatocarcinogenesis (9). Their findings include components of
the RAS/RAF/mitogen-extracellular activated protein kinase
(MEK)/extracellular signal-regulated kinase (ERK), and
phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of
rapamycin (mTOR) pathways that regulate cell proliferation,
apoptosis, and protein synthesis; receptor tyrosine kinases (RTKs),
including the epidermal growth factor receptor (EGFR),
KIT, FLT3 and RET, which transmit growth
factor signals to downstream intracellular pathways; and
proangiogenic factors that bind VEGFR and PDGFR which induce
angiogenic signaling via the RAS/RAF/MEK/ERK, PI3K/AKT/mTOR and Wnt
signal transduction pathways (6,10–12). A
thorough understanding of the mutations of genes associated with
molecular-targeted therapy is needed to screen compounds and
antibodies effective in HCC treatment.
Targeted sequencing allows accurate analysis of
multiple cancer genes (13–15). However, mutational profiling of
driver genes in Chinese patients with HCC has not been reported, to
date. In the present study, we detected multiple mutations in 45
genes in 100 patients with HCC using next-generation targeted
sequencing. These genes were categorized according to the following
biological processes or signaling pathways: RTKs, angiogenesis,
RAS/RAF/MEK/ERK and PI3K/AKT/mTOR. In particular, we identified
numerous novel somatic mutations in the driver genes and further
found that patients with TP53, RET and
sorafenib-targeted gene mutations, were associated with poor HCC
prognosis.
Materials and methods
Patients
We analyzed 100 patients who underwent HCC resection
between November 2009 and December 2011. Patients were subjected to
pathological assessment in order to establish histological
diagnosis and tumor cellularity. Only patients with a pathological
diagnosis of HCC and tumor nuclei ≥80% of the total cellular nuclei
were included. The present study was approved by the Ethics
Committee of The First Affiliated Hospital, School of Medicine,
Zhejiang University (Hangzhou, China). Signed informed consent
forms were obtained from patients before their participation in the
present study.
Exon capture array and deep
sequencing
A customized NimbleGen HD 2.1 Array was constructed
using the SeqCap v2 software. Target sequence capturing was
performed using the SeqCap EZ Reagent kit. The captured DNA was
randomly fragmented into an average size of 200–300 bp, and both
ends of the fragments were ligated with adaptors that bind to
different index primers. The enriched DNA fragments were eluted
from the array and amplified by ligation-mediated PCR. On average,
we sequenced the target exon regions of each sample to a mean depth
of 75x using the Illumina HiSeq 2000 platform.
Genome mapping and mutation
detection
Mapping and Assembly with Quality software
(http://maq.sourceforge.net) was used to
align the sequence reads to the referenced human genome (hg19). The
parameters used for the alignment were as follows: i) maximum
distance between sequences, 300; ii) maximum allowed sum of
qualities for 2-paired reads, 70; and iii) number of mismatches in
the first 24 bp of mismatches. Single nucleotide variations (SNVs)
of high quality were obtained using the following filtering
parameters: i) SNVs with depth ≥5; ii) consensus quality ≥30; iii)
3-bp flanking quality ≥40; iv) highest mapping quality ≥30; and v)
SNVs with variant depth ≥8. We defined the variant depth as 8 based
on the results of a previous study (13). The high quality SNVs were filtered
using the dbSNP (v.132) and 1K Genome databases to define the
mutations (16).
MassARRAY and Sanger sequencing
validation
Non-synonymous mutations were validated using the
iPLEX MassARRAY system (Sequenom Inc., San Diego, CA, USA) and
Sanger sequencing in HCC tumors and paired peritumoural liver
tissues to discriminate somatic and germline mutations. Both the
PCR and MassEXTEND® primers for each mutation were in
silico designed using the MassARRAY Assay design 4.0 software.
Multiplex PCR was performed using the GeneAmp PCR System 9700 Dual
384-Well Sample Block Module (Applied Biosystems, Foster City, CA,
USA), followed by dephosphorylation, single-base extension, and
desalting. The MassARRAY Nanodispenser RS1000 was used to spot
reactions with the 384 SpectroCHIP, which was loaded into a
MALDI-TOF mass spectrometer. Genotype calls by MassARRAY Type 4.0
were confirmed by examining the spectra for each assay and sample.
Mutations not detected by the iPLEX MassARRAY were reconfirmed by
Sanger sequencing.
Immunohistochemistry and tissue
microarray
In patients with multinodular tumors, samples were
obtained from the largest tumor. Rabbit anti-human monoclonal Ret
antibody (EPR2871, 1:500; Abcam, Cambridge, MA, USA) was used to
detect the protein expression of RET. The intensity of RET was
calculated based on mean area of positive staining. Tissues were
incubated with primary rabbit anti-human monoclonal Ret antibody
(EPR2871, 1:500; Abcam), then, treated with biotinylated goat
anti-rabbit secondary antibodies. Antibodies were visualized using
diaminobenzidine hydrogen peroxidase as the chromogen, and slides
were counterstained with 0.5% hematoxylin. In addition, we analyzed
another 90 independent samples to elucidate RET protein expression
using tissue microarray. Matched 90 pairs of primary HCC samples
and peritumoral liver tissues were used to prepare tissue
microarray (Shanghai Biochip Co., Ltd., Shanghai, China) as
previously described (17). The
intensity of RET was classified into high expression and low
expression based on the mean area of positive staining. High
expression was defined as ≥40% staining of a tumor section, and low
expression as <40%.
Statistical analysis
Statistical analyses were performed using SPSS
software (version 21.0; SPSS, Inc., Chicago, IL, USA). Age, gender,
tumor stage, number, size and grade, HBV infection, and
α-fetoprotein (AFP) were the covariates of clinical characteristics
included in the model. Chi-square and Fisher's exact tests were
applied to compare the frequencies between genetic and clinical
variables. The prognosis analyses of patients with gene mutation
status were summarized using Kaplan-Meier curves. Univariate
disease-free survival (DFS) and overall survival (OS) analyses were
carried out using log-rank tests, and multivariate analyses were
conducted using Cox's proportional hazards model. Postoperative
mortality was assessed, with deaths unrelated to tumor recurrence
considered censored observations at the time of death. P<0.05
was considered to indicate a statistically significant result.
