Introduction
Superantigens (SAgs) derived from bacterial or viral
products are known as potent activators, which bind to both MHC
class II molecules and specific Vβ regions of T cell receptors as
an unprocessed protein, resulting in the activation of more than
10–25% of the T-cell population (1). The activation of lymphocytes by
superantigen resulted in cytokine production, proliferation and
cytotoxicity, and could elicit systemic antitumor immunity in
vitro and in vivo (2–7). This
property of SAgs has been used in cancer immunotherapy. Antitumor
strategies of superantigen included antibody-targeted super antigen
(8–13), superantigen-transmembrane sequence
fusion protein (14), and gene
therapy (15). Staphylococcus
enterotoxin A (SEA) is a kind of superantigen and was widely used
in research of antitumor therapy (8–11,13,15).
In our previous study (15), SEA
was used for cancer gene therapy and induced efficient antitumor
effects. The low affinity T-cell receptor (TCR) interaction of SEA
in the absence of MHC class II antigens is sufficient for induction
of cytotoxicity but requires additional CD28/B7 signaling to result
in the proliferation of resting T cells (16). Previous studies have also shown that
SEA, in combination with B7 costimulatory molecule, induced a
stronger lymphocyte proliferation response and antitumor immunity
than either SEA or B7 alone (17).
In this study, SEA and B7 (CD80) genes were used to investigate
their antitumor effects.
In the gene therapy of tumors, recombinant
adenovirus is widely used for the transfer of foreign genes into
tumor cells. However, the utilization of these vectors requires the
specific and efficient expression of the transferred gene in tumor
cells because infections of adenovirus to cells lack tissue
specificity. One targeting strategy most often applied is the
expression of the transgene controlled by a tumor-specific
promoter, such as α-fetoprotein (AFP) promoter. However, true
tumor-specific promoter is rare, and often these promoters are
useful only for the particular types of cancers. Previously, we
constructed recombinant adenovirus carrying SEAtm gene driven by
AFP enhancer/promoter, which could only be used for hepatocellular
carcinoma. In vitro and in vivo studies have
demonstrated that the human telomerase reverse transcriptase TERT
(hTERT) promoter is highly active in 80–90% human cancer cells but
not in normal differentiated human cells. Therefore, hTERT promoter
has been widely used for targeted gene therapy to many types of
cancers (18). Similarly,
telomerase activities were at high levels in approximately 90% of
mouse cancers or tumor-derived cell lines through TERT
transcriptional upregulation (19).
In our previous study, we found the proximal 333-bp
fragment was the core promoter of the mTERT gene in the cancer
cells. The proximal 333-bp fragment of mTERT promoter was used to
drive SEA and CD80 gene in our constructed recombinant adenovirus
in order to be applied in various types of cancers. Myc family
protein, groups of the helix-loop-heilx/leucine zipper family,
forms heterodimers with a partner protein, Max. This Myc-Max
protein complex binds to the CACGTG sequence and activates
transcription (20,21). Myc and Max protein expression
increased in many kinds of cancers. Myc-Max response elements
(MMRE) were used to increase the hTERT promoter activity (22,23).
In the present study, to extend the applicability of
the gene therapy vectors for different kinds of tumors, we
constructed a recombinant adenovirus carrying SEAtm and mouse CD80
gene driven by the proximal 333-bp fragment of mTERT promoter,
upstream of which MMRE was used to increase the mTERT promoter
activity. Then, antitumor effects of recombinant adenovirus were
observed in hepatoma, colon cancer and melanoma in vitro and
in vivo.
Materials and methods
Cell lines
Mouse hepatoma cell line Hepa1-6, melanoma cell line
B16, colon cancer cell line CT26 and fibroblast cell line NIH3T3
were stored in our laboratory. All the cell lines were maintained
in Roswell Park Memorial Institute (RPMI)-1640 (Gibco-BRL) or high
glucose Dulbecco's modified Eagle's medium (DMEM), which were
supplemented with 10% fetal bovine serum and
penicillin/streptomycin in an atmosphere of 5% CO2
chamber at 37°C.
Antibodies
Phycoerythrin (PE)-conjugated anti-mouse CD80
(eBioscience, San Diego, CA, USA) and CD4 (eBioscience),
APC-conjugated anti-mouse CD8 (eBioscience), rabbit anti-SEA
(Abcam, Cambridge, MA, USA), fluorescein isothiocyanate
(FITC)-conjugated donkey anti-rabbit IgG (Biolegend) and anti-mouse
CD3 (eBioscience) were used in this study.
Recombinant adenovirus
preparation
We used the AdEasy vector system (Qbiogene Inc.),
that is, human adenovirus serotype 5 and rendered replication
defective by the deletion of the E1 and E3 gene. Preparations of
the recombinant adenoviruses were performed as previously described
(25). Proliferation, purification
and titering of adenovirus Ad(empty), Ad-MMRE-mTERT-CD80 (carrying
mouse CD80 gene only), Ad-MMRE-mTERT-SEAtm (carrying SEAtm gene
only) or Ad-MMRE-mTERT-BIS (carrying mouse CD80 and SEAtm genes)
were performed as described in the manufacturer's protocol. None of
the stocks of virus used in the experiments contained detectable
replication-competent viruses as evaluated by PCR assay, which used
two pairs of primers to detect adenoviral E1A DNA.
Adenovirus-mediated SEA and CD80
expression in different cell lines in vitro
Hepa1-6, B16, CT26 or NIH3T3 cells were plated at a
density of 5×105 cells/well in 6-well culture plates 24
h before the adenoviruses infection. Immediately before the
infection with Ad(empty), Ad-MMRE-mTERT-CD80, Ad-MMRE-mTERT-SEAtm
or Ad-MMRE-mTERT-BIS, culture medium was aspirated and the
adenoviruses were distributed over the cell monolayer at
multiplicity of infection (MOI) of 100. The ratio of the number of
adenovirus per cell was expressed as MOI. After 48 h of culture,
the cells were digested with 0.02% ethylenediaminetetraacetic acid,
washed in PBS containing 2% calf serum, incubated with a rabbit
anti-SEA polyclonal antibody and detected with FITC-labeled donkey
anti-rabbit immunoglobulin G (IgG) and/or with PE-conjugated
anti-mouse CD80. The analysis was performed by FCM.
The expression of SEA and CD80 on the surface of
Hepa1-6 and B16 cells was also visualized by in situ
immunofluorescent staining. Hepa1-6 or B16 cells were seeded at a
density of 1×104 cells/well onto glass coverslips and
grown for 48 h after infection with Ad-MMRE-mTERT-BIS at MOI 100.
Cells were stained with the Abs at 4°C as described above and fixed
with 4% paraformaldehyde in PBS for 30 min, and then washed twice
in PBS buffer. Finally, the coverslips were mounted onto glass
slides. Fluorescence distribution was analyzed using a laser
confocal scanning microscope.
