Introduction
Epithelial ovarian cancer (EOC) is one of the
leading causes of cancer death among women worldwide (1). Although continuous improvement in
surgical techniques and initial response to chemotherapy has been
made in the past few decades, recurrence still occurs in ~70% of
patients who underwent the first-line chemotherapy within 18 months
(2,3). The 5-year survival rate remains poor
at 30.6% for patients with advanced EOC (4,5). Thus,
it is of great clinical importance to develop effective
chemotherapy strategies for EOC.
Differentiation therapy has emerged as a potential
chemotherapy strategy against tumors (6). The clinical effectiveness of
differentiation therapy has been demonstrated in acute
promyelocytic leukemia (APL) using all-trans-retinoic acid
as an inducer (7). Recently,
arsenic trioxide (As2O3), a well-established
human carcinogen, has also proven to be a differentiation agent in
the treatment of APL patients (8).
Of interest, such effects are not reliably reproduced in solid
tumors. Differentiation agents for ovarian cancer remain
elusive.
Thrichostatin A (TSA), a hydroxamate-type histone
deacetylase inhibitor, can promote histone acetylation by
remodeling of chromatin architecture, and induce cell
differentiation (9). TSA also
catalyzes the acetylation of non-histone proteins, which may
regulate signaling related to tumorigenesis (10,11).
Accumulated evidence indicates that TSA can activate AKT signaling
pathway (12). Overexpression of
epidermal growth factor receptor (EGFR), the upstream effector of
AKT, has been associated with increased chemoresistance and poorer
clinical outcome (13,14). However, it is unclear whether TSA
transactivates the EGFR/AKT pathway in EOC, and whether the
blockage of this pathway can potentiate the effect of TSA on EOC
differentiation.
In this study, we demonstrate that TSA can induce
cellular differentiation in HO8910 ovarian cancer cells. We show
that TSA transiently activates EGFR/AKT signaling pathway, causing
expression of differentiation-related genes, and blocking this
pathway sensitizes HO8910 cells to TSA. This implies that a
combination of EGFR/AKT pathway inhibitors with TSA may represent a
better differentiation therapy strategy for ovarian cancer.
Materials and methods
Cell line and culture
Human ovarian cancer cell line, HO8910, was kindly
provided by Dr Qixiang Shao of Jiangsu University (Zhenjiang,
China). The HO8910 cells were derived from ascites of ovarian
adenocarcinoma patients. The cells were maintained for no longer
than 3 months as described previously (15).
Antibodies and reagents
EGFR, phospho-EGFR (Tyr992), AKT, phospho-AKT
(Ser473), FOXA2, and acetyl-histone H3K9 antibodies were purchased
from Cell Signaling Technology (Danvers, MA, USA). α-tubulin and
secondary antibodies were procured from Bioworld Technology
(Shanghai, China). OCT4, SOX2, p21Cip1, cyclin D1, and
histone H3 antibodies were obtained from Abcam (Cambridge, MA,
USA). The EGFR inhibitor AG1478, AKT inhibitor A6730, and HDAC
inhibitor TSA were procured from Sigma (St. Louis, MO, USA). TSA
and A6730 were dissolved in ethanol as stocks (6.6 mM) and dimethyl
sulfoxide as stocks (20 mM), respectively.
Drug treatment
In TSA experiments, HO8910 cells were incubated with
100, 200, and 400 nM TSA and harvested at 48 h. When the expression
of differentiation-related genes was examined, cells were harvested
at 24 h. For cotreatment of TSA and EGFR pathway inhibitors, cells
were incubated with 200 nM TSA in presence of AG1478 (10 µM) or
A6730 (20 µM) for 24 h.
Morphological evaluation
The morphological changes in TSA-treated cells were
observed during a 72-h time course with a light microscope
(Olympus, Tokyo, Japan), and images were obtained via a CCD camera
(Olympus).
Histone protein and whole-cell
extraction and western blotting
Histones were prepared using the bioepitope nuclear
and cytoplasmic extraction kit (Bioworld Technology) following the
manufacturer's protocol. Total cellular proteins were isolated
directly from cultures in 100-mm Petri dishes after being washed
with ice-cold PBS and the addition of 200 µl Cell and Tissue
Protein Extraction reagent (Kangchen Biotech, Shanghai, China),
containing protease inhibitor and phosphatase inhibitor cocktails
(Roche). Antibodies against OCT4 (1:5,000), SOX2 (1:5,000), FOXA2
(1:1,000), cyclin D1 (1:10,000), p21Cip1 (1:3,000), EGFR
(1:1,000), p-EGFR (1:600), AKT (1:1,000), p-AKT (1:800), α-tubulin
(1:1,000), and histone H3 (1:2,000) were used for western blot
analysis, which was performed as described previously (16).
Cell proliferation assays
The cell proliferation assays were performed using
CCK-8 and EdU. Briefly, HO8910 cells (5×103) were seeded
in 96-well plates, and then treated with TSA for 24, 48, and 72 h.
