Introduction
Breast cancer is the most frequently diagnosed
cancer and the leading cause of cancer death among females
worldwide, with an estimated 1.7 million cases and 521,900 deaths
in 2012. Especially, less developed countries account for
approximately one-half of all breast cancer cases and 62% of deaths
(1). Breast cancer is also the
leading cause of cancer death in women younger than 45 years in
China (2); most of these fatalities
are caused by metastatic disease. Approximately, 15–20% of breast
cancers are triple-negative breast cancer (TNBC) (3). TNBC is characterized by the absence of
estrogen receptor, progesterone receptor, and HER-2 expression;
this cancer results in high morbidity and mortality because of its
rapid growth rate, metastatic potential, and frequently acquired
treatment resistance (4). So far
there is no FDA (Food and Drug Administration)-approved targeted
therapy drug for TNBC patients. Considering that <30% of women
with metastatic TNBC survive 5 years (5), there is an urgent need to identify new
therapy targets and illustrate the molecular events of TNBC.
Cancerous inhibitor of protein phosphatase 2A
(CIP2A) is a cellular inhibitor of the important tumor-suppressor
protein, protein phosphatase 2A (PP2A) (6). PP2A functions as a serine/threonine
protein phosphatase that regulates several important oncogenic
proteins such as Akt, ERK, c-Myc, and p70S6K (7,8). When
PP2A is inhibited by CIP2A, PP2A-mediated dephosphorylation of
these oncoproteins is blocked, thus promoting anchorage-independent
cell growth and in vivo tumor formation. CIP2A functions as
an oncoprotein that promotes proliferation and aggressiveness of
several cancer types including head and neck squamous cell
carcinoma, oral squamous cell carcinoma, esophageal squamous cell
carcinoma, colon, gastric, breast, prostate, tongue, lung, and
cervical cancer, as well as acute myeloid leukemia (6,9–12). In
addition, aberrant expression, mutations, and somatic alterations
of the PP2A scaffold and regulatory subunits are frequently found
in human breast, lung, colon, and skin cancers (13). Therefore, reactivation of PP2A
activity based on its tumor suppressor properties through
inhibiting of CIP2A is considered to be an attractive therapeutic
strategy for human cancer treatment (14,15).
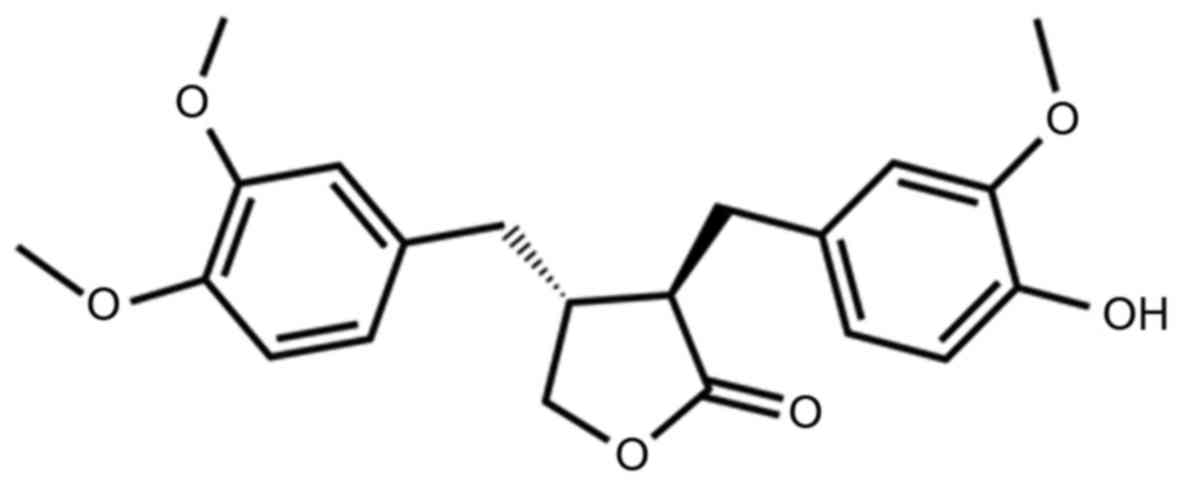
Arctigenin (Atn, Fig.