Proportion of CD4+ and
CD8+ T lymphocytes and cytokine production in
splenocytes induced by tumor cells infected with the recombinant
adenoviruses in vitro
Mouse splenocytes (107 cells/well) were
co-cultured with 106 inactivated Hepa1-6 or B16 cells
non-infected or infected with Ad(empty), Ad-MMRE-mTERT-CD80,
Ad-MMRE-mTERT-SEAtm or Ad-MMRE-mTERT-BIS in 24-well plate and
incubated for 48 h in a humidified chamber at 37°C, 5%
CO2. CD4+ and CD8+ T lymphocytes
in splenocytes were stained with FITC-conjugated anti-mouse CD3,
PE-conjugated anti-mouse CD4 and APC-conjugated anti-mouse CD8 and
analyzed by FCM. IL-2, TNF-α, and IFN-γ in cell culture supernatant
were measured using ELISA kits (eBioscience).
Mouse tumor preparation
Female C57BL/6 mice, 6–8 weeks of age, were obtained
from the Experimental Animal Center (Health Science Center, Peking
University, Beijing, China) under strictly controlled
specific-pathogen-free conditions. Principles of laboratory animal
care (NIH publication no. 85–23, revised 1985) were followed, as
well as the current version of the Chinese Law on the Protection of
Animals. Mice were held in accordance with the permission of the
responsible authority. Tumors were generated by subcutaneous
injection of 106 Hepa1-6 or B16 cells per mouse in 100
µl of PBS on the right hind limb of C57BL/6 mice. Visible tumors
developed at 7–9 days after tumor cell inoculation.
Tumor treatment in vivo
When the largest diameter of tumor exceeded 0.5 cm,
the Hepa1-6 hepatoma-bearing mice and B16 melanoma-bearing mice
were, respectively, randomized into the following groups, and ten
mice were included in each group: 1, B16-PBS (blank control); 2,
B16-Ad(empty); 3, B16-CD80; 4, B16-SEAtm; 5, B16-BIS; 6,
Hepa1-6-PBS (blank control); 7, Hepa1-6-Ad(empty); 8, Hepa1-6-CD80;
9, Hepa1-6-SEAtm; and 10, Hepa1-6-BIS. The mice in the control
group were intratumorally injected with 100 µl PBS per mouse. Mice
in Ad(empty), CD80, SEAtm and BIS group were, respectively,
injected intratumorally with 1×109 PFU of Ad(empty),
Ad-MMRE-mTERT-CD80, Ad-MMRE-mTERT-SEAtm or Ad-MMRE-mTERT-BIS in 100
µl PBS per mouse. The mice were injected twice a week for 2 weeks.
Five mice in each group were sacrificed on day 14 after the last
injection, and splenocytes were isolated for CTL activity and
IFN-γ-producing cell assay. The other 5 mice in each group were
monitored for survival (≤60 days). Tumor sizes were measured before
adenovirus injection and subsequently twice a week. The final tumor
volume was measured on day 30 after tumor cell inoculation (before
any deaths occurred) to ensure inclusion of the data from all the
mice. Linear calipers were used to measure the longest diameter (a)
and width (b). The tumor volume was calculated using the formula:
(ab2/2) and was plotted as the mean tumor volume of the
group (± standard error, SE) versus days post-tumor challenge.
Systemic antitumor cytotoxicity assays
of CTLs in vivo
The CytoTox 96 non-radioactive cytotoxicity assay
(Promega, Madison, WI, USA) was performed to measure the cytotoxic
activity of the splenocytes in mice bearing tumors injected with
PBS, Ad(empty), Ad-MMRE-mTERT-CD80, Ad-MMRE-mTERT-SEAtm or
Ad-MMRE-mTERT-BIS, according to the manufacturer's protocol.
Briefly, splenocytes of mice in each group were prepared 14 days
after the last injection. Hepa1-6 or B16 cells in RPMI-1640 medium
with 10% FBS were used as the targets for the CTL assays. Targets
(2×105 cells/well) were mixed with splenocytes at
effector: target (E:T) ratios of 50:1, 25:1, and 12.5:1, and
incubated for 4 h in a humidified incubator at 37°C, 5%
CO2. Lysis solution was added to a portion of the target
cells, prior to centrifugation, as a maximum LDH release control.
Supernatant (50 µl) was transferred to the enzymatic assay plate
after centrifugation, 50 µl of the substrate mix was added to each
well; the plate was covered to protect it from light, and incubated
for 30 min at room temperature. Stop solution (50 µl) was added to
each well and the absorbance was recorded at 490 nm. The percentage
of specific lysis was determined according to the following
formula: [A (experimental)-A (effector spontaneous)-A (target
spontaneous)x100)/[A (target maximum)-A (target spontaneous)].
IFN-γ producing cell frequencies in
vivo
Mouse IFN-γ ELISpot assay was performed in PVDF
bottomed 96-well plates by using a murine IFN-γ ELISpot kit
(Millipore, Bedford, MA, USA) according to the manufacturer's
instructions. Briefly, plates were coated overnight at 4°C with
anti-IFN-γ capture antibody and washed three times with PBST
(PBS+0.05% Tween-20). Plates were blocked for 2 h with 2% skimmed
dry milk/PBS. Splenocytes (1×106 cells/well) of mice
after treatment were then co-cultured with the MMC inactivated
Hepa1-6 cells or B16 (5×104/well) and incubated for 24 h
at 37°C. Only splenocytes were added into wells as negative
control. Cells were then removed and a biotinylated IFN-γ detection
antibody was added and incubated for 1 h. Following extensive
washing with PBST and PBS, the plates were incubated with
streptavidin-alkaline phosphatase for 1 h at 37°C. Spots were
visualized by the addition of the alkaline phosphatase substrate
BCIP/NBT. The number of dots in each well was counted by two
separate investigators in a blinded manner using a dissecting
microscope.
Statistical analysis
One-way analysis of variance (ANOVA) was performed
to determine differences of immune response among the various
treatment groups. Newman-Keuls tests were performed as post-hoc
analysis for one-way ANOVA. The antitumor effects were considered
statistically significant when the P-value was <0.05.
