Introduction
Colorectal cancer is the third most common cancer in
males and the second most common in females worldwide (1). In colorectal cancer, gain-of-function
mutations in KRas are detected in approximately 40% of the tumors
and play a critical role in malignant transformation (2). KRas mutations activate intracellular
signals and stimulate tumor cell growth, invasion, metastasis, and
drug resistance (3–6). Moreover, KRas mutations promote
cytokine secretion in tumor cells, which might contribute to cancer
initiation and progression (7–9).
In cancer tissues, several types of cells, such as
myofibroblasts, immune cells, and vascular cells, form the tumor
microenvironment and thus promote tumor progression (10). The tumor microenvironment
contributes to cancer progression by promoting tumor cell growth,
inhibiting apoptosis, enhancing angiogenesis, and suppressing
antitumor immunity (11,12). The tumor cells, in turn, regulate
stromal cells through paracrine signaling and thereby generate an
environment conducive to tumor progression (13,14).
Intestinal myofibroblasts (IMFs) localized subjacent
to the intestinal epithelium interact with epithelial cells and
play crucial roles in maintaining epithelial homeostasis (15,16).
Myofibroblasts regulate various diseases, such as inflammation,
fibrosis, and carcinogenesis. In cancer tissues, myofibroblasts
localize adjacent to cancer stem cells and participate in mutual
feedback signaling loops (14,17).
Myofibroblasts secrete cytokines such as interleukin-6 (IL-6),
hepatocyte growth factor (HGF), and heparin-binding epidermal
growth factor-like growth factor (HB-EGF) and thus promote tumor
progression (17–20). Conversely, tumor cells secrete
transforming growth factor-β (TGF-β) and platelet-derived growth
factor (PDGF), which regulate the migration, differentiation, and
cytokine secretion of myofibroblasts (18,21).
Although these interactions have gradually come to be regarded as
new therapeutic targets (22), the
mechanisms of interaction between KRas-mutated cancer cells and
myofibroblasts have remained unclear.
In this study, to elucidate how KRas-mutated cancer
cells interact with myofibroblasts, we investigated the molecular
mechanisms of the regulation of IMFs by KRas-mutated cells.
Materials and methods
Mice
Male C57BL/6J mice (4–12 weeks old) were purchased
from Charles River Japan (Yokohama, Japan) and maintained in
accordance with the guidelines of the Animal Care and Use Committee
of Yamaguchi University. Experimental protocols were approved by
the Yamaguchi University Animal Care and Use Committee.
Cell culture and conditioned medium
(CM) collection
Mouse IMFs were isolated as previously described
(23). LmcMF, a mouse colon
myofibroblast cell line, was established in our previous study
(23). An adult mouse colon
epithelial cell line, aMoC1, was provided by Dr Mamoru Totsuka
(Tokyo University) (24). IMFs and
LmcMF cells were cultured in Dulbecco's modified Eagle's medium
(DMEM; Thermo Fisher Scientific, Waltham, MA, USA) containing 10%
fetal bovine serum (FBS) and 1% antibiotic-antimycotic solution
(AA; Thermo Fisher Scientific), whereas aMoC1 cells were cultured
in DMEM containing 5% FBS, 1% insulin-transferrin-selenium-X
(Thermo Fisher Scientific), and 1% AA solution. CM was obtained by
collecting the supernatant from aMoC1 cell cultures in serum-free
medium at 24 h after plating and centrifuging for 10 min at 3,000 ×
g; aMoC1-CM was used at 50% concentration in all experiments.
Generation of KRasV12-expressing aMoC1
cells
FLAG-tagged human KRasV12 was PCR-amplified from a
human colorectal cancer cell line, SW620, that carries the KRasV12
mutation, and was subcloned into the EcoRI/NotI sites
of pLVSIN-EF1α-IRES-ZsGreen1 vector (Takara Bio, Shiga, Japan). The
empty vector was used as a control. Lentivirus was produced as
previously described (25), and
aMoC1 cells were treated with virus-containing supernatants for
12–24 h. ZsGreen-expressing cells were sorted using an SH800 flow
cytometer (Sony, Tokyo, Japan), and were used as mock-transfected
aMoC1 cells (Mock-aMoC1 cells) and KRasV12-expressing aMoC1 cells
(KRasV12-aMoC1 cells).
Western blotting
Western blotting was performed as previously
described (26). Briefly, cells
were lysed in a buffer containing 50 mM Tris-HCl (pH 8.0), 5 mM
EDTA (pH 8.0), 5 mM EGTA (pH 8.0), 1% Triton X-100, 1 mM
Na3VO4, 20 mM sodium pyrophosphate, and Roche
complete protease-inhibitor cocktail (Roche, Basel, Switzerland).
Proteins were quantified using a Bio-Rad DC protein assay kit
(Bio-Rad, Hercules, CA, USA) and then separated using sodium
dodecyl sulfate-polyacrylamide gel electrophoresis and transferred
onto nitrocellulose membranes (Wako Pure Chemical Industries,
Osaka, Japan). The membranes were blocked with 0.5% skim milk and
then probed with primary and secondary antibodies, and the stained
protein bands were detected using Western Lightning ECL Pro
(PerkinElmer, Waltham, MA, USA) and visualized using a LAS-3000
mini luminescence imager (Fujifilm, Tokyo, Japan). The primary
antibodies used were against these targets: c-Myc, GSK-3β
phospho-Ser9, ERK p44/42 phospho-Thr202/Tyr204, Akt1
phospho-Ser473, p38 MAPK phospho-Thr180/Tyr182, p70 S6 Kinase (S6K)
phospho-Thr389, Stat3α phospho-Tyr705, and Yap phospho-Ser127 (Cell
Signaling Technology, Danvers, MA, USA); JNK phospho-Thr183/Tyr185
(Promega, Madison, WI, USA); α-smooth muscle actin (α-SMA) and
vimentin (Sigma, St. Louis, MO, USA); actin (Santa Cruz
Biotechnology, Dallas, TX, USA); Ras (Merck Millipore, Darmstadt,
Germany); tubulin (Thermo Fisher Scientific); CD44 (Bethyl
Laboratories, Montgomery, TX, USA); Lgr5 (Abgent, San Diego, CA,
USA); and p97/VCP (GeneTex, Irvine, CA, USA). The secondary
HRP-conjugated antibodies used were goat anti-mouse IgG (R&D
Systems, Minneapolis, MN, USA), goat anti-rabbit IgG (Cayman
Chemical Co., Ann Arbor, MI, USA), and donkey anti-goat IgG (Bethyl
Laboratories) antibodies.
Cell proliferation assay
At 24 h (day 1) and 72 h (day 3) after cell seeding,
cell proliferation was analyzed by using a Cell Counting Kit-8
(Dojindo Laboratories, Kumamoto, Japan) as per the manufacturer's
instructions, and the proliferative rate was calculated as the day
3/day 1 rate. For IMFs, CM was added to the culture medium at 24 h
after seeding.
Boyden chamber assay
Boyden chamber migration assays were performed using
transwell chambers (Corning, Corning, NY, USA) as previously
described (27). For invasion
assays, 8-µm-pore-size polycarbonate membranes were coated with 10%
Matrigel, and then 800 µl of serum-free DMEM was added in the lower
chamber and 1×104 aMoC1 cells or 4×104 IMFs
in 200 µl of serum-free DMEM were added in the upper chamber. After
2 h, 5% FBS (for aMoC1 cells) or 50% CM (for IMFs) was added to the
lower chamber and the cells were cultured for 6 h (aMoC1 cells) or
12 h (IMFs). Membranes were fixed with methanol for 15 min and
stained with Giemsa (Muto Pure Chemicals, Tokyo, Japan). After
washing the membranes with Milli-Q water, non-migrated cells were
wiped off with a cotton-swab, and then the migrated cells were
counted in 3–5 randomly selected ×200 fields under a light
microscope and averaged.