For CCK-8 analysis, the cells were incubated with 1/10 volume of
CCK-8 for 2 h. The plates were then read at 450 nm with a Bio-Rad
model 680 microplate reader (Richmond, CA, USA). The cell
proliferation rate =
ODexperiment/ODcontrol×100%. For EdU assay,
HO8910 cells were treated with 200 nM TSA for 48 h, and then
incubated with EdU (50 mM) for 2 h, after which the nuclei were
stained with DAPI. The images were photographed by an Olympus
inverted microscope system.
Cell cycle analysis
The effect of TSA on HO8910 cell cycle phase
distribution was determined by flow cytometry. HO8910 cells were
fixed in 70% ethanol for 30 min at 4°C. The cells were then
incubated with propidium iodide (50 µg/ml) for 30 min at 37°C,
after which the fluorescence was measured with a flow cytometer (BD
Biosciences, Heidelberg, Germany).
Statistical analysis
Data are presented as the mean ± SEM. Statistical
significance was calculated by Student's t-test or ANOVA, and
values of P<0.05 were considered statistically significant.
Results
TSA induces morphological changes of
HO8910 cells
To elucidate the effect of TSA on ovarian cancer
cell differentiation, we examined the morphology of HO8910 cells
treated with or without TSA. Microscopic observation of control
cells revealed round, stellate-like appearance and growth in
clusters. Unlike the morphology of control, a majority of
TSA-treated cells exhibited spindle shape and much longer, fine,
tapering processes (Fig. 1A).
Treatment with TSA also caused a decrease in cell density (Fig. 1A), which suggests that TSA may
impede proliferation of HO8910 cells. Consistent with this
prediction, we found that TSA treatment caused a notable reduction