Results
Recombinant adenovirus-mediated
expression of SEA and CD80 on the tumor cells
Hepa1-6, B16, CT26 and NIH3T3 cells were infected
with the recombinant adenovirus Ad-MMRE-mTERT-BIS. Flow cytometric
analysis showed that SEAtm and CD80 proteins were efficiently
co-expressed on the surface of the infected Hepa1-6 and B16 cells,
the expression of SEAtm and CD80 proteins in Hepa1-6 was stronger
than that in B16. A few infected CT26 cells and none of infected
NIH3T3 cells expressed SEAtm and CD80 proteins (Table I). There were no significant
differences between SEAtm and CD80 expression among the infected
Hepa1-6 cells or B16 cells. To determine the distribution of CD80
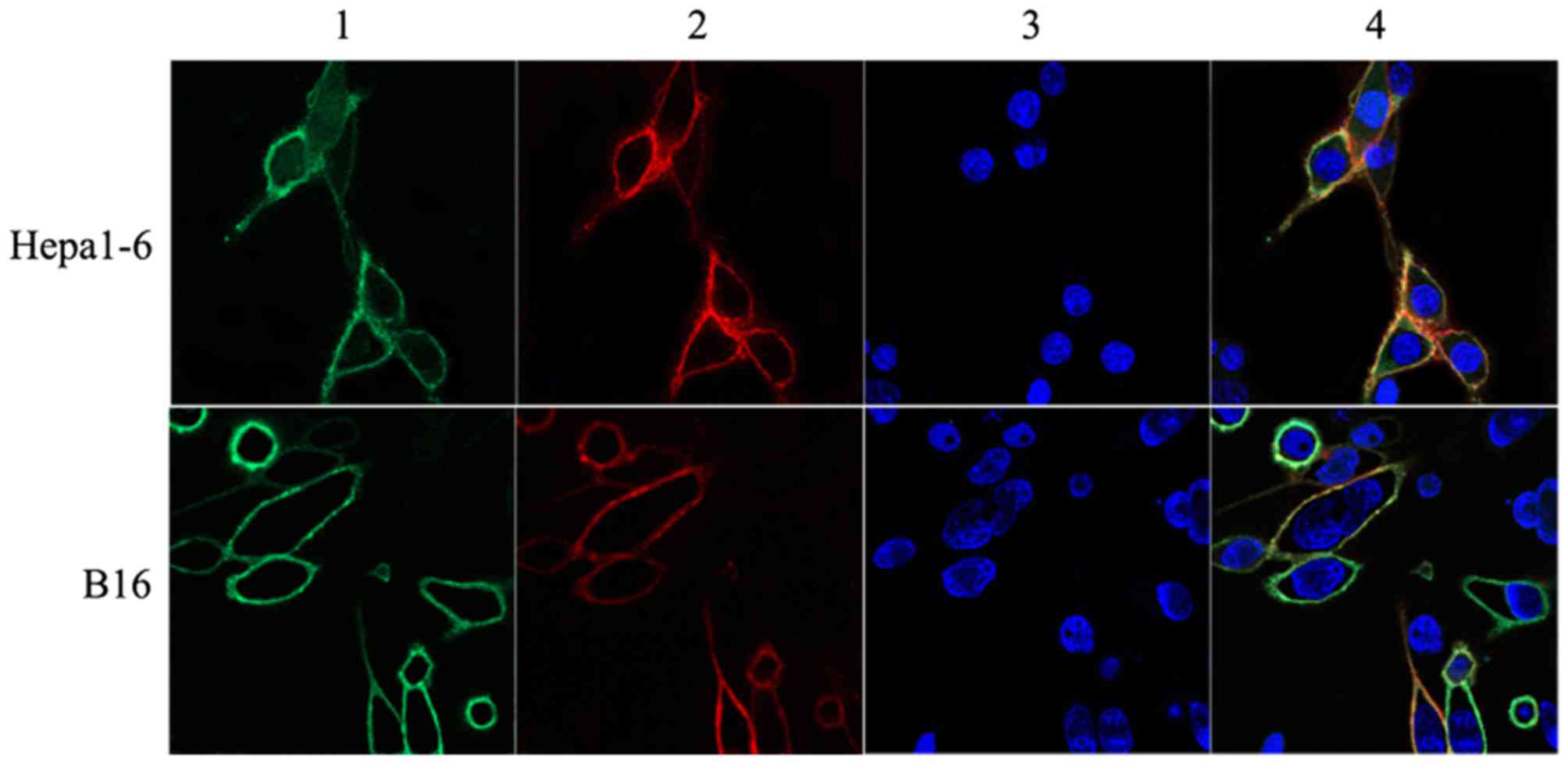
and SEAtm protein on the infected cells, the cell images were
visualized by laser confocal microscopy (Fig. 1). Since a few infected CT26 cells
expressed CD80 and SEAtm protein, we only observed CD80 and SEAtm
protein expression on the surface of B16 and Hepa1-6 cells under
laser confocal microscopy.
 | Table I.Recombinant adenovirus
Ad-MMRE-mTERT-BIS -mediated expression of SEA and CD80 on the tumor
cells and NIH3T3. |
Table I.
Recombinant adenovirus
Ad-MMRE-mTERT-BIS -mediated expression of SEA and CD80 on the tumor
cells and NIH3T3.
| Cell lines | CD80-positive cells
(%) | SEAtm-positive
cells (%) | CD80- and
SEA-positive cells (%) |
|---|
| NIH3T3 | 1.3±0.6 | 0.9±0.3 | 1.1±0.5 |
| CT26 | 5.6±0.9 | 5.7±1.1 | 5.3±0.8 |
| B16 | 41.2±1.9 | 39.3±2.1 | 39.1±1.5 |
| Hepa1–6 | 89.7±2.9 | 88.6±2.3 | 87.2±3.1 |
T lymphocyte sub-population
proliferation induced by tumor cells infected the recombinant
adenoviruses in vitro
The biological activity of CD80 and/or SEA expressed
on the surface of Hepa1-6 and B16 cells in vitro was
determined by the influence on the proportion of lymphocyte
sub-population. The results are shown in Table II. The numbers of CD3+,
CD3+ CD4+, and CD3+
CD8+ T lymphocytes in cultures induced by tumor cells
infected with Ad-MMRE-mTERT-BIS, Ad-MMRE-mTERT-CD80 or
Ad-MMRE-mTERT-SEAtm increased as compared with those induced by
non-infected tumor cells or tumor cells infected with Ad(empty)
(P<0.05). The numbers of CD3+ and CD3+
CD4+ T lymphocytes induced by tumor cells infected with
Ad-MMRE-mTERT-BIS were the highest among all groups (P<0.05).
The numbers of CD3+ and CD3+ CD4+
T lymphocytes in cultures induced by tumor cells infected with
Ad-MMRE-mTERT-BIS or Ad-MMRE-mTERT-SEAtm increased compared to that
in cultures induced by tumor cells infected with Ad-MMRE-mTERT-CD80
(P<0.05). There were no statistical differences in the number of
CD3+ CD8+ T lymphocytes among cultures with
tumor cells infected with Ad-MMRE-mTERT-BIS, Ad-MMRE-mTERT-SEAtm
and Ad-MMRE-mTERT-CD80 (P>0.05).
| Table II.The proportion of T lymphocyte
sub-populations in splenocytes co-cultured with tumor cells
infected with the recombinant adenoviruses in vitro. |
Table II.
The proportion of T lymphocyte
sub-populations in splenocytes co-cultured with tumor cells
infected with the recombinant adenoviruses in vitro.
|
| CD3+ T
cells (%) | CD3+
CD4+ T cells (%) | CD3+
CD8+ T cells (%) |
|---|
|
|
|
|
|
|---|
| Group | Hepa1-6 | B16 | Hepa1-6 | B16 | Hepa1-6 | B16 |
|---|
| Non-infection | 77.87±0.88 | 67.87±0.78 | 52.79±0.47 | 45.34±0.57 | 26.36±0.53 | 21.36±0.59 |
| Ad(empty) | 78.28±0.91 | 67.74±0.82 | 52.26±0.91 | 45.58±0.95 | 26.02±0.08 | 22.16±0.78 |
|
Ad-MMRE-mTERT-CD80 |
87.13±0.90a,b |
78.00±0.79a,b | 53.27±0.93 | 46.23±0.85 |
33.86±1.72a,b |
31.77±0.79a,b |
|
Ad-MMRE-mTERT-SEAtm |
90.41±1.20a,b,c |
87.93±0.95a,b,c |
57.27±0.77a,b,c |
57.17±0.97a,b,c |
33.14±1.14a,b |
30.76±0.96a,b |
|
Ad-MMRE-mTERT-BIS |
94.53±0.50a,b,c,d | 91.75±
0.92a,b,c,d |
61.86±0.99a,b,c,d |
60.46±0.89a,b,c,d | 32.67±
1.48a,b |
31.29±1.08a,b |
Cytokine production of splenocytes
induced by tumor cells infected by the recombinant adenoviruses in
vitro
Splenocytes were isolated from a mouse and
stimulated with inactivated Hepa1-6 or B16 cells non-infected or
infected with Ad(empty), Ad-MMRE-mTERT-CD80, Ad-MMRE-mTERT-SEAtm or
Ad-MMRE-mTERT-BIS and cultured for 48 h. The cytokines IL-2, IFN-γ
and TNF-α in cell culture supernatant were detected. The results
are shown in Table III. The
levels of the three cytokines in splenocytes induced by the tumor
cells infected with Ad-MMRE-mTERT-BIS were the highest in all the
groups. The levels of the three cytokines in splenocytes induced by
tumor cells infected with Ad-MMRE-mTERT-SEAtm, the levels of the
three cytokines in splenocytes induced by Hepa1-6 cells or IL-2 and
TNF-α in splenocytes induced by B16 cells after infection with
Ad-MMRE-mTERT-CD80 increased as compared with those induced by
non-infected tumor cells or infected with Ad(empty). The levels of
IFN-γ induced by Hepa1-6 cells and TNF-α induced by B16 cells
infected with Ad-MMRE-mTERT-SEAtm were higher than those induced by
the same cells infected with Ad-MMRE-mTERT-CD80.