Results
Genomic alteration landscape in HCC by
whole-exome sequencing
Whole exons of 45 genes were sequenced in 100
patients with HCC using array-based sequence capture and Illumina
HiSeq 2000 sequencing. Among them, 15 genes were associated with
RTKs, 8 with angiogenesis, 13 with the RAS/RAF/MEK/ERK pathway, and
9 with the PI3K/AKT/mTOR pathway. For each sample, we generated an
average of 33.75 Mb bases, 97.9% of which were well-mapped to the
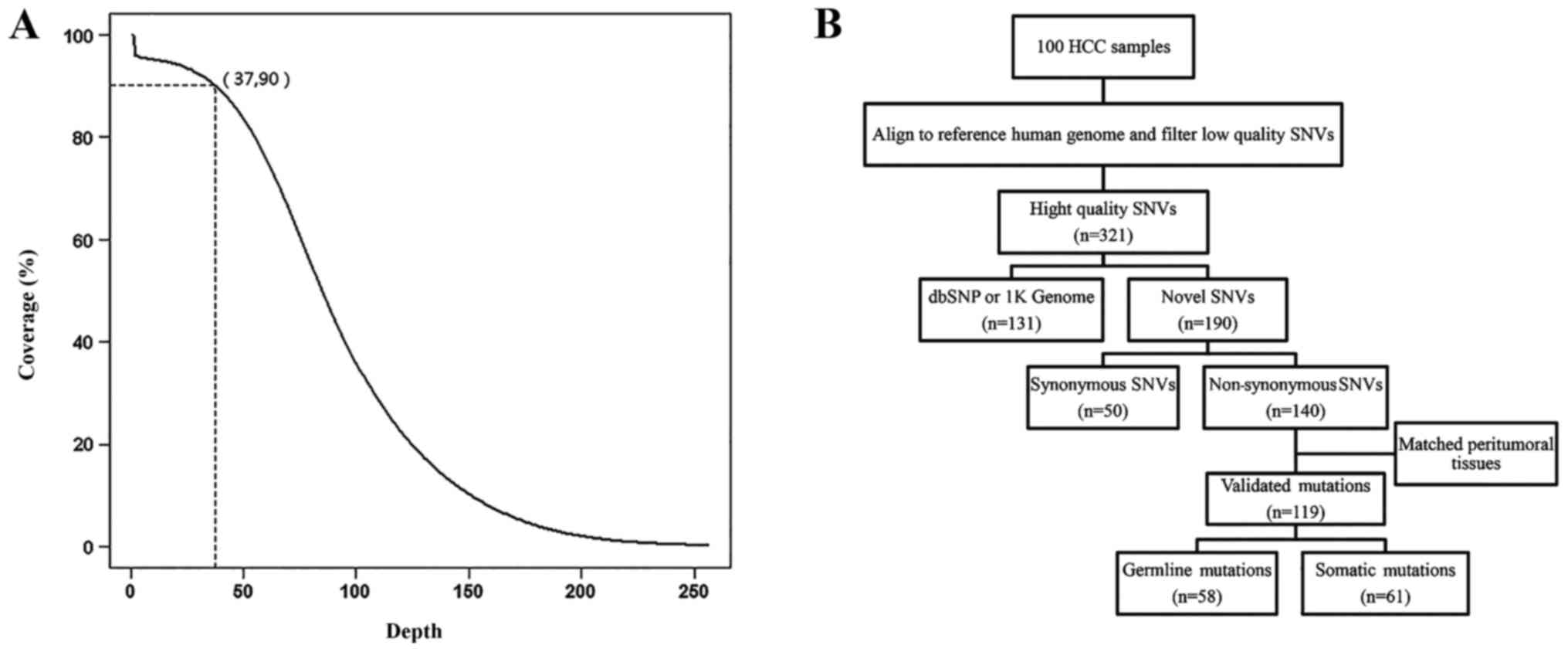
human genome. The average sequencing depth was 75x (Table I). We achieved a read coverage of
90% of the targeted exons at the sequencing depth of 37x (Fig. 1A). These results indicate high
quality targeted sequencing for mutation analysis.
 | Table I.Summary of sequencing coverage of the
100 HCC samples. |
Table I.
Summary of sequencing coverage of the
100 HCC samples.
| Category | Mean of 100 HCC
samples |
|---|
| Total reads | 393,273.06 |
| Total bases | 35,394,575.4 |
| Average read length
(bp) | 90 |
| Mappable reads | 385,309.01 |
| Mappable bases | 34,677,810.9 |
| Mappable base rates
(%) | 97.90299419 |
| Average sequencing
depth | 75x |
| Mean coverage over
target gene | 75x |
SNV identification, validation and
annotation
A computational pipeline was developed to discover
novel SNVs (Fig. 1B). A total of
321 exonic SNVs were identified in the 100 HCC patients.
Approximately 40.8% (131/321) of the SNVs were present after the
data were filtered using the dbSNP and 1K Genome databases. In most
SNVs identified we observed genetic polymorphisms, which confirmed
our sequencing data. The remaining 190 novel SNVs included 50
synonymous and 140 non-synonymous SNVs. In a random validation of
40 SNVs with MassARRAY and Sanger sequencing, the confirmation rate
was 90%. Furthermore, non-synonymous SNVs were validated in the
original HCC and paired peritumoural liver tissues using MassARRAY
and Sanger sequencing. We validated 119 mutations, of which 60 were
somatic mutations and 58 were germline mutations. In the somatic
mutations, 34 were novel mutations and 27 were recorded in COSMIC,
a public database of somatically acquired mutations in cancer
(18).
The annotations of somatic mutations observed in the
present study, are summarized in Table
II. The list of functional domains (identified using the NCBI
database) which harbor the mutations and molecular-targeted agents
of the mutated genes were identified using the DrugBank database
(Table II) (19). A few of the mutations were observed
in the protein kinase domains, particularly the tyrosine kinase
motifs of the target genes, which play a key role in the signaling
pathways that contribute to carcinogenesis.
| Table II.List of non-synonymous somatic
mutations and annotation of functional domains and
molecular-targeted agents. |
Table II.
List of non-synonymous somatic
mutations and annotation of functional domains and
molecular-targeted agents.
| Biological
classification | Gene | Nucleotide
(genomic) | Amino acid