in cell proliferation rate compared with the control in a dose- and
time-dependent manner (Fig. 1B and
C). The suppressive effect of TSA on cell proliferation was
further confirmed by 5-ethynyl-2′-deoxyuridine (EdU) assay
(Fig. 1D).
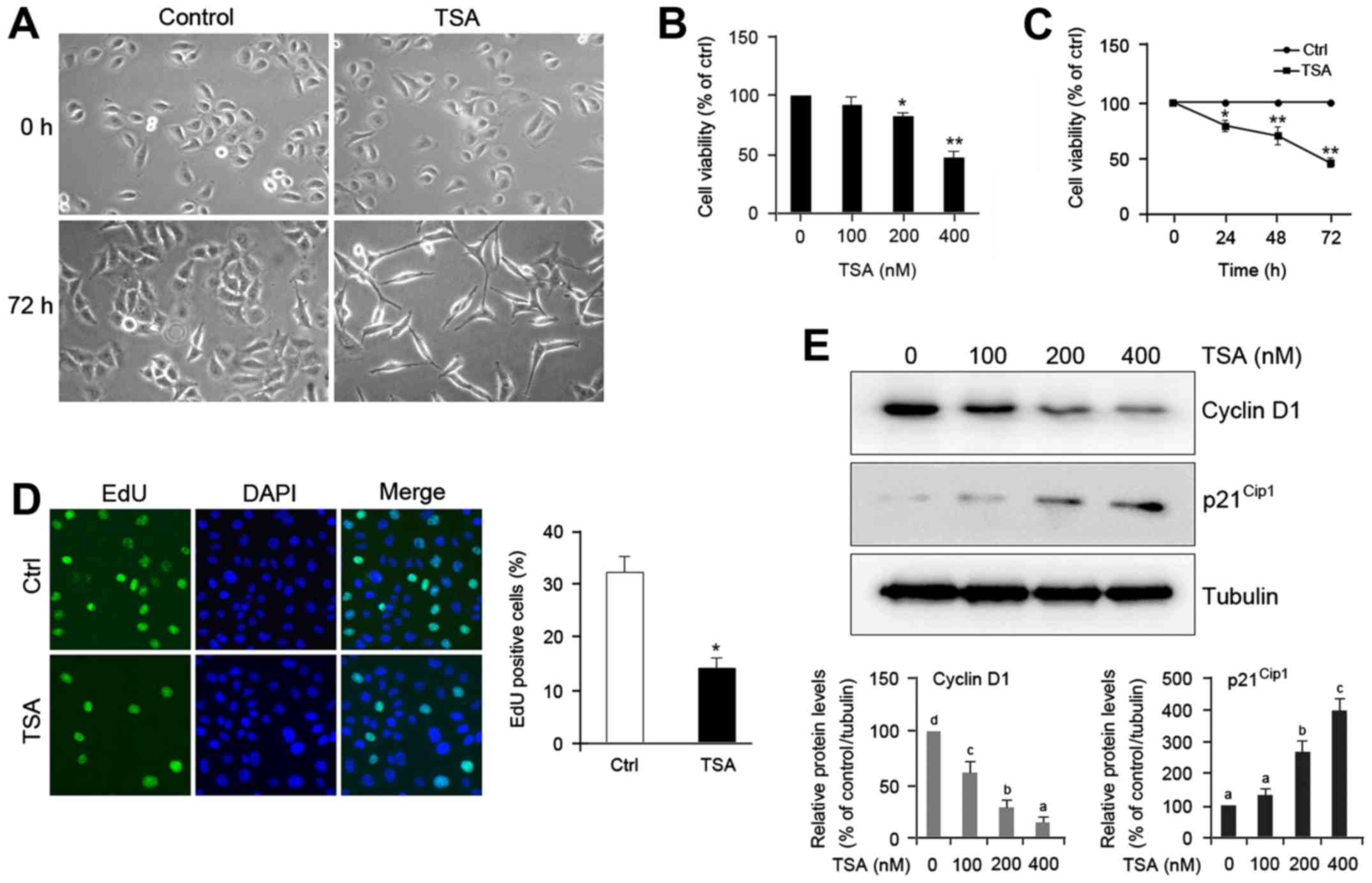 | Figure 1.TSA induces cell differentiation and
proliferation inhibition of HO8910 ovarian cancer cells. (A) Cells
were incubated without or with 200 nM thrichostatin A (TSA) for the
indicated times (magnification, ×200). (B) Cells were treated with
different doses of TSA for 48 h, and cell proliferation was
analyzed by CCK-8 assay. (C) Cells were treated with 200 nM TSA for
the indicated times, after which cell proliferation was detected
via CCK-8 assay. (D) Cells were treated without or with TSA 200 nM
TSA for 48 h, and then incubated with 50 mM
5-ethynyl-2′-deoxyuridine (EdU) for 2 h and the nuclei were stained
with DAPI. Representative micrographs (left panel, magnification,
×400) and quantification (right panel) of EdU-incorporating cells
in control and TSA-treated cells. Green, EdU; blue, DAPI. The error
bars represent mean ± SEM (n=3). Statistical differences compared
with the controls are given as *P<0.05 and **P<0.01
(Student's t-test). (E) Cells were treated with different doses of
TSA for 48 h. Total protein was extracted and blotted with
antibodies against cyclin D1, p21Cip1, and α-tubulin.
The error bars represent the mean ± SEM (n=3). Values within the
same row with different superscripted letters are significantly
different, P<0.05 (one-way ANOVA). |
To identify the mechanism of action of TSA in
decreasing cell proliferation, we undertook cell cycle analysis
with propidium iodide to label DNA. Table I reveals that treatment with TSA for