| Table III.Cytokine production of splenocytes
induced by tumor cells infected by the recombinant adenoviruses
in vitro. |
Table III.
Cytokine production of splenocytes
induced by tumor cells infected by the recombinant adenoviruses
in vitro.
|
| IL-2 (pg/ml) | IFN-γ (pg/ml) | TNF-α (pg/ml) |
|---|
|
|
|
|
|
|---|
| Group | Hepa1-6 | B16 | Hepa1-6 | B16 | Hepa1-6 | B16 |
|---|
| Non-infection | 7.9±0.7 | 7.3±1.1 | 37.6±6.7 | 59.7±11.6 | 28.3±1.5 | 35.9±0.9 |
| Ad(empty) | 8.2±1.5 | 7.3±0.7 | 34.6±13.9 | 60.8±7.1 | 28.2±2.5 | 35.6±1.8 |
|
Ad-MMRE-mTERT-CD80 |
22.6±4.2a,b |
12.6±1.0a,b |
206.5±51.8a,b | 65.8±12.9 |
83.5±5.9a,b |
69.9±3.5a,b |
|
Ad-MMRE-mTERT-SEAtm |
19.8±6.7a,b |
13.3±0.9a,b |
352.4±26.7a,b,c |
74.8±7.0a,b |
91.6±6.6a,b |
89.1±6.8a,b,c |
|
Ad-MMRE-mTERT-BIS |
162.6±5.9a,b,c,d |
70.7±3.9a,b,c,d |
444.0±36.2a,b,c,d |
349.3±24.5a,b,c,d |
107.4±15.6a,b,c,d |
159.0±7.0a,b,c,d |
IFN-γ-producing cell frequencies of
splenic lymphocytes from the mice
Spleen lymphocytes were isolated from the mice (in
triplicate) 14 days after the last injection, co-cultured with
inactivated Hepa1-6 or B16 cells (treated with MMC, 100 µg/ml at
37°C for 1 h) for 24 h. Cells were removed and IFN-γ-producing cell
frequency was determined for each group of mice with different
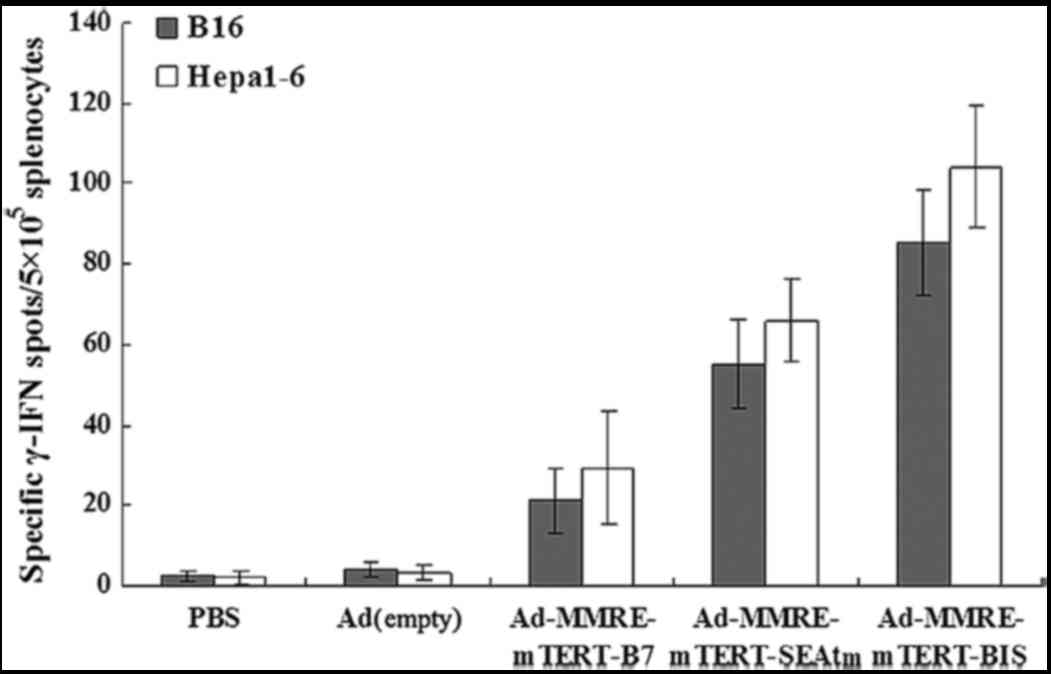
treatments. As shown in Fig. 2,
IFN-γ-producing cell frequencies of lymphocytes in Hepa1-6
hepatoma-bearing mice or B16 myeloma-bearing mice injected with
Ad-MMRE-mTERT-BIS was the highest among all groups. The
IFN-γ-producing cell frequencies in the mice injected with
Ad-MMRE-mTERT-CD80 and Ad-MMRE-mTERT-SEAtm were much higher than
those in mice injected with Ad(empty) and PBS (P<0.05).