1) is a bioactive lignan isolated from the seeds of Arctium
lappa L. Atn demonstrates anti-inflammatory effects by
inhibiting nuclear transcription factor-κ B (NF-κB) (16). Atn can also enhance the
chemosensitivity of several cancer cells (HepG2, HeLa, and K562),
to cisplatin by inhibiting the STAT3 signaling pathway at high
doses (17). As we showed in our
previous study (18), Atn inhibited
cell growth and induced apoptosis in TNBCs by inhibiting STAT3. We
used computational docking and molecular dynamics simulation showed
that Atn had high-affinity interaction with the SH2 domain of STAT3
with residues (Arg688, Pro689, and Pro695). Atn inhibited STAT3
binding to genomic DNA by disrupting hydrogen bond binding between
DNA and STAT3. However, whether Atn influences other pathways
correlated to tumor aggressiveness and whether Atn functions as a
multi-targeted drug requires further study.
In this study, we tested the effects of Atn on CIP2A
expression in TNBC cells, and showed that Atn inhibits metastasis
of TNBC cells with inhibition of CIP2A expression to reactivate
PP2A activation. These data indicate that Atn is a potential
multi-targeted drug for the treatment of TNBC.
Materials and methods
Reagents
Arctigenin (Atn) with a purity of ≤98% was purchased
from Shanghai Yuanye Bio-Technology Co., Ltd. (Shanghai, China).
Atn was dissolved in DMSO (Sigma-Aldrich, St. Louis, MO, USA) at a
stock solution of 50 mM and stored at −20°C. 3-(4,5)-dimethylthiazol(−z-y1)-3,
5-di-phenytetrazolium bromide (MTT) and okadaic acid (OA) were
purchased from Sigma-Aldrich.
Cell culture
Human TNBC lines MDA-MB-231 and MDA-MB-468 were
obtained from American Type Culture Collection (ATCC, Manassas, VA,
USA), and maintained in Leibovitz's L-15 (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% FBS
(Hyclone, GE Healthcare Life Sciences, Chalfont, UK) and
antibiotics and incubated in a humidified atmosphere without
CO2 at 37°C.
Western blotting
Cell pellets were lysed in RIPA buffer containing 50
mM Tris pH 8.0, 150 mM NaCl, 0.1% SDS, 0.5% deoxycholate, 1% NP-40,
1 mM DTT, 1 mM NaF, 1 mM sodium vanadate, 1 mM PMSF (Sigma-Aldrich,
Merck Millipore, Darmstadt, Germany), and 1% protease inhibitors
cocktail (Merck, Millipore). Lysates were normalized for total
protein (25 µg) and loaded on 8–12% sodium dodecyl sulfate
polyacrylamide gel, electrophoresed, and transferred to a PVDF
membrane (Millipore, Kenilworth, NJ, USA), followed by blocking
with 5% skimmed milk at room temperature for 1 h. The membrane was
incubated with primary antibodies overnight at 4°C and rinsed with
Tris-buffered saline with Tween-20. The primary antibodies used
were anti-phospho-ERK1/2 (Thr202/Tyr204) (1:1,000 dilation; cat.
no. 9010), anti-ERK1/2 (1:1,000 dilation; cat. no. 9102),
anti-PP2Ac (1:1,000 dilation; cat. no. 2038, anti-caspase-3
(1:1,000 dilation; cat. no. 9662), anti-caspase-9 (1:1,000
dilation; cat. no. 9508), anti-PARP (1:1,000 dilation; cat. no.
9542) (Cell Signaling Technology, Danvers, MA, USA), anti-CIP2A
(1:500 dilation; cat. no. sc-80662), anti-phospho-Akt (Ser473)
(1:500 dilation; cat. no. sc-7985), anti-Akt (1:500 dilation; cat.
no. sc-8312), anti-CDK2 (1:500 dilation; cat. no. sc-6248) (Santa
Cruz Biotechnology, Santa Cruz, CA, USA), and anti-GAPDH (1:5,000
dilation; cat. no. M20006; Abmart, Shanghai, China) antibodies. The
blots were then washed and incubated with horseradish peroxidase
(HRP)-conjugated secondary antibody (1:10,000 dilation; cat. no.