48 h led to an accumulation of HO8910 cells in the
G0/G1 phase (60.2 vs. 82.8%, P<0.05) and a
concomitant decrease in the S-phase cell fraction (26.7 vs. 3.8%,
P<0.01). The TSA-induced cell cycle arrest was further confirmed
by examining the expression of cell cycle regulatory protein cyclin
D1 in HO8910 cells. Western blot analysis indicated that TSA
treatment exhibited decrease of cyclin D1 levels and increase of
p21Cip1 protein (Fig.
1E). These data suggest that TSA induces cell differentiation
and proliferation inhibition of HO8910 cells.
 | Table I.TSA causes cell cycle arrest in
G0/G1 phase in HO8910 cells. |
Table I.
TSA causes cell cycle arrest in
G0/G1 phase in HO8910 cells.
|
| Control | TSA (nM) |
|---|
|
|
|
|---|
| Groups | 0 | 200 |
|---|
|
G0/G1 | 60.2±1.8 |
82.8±1.7a |
| S | 26.7±1.3 |
3.8±0.6b |
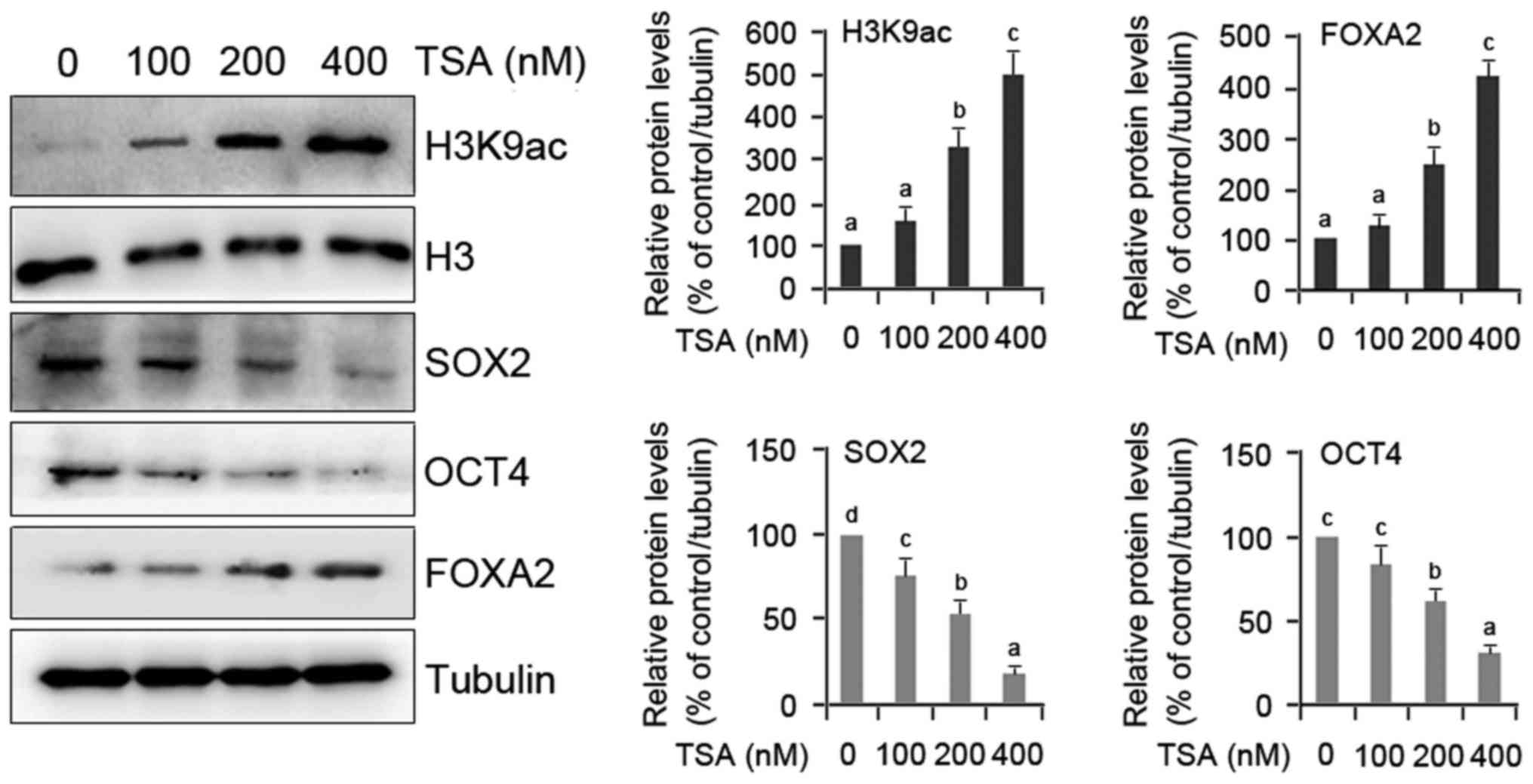
Molecular evidence of TSA-induced
differentiation of HO8910 cells
To verify the role of TSA in HO8910 cell
differentiation, two groups of genes including FOXA2 as
differentiation marker gene, and SOX2 and OCT4 as pluripotency
marker genes were evaluated quantitatively. FOXA2 gene was
evaluated because it plays important roles in regulating the
expression of genes involved in cell differentiation in a number of
different organs (17), and is
essential for differentiation and development of glands in mouse
uterus (18). A dose-dependent
upregulation of FOXA2 protein and H3K9 acetylation was observed in
TSA-treated cells compared with the controls (Fig. 2). In contrast, the expression of
SOX2 and OCT4 proteins was notably restrained by TSA (Fig. 2). This molecular evidence confirms
that TSA actually induces the differentiation of HO8910 cells.