CTL activity of splenocytes of the
mice
Splenocytes isolated from mice in different groups
14 days after the last injection and used as CTL effector cells,
and tested against Hepa1-6 or B16 cells as target cells, CTL
activities were determined at effector:target (E:T) ratios of
12.5:1, 25:1, and 50:1 by a standard CytoTox 96 non-radioactive
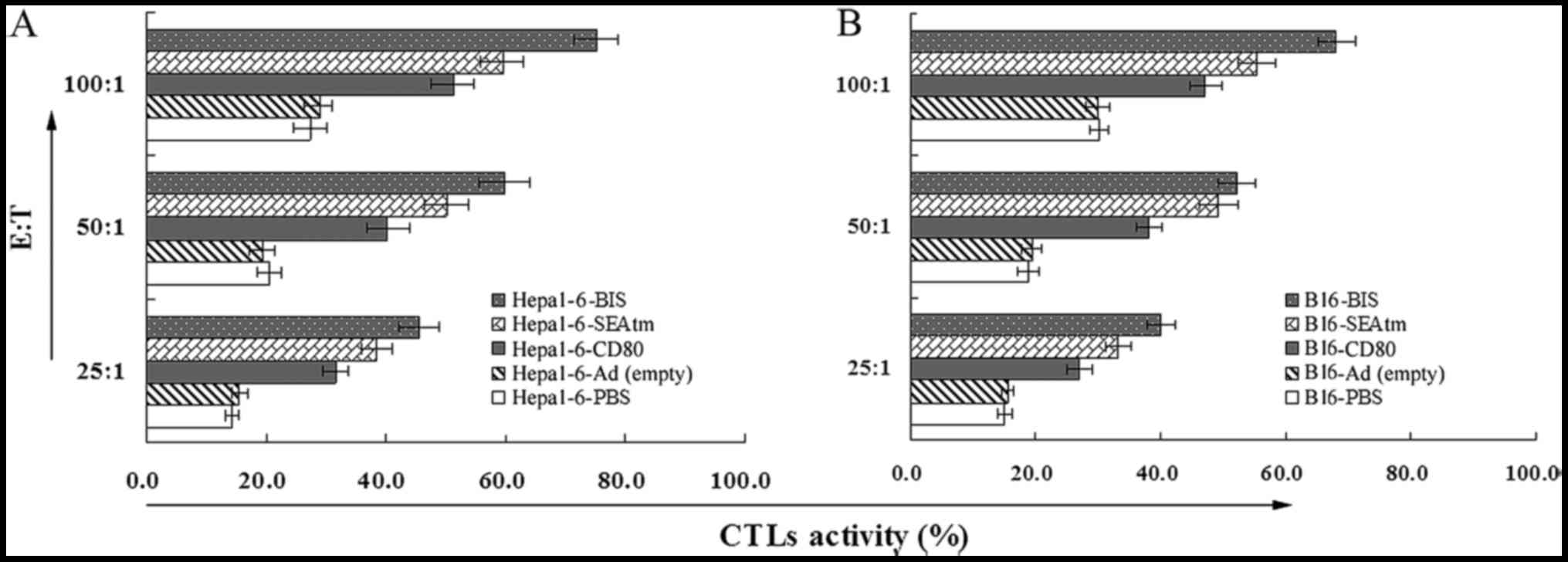
cytotoxicity assay. As shown in Fig. 3A
and B, lymphocytes derived from the mice treated with the
Ad-MMRE-mTERT-BIS showed the highest CTL activities in all the
groups. The CTL activities of the mice treated with
Ad-MMRE-mTERT-CD80 and Ad-MMRE-mTERT-SEAtm were much higher than
those of mice treated with Ad(empty) and PBS (P<0.05).
Antitumor effects of the recombinant
adenoviruses in vivo
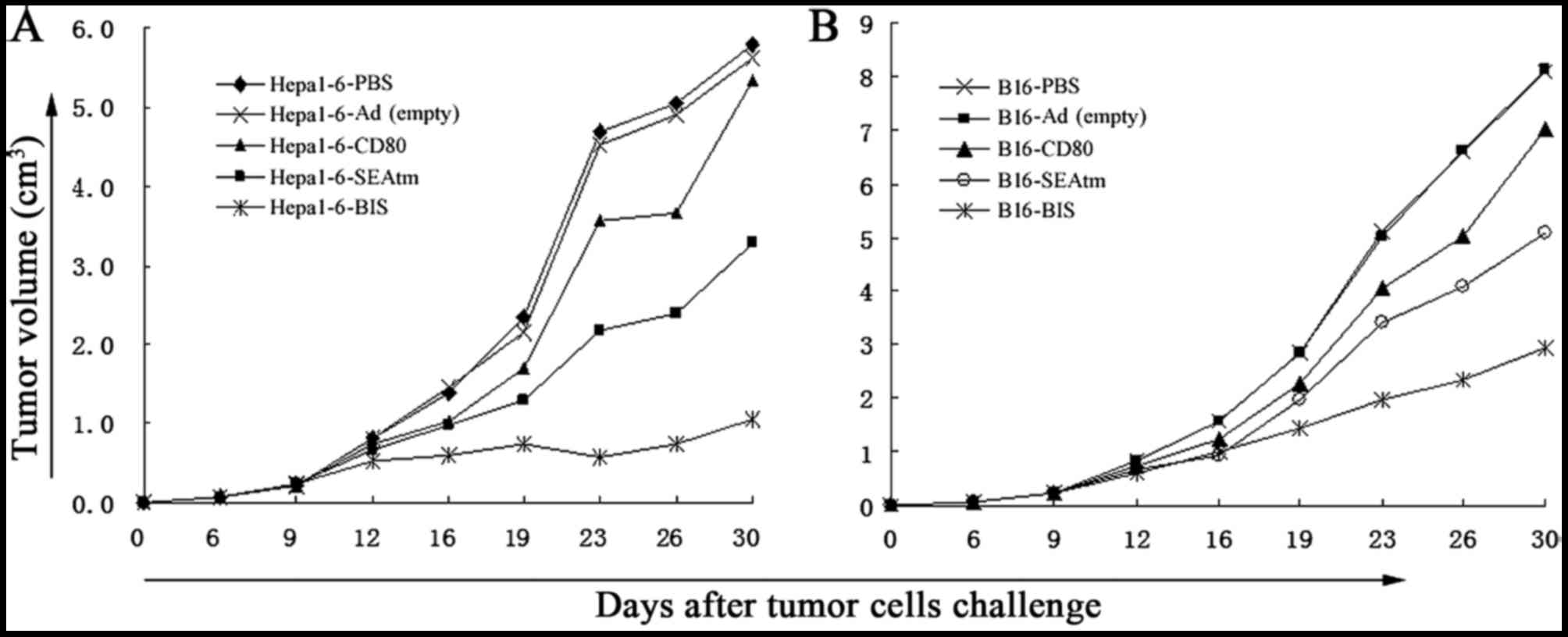
The results in Fig.