change | Mutation type | No. of samples | Mutation in
domain | Molecular-targeted
agents |
|---|
| RTKs | ERBB1 (EGFR) |
g.chr7:55225446A>T | E.11:H433L | M | 1 | Approximate | Cetuximab,
trastuzumab, lidocaine, gefitinib, erlotinib, lapatinib,
panitumumab, vandetanib, afatinib |
|
| ERBB2 |
g.chr17:37866722A>T | E.6:T297S | M | 1 |
| Trastuzumab,
lapatinib, ado-trastuzumab emtansine, pertuzumab, afatinib |
|
| ERBB3 |
g.chr12:56478815A>T | E.3:M91L | M | 1 | Recep_L_domain |
|
|
| ERBB3 |
g.chr12:56495385A>G | E.28:D1192G | M | 1 |
|
|
|
| FGFR2 |
g.chr10:123260441A>T | E.10:L488Q | M | 1 |
| Palifermin,
thalidomide, regorafenib, ponatinib |
|
| C-FMS (CSF1R) |
g.chr5:149449842A>T | E.8:W408R | M | 1 |
| Imatinib,
sunitinib |
|
| FLT3 |
g.chr13:28608267A>G | E.14:Y597H | M | 1 |
| Sorafenib,
sunitinib, ponatinib |
|
| NTRK2 |
g.chr9:87285754G>A | E.1:A31T | M | 1 | Sig_peptide | Amitriptyline |
|
| RET |
g.chr10:43595961A>T | E.2:D43V | M | 1 |
| Sorafenib,
cabozantinib, |
|
| RET |
g.chr10:43595967C>T | E.2:A45V | M | 1 |
| regorafenib,
ponatinib |
|
| RET |
g.chr10:43607598G>A | E.8:R525Q | M | 1 |
|
|
|
| RET |
g.chr10:43608385T>G | E.9:I578S | M | 1 |
|
|
|
| RET |
g.chr10:43612043G>T | E.12:K716N | M | 1 |
|
|
|
| RET |
g.chr10:43615096C>A | E.14:S837Y | M | 1 | PTKc_RET,
Pkinase_Tyr |
|
| Angiogenesis | VEGFR1 (FLT1) |
g.chr13:28893583A>T |
E.24:L1088a | N | 1 | PTKc_VEGFR,
Pkinase_Tyr | Sorafenib,
sunitinib, pazopanib, axitinib, regorafenib |
|
| VEGFR2 (KDR) |
g.chr4:55979579T>C | E.7:S290G | M | 1 | V-set,
Ig1_VEGFR | Sorafenib,
sunitinib, pazopanib, |
|
| VEGFR2 (KDR) |
g.chr4:55974048A>T | E.10:I423N | M | 1 |
| axitinib,
cabozantinib, |
|
| VEGFR2 (KDR) |
g.chr4:55955069T>C | E.26:H1159R | M | 1 | Pkinase_Tyr | regorafenib,
ponatinib |
|
| VEGFR3 (FLT4) |
g.chr5:180048252G>T | E.14:A674D | M | 1 |
| Sorafenib,
sunitinib, pazopanib, axitinib, cabozantinib, regorafenib,
ponatinib |
|
| PDGFRA |
g.chr4:55133573C>A | E.5:R293S | M | 1 | Ig1_PDGFR-αβ | Becaplermin,
imatinib, sunitinib, pazopanib, regorafenib, ponatinib |
|
| PDGFRB |
g.chr5:149515249T>G | E.2:D78A | M | 1 |
| Becaplermin,
sorafenib, imatinib, dasatinib, sunitinib, pazopanib,
regorafenib |
|
| TIE1 |
g.chr1:43779006G>A | E.13:A113T | M | 1 | Interdomain
contacts, FN3 |
|
| RAS/RAF/MEK/ERK
pathway | RASSF1 |
g.chr3:50369548T>A | E.3:Q62L | M | 1 |
|
|
|
| MAP2 |
g.chr2:210574644A>T | E.7:E281V | M | 1 |
| Estramustine,
paclitaxel, |
|
| MAP2 |
g.chr2:210594945G>A | E.11:A502T | M | 1 |
| docetaxel |
|
| PLCE1 |
g.chr10:96005789A>T | E.7:Q836L | M | 1 |
|
|
|
| PLCE1 |
g.chr10:96006158A>G | E.7:Q959R | M | 1 |
|
|
|
| PLCE1 |
g.chr10:96022320A>T | E.13:Q1295L | M | 1 |
|
|
|
| PLCE1 |
g.chr10:96053334G>T | E.22:G1702V | M | 1 | Required for
activation by RHOA, RHOB |
|
|
| PLCE1 |
g.chr10:96058211T>A | E.23:I1748N | M | 1 | Required for
activation by RHOA, RHOB |
|
|
| SHC1 |
g.chr1:154938476C>T | E.10:G116R | M | 1 |
|
|
| PI3K/PTEN/AKT/mTOR
pathway | TP53 |