E030120-01 and E030110-01; EarthOx, LLC, San Francisco, CA, USA) at
room temperature for 1.5 h. Detection was performed by using a
SuperSignal® West Pico Trial kit (cat. no. QA210131;
Pierce Biotechnology, Inc., Rockford, IL, USA) (19). The defined sections of the film were
scanned for image capture and quantification using Adobe Photoshop
software (CS4, Adobe Systems Inc., USA) and ImageJ software
(National Institutes of Health, Bethesda, MD, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Expression of the CIP2A gene was examined by
real-time polymerase chain reaction (RT-PCR) normalized to
expression of GAPDH. Total RNA was extracted from cells using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. RT-qPCR analysis of
CIP2A was performed with 2 µg of total RNA and ReverTra Ace
qPCR RT kit (Toyobo Co., Ltd. Life Science Department, Osaka
Japan). Mixed 2 µg RNA, 4 µl 5X RT buffer, 1 µl RT enzyme mix, 1 µl
primer mix, and nuclease-free water ≤20 µl volume. The reverse
transcription step was: 37°C for 15 min; 98°C for 5 min, then
stored at −20°C. For RT-qPCR, we used CIP2A gene forward
primer 5′-5′-TGCGGCACTTGGAGGTAATTTC-3′, CIP2A gene reverse
primer 5′-AGCTCTACAAGGCAACTCAAGC-3′; GAPDH forward primer
5′-TGTTGCCATCAATGACCCCTT-3′, GAPDH reverse primer
5′-CTCCACGACGTACTCAGCG-3′. RT-qPCR was performed in an ABI
StepOnePlus™ Real-Time PCR system (ABI; Thermo Fisher Scientifc,
Inc.) using SYBR® Green Real-Time PCR Master Mix (Toyobo
Co., Ltd. Life Science Department). Mixed SYBR Green PCR Master Mix
10 µl, forward and reverse primers 200 nM, cDNA template 100 ng,
and ddH2O ≤20 µl volume. PCR conditions consisted of the
following: 95°C for 3 min for denaturation; 95°C for 15 sec for
annealing; and 60°C for 1 min for extension, for 40 cycles. The
threshold cycle for each sample was selected from the linear range
and converted to a starting quantity by interpolation from a
standard curve generated on the same plate for each set of primers.
The CIP2A mRNA levels were normalized for each well to the GAPDH
mRNA levels using the 2−∆∆Cq method (20). Each experiment was repeated three
times.
PP2A activity assay
PP2A immunoprecipitation phosphatase assay kit
(Upstate, Temecula, CA, USA) was used to measure phosphate release
as an index of phosphatase activity according to the manufacturer's
instructions. Briefly, 100 µg protein isolated from cells was
incubated with 4 µg anti-PP2A monoclonal antibody overnight.
Protein A agarose beads (40 µl) were added and the mixture was
incubated at 4°C for 2 h. Subsequently, the beads were collected
and washed three times with 700 µl of ice-cold TBS and one time
with 500 µl Ser/Thr assay buffer. The beads were further incubated
with 750 mM phosphopeptide in assay buffer for 10 min at 30°C with
constant agitation. Malachite Green Phosphate Detection solution
(100 µl) was added and the absorbance at 650 nm was measured on a
microplate reader (21).
Cytotoxic assay and cell
viability
Cells were seeded into 96-well plate and
pre-cultured for 24 h, then treated with Atn for 24 h. Cell
cytotoxicity was determined by MTT assay. The absorbance was
measured at 570 nm by automated microplated reader (Bio-Tek, VT,
USA), and the cell death rate was calculated as follows: Inhibition
rate (%) = (average A570 of the control group - average
A570 of the experimentalgroup) / (average
A570 of the control group - average A570 of
the blank group) × 100%. Cell viability was estimated by trypan
blue dye exclusion (19).
Transfection of siRNA
Two siRNAs targeting CIP2A were designed and
synthesized by Shanghai GenePharma Co., Ltd., (Shanghai, China)
referred to as siRNA1 and siRNA2. The siRNA sequences were as
follows: 5′-CUGUGGUUGUGUUUGCACUTT-3′ (CIP2A siRNA1),
5′-ACCAUUGAUAUCCUUAGAATT-3′ (CIP2A siRNA2),
5′-UUCUCCGAACGUGUCACGUTT-3′ [negative control (NC) siRNA]. Using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol, TNBC
cells were transfected with 100 nM siRNA. Also, 48 h after
transfection the cells were then harvested for western blotting,
and cell viability.