4 showed that tumor growth in mice treated with recombinant
adenoviruses Ad-MMRE-mTERT-CD80, Ad-MMRE-mTERT-SEA or
Ad-MMRE-mTERT-BIS was markedly inhibited as compared with that in
mice treated with PBS or Ad(empty) from day 16 in Hepa1-6
tumor-bearing mice (Fig. 4A) and
from day 19 in B16 tumor-bearing mice (Fig. 4B; P<0.05), the inhibition of the
dual-gene therapy was significantly stronger than that of single
gene therapy (P<0.05), there were no significant differences in
tumor growth inhibition between the groups treated with
Ad-MMRE-mTERT-CD80 and Ad-MMRE-mTERT-SEA (P>0.05). Five
tumor-bearing mice in each group were monitored for their survival
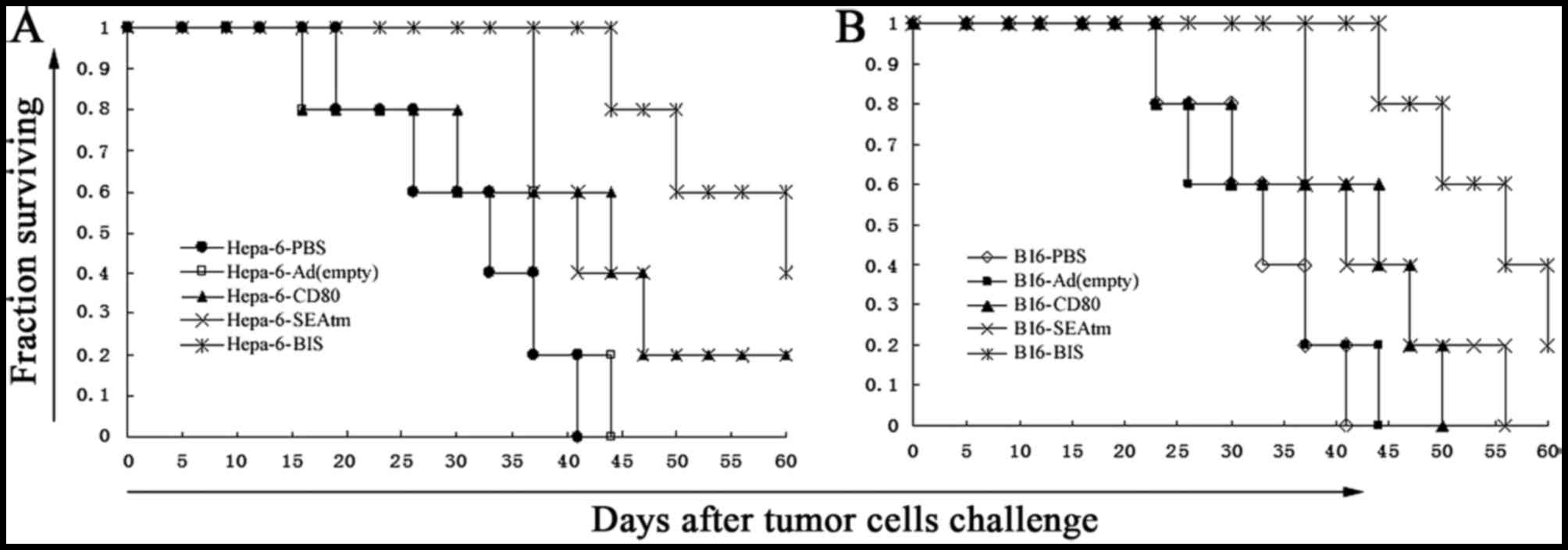
period. The results in Fig. 5 show
that Hepa1-6 (Fig. 5A) or B16
(Fig. 5B) tumor-bearing mice
treated with Ad-MMRE-mTERT-CD80, Ad-MMRE-mTERT-SEA or
Ad-MMRE-mTERT-BIS survived longer than mice treated with PBS and
Ad(empty) (P<0.05), tumor-bearing mice treated with
Ad-MMRE-mTERT-BIS survived the longest of all the groups, there was
no significant difference in the survival period between the mice
treated with Ad-MMRE-mTERT-CD80 or Ad-MMRE-mTERT-SEA (P>0.05),
two mice in Hepa1-6-BIS group and one mouse in B16-BIS group had a
survival period exceeding 60 days.
Discussion
As a SAg, SEA is a powerful immunostimulant.
Previous studies have demonstrated that SEA anchoring onto
MHC-II-negative tumor cells through antibodies directs T
cell-mediated cytotoxicity against these tumors with reduced
toxicity against normal MHC-II+ cells (24–27).
In addition, genetically engineered fusion protein of SEA with the
transmembrane region sequence of c-erb-B2 could anchor on the
surface of tumor and was capable of eliciting systemic antitumor
immunity without any measured toxicity (28). These results indicated that the SAg
anchoring on MHC-II-negative tumor cells assumes T cell
stimulation, but it circumvents conventionally defined MHC
‘presentation’. Furthermore, the anchored SAg showed a greater
reduction in MHC class II binding compared to native forms and
could elicit MHC-II-independent T cell stimulation in vitro
as long as co-stimulatory signals were provided (29,30).
To decrease systemic activation as a consequence of SAg-MHC class
II interaction with monocytes and B cells, and to localize the
cytotoxic capabilities of SAg-activated T cells to tumor sites, the
membrane-expressing SEA (31) was
used in our study. To efficiently stimulate T cells, CD80 was
simultaneously transduced into tumor cells. CD80 binds CD28 of T
cells and provides the second signal for activation of T cells.
Pericuesta et al (32) generated the construct mTERT-GFP
using the mTER gene promoter of 1, 2 or 5 kb upstream of the first
ATG of the open reading frame of mTERT gene to promote the
expression of EGFP. In their transgenic model, no fluorescent
expression of the mTERT-EGFP construct could be identified in adult
tissues. This suggests that although telomerase activity exists in
colon, liver, ovary, and testis, there is a tight repression system
of mTERT gene promoter in adult mouse tissues in physiological
conditions. In our previous study (33), we found the proximal 333-bp fragment
was the core promoter of the mTERT gene in the cancer cells. The
proximal 333-bp fragment was able to make SEA express on the
surface of hepatoma cell line Hepa1-6, melanoma cell line B16,
colon cancer cell line CT26, but not in the fibroblast NIH3T3
cells. In our study, SEAtm and CD80 gene driven by mTERT promoter
in the recombinant adenovirus were constructed. The results showed
that SEAtm and CD80 were co-expressed in 87.2% of Hepa1-6 cells,
39.1% of B16 cells and 5.3% of CT26 cells, but not in fibroblast
cell line NIH3T3 cells after infection with the recombinant
adenovirus Ad-MMRE-mTERT-BIS. The positive rates of SEA and CD80 in
Hepa1-6 cells, B16 cells and CT26 cells were different. The
possible reasons are that the infection efficiency and expression
efficiency of the same recombinant adenovirus Ad-MMRE-mTERT-BIS are
different in different tumor cells. Ad-MMRE-mTERT-BIS was not used
to treat CT26 colon cancer because the positive rate of SEA and
CD80 in CT26 cells was very low after infection with
Ad-MMRE-mTERT-BIS.
SEA are known as potent activators of T lymphocytes
and effcient inducers of cytokine production (34,35).
Various cytokines, such as interleukin, interferon and tumor
necrosis factor, can destroy vascular endothelial cells of the
tumors, promote thrombosis and reduce blood supply to the tumor
tissues, resulting in tumor cell necrosis and apoptosis. Cytokines
also stimulate proliferation and differentiation of T cells, which
will in return produce more cytokines, so as to form endogenously
circulating biological effects and speed up the apoptosis of cancer
cells. Cytokines can suppress tumor growth both directly and
synergistically (35). In addition,
cytokines can induce LAK activity, activate natural killer cells
and macrophages (10,36,37),
facilitate penetration of high molecular weight proteins and
upregulate cell adhesion molecules and MHC molecule expression on
tumor cells. In our results, SEA expressed on the surface of
Hepa1-6 and B16 cells was biologically active in vitro, as
shown by the ability of Hepa1-6 and B16 cells infected by
Ad-MMRE-mTERT-BIS to elicit proliferation and cytokine production
of splenocytes. After stimulation with Hepa1-6 and B16 infected by
Ad-MMRE-mTERT-BIS, Ad-MMRE-mTERT-SEAtm or Ad-MMRE-mTERT-CD80, the
proportion of CD3+, CD8+ or/and
CD4+ T cells in splenocytes and the level of IL-2, IFN-γ
and TNF-α produced by splenocytes increased. SEA and CD80
co-expressed tumor cells could more effectively activate
splenocytes than SEA or CD80 expressed tumor cells.