g.chr17:7579355A>G | E.4:L111P | M | 2 | P53 DNA-binding
domain |
|
|
| TP53 |
g.chr17:7578542G>A | E.5:L130F | M | 1 | P53 DNA-binding
domain |
|
|
| TP53 |
g.chr17:7578535T>A | E.5:K132M | M | 1 | P53 DNA-binding
domain |
|
|
| TP53 |
g.chr17:7578515T>C | E.5:K139E | M | 1 | P53 DNA-binding
domain |
|
|
| TP53 |
g.chr17:7578457C>A | E.5:R158L | M | 1 |
|
|
|
| TP53 |
g.chr17:7578436T>G | E.5:Q165P | M | 1 |
|
|
|
| TP53 |
g.chr17:7578394T>A | E.5:H179L | M | 1 |
|
|
|
| TP53 |
g.chr17:7578226T>A | E.6:D208V | M | 1 | P53_tetramer |
|
|
| TP53 |
g.chr17:7578206T>C | E.6:S215G | M | 1 | P53_tetramer |
|
|
| TP53 |
g.chr17:7578177C>A | E.6:E224D | M | 1 | P53_tetramer |
|
|
| TP53 |
g.chr17:7577586A>T | E.7:I232N | M | 1 |
|
|
|
| TP53 |
g.chr17:7577523G>A | E.7:T253I | M | 1 |
|
|
|
| TP53 |
g.chr17:7577130A>C | E.8:F270V | M | 1 |
|
|
|
| TP53 |
g.chr17:7577114C>A | E.8:C275F | M | 1 |
|
|
|
| TP53 |
g.chr17:7577105G>C | E.8:P278R | M | 1 |
|
|
|
| TP53 |
g.chr17:7577098T>A | E.8:R280S | M | 1 |
|
|
|
| TP53 |
g.chr17:7577097C>G | E.8:D281H | M | 1 |
|
|
|
| TP53 |
g.chr17:7577095G>C | E.8:D281E | M | 1 |
|
|
|
| TP53 |
g.chr17:7577082C>T | E.8:E286K | M | 1 |
|
|
|
| TP53 |
g.chr17:7574006A>C | E.10:F341V | M | 1 |
|
|
|
| TP53 |
g.chr17:7574003G>A |
E.10:R342a | N | 1 |
|
|
|
| PIK3CA |
g.chr3:178917513A>T | E.2:M130L | M | 1 |
|
|
|
| PIK3CA |
g.chr3:178947074G>T | E.17:G837V | M | 1 | PI3Kc_IA_α,
PI3Kc |
|
|
| PTEN |
g.chr10:89692781C>A | E.5:P89T | M | 1 | PTPc |
|
|
| PTEN |
g.chr10:89692948A>T | E.5:K144N | M | 1 | PTPc |
|
|
| PTEN |
g.chr10:89711875G>T |
E.6:G165a | N | 1 | PTPc |
|
|
| PTEN |
g.chr10:89717630C>T |
E.7:Q219a | N | 1 | PTEN_C2 |
|
|
| MTOR |
g.chr1:11298522T>A | E.11:T647S | M | 1 | HEAT 2 | Pimecrolimus,
sirolimus, everolimus, temsirolimus |
|
| MTOR |
g.chr1:11288742C>A | E.18:D1005Y | M | 1 | HEAT 4,
DUF3385 |
|
|
| STK11 |
g.chr19:1221966C>T | E.7:P294L | M | 1 | S_TKc, PKc |
|
Mutation frequency of each gene
distributed in 4 biological categories
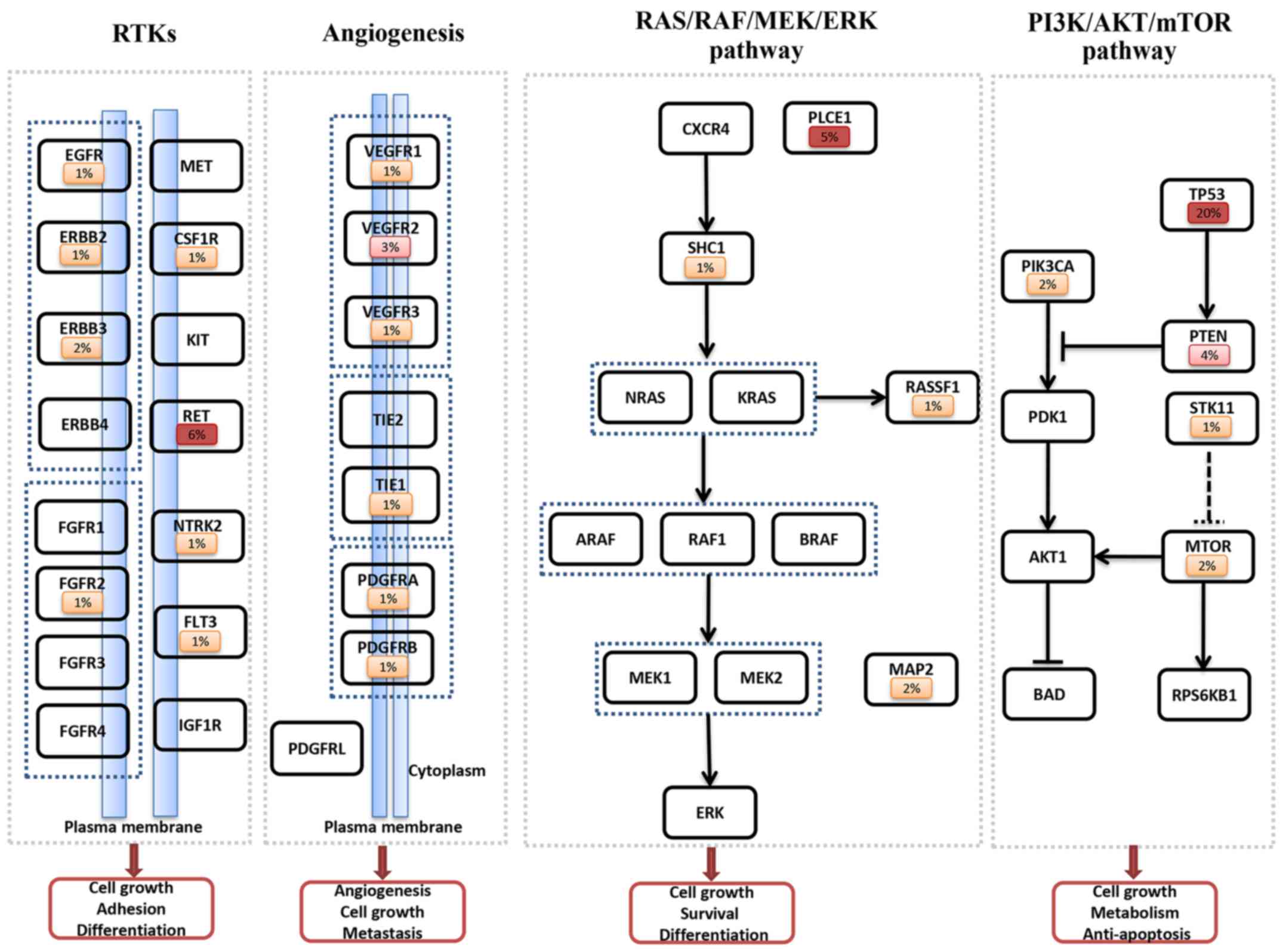
A total of 60 somatic mutations occurred within 45
genes, with an average mutation frequency of 0.62/affected
individual. The number of somatic mutations ranged from 0 to 3 in
the HCC patients. Fig. 2 shows the
complete somatic mutation frequency of each gene in the 4
categories including RTKs, angiogenesis, the RAS/RAF/MEK/ERK and
the PI3K/AKT/mTOR pathways.
There were 14 somatic mutations in the RTK genes
(Fig. 2), with 4 mutations
identified in the EGFR family, including 1 in EGFR, 1
in ERBB2 and 2 in ERBB3. RET was found with a
recurrent somatic mutation of 6% in HCC, which was validated using
MassARRAY and Sanger sequencing. The RET protein is composed of an
extracellular ligand-binding domain, a hydrophobic transmembrane
domain and a cytoplasmic part with a protein tyrosine kinase domain
(TK domain) (20).
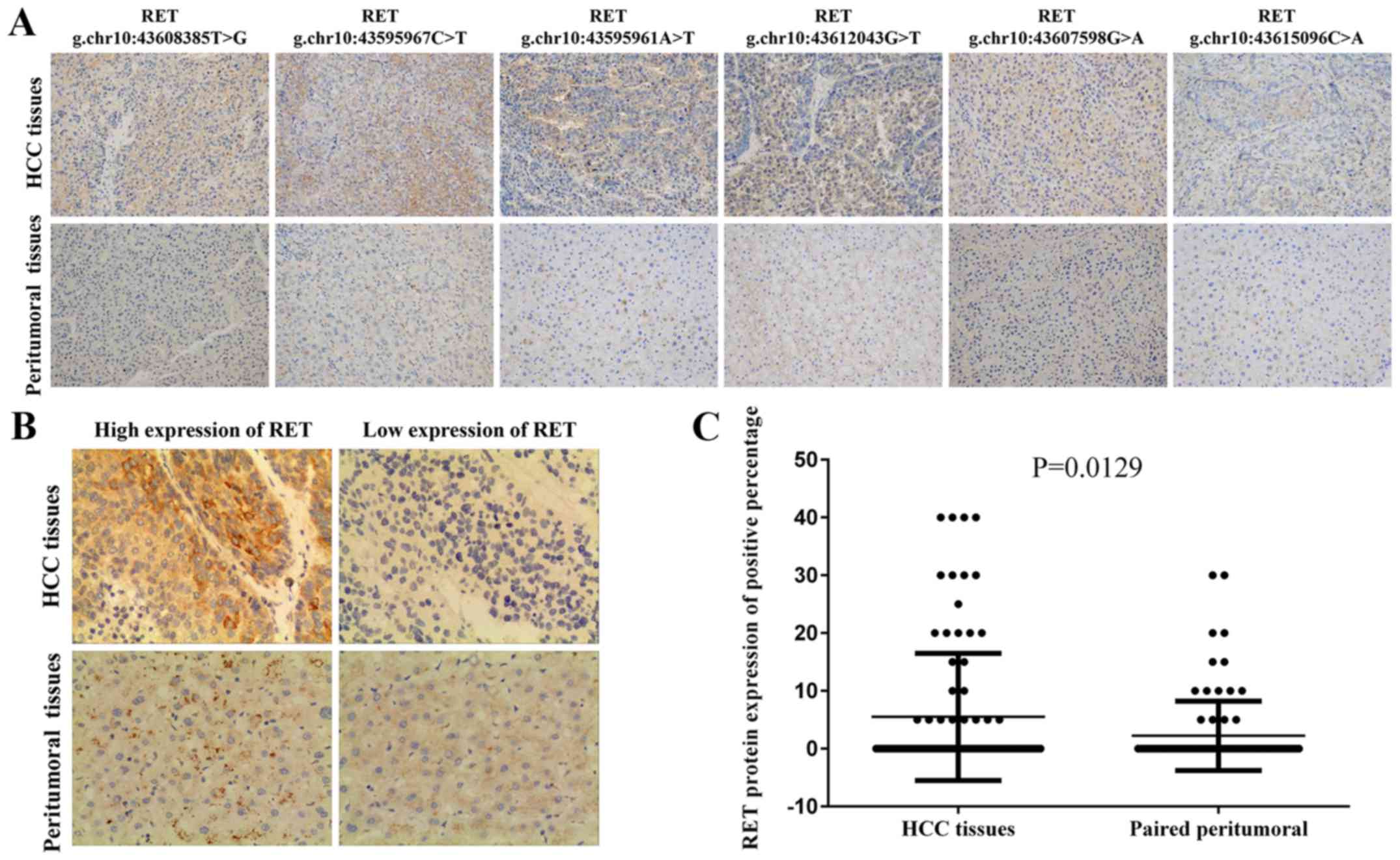
Immunohistochemical staining of HCC with RET mutation
further revealed a significant (Fig.