Soft-agar colony-formation assay
Into each well of 6-well plates, 2.5 ml of 0.75%
agarose in DMEM containing 2.8% NaHCO3, 10% FBS, and 1%
AA was poured and allowed to solidify at 4°C for 8 min. After
solidification, 1,000 cells were suspended in 1 ml of 0.36% agarose
in the same medium and then plated on top of the base layer. The
cells were cultured for 2 weeks, and colonies were stained with
crystal violet.
Wound-healing migration assay
Cells were cultured in 35-mm dishes as confluent
monolayers. The monolayers were cultured in serum-free medium
overnight, wounded in a line across the well by using a 200-µl
pipette tip, and incubated in fresh serum-free medium containing CM
and/or cytokines (10 ng/ml) for 6 h (LmcMF cells) or 12 h (primary
IMFs). Cells were pretreated with inhibitors for 30 min. Wound
areas were measured using ImageJ software (National Institutes of
Health, Bethesda, MD, USA).
Quantitative real-time PCR
Total RNA was extracted from cells by using TRIzol
(Thermo Fisher Scientific) as per the manufacturer's protocol. RNA
concentration was adjusted to 0.5 µg/µl, and quantitative real-time
PCR was performed using a QuantiTect Reverse Transcription kit, a
QuantiTect SYBR I kit (Qiagen, Hilden, Germany), and CFX96 Touch™
Real-time PCR Detection System (Bio-Rad), following manufacturer
protocols. The ∆∆Cq method was used for quantification (28). The specific primers used for mouse
epidermal growth factor (EGF), HB-EGF, insulin-like growth factor-1
(IGF-1), IL-6, TGF-β1, tumor necrosis factor-α (TNF-α), and β-actin
were the following: EGF, forward CCCAGGCAACGTATCAAAGT, reverse
GGTCATACCCAGGAAAGCAA; HB-EGF, forward GACCCATGCCTCAGGAAATA, reverse
TGAGAAGTCC CACGATGACA; IGF-1, forward TTCAGTTCGTGTGTGG ACCGAG,
reverse TCCACAATGCCTGTCTGAGGTG; IL-6, forward CACGGCCTTCCCTACTTCAC,
reverse TGCAAGTGCATCATCGTTGT; TGF-β1, forward TTGC
TTCAGCTCCACAGAGA, reverse TGGTTGTAGAGGGC AAGGAC; TNF-α, forward
AGACCCTCACACTCAGATC ATCTTC, reverse TTGCTACGACGTGGGCTACA; and
β-actin, forward GATTACTGCTCTGGCTCCTAGC, reverse
GACTCATCGTACTCCTGCTTGC.
Reagents
The cytokines used were: HB-EGF (Sigma); EGF, IL-6
(PeproTech, Rocky Hill, NJ, USA); TGF-β1 (AMBiS, Okinawa, Japan);
TNF-α (Wako Pure Chemical Industries); and IGF-1 (ProSpec-Tany
TechnoGene Ltd., Rehovot, Israel). The inhibitors used were
dacomitinib (Wako Pure Chemical Industries), FR180204 (Sigma), and
SP600125 (Cayman Chemical Co.).
Statistical analysis
Results are expressed as means ± SEM. Student's
t-test was used for comparison between two groups. Groups of three
or more were compared using one-way analysis of variance, followed
by Tukey's test. In all analyses, P<0.05 was considered
statistically significant.
Results
KRasV12 overexpression converts aMoC1
cells into tumor cells
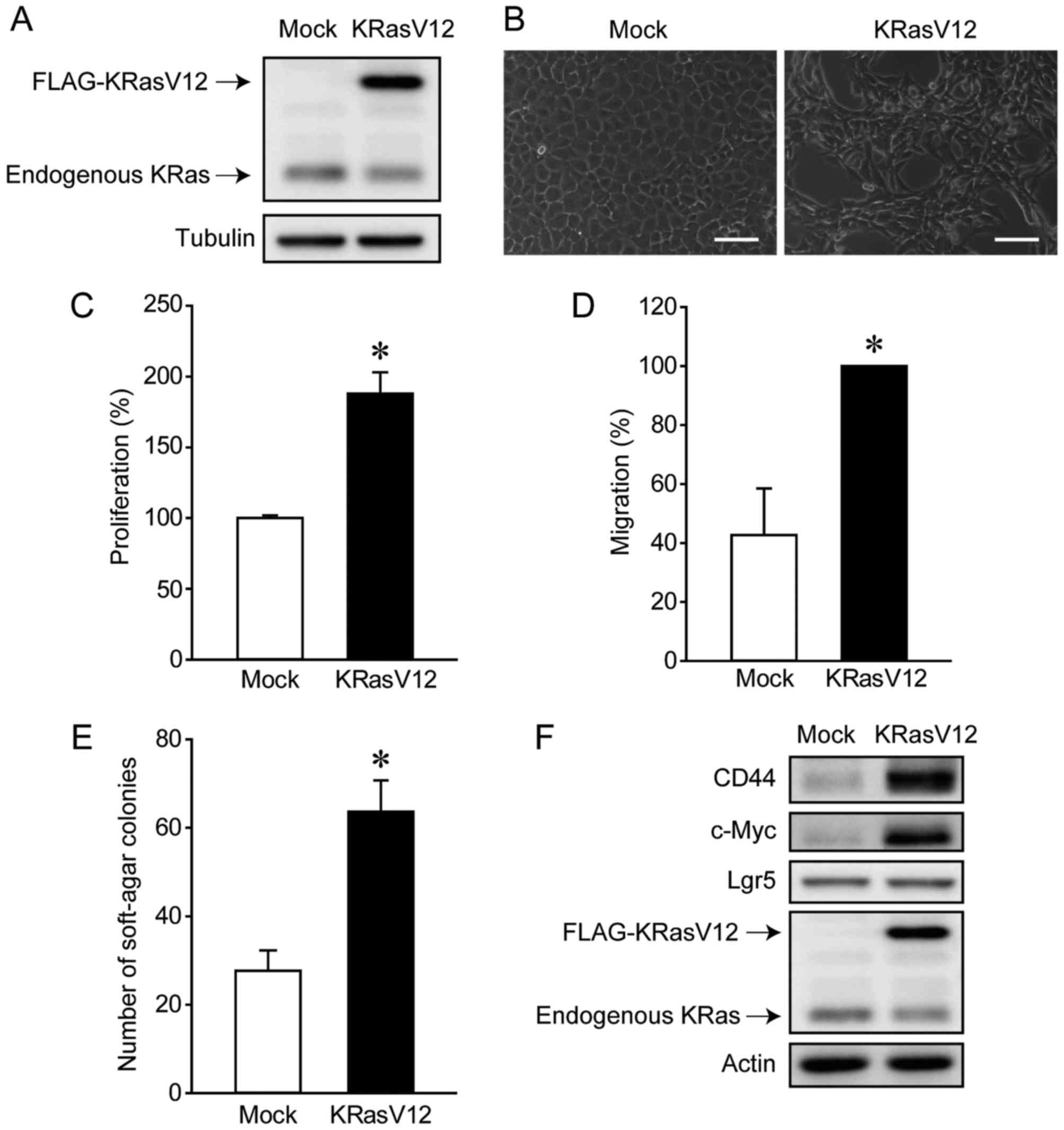
We first analyzed the oncogenic effects of KRasV12
overexpression in aMoC1 cells. KRasV12 was expressed at higher
levels in KRasV12-overexpressing aMoC1 cells (KRasV12-aMoC1 cells)
than in control cells transfected with the empty vector (Mock-aMoC1
cells) (Fig. 1A). KRasV12
overexpression altered the morphology of aMoC1 cells, leading to
the formation of atypically shaped cells (Fig. 1B). We further confirmed that KRasV12
overexpression enhanced the proliferation, migration, and
anchorage-independent growth of aMoC1 cells (Fig. 1C-E). Since KRas mutations have been
reported to enhance stemness (29,30),
we examined the expression of the cancer stem cell markers, CD44,
c-Myc, and Lgr5. KRasV12 overexpression increased the expression of
CD44 and c-Myc but not Lgr5 (Fig.