Endogenously produced IFN-γ protects the host from
not only growing transplanted tumors, but also the formation of
primary chemically-induced and spontaneous tumors (38–42)
and plays a crucial role for the eradication of tumors in
vivo (43). Injection of
neutralizing mAbs for IFN-γ into mice bearing transplanted,
established Meth A tumors blocked LPS-induced tumor rejection
(38). In addition, transplanted
fibrosarcomas grew faster and more efficiently in mice treated with
IFN-γ specific mAbs. Moreover, IFN-γ was shown to be involved in
the antitumor effects of antibody-targeted superantigens (44). ELISpot is a sensitive functional
assay used to measure INF-γ production at the single cell level.
The ELISpot showed there were much more tumor-specific
INF-γ-producing cells in the mice bearing hepa1-6 hepatoma and B16
melanoma treated with Ad-MMRE-mTERT-BIS compared with those in
other groups. CD8+ CTLs are one of the most crucial
components among antitumor effectors (45). In our study, the results showed
higher tumor-specific CTL activity was induced in the mice bearing
hepa1-6 hepatoma and B16 melanoma treated with the recombinant
adenoviruses compared with that in other groups. The ELISpot and
cytotoxicity assays indicate Ad-MMRE-mTERT-BIS could induce
stronger systemic antitumor immunity than either
Ad-MMRE-mTERT-SEAtm or Ad-MMRE-mTERT-CD80. The survival period of
the mice bearing hepa1-6 hepatoma or B16 melanoma treated with
Ad-MMRE-mTERT-BIS was significantly longer and their tumors grew
more slowly than those of mice in other groups. The regression of
tumor indicates local antitumor immunity induced by
Ad-MMRE-mTERT-BIS treatment.
In summary, our findings show that tumor cells
infected with the recombinant adenovirus bearing SEA and CD80 genes
can generate stronger antitumor immunity than the recombinant
adenovirus bearing a single gene in vitro and in
vivo, indicating that SEAtm and CD80 are able to stimulate
antitumor immune responses synergistically. The same recombinant
adenovirus bearing foreign gene controlled by TERT promoter may be
used for targeting gene therapy of different kinds of tumors. The
results provided experimental evidence that supports the
feasibility and effectiveness of this novel approach in cancer
immunotherapy. Additional underlying mechanisms need to be further
studied.
Acknowledgements
This study was supported by the National Natural
Sciences Foundation of China (no. 30772524).
References
|
1
|
Kotb M: Bacterial pyrogenic exotoxins as
superantigens. Clin Microbiol Rev. 8:411–426. 1995.PubMed/NCBI
|
|
2
|
Akbari A, Farahnejad Z, Akhtari J,
Abastabar M, Mobini GR and Mehbod AS: Staphylococcus aureus
enterotoxin B down-regulates the expression of transforming growth
factor-beta (TGF-β) signaling transducers in human glioblastoma.
Jundishapur J Microbiol. 9:e272972016.PubMed/NCBI
|
|
3
|
Akbari A, Mobini GR, Maghsoudi R, Akhtari
J, Faghihloo E and Farahnejad Z: Modulation of transforming growth
factor-β signaling transducers in colon adenocarcinoma cells
induced by staphylococcal enterotoxin B. Mol Med Rep. 13:909–914.
2016.PubMed/NCBI
|
|
4
|
Zhao W, Li Y, Liu W, Ding D, Xu Y, Pan L
and Chen S: Transcytosis, antitumor activity and toxicity of
staphylococcal enterotoxin C2 as an oral administration protein
drug. Toxins (Basel). 8:E1852016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang G, Xu M, Song Y, Su Z, Zhang H and
Zhang C: TNF-α produced by SEC2 mutant (SAM-3)-activated human T
cells induces apoptosis of HepG2 cells. Appl Microbiol Biotechnol.
100:2677–2684. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hosseini Mahmoodzadeh H, Soleimanirad J,
Mehdizadeh Aghdam E, Amin M and Imani Fooladi AA: Texosome-anchored
superantigen triggers apoptosis in original ovarian cancer cells.
Med Oncol. 32:4092015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Terman DS, Serier A, Dauwalder O, Badiou
C, Dutour A, Thomas D, Brun V, Bienvenu J, Etienne J, Vandenesch F,
et al: Staphylococcal entertotoxins of the enterotoxin gene cluster
(egcSEs) induce nitrous oxide- and cytokine dependent tumor cell
apoptosis in a broad panel of human tumor cells. Front Cell Infect
Microbiol. 3:382013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kato M, Nakamura Y, Suda T, Ozawa Y, Inui
N, Seo N, Nagata T, Koide Y, Kalinski P, Nakamura H, et al:
Enhanced anti-tumor immunity by superantigen-pulsed dendritic
cells. Cancer Immunol Immunother. 60:1029–1038. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gong Z, Han C, Hao L, Yang J, Tang W and
Teng G: Preparation and in-vitro bioactivity of a novel
superantigen conjugate targeting bladder carcinoma. J Pharm
Pharmacol. 61:869–875. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dohlsten M, Hedlund G, Akerblom E, Lando
PA and Kalland T: Monoclonal antibody-targeted superantigens: A
different class of anti-tumor agents. Proc Natl Acad Sci USA.
88:9287–9291. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Litton MJ, Dohlsten M, Lando PA, Kalland
T, Ohlsson L, Andersson J and Andersson U: Antibody-targeted
superantigen therapy induces tumor-infiltrating lymphocytes,
excessive cytokine production, and apoptosis in human colon
carcinoma. Eur J Immunol. 26:1–9. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gidlöf C, Dohlsten M, Lando P, Kalland T,
Sundström C and Tötterman TH: A superantigen-antibody fusion
protein for T-cell immunotherapy of human B-lineage malignancies.
Blood. 89:2089–2097. 1997.PubMed/NCBI
|
|
13
|
Ueno A, Arakawa F, Abe H, Matsumoto H,
Kudo T, Asano R, Tsumoto K, Kumagai I and Kuroki M and Kuroki M:
T-cell immunotherapy for human MK-1-expressing tumors using a
fusion protein of the superantigen SEA and anti-MK-1 scFv antibody.
Anticancer Res. 22:769–776. 2002.PubMed/NCBI
|
|
14
|
Wahlsten JL, Mills CD and Ramakrishnan S:
Antitumor response elicited by a superantigen-transmembrane
sequence fusion protein anchored onto tumor cells. J Immunol.
161:6761–6767. 1998.PubMed/NCBI
|
|
15
|
Si S, Sun Y, Li Z, Ge W, Zhang X, Hu P,
Huang Y, Chen G, Song H, Huang Y, et al: Gene therapy by
membrane-expressed superantigen for alpha-fetoprotein-producing
hepatocellular carcinoma. Gene Ther. 13:1603–1610. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lando PA, Dohlsten M, Hedlund G, Brodin T,
Sansom D and Kalland T: Co-stimulation with B7 and targeted
superantigen is required for MHC class II-independent T-cell
proliferation but not cytotoxicity. Immunology. 80:236–241.
1993.PubMed/NCBI
|
|
17
|
Si SY, Hu PZ, Huang YY, Ye J, Huang Y, Li
ZS, Ge W, Li X, Qu P, Zhang XM, et al: Tumor cells with B7.1 and
transmembrane anchored staphylococcal enterotoxin A generate
effective antitumor immunity. Biochem Biophys Res Commun.