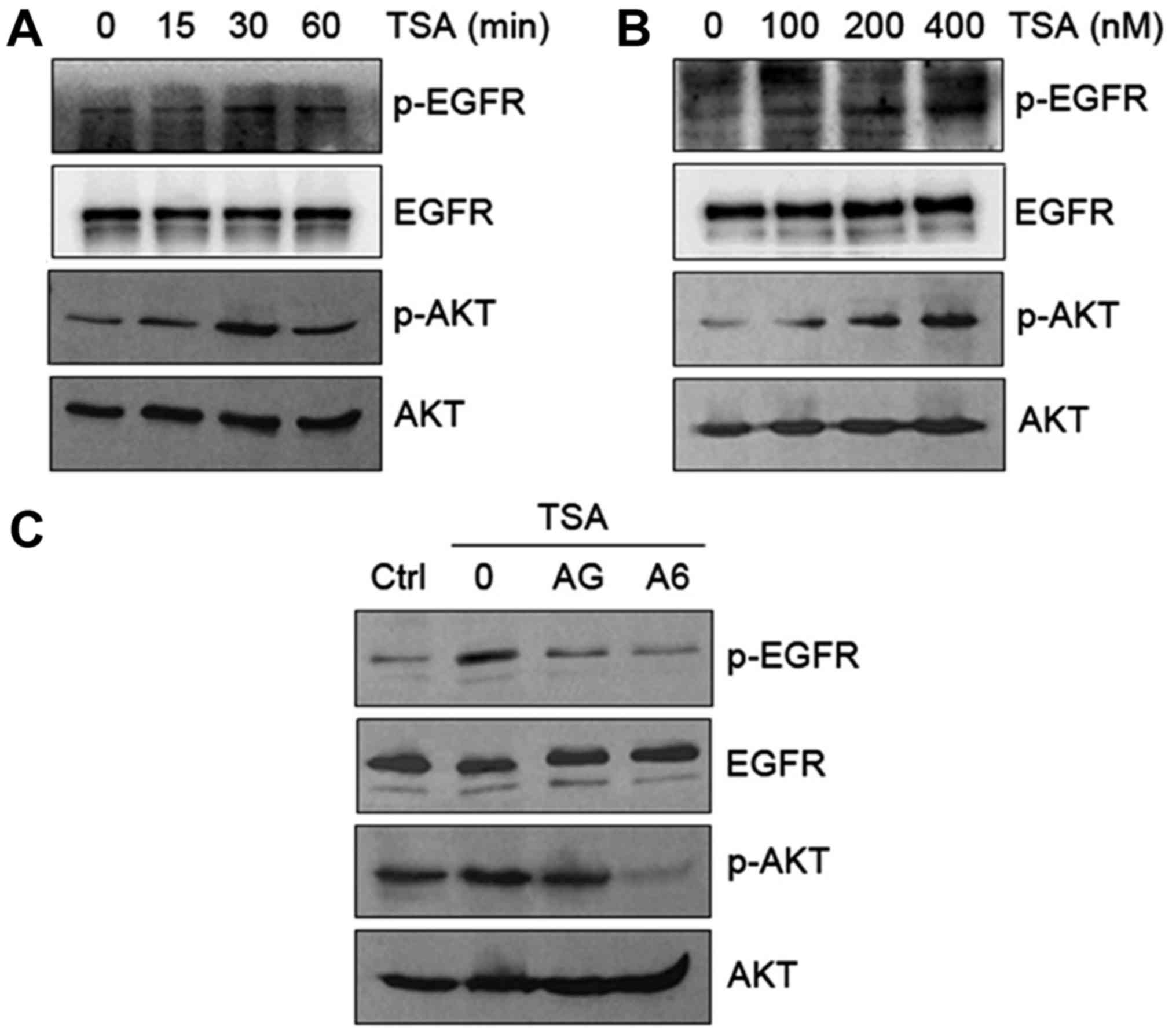
Transactivation of EGFR/AKT pathway by
TSA in HO8910 cells
Since EGFR expression strongly correlates with tumor
resistance to cytotoxic agents (19), EGFR could be a potential target for
anticancer therapy. To decipher whether TSA transactivates EGFR in
HO8910 cells, we first examined phosphorylation of EGFR and AKT in
response to TSA. Our results indicated that TSA caused a time- and
dose-dependent increase in phosphorylation of EGFR (Tyr992) and AKT
(Ser473) (Fig. 3A and B). The
TSA-induced phosphorylation of EGFR and AKT occurred at 15 min and
peaked at 30 min (Fig. 3A). The
observed changes in EGFR and AKT phosphorylation were reversed in
HO8910 cells in response to EGFR inhibitor AG1478 and AKT inhibitor
A6730 (Fig. 3C). These data suggest
that TSA activates EGFR phosphorylation and subsequent activation
of downstream AKT signaling.
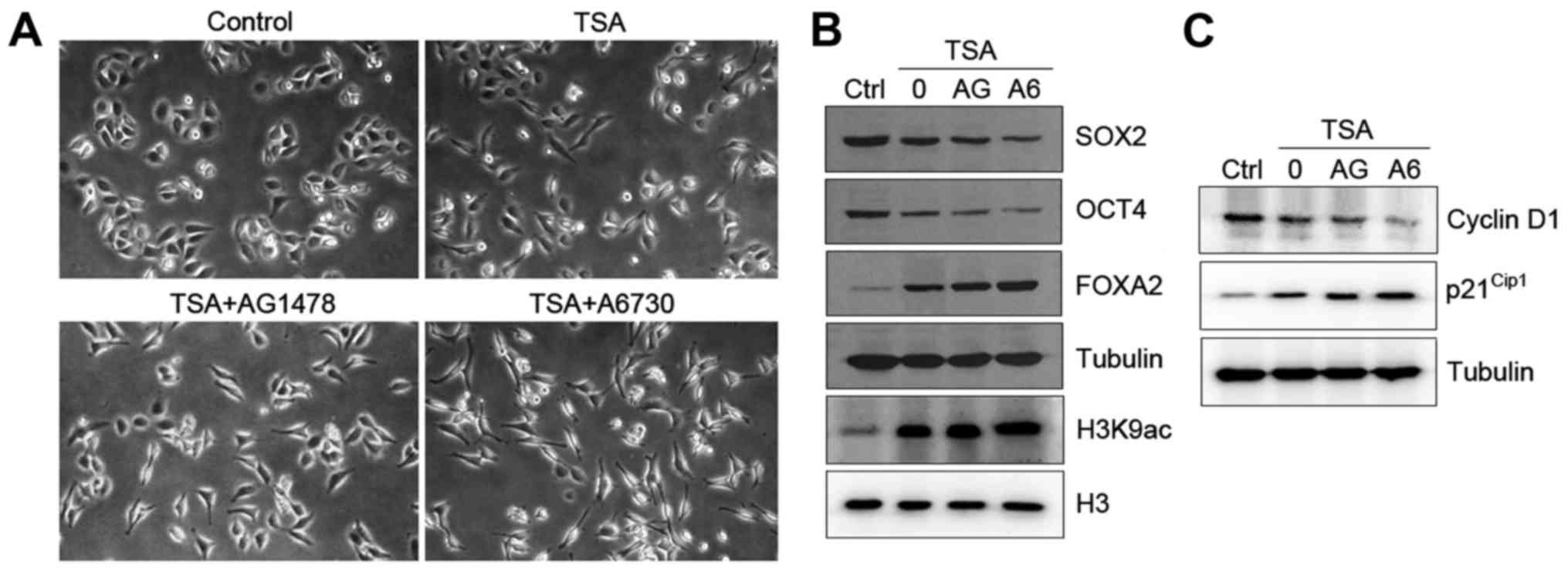
Inhibition of EGFR/AKT pathway
promotes TSA-induced differentiation of HO8910 cells
The above data indicate that TSA induces ovarian
cancer cell differentiation prompted us to test whether the
EGFR/AKT pathway was involved in this process. HO8910 cells treated
with either AG1478 or A6730 markedly promoted TSA-induced cell
differentiation with morphological changes (Fig. 4A). To determine the mechanism by
which inhibition of the EGFR/AKT pathway enhances cell
differentiation, we tested the expression of
differentiation-related genes in HO8910 cells. As expected,
combination of TSA and AG1478 or A6730 drastically reduced the
levels of SOX2 and OCT4 and increased the expression of FOXA2 in
the cells (Fig. 4B). Both
inhibitors also caused an increase in global levels of H3K9
acetylation (Fig. 4B).