3A, the left 4 panels) or slight increase (Fig. 3A, the right 2 panels) in the
expression of RET in tumor tissues compared with peritumoral
tissues. We also performed a tissue microarray study of a cohort
containing another 90 HCC patients. The protein expression tendency
revealed that RET protein levels were higher in HCC tissues than in
paired peritumoral liver tissues using Student's t-test (P=0.012,
Fig. 3B and C). The group with the
high expression of RET included 28.9% (26/90) of the patients.
FGFR2, CSF1R, FLT3 and NTRK2 had one
mutation each, while there was no mutation for ERBB4,
FGFR1, FGFR3, FGFR4, IGF1R, MET
and KIT.
Eight somatic mutations were identified within genes
associated with angiogenesis (Fig.
2). Of these mutations, 5 were identified in members of the
VEGFR family, including one mutation in VEGFR1
(FLT1), 3 mutations in VEGFR2 (KDR), and 1
mutation in VEGFR3 (FLT4). PDGFRA,
PDGFRB and TIE1 had one mutation each, while no
mutation was detected in PDGFRL and TEK
(TIE2).
In the RAS/RAF/MEK/ERK pathway, 9 somatic mutations
were detected (Fig. 2). Somatic
mutations occurred mostly in PLCE1 (5%). Another recurrent
mutated gene was MAP2 (2%), and RASSF1 and
SHC1 exhibited one somatic mutation each. No somatic
mutation was observed in ERK, RAF1, ARAF,
BRAF, NRAS, MEK1, MEK2, CXCR4
and KRAS.
Twenty-nine somatic mutations were detected within 9
genes associated with the PI3K/AKT/mTOR pathway, including
recurrent mutations in TP53 (20%), PTEN (4%),
mTOR (2%), PIK3CA (2%), and a single mutation in
STK11 (Fig. 2). TP53
was the highest mutated gene. No somatic mutation was observed in
BAD, PDK1, AKT1 and RPS6KB1.
Analysis of clinical characterization
and prognosis
The clinicopathological characteristics of patients,
including age, gender, tumor stage (American Joint Committee on
Cancer; ver. 7), number, size, grade, serological HBV concentration
and presence of the tumor marker AFP are summarized in Table III. The median follow-up of cases
was 31.6 months (range, 1.8–48.1). A total of 39% of the patients
died, with a median OS of 41.5 months and a 3-year OS of 64.0% as
estimated by Kaplan-Meier analysis. During follow-up, 62 cases with
recurrence were identified for a median DFS of 21.2 months.
Correlation analysis of the clinical characteristics was based on
our data. AFP-positive patients demonstrated a higher rate of
RET mutations compared to those who were AFP-negative (11.1
vs. 0%; P=0.039), without correlation to other genes.
| Table III.Patient characteristics. |
Table III.
Patient characteristics.
| Factors | Total no. of
patients (N=100) |
|---|
| Age, years |
|
|
Median | 55 |
|
Standard deviation | 11 |
| Gender |
|
|
Male | 84 |
|
Female | 16 |
| HBV-DNA |
|
|
Positive | 64 |
|
Negative | 36 |
| Stage |
|
| I | 65 |
| II | 23 |
|
IIIA | 7 |
|
IIIB | 4 |
|
IIIC | 1 |
| AFP (ng/ml) |
|
|
Positive >20 | 60 |
|
Negative ≤20 | 40 |
| Tumor grade
(differentiation) |
|
|
Well | 6 |
|
Moderately | 44 |
|
Poorly | 50 |
| Tumor number |
|
|
Solitary | 90 |
|
Multifocal | 10 |
| Tumor size
(cm) |
|
|
Median | 5 |
|
Standard deviation | 3 |
The univariate analysis of DFS indicated that the
significant predictors of DFS were the somatic mutation status of
RET (P=0.028), tumor size (P<0.001), tumor stage
(P<0.001), and tumor marker AFP concentration (P=0.030)
(Table IV and Fig. 4A). The somatic mutation status of
TP53 was associated with decreased DFS without statistical
significance (Table IV and
Fig. 4C). Meanwhile, the univariate
analysis of OS suggested that the somatic mutation status of
RET (P=0.001) and TP53 (P=0.002), tumor size
(P=0.002), tumor stage (P=0.009) and AFP concentration(P=0.007)
were associated with the OS obtained from the follow-up (Table IV, Fig.
4B and D). Furthermore, the mutation status of sorafenib-target
genes were associated with decreased DFS (P=0.039) and decreased OS
(P=0.15) without statistical significance, which suggest poor
prognosis in these patients (Fig. 4E
and F).
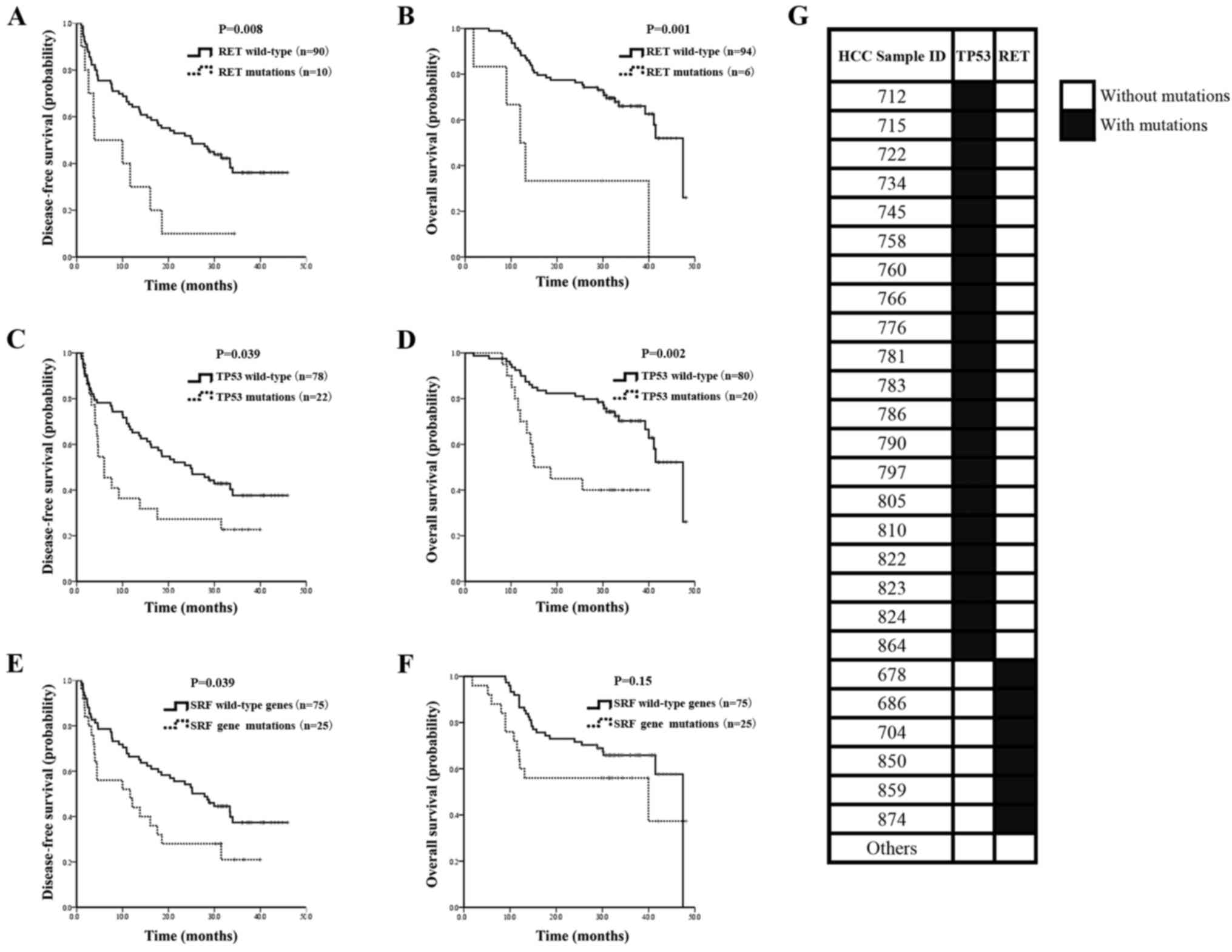 | Figure 4.Kaplan-Meier survival estimates
according to any mutations in TP53, RET and
sorafenib-target genes, and mutation status of TP53 and
RET in HCC patients. (A) Data are shown for the DFS of
patients with and without mutations in RET (median DFS,
3.700 vs. 24.833 months, respectively, P=0.028). (B) Data are shown
for the OS of patients with and without mutations in RET
(median OS, 12.000 vs. 47.433 months, respectively, P=0.001). (C)
Data are shown for the DFS of patients with and without mutations
in TP53 (median DFS, 24.172 vs. 24.833 months, respectively,
P=0.133). (D) Data are shown for the OS of patients with and
without mutations in TP53 (median OS, 14.967 vs. 47.433
months, respectively, P=0.002). (E) Data are shown for the DFS
(median DFS, 11.667 vs. 27.833 months, respectively, P=0.039) of
patients with and without mutations in sorafenib-target genes. (F)
Data are shown for the OS (median OS, 39.967 vs. 47.433 months,
respectively, P=0.15) of patients with and without mutations in
sorafenib-target genes. (G) Mutations of TP53 and RET are
mutually exclusive. Black indicates patients with mutations and
white indicates patients without mutations. SRF, sorafenib; HCC,
hepatocellular carcinoma; DFS, disease-free survival; OS, overall
survival. |
| Table IV.Predictors of disease-free and
overall survival. |
Table IV.