1F). These results suggest that KRasV12 overexpression converts
colorectal epithelial cells into tumor cells.
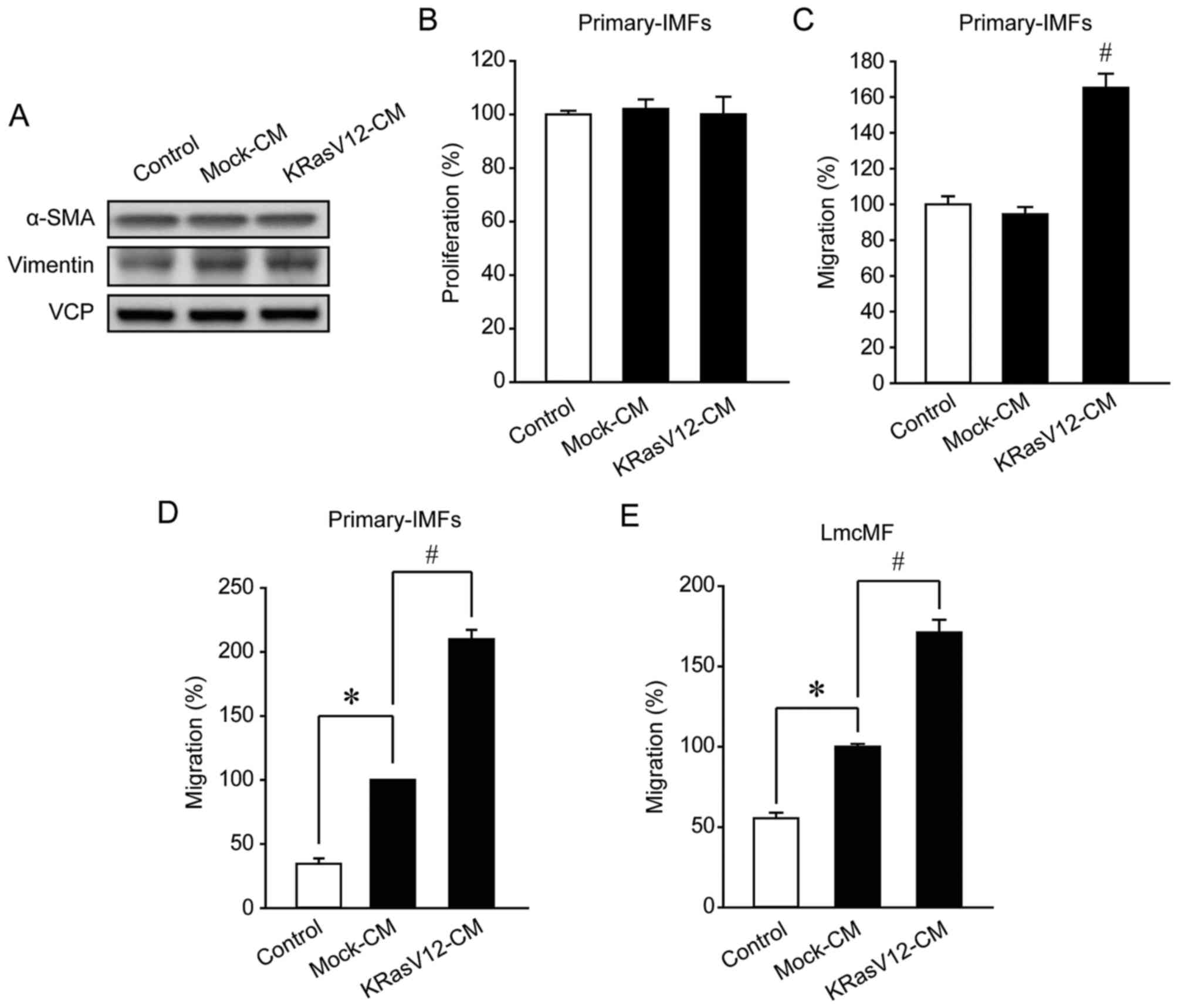
KRasV12-aMoC1 CM promotes IMF
migration
To investigate how KRasV12 overexpression in
epithelial cells affects the functions of IMFs, we treated IMFs
with the CM from Mock-aMoC1 cells (Mock-CM) or KRasV12-aMoC1 cells
(KRasV12-CM) and then examined IMF differentiation, proliferation,
and migration. IMF differentiation was evaluated based on the
expression level of myofibroblast markers (α-SMA and vimentin) as
previously described (23). In
primary IMFs, KRasV12-CM exerted little effect on differentiation
and proliferation (Fig. 2A and B).
By contrast, primary-IMF migration was promoted by KRasV12-CM
compared with Mock-CM (Fig. 2C).
Furthermore, in Boyden chamber assays, IMF migration was weakly
promoted by Mock-CM but strongly promoted by KRasV12-CM (Fig. 2D). Similarly, in the IMF cell line
LmcMF, Mock-CM and KRasV12-CM weakly and strongly promoted
migration, respectively (Fig. 2E).
These results indicate that KRas-mutated colorectal cancer cells
promote IMF migration but do not influence IMF differentiation and
proliferation.