Next, we tested the effect of TSA on cell cycle
regulation when EGFR/AKT signaling pathway was inhibited. Treatment
with AG1478 or A6730 decreased the levels of cyclin D1 and
increased the expression of p21Cip1 in HO8910 cells in
response to TSA (Fig. 4C).
Furthermore, combination treatment with TSA and AG1478 or A6730 led
to an accumulation of cells in G0/G1 phase in
comparison to TSA alone (Table
II). Collectively, our data suggest that TSA plays a role in
cell differentiation and that blockage of EGFR/AKT signaling
pathway enhances TSA-induced differentiation.
| Table II.Inhibition of EGFR/AKT signaling
pathway enhances TSA-induced G0/G1-phase cell
cycle arrest in HO8910 cells. |
Table II.
Inhibition of EGFR/AKT signaling
pathway enhances TSA-induced G0/G1-phase cell
cycle arrest in HO8910 cells.
| Groups | Control | TSA | TSA+AG1478 | TSA+A6730 |
|---|
|
G0/G1 |
58.3±1.2c |
79.7±2.4b |
85.6±1.1a |
89.5±2.5a |
| S |
27.1±1.3a |
3.9±0.7b |
3.8±1.6b |
2.6±1.8b |
Discussion
Epigenetic regulation of gene expression represents
a promising strategy for differentiation-based therapy (20). Histone acetylation is determined by
antagonistic actions of histone acetyltransferases (HATs) and HDACs
(21,22). A variety of HDAC inhibitors have
been demonstrated to induce differentiation in some solid tumors
(23). In this study, we
characterized the effect of TSA, one of the potent HDAC inhibitors,
on cell differentiation of ovarian tumors. We show that TSA
increases the levels of H3K9 acetylation, and induces cell
differentiation in HO8910 ovarian cancer cells with marked
morphological transformation. Molecular evidence for
differentiation is demonstrated by decreased expression of SOX2,
OCT4, and cyclin D1, and increased expression of FOXA2 and
p21Cip1. Significantly, blockage of EGFR/AKT signaling
pathway enhances the TSA-induced differentiation of HO8910
cells.
Cancer stem cells (CSCs) that retain both the
self-renewal and differentiation, contributes to tumorigenesis and
chemotherapy resistance in malignancies (24,25).
The poor clinical outcome in patients with ovarian cancer may be
due to the existence of CSCs (24).
Overexpression of OCT4 and SOX2 is detected more frequently in
poorly differentiated cancers than in well-differentiated ones, and
is not detected in paraneoplastic tissues and benign tumors
(26,27). OCT4 is also overexpressed in CSCs
and is closely related to chemotherapy resistance (28), suggesting that OCT4 is a
pluripotency marker for CSCs. In addition, FOXA2 is a sensitive
marker of CSC and cancer cell differentiation (29,30),
and it was reported that apicidin, another HDAC inhibitor,
upregulates FOXA2 expression, leading to enhanced differentiation
of dopaminergic neurons (29). Our
current data demonstrated that TSA may promote cell differentiation
through upregulation of FOXA2 expression and downregulation of OCT4
and SOX2 expression. Strikingly, inhibition of EGFR/AKT signaling
pathway enhances the induction of cell differentiation and FOXA2
expression in response to TSA. These data suggest that the EGFR
signaling may overcome the TSA-induced cell differentiation through
downregulation of FOXA2 expression.
The Cdk inhibitor p21Cip1 plays a
critical role in mediating proliferation inhibition and acts as a
cell cycle arrest point (31).
There is accumulating evidence linking p21Cip1 to
carcinogenesis in many tumors, including ovarian cancer (32–34).
The increase in p21Cip1 could be associated with
increased cell cycle suppression by TSA in our study. The
concomitant reduction in cyclin D1 expression may cooperate with
induction of p21Cip1 to arrest the cell cycle, and thus
contribute to proliferation inhibition induced by TSA. The
upregulation of p21Cip1 might lead to preferential cell
cycle arrest upon TSA treatment.
HDACi in combination with chemotherapy has been
confirmed to give better clinical outcome than chemotherapy alone
(35,36). One potential strategy to increase
treatment efficacy is the combination of HDACi with other novel
agents. A recent study found that inhibition of EGFR/PI3K signaling
pathway enhanced TSA-induced cell death and inhibited cell
migration (37). The EGFR/PI3K
pathway also mediates Lewis(y)-induced cell proliferation (38). In this study, the reduction of cell
proliferation was additionally enhanced when TSA was combined with
the EGFR/AKT pathway inhibitors AG1478 or A6730, in accordance with
a previous report (37). This
finding suggests that EGFR signaling pathway could be a target for
improving therapeutic efficacy of TSA.