Predictors of disease-free and
overall survival.
|
| Univariate
analysis | Multivariate
analysis |
|---|
|
|
|
|
|---|
|
| Disease-free
survival | Overall
survival | Disease-free
survival | Overall
survival |
|---|
|
|
|
|
|
|
|---|
| Factor | Median DFS (months)
(95% CI) | P-value | Median OS (months)
(95% CI) | P-value | HR (95% CI) | P-value | HR (95% CI) | P-value |
|---|
| TP53 |
| 0.133 |
| 0.002 | 1.843
(0.982–3.459) | 0.057 | 4.101
(1.941–8.668) | <0.001 |
| No
mutation | 24.833
(14.899–34.767) |
| 47.433
(40.041–54.826) |
|
|
|
|
|
|
Mutation | 24.172
(0–11.314) |
| 14.967
(6.347–23.586) |
|
|
|
|
|
| RET |
| 0.028 |
| 0.001 | 3.592
(1.331–9.693) | 0.012 | 4.270
(1.511–12.066) | 0.006 |
| No
mutation | 24.833
(15.402–34.265) |
| 47.433
(40.063–54.803) |
|
|
|
|
|
|
Mutation | 3.700
(1.219–6.181) |
| 12.000
(7.039–16.961) |
|
|
|
|
|
| Age (years) |
| 0.075 |
| <0.001 | 1.029
(1.003–1.055) | 0.027 |
|
|
| Tumor number |
| 0.176 |
| 0.550 |
|
|
|
|
|
Solitary | 24.833
(13.773–35.894) |
| 41.467
(37.317–45.616) |
|
|
|
|
|
|
Multifocal | 10.900
(0–25.362) |
| 41.100
(17.999–64.201) |
|
|
|
|
|
| Tumor size |
| <0.001 |
| 0.002 | 1.090
(0.999–1.188) | 0.053 | 1.145
(1.042–1.258) | 0.005 |
| HBV-DNA |
| 0.184 |
| 0.711 |
|
|
|
|
|
Negative | 31.467
(20.117–42.816) |
| NA |
|
|
|
|
|
|
Positive | 17.567
(10.319–24.815) |
| 41.467
(37.465–45.469) |
|
|
|
|
|
| Tumor grade
(differentiated) |
| 0.237 |
| 0.196 |
|
|
|
|
|
Well | NA |
| NA |
|
|
|
|
|
|
Moderately | 25.133
(12.471–37.796) |
| NA |
|
|
|
|
|
|
Poorly | 13.600
(5.669–21.531) |
| NA |
|
|
|
|
|
| Gender |
| 0.901 |
| 0.892 |
|
|
|
|
|
Male | 21.200
(12.373–30.027) |
| 47.433
(38.739–56.128) |
|
|
|
|
|
|
Female | 18.567
(0–51.495) |
| 41.100
(25.428–56.772) |
|
|
|
|
|
| Stage |
| <0.001 |
| 0.009 | 1.624
(1.254–2.101) | <0.001 |
|
|
| I | 33.367
(22.994–43.740) |
| NA |
|
|
|
|
|
| II | 13.767
(0–45.259) |
| NA |
|
|
|
|
|
|
IIIA | 4.000
(1.605–6.395) |
| NA |
|
|
|
|
|
|
IIIB | 1.400
(1.302–1.498) |
| NA |
|
|
|
|
|
|
IIIC | NA |
| NA |
|
|
|
|
|
| AFP (ng/ml) |
| 0.030 |
| 0.007 | 1.000
(1.000–1.000) | 0.034 |
|
|
|
Positive >20 | 33.433
(26.512–40.355) |
| 39.967
(30.592–49.342) |
|
|
|
|
|
|
Negative ≤20 | 13.767
(8.037–19.496) |
| NA |
|
|
|
|
|
The conditional multivariable analysis of DFS
revealed that the somatic mutation status of RET (hazard
ratio (HR)=3.592; 95% confidence interval (CI), 1.331–9.693;
P=0.012), age of patients (HR=1.029; 95% CI, 1.003–1.055; P=0.027),
tumor size (HR=1.090; 95% CI, 0.999–1.188; P=0.053), tumor stage
(HR=1.624; 95% CI, 1.254–1.101; P<0.001), and tumor marker AFP
concentration (HR=1.000; 95% CI, 1.000–1.000; P=0.034) (Table IV) were significant predictors of
DFS. Conditional multivariable survival analysis demonstrated that
the independent predictors of OS were the somatic mutation status
of TP53 (HR=4.101; 95% CI, 1.941–8.668; P<0.001),
RET (HR=4.270; 95% CI, 1.511–12.066; P=0.006), and tumor
size (HR=1.145; 95% CI, 1.042–1.258; P=0.005) (Table IV). In addition, mutual exclusion
of TP53 and RET mutations was observed in the present
study (Fig. 4G). These results
suggest that both TP53 and RET are significant
biomarkers in the prognosis of HCC.
Discussion
To date, sorafenib, an oral multi-kinase inhibitor
of BRAF, RAF1, FLT3, KIT, VEGFR
and PDGFR, is the main clinical treatment used in advanced
HCC. With the development of findings in new target driver genes
and molecular-targeted therapies in HCC, RTKs, angiogenesis,
RAS/RAF/MEK/ERK and PI3K/AKT/mTOR pathways have been reported to be
involved in hepatocarcinogenesis (6,10–12).