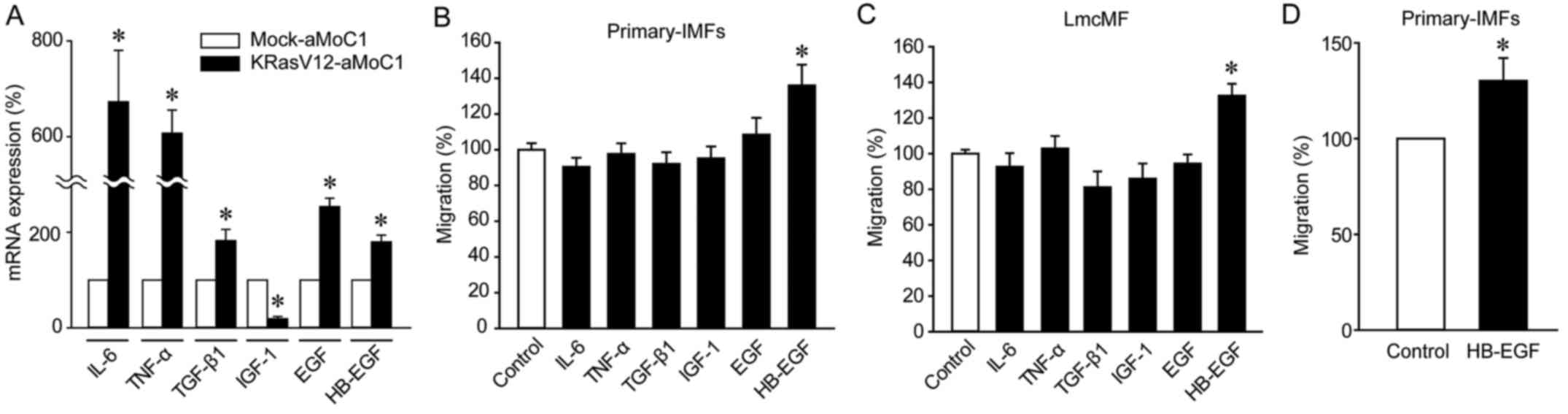
HB-EGF is upregulated in KRasV12-aMoC1
cells and mediates IMF migration
To clarify the mechanisms of KRasV12-CM-mediated
promotion of IMF migration, we compared cytokine expression levels
in Mock-aMoC1 and KRasV12-aMoC1 cells: Quantitative real-time PCR
analyses revealed that IL-6, TNF-α, TGF-β1, EGF, and HB-EGF were
expressed at higher levels in KRasV12-aMoC1 cells than in
Mock-aMoC1 cells, whereas IGF-1 was expressed at a lower level in
KRasV12-aMoC1 cells (Fig. 3A).
Among these cytokines, HB-EGF markedly promoted the migration of
primary IMFs and LmcMF cells (Fig. 3B
and C). We further confirmed that HB-EGF promoted IMF migration
by performing Boyden chamber assays (Fig. 3D). These results imply that
KRas-mutated cancer cells promote IMF migration through HB-EGF
upregulation.
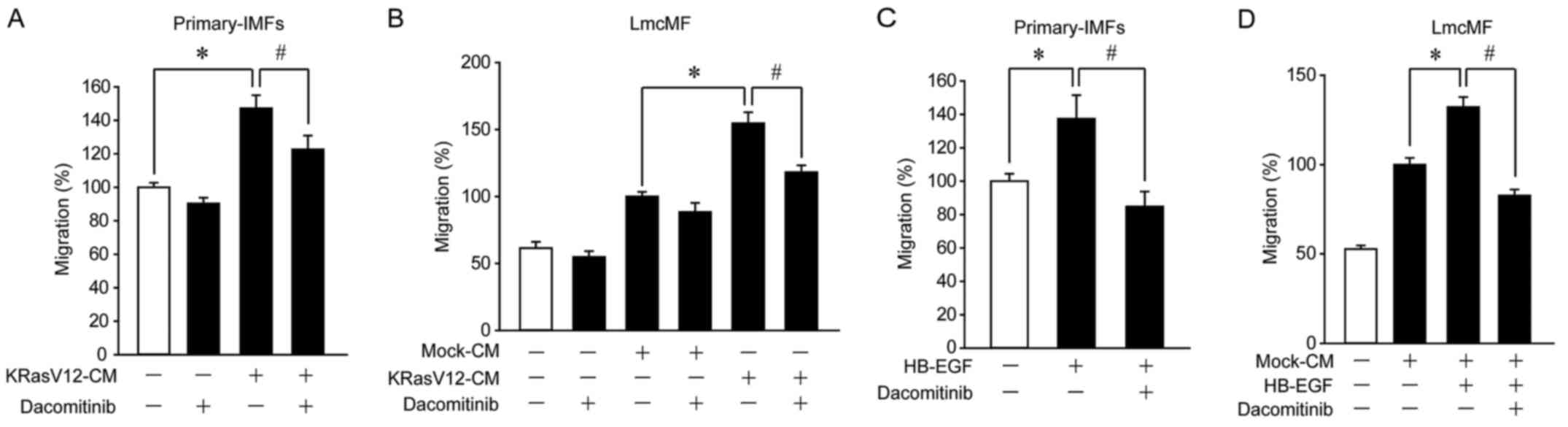
KRasV12-aMoC1 cells promote IMF
migration through activation of HB-EGF receptors
Since HB-EGF is recognized as a ligand of ErbB1 and
ErbB4 (31), we analyzed the
involvement of these receptors in KRasV12-CM-induced IMF migration.
In primary IMFs and LmcMF cells, KRasV12-CM-induced migration was
suppressed by dacomitinib, an inhibitor of ErbB1, ErbB2, and ErbB4
(Fig. 4A and B). Furthermore,
dacomitinib suppressed HB-EGF-induced migration in primary IMFs and
LmcMF cells (Fig. 4C and D). These
results suggest that KRas-mutated cancer cells promote IMF
migration through activation of HB-EGF receptors.
HB-EGF promotes IMF migration through
activation of ERK1/2 and JNK signals
Lastly, we investigated the mechanisms underlying
HB-EGF-mediated promotion of IMF migration by examining the effects
of KRasV12-CM and HB-EGF on the activation of various cellular
signaling pathways. Western blot analyses revealed that whereas
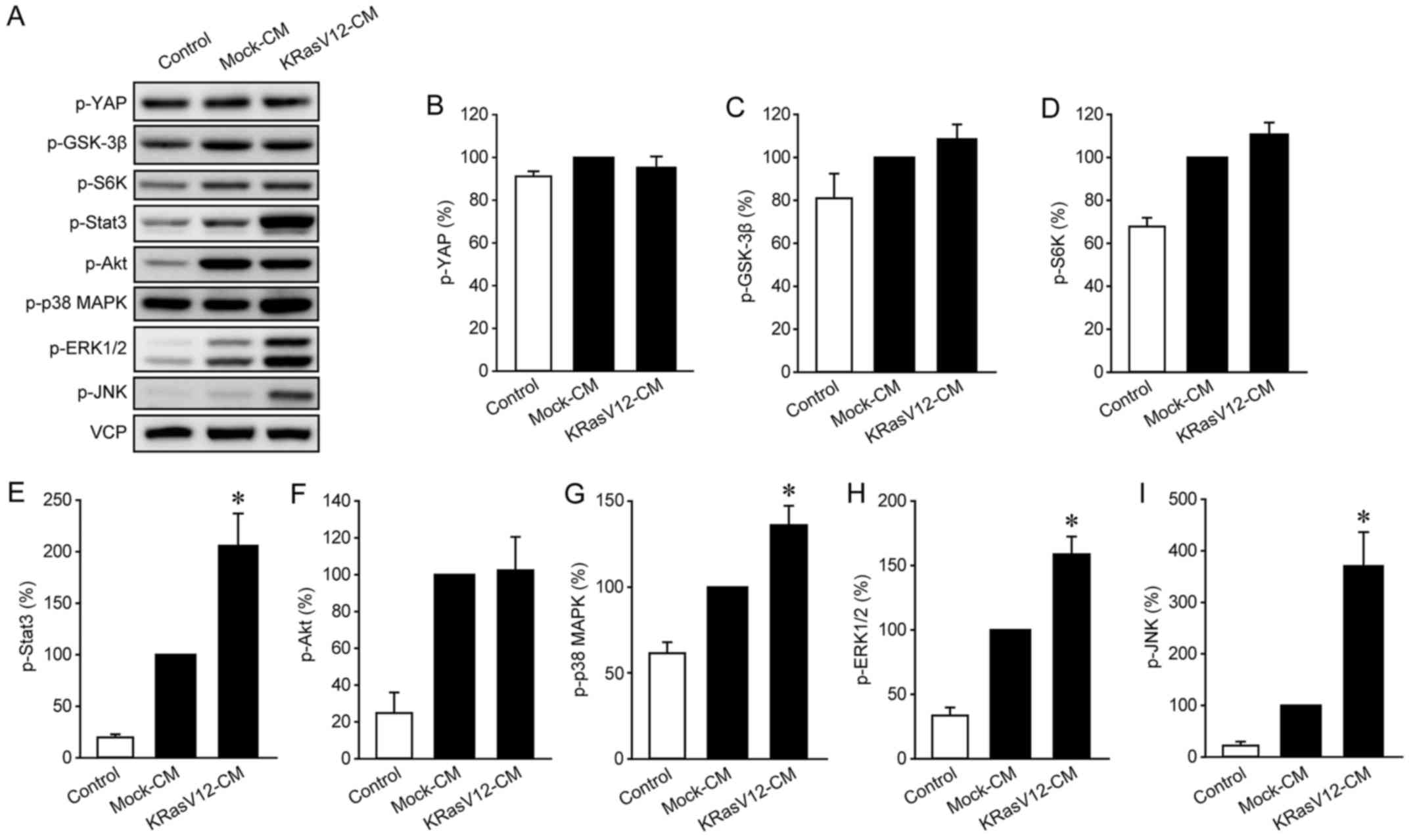
KRasV12-CM increased the phosphorylation of Stat3, p38 MAPK,
ERK1/2, and JNK in LmcMF cells (Fig.