Transfection of DNA
The pOTENT-1-CIP2A expression plasmid was purchased
from Youbio Co. Ltd. (Changcha, China). Transfection of the the
pOTENT-1-CIP2A plasmid into TNBC cells were carried out using
Lipofectamine® 3000 transfection reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) following the manufacturer's
protocol.
Flow cytometric assays for Annexin V
(AV)
Cell apoptosis was evaluated by AV detection using
an AV-FITC kit (BD Biosciences, San Jose, CA, USA), according to
the manufacturer's instructions (22).
Invasion assay
An invasion assay was carried out using 24-well
plate (Corning). A polyvinyl-pyrrolidone-free polycarbonate filter
(8-µm pore size) (Corning) was coated with Matrigel (BD). The lower
chamber was filled with medium containing 20% FBS as
chemoattractant. The coated filter and upper chamber were laid over
the lower chamber. Cells (1×104 cells/well) were
preincubated with Atn for 30 min at room temperature, and then cell
suspension containing Atn was seeded onto the upper chamber wells.
After incubation for 20 h at 37°C, the filter was fixed and stained
with 2% ethanol containing 0.2% crystal violet (15 min). After
being dried, the stained cells were enumerated under a light
microscope at ×10 objective. For quantification, the invaded
stained cells on the other side of the membrane were extracted with
33% acetic acid. The absorbance of the eluted stain was determined
at 570 nm.
Wound healing assay
Cells (4×105 cells/2 ml) were seeded in a
6-well plate and incubated at 37°C until 90–100% confluence. Then
the confluent cells were scratched with a 200-µl pipette tip,
followed by washing with PBS, and then treated with Atn in a basic
medium. After 24 h of incubation, the cells were fixed and stained
with 2% ethanol containing 0.2% crystal violet powder (15 min), and
randomly chosen fields were photographed under a light microscope
at 4X objective. The number of cells migrated into the scratched
area was calculated.