347:208–214. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wirth T, Kühnel F and Kubicka S:
Telomerase-dependent gene therapy. Curr Mol Med. 5:243–251. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Autexier C and Greider CW: Telomerase and
cancer: Revisiting the telomere hypothesis. Trends Biochem Sci.
21:387–391. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Blackwood EM and Eisenman RN: Max: A
helix-loop-helix zipper protein that forms a sequence-specific
DNA-binding complex with Myc. Science. 251:1211–1217. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kumagai T, Tanio Y, Osaki T, Hosoe S,
Tachibana I, Ueno K, Kijima T, Horai T and Kishimoto T: Eradication
of Myc-overexpressing small cell lung cancer cells transfected with
herpes simplex virus thymidine kinase gene containing Myc-Max
response elements. Cancer Res. 56:354–358. 1996.PubMed/NCBI
|
|
22
|
Song JS: Adenovirus-mediated suicide SCLC
gene therapy using the increased activity of the hTERT promoter by
the MMRE and SV40 enhancer. Biosci Biotechnol Biochem. 69:56–62.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Si SY, Song SJ, Xu BX, Zhao G, Tan XQ, Liu
JL, Zhang JZ and Liu ZG: Construction of recombinant adenovirus of
SEA and CD80 genes co-expression regulated by mouse TERT promoter
and identification of its expression in hepatoma cells. Xi Bao Yu
Fen Zi Mian Yi Xue Za Zhi. 27:717–720. 2011.(In Chinese).
PubMed/NCBI
|
|
24
|
Nielsen SE, Zeuthen J, Lund B, Persson B,
Alenfall J and Hansen HH: Phase I study of single, escalating doses
of a superantigen-antibody fusion protein (PNU-214565) in patients
with advanced colorectal or pancreatic carcinoma. J Immunother.
23:146–153. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cheng JD, Babb JS, Langer C, Aamdal S,
Robert F, Engelhardt LR, Fernberg O, Schiller J, Forsberg G,
Alpaugh RK, et al: Individualized patient dosing in phase I
clinical trials: The role of escalation with overdose control in
PNU-214936. J Clin Oncol. 22:602–609. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ihle J, Holzer U, Krull F, Dohlsten M,
Kalland T, Niethammer D and Dannecker GE: Antibody-targeted
superantigens induce lysis of major histocompatibility complex
class II-negative T-cell leukemia lines. Cancer Res. 55:623–628.
1995.PubMed/NCBI
|
|
27
|
Holzer U, Bethge W, Krull F, Ihle J,
Handgretinger R, Reisfeld RA, Dohlsten M, Kalland T, Niethammer D
and Dannecker GE:
Superantigen-staphylococcal-enterotoxin-A-dependent and
antibody-targeted lysis of GD2-positive neuroblastoma cells. Cancer
Immunol Immunother. 41:129–136. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ma W, Yu H, Wang Q, Jin H, Solheim J and
Labhasetwar V: A novel approach for cancer immunotherapy: Tumor
cells with anchored superantigen SEA generate effective antitumor
immunity. J Clin Immunol. 24:294–301. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Green JM, Turka LA, June CH and Thompson
CB: CD28 and staphylococcal enterotoxins synergize to induce
MHC-independent T-cell proliferation. Cell Immunol. 145:11–20.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Taub DD and Rogers TJ: Direct activation
of murine T cells by staphylococcal enterotoxins. Cell Immunol.
140:267–281. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lu SY, Sui YF, Li ZS, Pan CE, Ye J and
Wang WY: Construction of a regulable gene therapy vector targeting
for hepatocellular carcinoma. World J Gastroenterol. 9:688–691.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pericuesta E, Ramírez MA, Villa-Diaz A,
Relaño-Gines A, Torres JM, Nieto M, Pintado B and Gutiérrez-Adán A:
The proximal promoter region of mTert is sufficient to regulate
telomerase activity in ES cells and transgenic animals. Reprod Biol
Endocrinol. 4:5–16. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Si SY, Song SJ, Zhang JZ, Liu JL, Liang S,
Feng K, Zhao G and Tan XQ: Cloning of mouse telomerase reverse
transcriptase gene promoter and identification of proximal core
promoter sequences essential for the expression of transgenes in
cancer cells. Oncol Rep. 26:377–382. 2011.PubMed/NCBI
|
|
34
|
Tian XL, Yan Z, Chen J, Zhao WH and Guo W:
Clinical application of highly agglutinative staphylococcin in
cancer treatment updates of the literature. Eur Rev Med Pharmacol
Sci. 20:2718–2725. 2016.PubMed/NCBI
|
|
35
|
Dohlsten M, Sundstedt A, Björklund M,
Hedlund G and Kalland T: Superantigen-induced cytokines suppress
growth of human colon-carcinoma cells. Int J Cancer. 54:482–488.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lando PA, Dohlsten M, Hedlund G, Akerblom
E and Kalland T: T cell killing of human colon carcinomas by
monoclonal-antibody-targeted superantigens. Cancer Immunol
Immunother. 36:223–228. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lando PA, Hedlund G, Dohlsten M and
Kalland T: Bacterial superantigens as anti-tumour agents: Induction
of tumour cytotoxicity in human lymphocytes by staphylococcal
enterotoxin A. Cancer Immunol Immunother. 33:231–237. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dighe AS, Richards E, Old LJ and Schreiber
RD: Enhanced in vivo growth and resistance to rejection of tumor
cells expressing dominant negative IFN-γ receptors. Immunity.
1:447–456. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kaplan DH, Shankaran V, Dighe AS, Stockert
E, Aguet M, Old LJ and Schreiber RD: Demonstration of an interferon
gamma-dependent tumor surveillance system in immunocompetent mice.
Proc Natl Acad Sci USA. 95:7556–7561. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shankaran V, Ikeda H, Bruce AT, White JM,
Swanson PE, Old LJ and Schreiber RD: IFNgamma and lymphocytes
prevent primary tumour development and shape tumour immunogenicity.
Nature. 410:1107–1111. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Street SE, Cretney E and Smyth MJ:
Perforin and interferon-gamma activities independently control
tumor initiation, growth, and metastasis. Blood. 97:192–197. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Street SE, Trapani JA, MacGregor D and
Smyth MJ: Suppression of lymphoma and epithelial malignancies
effected by interferon gamma. J Exp Med. 196:129–134. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nishimura T, Nakui M, Sato M, Iwakabe K,
Kitamura H, Sekimoto M, Ohta A, Koda T and Nishimura S: The
critical role of Th1-dominant immunity in tumor immunology. Cancer
Chemother Pharmacol. 46:(Suppl). S52–S61. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Rosendahl A, Kristensson K, Hansson J,
Riesbeck K, Kalland T and Dohlsten M: Perforin and IFN-gamma are
involved in the antitumor effects of antibody-targeted
superantigens. J Immunol. 160:5309–5313. 1998.PubMed/NCBI
|
|
45
|
Melief CJ and Kast WM: T-cell
immunotherapy of tumors by adoptive transfer of cytotoxic T
lymphocytes and by vaccination with minimal essential epitopes.
Immunol Rev. 145:167–177. 1995. View Article : Google Scholar : PubMed/NCBI
|