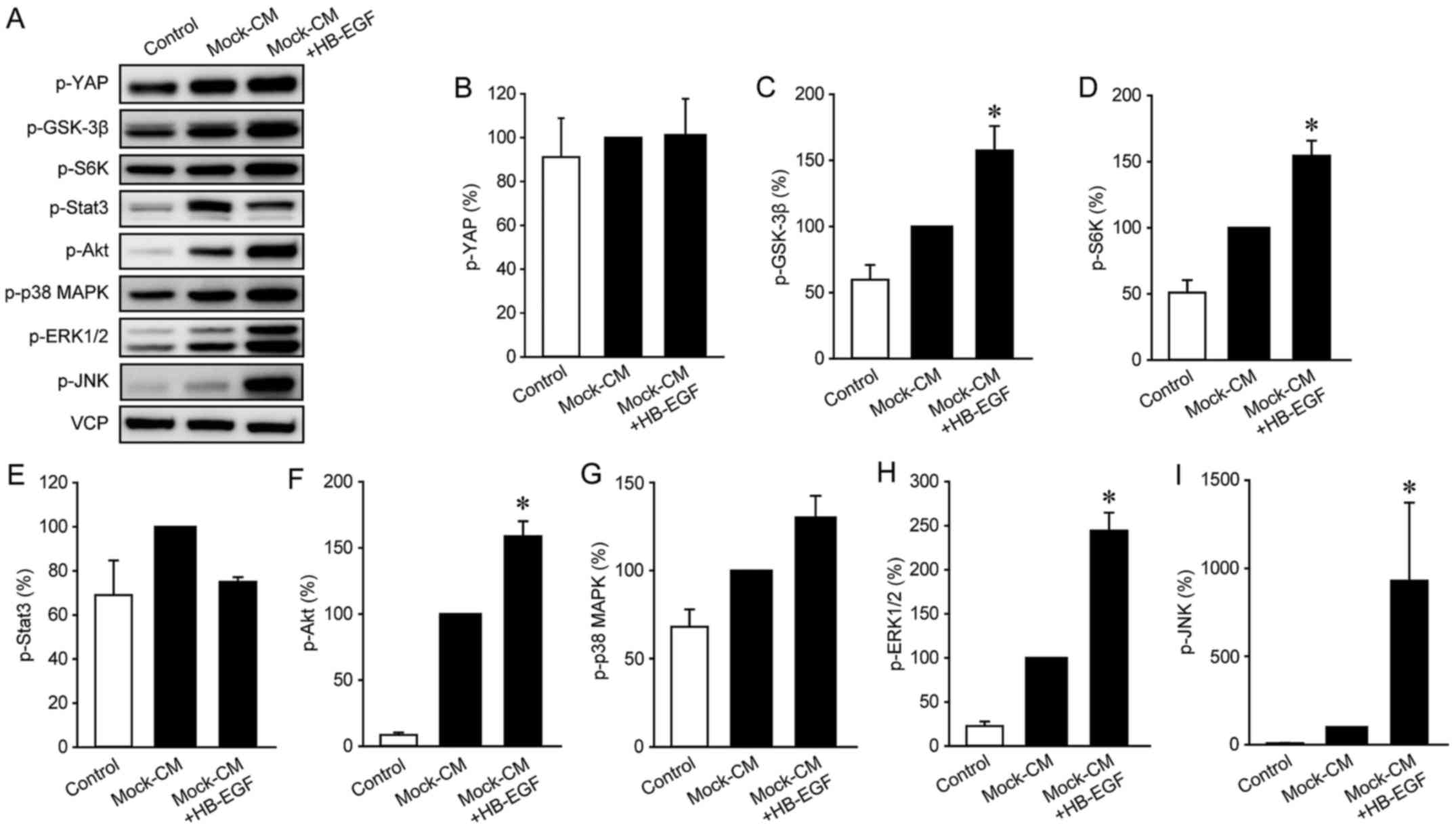
5), HB-EGF increased the phosphorylation of GSK-3β, S6K, Akt,
ERK1/2, and JNK in these cells (Fig.