Statistical analysis
All experiments were repeated at least three times
and the data are presented as the mean ± SD unless noted otherwise.
Differences between data groups were evaluated for significance
using Student's t-test of unpaired data or one-way analysis of
variance and Bonferroni post-test. P-values <0.05 indicate
statistical significance.
Results
Atn downregulates CIP2A transcription
and induces CIP2A proteolysis
In our previous study, we reported that CIP2A plays
a critical role in the proliferation and aggressiveness of TNBC
(23), and a natural compound
oridonin is able to downregulate CIP2A in lung cancer cells
(12). In this study, we found
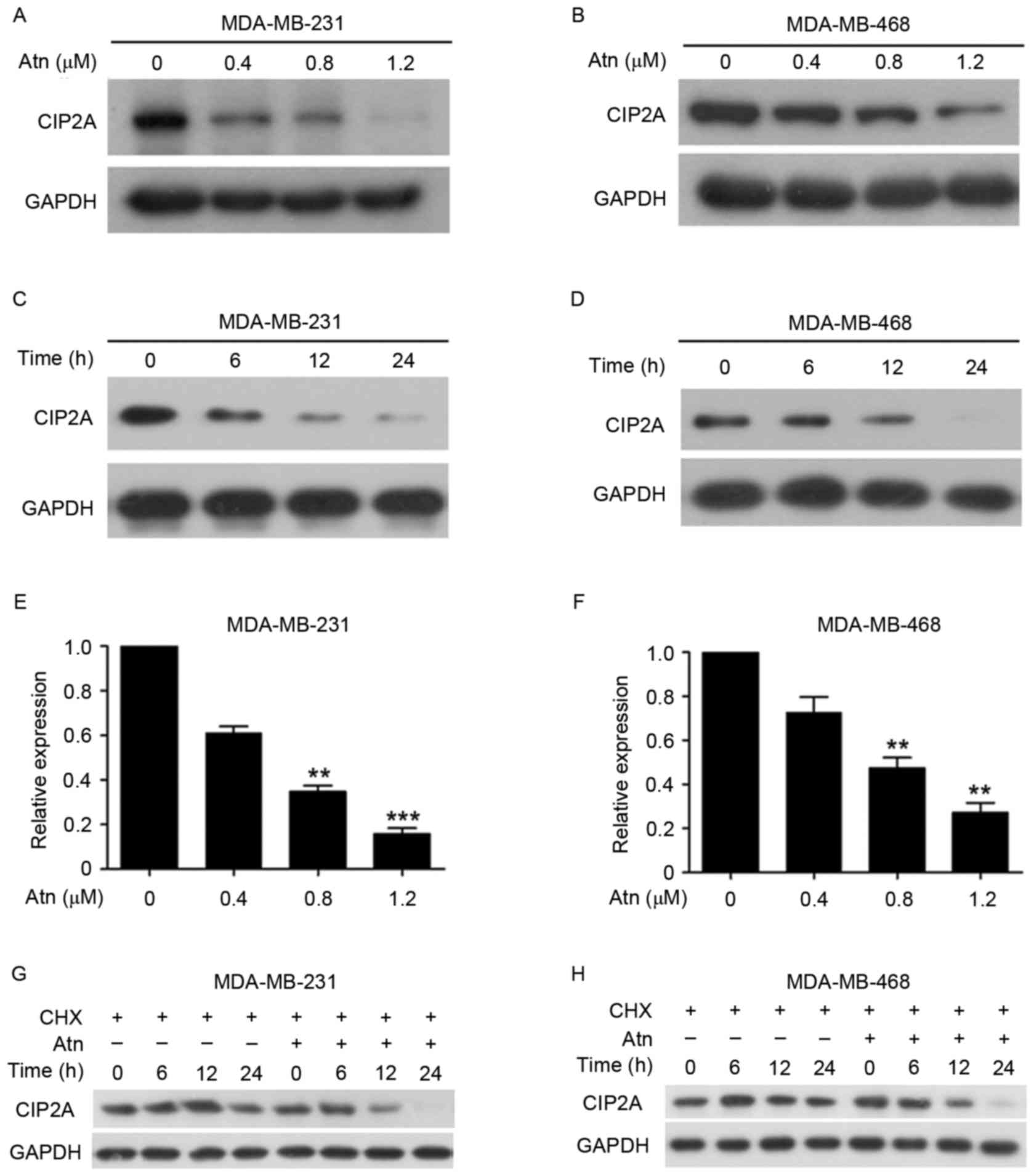
that, treatment with Atn at 0.4–1.2 µM for 24 h could downregulate
CIP2A expression in TNBC cell lines MDA-MB-231 and MDA-MB-468
(Fig. 2A and B). As shown in
Fig. 2A, the expression of CIP2A
protein was reduced in MDA-MB-231 cells exposed to Atn at 0.4 µM
and became undetectable in cells treated with Atn at 1.2 µM for 24
h. Also, treatment of MDA-MB-468 cells with Atn at 0.8 µM resulted
in an apparent reduction of CIP2A (Fig.
2B). We further showed that Atn caused downregulation of CIP2A
in a time-dependent manner (Fig. 2C and
D). We next investigated whether Atn affected CIP2A gene
transcription by RT-qPCR. We found that in response to Atn
treatment for 24 h, CIP2A was decreased drastically (Fig. 2E and F). These data indicate that
Atn decreases CIP2A transcription. Since Atn caused a
relatively rapid decrease of CIP2A, we hypothesized that Atn might
also affect CIP2A stability. Thus, next we used a protein synthesis
inhibitor cycloheximide (CHX) to block protein synthesis and then
we found that CIP2A was stable under treatment with CHX for >12
h. However, it was downregulated in 6 h in cells coincubated with
CHX and Atn (Fig. 2G and H). In
conclusion, these results indicate that Atn downregulates
CIP2A transcription and induce CIP2A proteolysis.