However, mutational profiling of these driver genes in Chinese
patients with HCC has not been reported, to date. In the present
study, targeted deep sequencing was used to conduct the
simultaneous analysis of 45 driver genes in 100 patients with HCC,
which were categorized according to the following biological
processes or signaling pathways: RTKs, angiogenesis,
RAS/RAF/MEK/ERK and PI3K/AKT/mTOR. To the best of our knowledge,
this is the first comprehensive analysis of driver genes in Chinese
patients with HCC.
In the present study, 61 non-synonymous somatic
mutations were identified in 43% of the HCC patients. Among members
of RTKs, we found somatic mutations in the EGFR family, of which
FLT3, C-FMS and FGFR2 are the targets of
multikinase inhibitors, including sorafenib, sunitinib and
regorafenib, which exhibit effective results in HCC (21). Somatic mutations were also observed
in angiogenesis-associated genes including VEGFR1,
VEGFR2, VEGFR3, PDGFRA and PDGFRB,
particularly non-synonymous mutations of L1088* (VEGFR1) and
H1159R (VEGFR2) located in the catalytic domain of TK.
Sorafenib targets both VEGFR2 and VEGFR3 with encouraging outcomes
in advanced HCC (7,22). The dual inhibition of VEGF and PDGF
signaling demonstrated marked anti-angiogenic effects in
vivo (23). Linifanib (ABT-869)
is a novel selective inhibitor of VEGF and PDGF RTK families in a
phase III clinical trial for HCC treatment (24). Recurrent mutations in PLCE1
were also observed, since the RAS/RAF/MEK/ERK pathway participates
in HCC growth and progression (6).
Moreover, several studies have reported that the genetic variations
in PLCE1 are associated with esophageal squamous cell
carcinoma and gastric adenocarcinoma (25,26).
Thus, PLCE1 may be further studied for HCC treatment. In
addition, mutations of PIK3CA, PTEN and mTOR
in the PI3K/AKT/mTOR signaling pathway were observed. PIK3CA
mutations have been reported to sensitize cancer cells to mTOR
inhibitor everolimus (27). In the
present study, in our findings, somatic mutation of G837V was
identified in the catalytic subunit of PIK3CA, and both
G165* and Q219* mutations of PTEN were truncating mutations
which destroyed the function of PTEN. PTEN (G165*, Q219*)
mutations may serve as molecular markers for mTOR
inhibitor-targeted therapy.
As shown in Fig. 2,
the most frequent mutations were: TP53 (20%), RET
(6%), PLCE1 (5%), PTEN (4%) and VEGFR2 (3%).
Genome-wide sequencing analyses have revealed many mutant genes,
such as TP53 and β-catenin (CTNNB1) in HCC (28,29).
The recurrent mutations of TP53 (20%) identified in the
present study were similar with earlier studies, which confirmed
the reliability of our sequencing data. Further prognostic analysis
which revealed that patients with mutations in TP53 had
lower overall survival (OS) than those without mutations was
consistent with earlier studies (28,30).
Dysregulation of RET activity is an important contributor to
several human types of cancer including thyroid, lung, breast and
pancreatic tumors (31–34), suggesting that RET is an
important target for therapeutic intervention in many diseases
(35). Genetic aberrations,
including rearrangement, germline and somatic activation mutations,
are responsible for a fraction of papillary thyroid carcinoma,
medullary thyroid carcinoma, and a small subset of non-small cell
lung cancer (7,32,36–38).
In addition, the prognostic study further revealed that HCC
patients with RET somatic mutations had poorer DFS and OS
compared with wild-type patients in the present study. In
particular, the mutual exclusion of TP53 and RET
mutations were observed. All aforementioned results indicated that
both TP53 and RET are significant biomarkers in the
prognosis of HCC.
With the development of genome technology and lower
costs for sequencing, the simultaneous analysis of genetic
variation of the set of driver genes in tumor tissue after
resection or biopsy is available. Presently, personalized medicine
is emerging with genomic technology applied in clinical oncology.
In addition, finding subpopulations of patients who may benefit
from molecular-targeted therapy is important. Identification of
specific genetic variations in individual patients may serve as a
guide to develop effective drugs used in the treatment of HCC
patients. In the present study, we conducted the mutation profiling
of driver genes and clinical prognostic analysis, which indicated
that TP53 and RET mutations may serve as biomarkers
for targeted therapy in HCC. However, the present study has certain
limitations. Firstly, we only analyzed a small group of patients,
and a perspective analysis of frequent mutations may be carried out
in a large-scale study of patients with HCC to confirm our results.
Secondly, numerous genetic mutations were identified; however, not
all mutations were functional, and may serve as passenger
mutations. Thus, functional analysis of mutations is needed to
further illustrate the carcinogenic mechanism in HCC. Although,
further studies are needed to guide molecular-targeted therapy in
HCC, in the present study we identified TP53 and RET
mutations to be suitable markers for prognostic evaluation and
targeted therapy in HCC.
Acknowledgements
We would like to thank Leiming Chen and Guoqiang Cao
for their expert technical assistant in specimen collection. The
present study was supported by the National High Technology
Research and Development Program of China (863 Program, no.
2012AA02A205), the Science Technology Department of Zhejiang
Province (nos. 2016C33116 and 2011C23088), the National Natural
Science Fundation of China (no. J20121214), the CSCO Merck Serono
Oncology Research Fund, SCORE (no. Y-MX2015-038), the Health Bureau
of Zhejiang Province (no. 201484382), and the Key Research Project
of Science Technology Department of Zhejiang Province (no.
2015C03030).
References
|
1
|
Mittal S and El-Serag HB: Epidemiology of
hepatocellular carcinoma: Consider the population. J Clin
Gastroenterol. 47:(Suppl). S2–S6. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
El-Serag HB: Epidemiology of viral
hepatitis and hepatocellular carcinoma. Gastroenterology.
142:1264–1273. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tanaka M, Katayama F, Kato H, Tanaka H,
Wang J, Qiao YL and Inoue M: Hepatitis B and C virus infection and
hepatocellular carcinoma in China: A review of epidemiology and
control measures. J Epidemiol. 21:401–416. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rampone B, Schiavone B, Martino A, Viviano
C and Confuorto G: Current management strategy of hepatocellular
carcinoma. World J Gastroenterol. 15:3210–3216. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Villanueva A and Llovet JM: Targeted
therapies for hepatocellular carcinoma. Gastroenterology.
140:1410–1426. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S,
Kim JS, Luo R, Feng J, Ye S, Yang TS, et al: Efficacy and safety of
sorafenib in patients in the Asia-Pacific region with advanced
hepatocellular carcinoma: A phase III randomised, double-blind,
placebo-controlled trial. Lancet Oncol. 10:25–34. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Germano D, Tinessa V, Barletta E, Cannella
L and Daniele B: Targeted therapy for advanced hepatocellular
cancer in the elderly: Focus on sorafenib. Drugs Aging. 30:887–892.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhu AX: Molecularly targeted therapy for
advanced hepatocellular carcinoma in 2012: Current status and
future perspectives. Semin Oncol. 39:493–502. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kudo M: Current status of molecularly
targeted therapy for hepatocellular carcinoma: Clinical practice.