6). Since ERK1/2 and JNK were activated by both KRasV12-CM and
HB-EGF, we hypothesized that ERK1/2 and JNK are involved in
KRasV12-CM-induced IMF migration. To test this, we measured
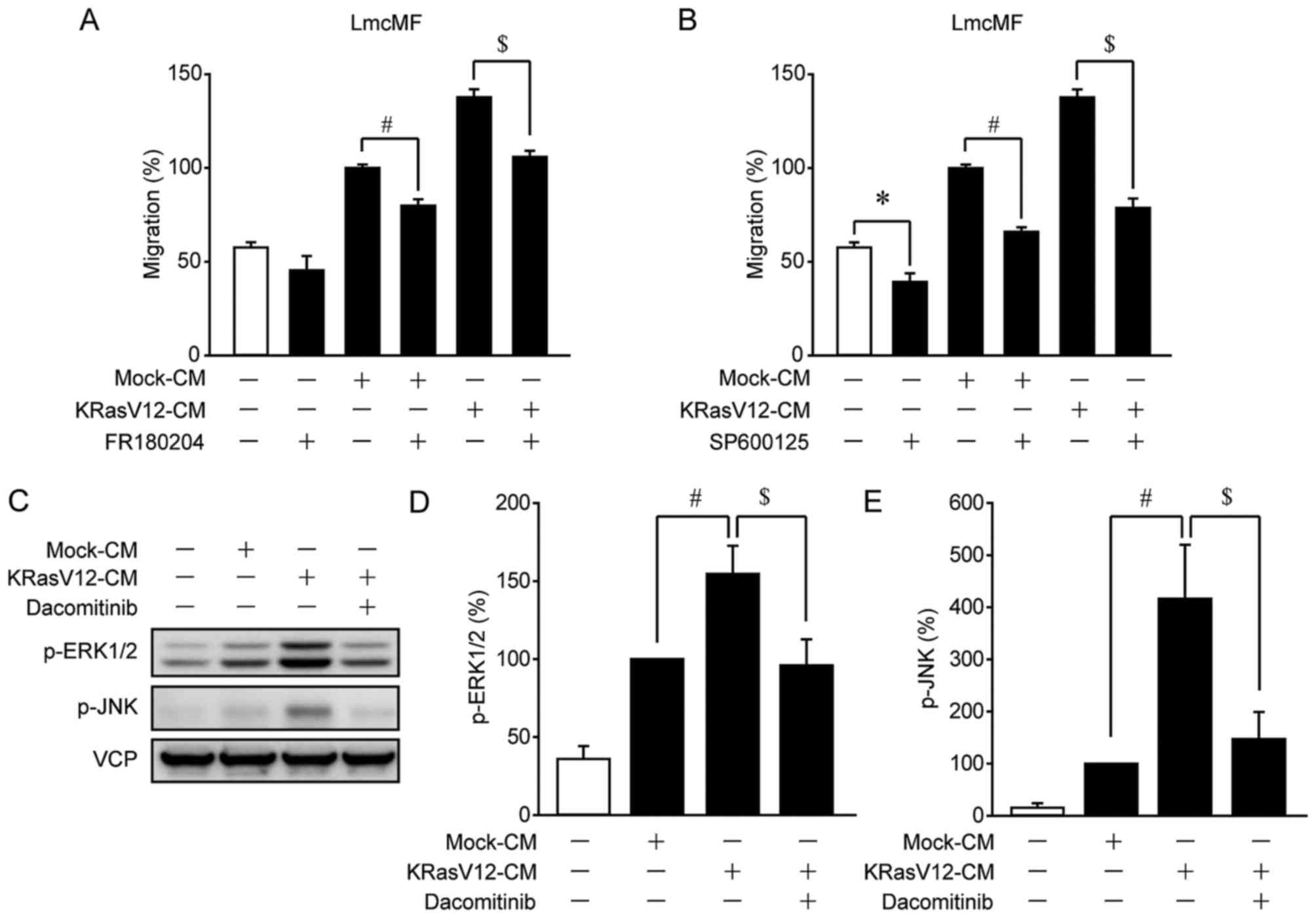
KRasV12-CM-induced IMF migration after treatment with the ERK1/2
inhibitor FR180204 or the JNK inhibitor SP600125: Both compounds
strongly inhibited KRasV12-CM-induced migration in LmcMF cells, but
partially suppressed basal and Mock-CM-induced migration (Fig. 7A and B). Moreover, our results
confirmed that the activation of ERK1/2 and JNK by KRasV12-CM was
blocked by dacomitinib in LmcMF cells (Fig. 7C-E). These results suggest that
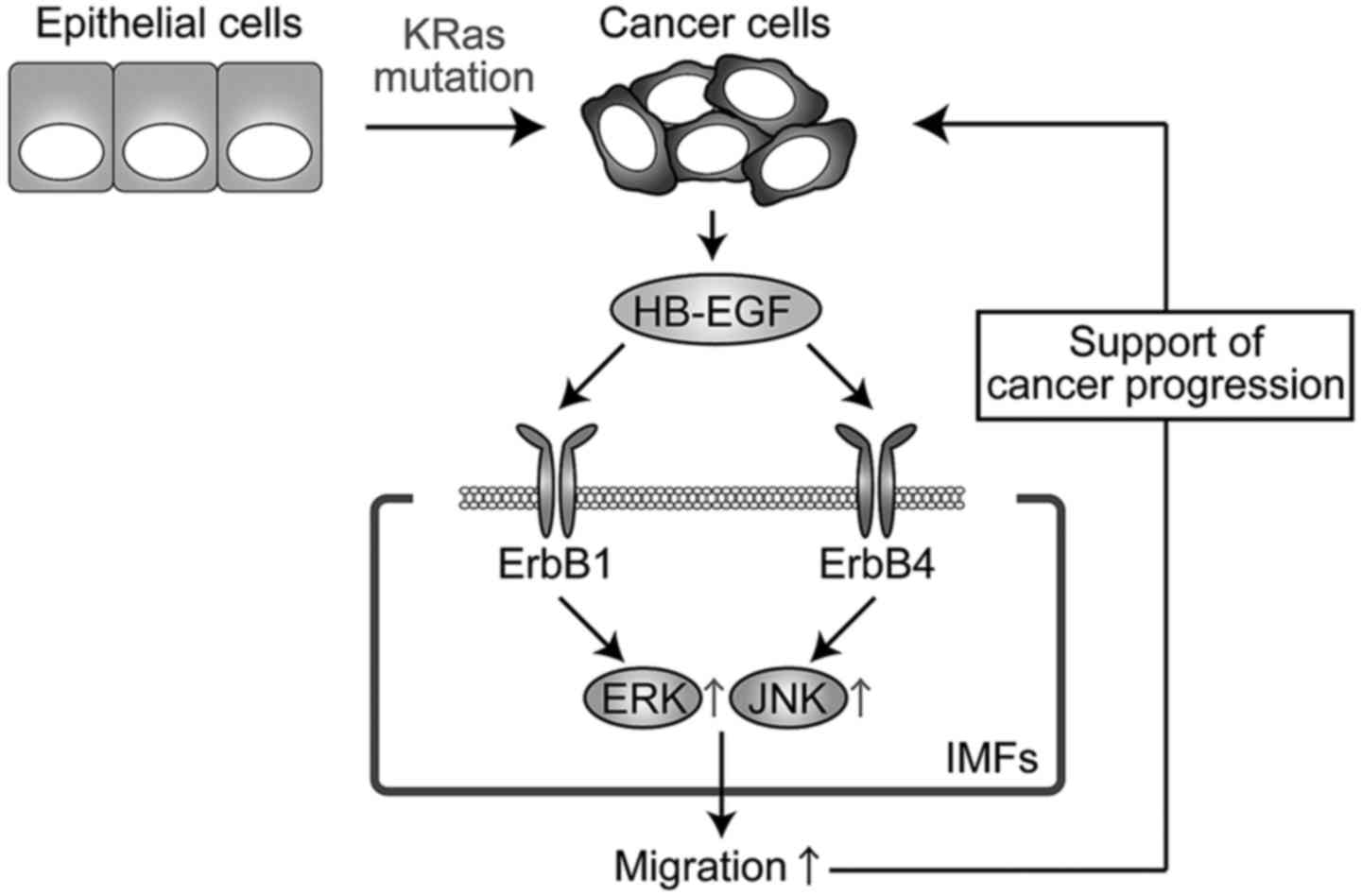
HB-EGF secreted from KRasV12-mutated cancer cells promotes IMF
migration through activation of ERK1/2 and JNK signals (Fig. 8).
Discussion
In colorectal cancer, KRas mutations occur
frequently and are known to play key roles in carcinogenesis.
However, the molecular mechanisms by which KRas mutations in
epithelial cells affect the functions of myofibroblasts have
remained unclear. We demonstrated here, for the first time, that
HB-EGF secreted from KRas-mutated colorectal cancer cells promotes
IMF migration through the activation of ERK and JNK signals.
We used KRasV12-aMoC1 cells as a model of colorectal
cancer cells for investigating the functions of KRas-mutated
colorectal cancer cells. Consistent with previous reports (3,6,29),
KRasV12 expression transformed aMoC1 cells, which was demonstrated
by our results showing that KRasV12 promoted aMoC1 cell
proliferation, migration, and anchorage-independent growth
(Fig. 1). Moreover, KRasV12
upregulated the expression of CD44 and c-Myc, which are involved in
the stemness of colorectal cancer cells (32,33).