Downregulation of CIP2A leads to
increase of activated PP2A and decrease of phosphorylated Akt in
TNSC cells
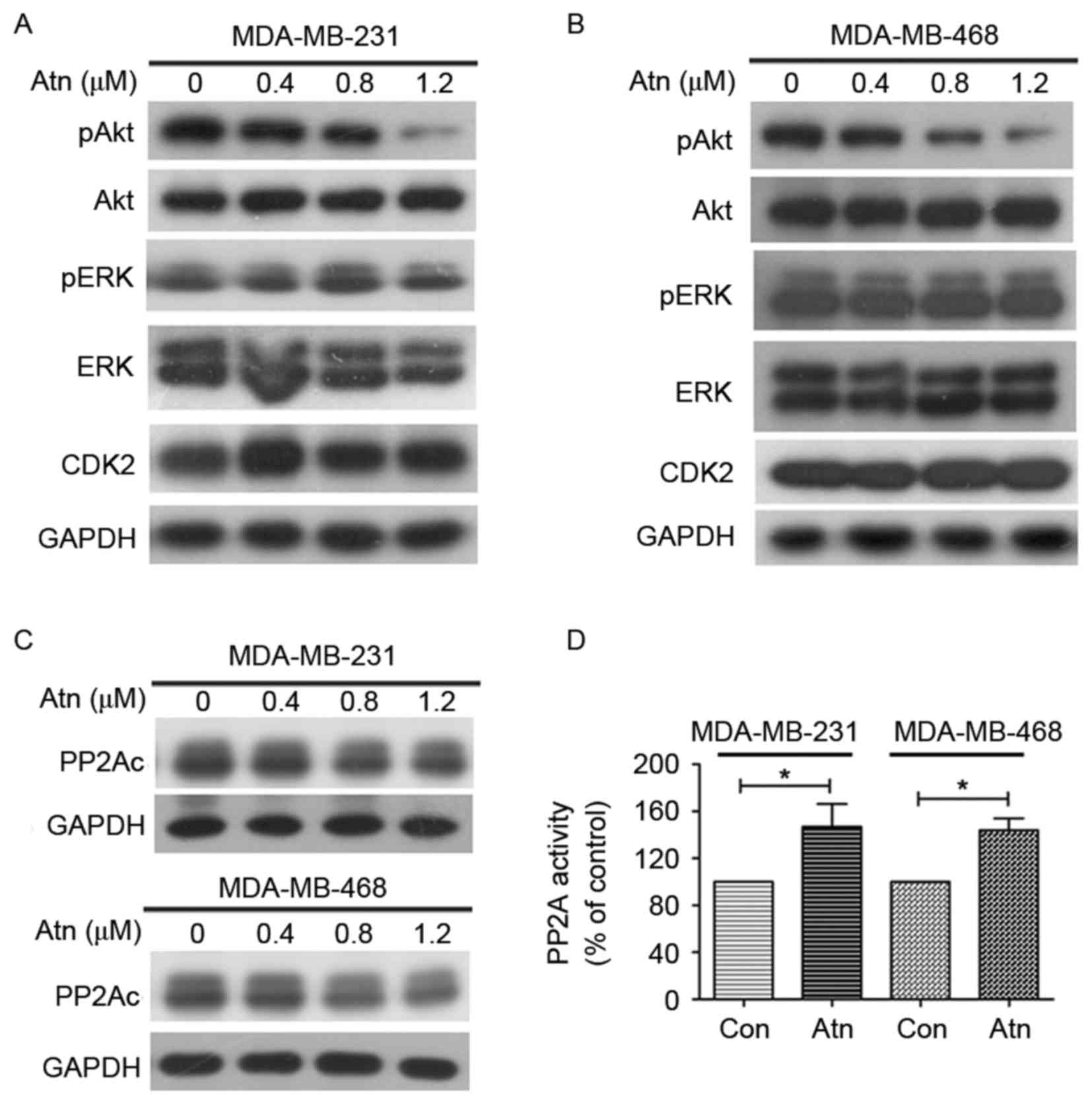
We next tested the expression of CIP2A downstream
molecule Akt and found that Atn treatment downregulated Akt
phosphorylation (pAkt) in both MDA-MB-231 (Fig. 3A) and MDA-MB-468 (Fig. 3B) cells, indicating Akt
inactivation. CIP2A is an endogenous inhibitor of PP2A and the
dephosphorylation of Akt is widely regulated by PP2A, we therefore
tested whether Atn could influence PP2A activity. As shown in
Fig. 3C, Atn treatment has no
effect on the expression of PP2Ac (catalytic subunit). We also
tested another downstream molecule of PP2A, ERK, and found that Atn
has no effect on ERK phosphorylation (pERK). Furthermore, we
investigated PP2A activity and found that PP2A was upregulated in
cells treated with Atn (Fig. 3D),
suggesting that Atn may affect CIP2A/PP2A/pAkt signaling axis.