Int J Clin Oncol. 15:242–255. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tanaka S and Arii S: Current status of
molecularly targeted therapy for hepatocellular carcinoma: Basic
science. Int J Clin Oncol. 15:235–241. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Finn RS: Emerging targeted strategies in
advanced hepatocellular carcinoma. Semin Liver Dis. 33:(Suppl 1).
S11–S19. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zang ZJ, Ong CK, Cutcutache I, Yu W, Zhang
SL, Huang D, Ler LD, Dykema K, Gan A, Tao J, et al: Genetic and
structural variation in the gastric cancer kinome revealed through
targeted deep sequencing. Cancer Res. 71:29–39. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nikiforova MN, Wald AI, Roy S, Durso MB
and Nikiforov YE: Targeted next-generation sequencing panel
(ThyroSeq) for detection of mutations in thyroid cancer. J Clin
Endocrinol Metab. 98:E1852–E1860. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cottrell CE, Al-Kateb H, Bredemeyer AJ,
Duncavage EJ, Spencer DH, Abel HJ, Lockwood CM, Hagemann IS, O'Guin
SM, Burcea LC, et al: Validation of a next-generation sequencing
assay for clinical molecular oncology. J Mol Diagn. 16:89–105.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Abecasis GR, Auton A, Brooks LD, DePristo
MA, Durbin RM, Handsaker RE, Kang HM, Marth GT and McVean GA: 1000
Genomes Project Consortium: An integrated map of genetic variation
from 1,092 human genomes. Nature. 491:56–65. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhu XD, Zhang JB, Zhuang PY, Zhu HG, Zhang
W, Xiong YQ, Wu WZ, Wang L, Tang ZY and Sun HC: High expression of
macrophage colony-stimulating factor in peritumoral liver tissue is
associated with poor survival after curative resection of
hepatocellular carcinoma. J Clin Oncol. 26:2707–2716. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Forbes SA, Bindal N, Bamford S, Cole C,
Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al:
COSMIC: Mining complete cancer genomes in the Catalogue of Somatic
Mutations in Cancer. Nucleic Acids Res. 39:D945–D950. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Knox C, Law V, Jewison T, Liu P, Ly S,
Frolkis A, Pon A, Banco K, Mak C, Neveu V, et al: DrugBank 3.0: A
comprehensive resource for omics research on drugs. Nucleic Acids
Res. 39:D1035–D1041. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fujimoto A, Totoki Y, Abe T, Boroevich KA,
Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F, et al:
Whole-genome sequencing of liver cancers identifies etiological
influences on mutation patterns and recurrent mutations in
chromatin regulators. Nat Genet. 44:760–764. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dekervel J, van Pelt J and Verslype C:
Advanced unresectable hepatocellular carcinoma: New biologics as
fresh ammunition or clues to disease understanding? Curr Opin
Oncol. 25:409–416. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al: SHARP Investigators Study Group: Sorafenib in advanced
hepatocellular carcinoma. N Engl J Med. 359:378–390. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kuhnert F, Tam BY, Sennino B, Gray JT,
Yuan J, Jocson A, Nayak NR, Mulligan RC, McDonald DM and Kuo CJ:
Soluble receptor-mediated selective inhibition of VEGFR and
PDGFRbeta signaling during physiologic and tumor angiogenesis. Proc
Natl Acad Sci USA. 105:10185–10190. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chiu YL, Carlson DM, Pradhan RS and Ricker
JL: Exposure-response (safety) analysis to identify linifanib dose
for a Phase III study in patients with hepatocellular carcinoma.
Clin Ther. 35:1770–1777. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bye H, Prescott NJ, Lewis CM, Matejcic M,
Moodley L, Robertson B, Rensburg C, Parker MI and Mathew CG:
Distinct genetic association at the PLCE1 locus with oesophageal
squamous cell carcinoma in the South African population.
Carcinogenesis. 33:2155–2161. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Palmer AJ, Lochhead P, Hold GL, Rabkin CS,
Chow WH, Lissowska J, Vaughan TL, Berry S, Gammon M, Risch H, et
al: Genetic variation in C20orf54, PLCE1 and
MUC1 and the risk of upper gastrointestinal cancers in
Caucasian populations. Eur J Cancer Prev. 21:541–544. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Di Nicolantonio F, Arena S, Tabernero J,
Grosso S, Molinari F, Macarulla T, Russo M, Cancelliere C, Zecchin
D, Mazzucchelli L, et al: Deregulation of the PI3K and KRAS
signaling pathways in human cancer cells determines their response
to everolimus. J Clin Invest. 120:2858–2866. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cleary SP, Jeck WR, Zhao X, Chen K,
Selitsky SR, Savich GL, Tan TX, Wu MC, Getz G, Lawrence MS, et al:
Identification of driver genes in hepatocellular carcinoma by exome
sequencing. Hepatology. 58:1693–1702. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kan Z, Zheng H, Liu X, Li S, Barber TD,
Gong Z, Gao H, Hao K, Willard MD, Xu J, et al: Whole-genome
sequencing identifies recurrent mutations in hepatocellular
carcinoma. Genome Res. 23:1422–1433. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Woo HG, Wang XW, Budhu A, Kim YH, Kwon SM,
Tang ZY, Sun Z, Harris CC and Thorgeirsson SS: Association of
TP53 mutations with stem cell-like gene expression and
survival of patients with hepatocellular carcinoma.
Gastroenterology. 140:1063–1070. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Romei C and Elisei R: RET/PTC
translocations and clinico-pathological features in human papillary
thyroid carcinoma. Front Endocrinol. 3:542012. View Article : Google Scholar
|
|
32
|
Kohno T, Ichikawa H, Totoki Y, Yasuda K,
Hiramoto M, Nammo T, Sakamoto H, Tsuta K, Furuta K, Shimada Y, et
al: KIF5B-RET fusions in lung adenocarcinoma. Nat Med.
18:375–377. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zeng Q, Cheng Y, Zhu Q, Yu Z, Wu X, Huang
K, Zhou M, Han S and Zhang Q: The relationship between
overexpression of glial cell-derived neurotrophic factor and its
RET receptor with progression and prognosis of human pancreatic
cancer. J Int Med Res. 36:656–664. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Plaza-Menacho I, Morandi A, Robertson D,
Pancholi S, Drury S, Dowsett M, Martin LA and Isacke CM: Targeting
the receptor tyrosine kinase RET sensitizes breast cancer cells to
tamoxifen treatment and reveals a role for RET in endocrine
resistance. Oncogene. 29:4648–4657. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Phay JE and Shah MH: Targeting RET
receptor tyrosine kinase activation in cancer. Clin Cancer Res.
16:5936–5941. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fusco A, Grieco M, Santoro M, Berlingieri
MT, Pilotti S, Pierotti MA, Porta G Della and Vecchio G: A new
oncogene in human thyroid papillary carcinomas and their
lymph-nodal metastases. Nature. 328:170–172. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Grieco M, Santoro M, Berlingieri MT,
Melillo RM, Donghi R, Bongarzone I, Pierotti MA, Porta G Della,
Fusco A and Vecchio G: PTC is a novel rearranged form of the
ret proto-oncogene and is frequently detected in vivo in
human thyroid papillary carcinomas. Cell. 60:557–563. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Takeuchi K, Soda M, Togashi Y, Suzuki R,
Sakata S, Hatano S, Asaka R, Hamanaka W, Ninomiya H, Uehara H, et
al: RET, ROS1 and ALK fusions in lung cancer. Nat Med. 18:378–381.
2012. View Article : Google Scholar : PubMed/NCBI
|