Our findings suggest that KRasV12 overexpression converts
colorectal epithelial cells into tumor cells.
KRasV12 overexpression in aMoC1 cells increased the
expression of TGF-β1, EGF, and TNF-α (Fig. 3A), which are recognized as major
regulators of myofibroblast differentiation and proliferation
(34–36), but KRasV12-CM exerted little effect
on the differentiation and proliferation of IMFs (Fig. 2A and B). However, KRasV12
overexpression decreased the levels of IGF-1, which is also
involved in the differentiation and proliferation of myofibroblasts
(34–36). Based on these results, we propose
that IGF-1 downregulation might suppress the IMF proliferation and
differentiation induced by the other cytokines examined here. In
contrast to these results, we found that KRasV12-CM increased IMF
migration and chemotaxis as compared with Mock-CM (Fig. 2C-E). These findings suggest that
KRas-mutated cancer cells attract IMFs mainly through migration and
thus generate a cancer microenvironment.
HB-EGF is an EGF-family molecule that regulates cell
proliferation and differentiation (31). Moreover, in cancer progression,
HB-EGF promotes tumor cell growth/survival and angiogenesis
(31,37), whereas, HB-EGF promotes the
migration of epithelial cells and fibroblasts (38–40).
Herein, HB-EGF expression was upregulated in KRasV12-aMoC1 cells,
and HB-EGF enhanced IMF migration (Fig.
3). Moreover, KRasV12-CM- and HB-EGF-induced IMF migration was
inhibited by dacomitinib (Fig. 4).
These results suggest that KRasV12-aMoC1 cells promoted IMF
migration through HB-EGF/ErbB signaling. However, dacomitinib did
not completely inhibit KRasV12-CM-induced IMF migration. Thus,
several other factors present in KRasV12-CM besides HB-EGF might
also be involved in promoting IMF migration, such as PDGF, which is
recognized to promote myofibroblast migration (41). Nevertheless, our results support the
conclusion that HB-EGF is one of the major factors in KRasV12-CM
that promotes IMF migration.
The results of this study showed that HB-EGF and
KRasV12-CM activate ERK1/2 and JNK signals (Figs. 5 and 6). ERK and JNK signaling pathways are
widely recognized to be involved in cell migration (42,43).
Accordingly, we found that inhibition of ERK1/2 and JNK suppressed
KRasV12-CM-induced IMF migration (Fig.
7A and B). Given that KRasV12-CM-induced activation of ERK1/2
and JNK was inhibited by dacomitinib (Fig. 7C-E), our results indicate that the
activation of these signals by KRasV12-CM is mediated by HB-EGF
receptors.
Myofibroblasts are components of the cancer
microenvironment and they promote cancer progression (44). The findings of this study suggest
that KRas-mutated colorectal cancer cells enhance IMF migration and
attract IMFs toward the cancer cells, which might contribute to
cancer progression. Since these mechanisms of IMF migration might
be essential for the generation or maintenance of the cancer
microenvironment, HB-EGF, due to its ability to regulate the cancer
microenvironment, could potentially emerge as a novel therapeutic
target in the treatment of patients with KRas-mutated colorectal
cancer.
In conclusion, our results suggest that HB-EGF
secreted from KRas-mutated colorectal cancer cells promotes IMF
migration through ErbB receptors, ERK1/2, and JNK and thereby
generates a tumor microenvironment that favors cancer progression.
Further investigation into the interaction between KRas-mutated
cells and IMFs could contribute to research on the cancer
microenvironment and the development of new therapies targeting
IMFs.
Acknowledgements
This work was partly supported by a Grant-in-Aid for
Scientific Research from the Japanese Ministry of Education,
Culture, Sports, Science and Technology (grant nos.: 15K14855,
K.S.; 14J06412, H.K.). We thank Dr Mamoru Totsuka (Tokyo
University) for providing the aMoC1 cells.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chang YY, Lin PC, Lin HH, Lin JK, Chen WS,
Jiang JK, Yang SH, Liang WY and Chang SC: Mutation spectra of RAS
gene family in colorectal cancer. Am J Surg. 212:537–544.e3. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pollock CB, Shirasawa S, Sasazuki T, Kolch
W and Dhillon AS: Oncogenic K-RAS is required to maintain changes
in cytoskeletal organization, adhesion, and motility in colon
cancer cells. Cancer Res. 65:1244–1250. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bäumer S, Bäumer N, Appel N, Terheyden L,
Fremerey J, Schelhaas S, Wardelmann E, Buchholz F, Berdel WE and
Müller-Tidow C: Antibody-mediated delivery of anti-KRAS-siRNA in
vivo overcomes therapy resistance in colon cancer. Clin Cancer Res.
21:1383–1394. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pereira AA, Rego JF, Morris V, Overman MJ,
Eng C, Garrett CR, Boutin AT, Ferrarotto R, Lee M, Jiang ZQ, et al:
Association between KRAS mutation and lung metastasis in advanced
colorectal cancer. Br J Cancer. 112:424–428. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Feng Y, Bommer GT, Zhao J, Green M, Sands
E, Zhai Y, Brown K, Burberry A, Cho KR and Fearon ER: Mutant KRAS
promotes hyperplasia and alters differentiation in the colon
epithelium but does not expand the presumptive stem cell pool.
Gastroenterology. 141:11003–1013.e1. –10. 2011. View Article : Google Scholar
|
|
7
|
Yoshida M, Taguchi A, Kawana K, Adachi K,
Kawata A, Ogishima J, Nakamura H, Fujimoto A, Sato M, Inoue T, et
al: Modification of the tumor microenvironment in KRAS or
c-MYC-induced ovarian cancer-associated peritonitis. PLoS One.
11:e01603302016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sparmann A and Bar-Sagi D: Ras-induced
interleukin-8 expression plays a critical role in tumor growth and
angiogenesis. Cancer Cell. 6:447–458. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller
G and Bar-Sagi D: Oncogenic Kras-induced GM-CSF production promotes
the development of pancreatic neoplasia. Cancer Cell. 21:836–847.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Quante M, Varga J, Wang TC and Greten FR:
The gastrointestinal tumor microenvironment. Gastroenterology.
145:63–78. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Augsten M, Hägglöf C, Peña C and Ostman A:
A digest on the role of the tumor microenvironment in
gastrointestinal cancers. Cancer Microenviron. 3:167–176. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hanahan D and Coussens LM: Accessories to
the crime: Functions of cells recruited to the tumor
microenvironment. Cancer Cell. 21:309–322. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tahara E: Abnormal growth factor/cytokine
network in gastric cancer. Cancer Microenviron. 1:85–91. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Avgustinova A, Iravani M, Robertson D,
Fearns A, Gao Q, Klingbeil P, Hanby AM, Speirs V, Sahai E, Calvo F,
et al: Tumour cell-derived Wnt7a recruits and activates fibroblasts
to promote tumour aggressiveness. Nat Commun. 7:103052016.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chivukula RR, Shi G, Acharya A, Mills EW,
Zeitels LR, Anandam JL, Abdelnaby AA, Balch GC, Mansour JC, Yopp
AC, et al: An essential mesenchymal function for miR-143/145 in
intestinal epithelial regeneration. Cell. 157:1104–1116. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Powell DW, Adegboyega PA, Di Mari JF and
Mifflin RC: Epithelial cells and their neighbors I. Role of
intestinal myofibroblasts in development, repair, and cancer. Am J
Physiol Gastrointest Liver Physiol. 289:G2–G7. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vermeulen L, De Sousa E, Melo F, van der
Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M,
Merz C, Rodermond H, et al: Wnt activity defines colon cancer stem
cells and is regulated by the microenvironment. Nat Cell Biol.