Here, we provide a link between the inhibition of
EGFR signaling and the differentiation effect of TSA on HO8910
cells. Our data indicate that TSA transactivates the EGFR signaling
pathway, which overcomes the TSA-induced differentiation of HO8910
cells via regulation of differentiation-related genes. Blockage of
the EGFR signaling pathway by AG1478 or A6730 enhances the
TSA-mediated cell differentiation. Although TSA is an epigenetic
regulator, the present study did not establish a mechanistic link
between TSA and epigenetic regulation of cell differentiation in
HO8910 cells. Further studies are needed to determine how TSA
activates the EGFR pathway in an epigenetic manner and how such
activation affects ovarican cancer cell differentiation.
Acknowledgements
This study was supported by National Natural Science
Foundation of China (81170573).
References
|
1
|
Romero I and Bast RC Jr: Minireview: human
ovarian cancer: biology, current management, and paths to
personalizing therapy. Endocrinology. 153:1593–1602. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lopez J, Banerjee S and Kaye SB: New
developments in the treatment of ovarian cancer - future
perspectives. Ann Oncol. 24:(Suppl 10). X69–X76. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim A, Ueda Y, Naka T and Enomoto T:
Therapeutic strategies in epithelial ovarian cancer. J Exp Clin
Cancer Res. 31:142012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ferlay J, Parkin DM and Steliarova-Foucher
E: Estimates of cancer incidence and mortality in Europe in 2008.
Eur J Cancer. 46:765–781. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Leitao MM Jr and Chi DS: Surgical
management of recurrent ovarian cancer. Semin Oncol. 36:106–111.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Leszczyniecka M, Roberts T, Dent P, Grant
S and Fisher PB: Differentiation therapy of human cancer: Basic
science and clinical applications. Pharmacol Ther. 90:105–156.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang ME, Ye YC, Chen SR, Chai JR, Lu JX,
Zhoa L, Gu LJ and Wang ZY: Use of all-trans retinoic acid in the
treatment of acute promyelocytic leukemia. Blood. 72:567–572.
1988.PubMed/NCBI
|
|
8
|
Kinjo K, Kizaki M, Muto A, Fukuchi Y,
Umezawa A, Yamato K, Nishihara T, Hata J, Ito M, Ueyama Y, et al:
Arsenic trioxide (As2O3)-induced apoptosis
and differentiation in retinoic acid-resistant acute promyelocytic
leukemia model in hGM-CSF-producing transgenic SCID mice. Leukemia.
14:431–438. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yamashita Y, Shimada M, Harimoto N,
Rikimaru T, Shirabe K, Tanaka S and Sugimachi K: Histone
deacetylase inhibitor trichostatin A induces cell-cycle
arrest/apoptosis and hepatocyte differentiation in human hepatoma
cells. Int J Cancer. 103:572–576. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rosato RR and Grant S: Histone deacetylase
inhibitors in clinical development. Expert Opin Investig Drugs.
13:21–38. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Buchwald M, Krämer OH and Heinzel T: HDACi
- targets beyond chromatin. Cancer Lett. 280:160–167. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Eickhoff B, Germeroth L, Stahl C, Köhler
G, Rüller S, Schlaak M and van der Bosch J: Trichostatin A-mediated
regulation of gene expression and protein kinase activities:
Reprogramming tumor cells for ribotoxic stress-induced apoptosis.
Biol Chem. 381:1127–1132. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Freudlsperger C, Burnett JR, Friedman JA,
Kannabiran VR, Chen Z and Van Waes C: EGFR-PI3K-AKT-mTOR signaling
in head and neck squamous cell carcinomas: Attractive targets for
molecular-oriented therapy. Expert Opin Ther Targets. 15:63–74.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lassus H, Sihto H, Leminen A, Joensuu H,
Isola J, Nupponen NN and Butzow R: Gene amplification, mutation,
and protein expression of EGFR and mutations of ERBB2 in serous
ovarian carcinoma. J Mol Med (Berl). 84:671–681. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shao G, Wang J, Li Y, Liu X, Xie X, Wan X,
Yan M, Jin J, Lin Q, Zhu H, et al: Lysine-specific demethylase 1
mediates epidermal growth factor signaling to promote cell
migration in ovarian cancer cells. Sci Rep. 5:153442015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li Y, Wan X, Wei Y, Liu X, Lai W, Zhang L,
Jin J, Wu C, Shao Q, Shao G, et al: LSD1-mediated epigenetic
modification contributes to ovarian cancer cell migration and
invasion. Oncol Rep. 35:3586–3592. 2016.PubMed/NCBI
|
|
17
|
Kaestner KH: The FoxA factors in
organogenesis and differentiation. Curr Opin Genet Dev. 20:527–532.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jeong JW, Kwak I, Lee KY, Kim TH, Large
MJ, Stewart CL, Kaestner KH, Lydon JP and DeMayo FJ: Foxa2 is
essential for mouse endometrial gland development and fertility.