12:468–476. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Murata T, Mizushima H, Chinen I, Moribe H,
Yagi S, Hoffman RM, Kimura T, Yoshino K, Ueda Y, Enomoto T, et al:
HB-EGF and PDGF mediate reciprocal interactions of carcinoma cells
with cancer-associated fibroblasts to support progression of
uterine cervical cancers. Cancer Res. 71:6633–6642. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhu L, Cheng X, Ding Y, Shi J, Jin H, Wang
H, Wu Y, Ye J, Lu Y, Wang TC, et al: Bone marrow-derived
myofibroblasts promote colon tumorigenesis through the
IL-6/JAK2/STAT3 pathway. Cancer Lett. 343:80–89. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Clapéron A, Mergey M, Aoudjehane L,
Ho-Bouldoires TH, Wendum D, Prignon A, Merabtene F, Firrincieli D,
Desbois-Mouthon C, Scatton O, et al: Hepatic myofibroblasts promote
the progression of human cholangiocarcinoma through activation of
epidermal growth factor receptor. Hepatology. 58:2001–2011. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lewis MP, Lygoe KA, Nystrom ML, Anderson
WP, Speight PM, Marshall JF and Thomas GJ: Tumour-derived TGF-beta1
modulates myofibroblast differentiation and promotes
HGF/SF-dependent invasion of squamous carcinoma cells. Br J Cancer.
90:822–832. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Martin M, Wei H and Lu T: Targeting
microenvironment in cancer therapeutics. Oncotarget. 7:52575–52583.
2016.PubMed/NCBI
|
|
23
|
Kawasaki H, Ohama T, Hori M and Sato K:
Establishment of mouse intestinal myofibroblast cell lines. World J
Gastroenterol. 19:2629–2637. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Iwamoto T, Yamada K, Shimizu M and Totsuka
M: Establishment of intestinal epithelial cell lines from adult
mouse small and large intestinal crypts. Biosci Biotechnol Biochem.
75:925–929. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yabe R, Miura A, Usui T, Mudrak I, Ogris
E, Ohama T and Sato K: Protein phosphatase methyl-esterase PME-1
protects protein phosphatase 2A from ubiquitin/proteasome
degradation. PLoS One. 10:e01452262015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fujiwara N, Kawasaki H, Yabe R,
Christensen DJ, Vitek MP, Mizuno T, Sato K and Ohama T: A potential
therapeutic application of SET/I2PP2A inhibitor OP449 for canine
T-cell lymphoma. J Vet Med Sci. 75:349–354. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Usui T, Morita T, Okada M and Yamawaki H:
Histone deacetylase 4 controls neointimal hyperplasia via
stimulating proliferation and migration of vascular smooth muscle
cells. Hypertension. 63:397–403. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Le Rolle AF, Chiu TK, Zeng Z, Shia J,
Weiser MR, Paty PB and Chiu VK: Oncogenic KRAS activates an
embryonic stem cell-like program in human colon cancer initiation.
Oncotarget. 7:2159–2174. 2016.PubMed/NCBI
|
|
30
|
Hammond DE, Mageean CJ, Rusilowicz EV,
Wickenden JA, Clague MJ and Prior IA: Differential reprogramming of
isogenic colorectal cancer cells by distinct activating KRAS
mutations. J Proteome Res. 14:1535–1546. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vinante F and Rigo A: Heparin-binding
epidermal growth factor-like growth factor/diphtheria toxin
receptor in normal and neoplastic hematopoiesis. Toxins (Basel).
5:1180–1201. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rao GH, Liu HM, Li BW, Hao JJ, Yang YL,
Wang MR, Wang XH, Wang J, Jin HJ, Du L, et al: Establishment of a
human colorectal cancer cell line P6C with stem cell properties and
resistance to chemotherapeutic drugs. Acta Pharmacol Sin.
34:793–804. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rowehl RA, Burke S, Bialkowska AB, Pettet
DW III, Rowehl L, Li E, Antoniou E, Zhang Y, Bergamaschi R, Shroyer
KR, et al: Establishment of highly tumorigenic human colorectal
cancer cell line (CR4) with properties of putative cancer stem
cells. PLoS One. 9:e990912014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hung CF, Rohani MG, Lee SS, Chen P and
Schnapp LM: Role of IGF-1 pathway in lung fibroblast activation.
Respir Res. 14:1022013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Theiss AL, Simmons JG, Jobin C and Lund
PK: Tumor necrosis factor (TNF) alpha increases collagen
accumulation and proliferation in intestinal myofibroblasts via TNF
receptor 2. J Biol Chem. 280:36099–36109. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jobson TM, Billington CK and Hall IP:
Regulation of proliferation of human colonic subepithelial
myofibroblasts by mediators important in intestinal inflammation. J
Clin Invest. 101:2650–2657. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yotsumoto F, Tokunaga E, Oki E, Maehara Y,
Yamada H, Nakajima K, Nam SO, Miyata K, Koyanagi M, Doi K, et al:
Molecular hierarchy of heparin-binding EGF-like growth
factor-regulated angiogenesis in triple-negative breast cancer. Mol
Cancer Res. 11:506–517. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yahata Y, Shirakata Y, Tokumaru S, Yang L,
Dai X, Tohyama M, Tsuda T, Sayama K, Iwai M, Horiuchi M, et al: A
novel function of angiotensin II in skin wound healing. Induction
of fibroblast and keratinocyte migration by angiotensin II via
heparin-binding epidermal growth factor (EGF)-like growth
factor-mediated EGF receptor transactivation. J Biol Chem.
281:13209–13216. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim JM, Bak EJ, Chang JY, Kim ST, Park WS,
Yoo YJ and Cha JH: Effects of HB-EGF and epiregulin on wound
healing of gingival cells in vitro. Oral Dis. 17:785–793. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Faria JA, de Andrade C, Goes AM, Rodrigues
MA and Gomes DA: Effects of different ligands on epidermal growth
factor receptor (EGFR) nuclear translocation. Biochem Biophys Res
Commun. 478:39–45. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kastanis GJ, Hernandez-Nazara Z, Nieto N,
Rincón-Sanchez AR, Popratiloff A, Dominguez-Rosales JA, Lechuga CG
and Rojkind M: The role of dystroglycan in PDGF-BB-dependent
migration of activated hepatic stellate cells/myofibroblasts. Am J
Physiol Gastrointest Liver Physiol. 301:G464–G474. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shi H, Lin B, Huang Y, Wu J, Zhang H, Lin
C, Wang Z, Zhu J, Zhao Y, Fu X, et al: Basic fibroblast growth
factor promotes melanocyte migration via activating
PI3K/Akt-Rac1-FAK-JNK and ERK signaling pathways. IUBMB Life.
68:735–747. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chun J and Kim YS: Platycodin D inhibits
migration, invasion, and growth of MDA-MB-231 human breast cancer
cells via suppression of EGFR-mediated Akt and MAPK pathways. Chem
Biol Interact. 205:212–221. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ong BA, Vega KJ and Houchen CW: Intestinal
stem cells and the colorectal cancer microenvironment. World J
Gastroenterol. 20:1898–1909. 2014. View Article : Google Scholar : PubMed/NCBI
|