Biol Reprod. 83:396–403. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Akimoto T, Hunter NR, Buchmiller L, Mason
K, Ang KK and Milas L: Inverse relationship between epidermal
growth factor receptor expression and radiocurability of murine
carcinomas. Clin Cancer Res. 5:2884–2890. 1999.PubMed/NCBI
|
|
20
|
Wang LT, Liou JP, Li YH, Liu YM, Pan SL
and Teng CM: A novel class I HDAC inhibitor, MPT0G030, induces cell
apoptosis and differentiation in human colorectal cancer cells via
HDAC1/PKCδ and E-cadherin. Oncotarget. 5:5651–5662. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu H, Wu H, Wang Y, Wang Y, Wu X, Ju S
and Wang X: Inhibition of class II histone deacetylase blocks
proliferation and promotes neuronal differentiation of the
embryonic rat neural progenitor cells. Acta Neurobiol Exp (Wars).
72:365–376. 2012.PubMed/NCBI
|
|
22
|
Kretsovali A, Hadjimichael C and
Charmpilas N: Histone deacetylase inhibitors in cell pluripotency,
differentiation, and reprogramming. Stem Cells Int.
2012:1841542012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cruz FD and Matushansky I: Solid tumor
differentiation therapy - is it possible? Oncotarget. 3:559–567.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ciurea ME, Georgescu AM, Purcaru SO,
Artene SA, Emami GH, Boldeanu MV, Tache DE and Dricu A: Cancer stem
cells: Biological functions and therapeutically targeting. Int J
Mol Sci. 15:8169–8185. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kwon MJ and Shin YK: Regulation of ovarian
cancer stem cells or tumor-initiating cells. Int J Mol Sci.
14:6624–6648. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li X, Wang J, Xu Z, Ahmad A, Li E, Wang Y,
Qin S and Wang Q: Expression of Sox2 and Oct4 and their clinical
significance in human non-small-cell lung cancer. Int J Mol Sci.
13:7663–7675. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ge N, Lin HX, Xiao XS, Guo L, Xu HM, Wang
X, Jin T, Cai XY, Liang Y, Hu WH, et al: Prognostic significance of
Oct4 and Sox2 expression in hypopharyngeal squamous cell carcinoma.
J Transl Med. 8:942010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lin H, Sun LH, Han W, He TY, Xu XJ, Cheng
K, Geng C, Su LD, Wen H, Wang XY, et al: Knockdown of OCT4
suppresses the growth and invasion of pancreatic cancer cells
through inhibition of the AKT pathway. Mol Med Rep. 10:1335–1342.
2014.PubMed/NCBI
|
|
29
|
Bang SY, Kwon SH, Yi SH, Yi SA, Park EK,
Lee JC, Jang CG, You JS, Lee SH and Han JW: Epigenetic activation
of the Foxa2 gene is required for maintaining the potential of
neural precursor cells to differentiate into dopaminergic neurons
after expansion. Stem Cells Dev. 24:520–533. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang X, Lu F, Wang J, Yin F, Xu Z, Qi D,
Wu X, Cao Y, Liang W, Liu Y, et al: Pluripotent stem cell protein
Sox2 confers sensitivity to LSD1 inhibition in cancer cells. Cell
Rep. 5:445–457. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sherr CJ and Roberts JM: CDK inhibitors:
Positive and negative regulators of G1-phase progression. Genes
Dev. 13:1501–1512. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Weiss RH: p21Waf1/Cip1 as a
therapeutic target in breast and other cancers. Cancer Cell.
4:425–429. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jackson RJ, Adnane J, Coppola D, Cantor A,
Sebti SM and Pledger WJ: Loss of the cell cycle inhibitors
p21(Cip1) and p27(Kip1) enhances tumorigenesis in knockout mouse
models. Oncogene. 21:8486–8497. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bali A, O'Brien PM, Edwards LS, Sutherland
RL, Hacker NF and Henshall SM: Cyclin D1, p53, and
p21Waf1/Cip1 expression is predictive of poor clinical
outcome in serous epithelial ovarian cancer. Clin Cancer Res.
10:5168–5177. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ahrens TD, Timme S, Hoeppner J, Ostendorp
J, Hembach S, Follo M, Hopt UT, Werner M, Busch H, Boerries M, et
al: Selective inhibition of esophageal cancer cells by combination
of HDAC inhibitors and Azacytidine. Epigenetics. 10:431–445. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shi YK, Li ZH, Han XQ, Yi JH, Wang ZH, Hou
JL, Feng CR, Fang QH, Wang HH, Zhang PF, et al: The histone
deacetylase inhibitor suberoylanilide hydroxamic acid induces
growth inhibition and enhances taxol-induced cell death in breast
cancer. Cancer Chemother Pharmacol. 66:1131–1140. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhou C, Qiu L, Sun Y, Healey S, Wanebo H,
Kouttab N, Di W, Yan B and Wan Y: Inhibition of EGFR/PI3K/AKT cell
survival pathway promotes TSA's effect on cell death and migration
in human ovarian cancer cells. Int J Oncol. 29:269–278.
2006.PubMed/NCBI
|
|
38
|
Liu JJ, Lin B, Hao YY, Li FF, Liu DW, Qi
Y, Zhu LC, Zhang SL and Iwamori M: Lewis(y) antigen stimulates the
growth of ovarian cancer cells via regulation of the epidermal
growth factor receptor pathway. Oncol Rep. 23:833–841.
2010.PubMed/NCBI
|