Targeting CIP2A/PP2A/pAkt axis
promotes molecular mechanism of Atn-induced TNBC cell
apoptosis
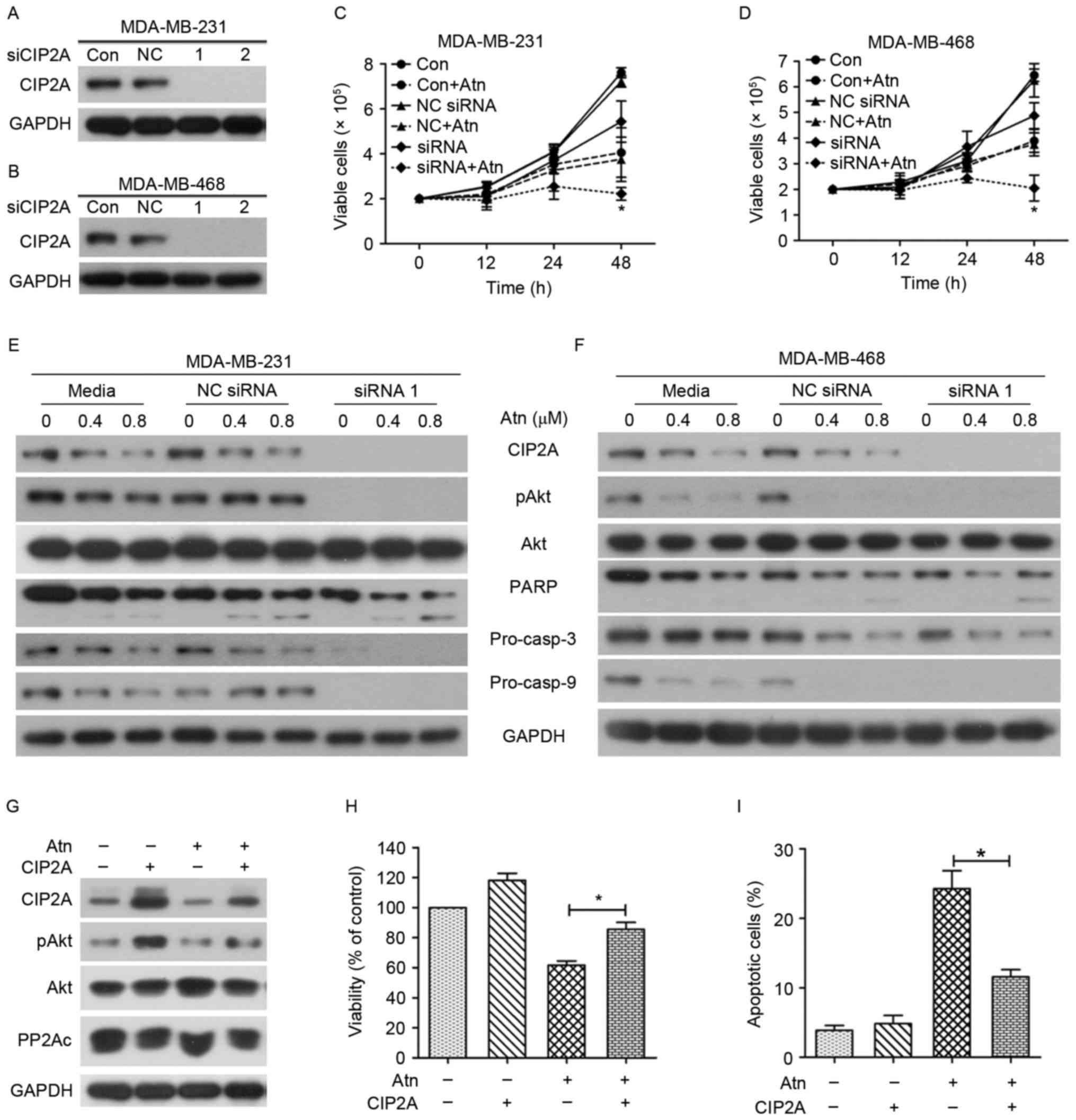
We subsequently examined whether CIP2A knockdown
would alter cellular sensitivity to Atn. Two siRNAs targeting CIP2A
were synthesized and applied to MDA-MB-231 and MDA-MB-468 cells
with different Atn concentrations. Both siRNA1 and siRNA2 knockdown
considerably decreased the expression of the CIP2A protein in each
TNBC cell line (Fig. 4A and B).
This finding validated the specificity and effectiveness of CIP2A
siRNAs. To evaluate the role of CIP2A in Atn-induced proliferation
inhibition and apoptosis, we transfected MDA-MB-231 and MDA-MB-468
cells with siRNA1 or siRNA2 targeting CIP2A and then subjected to
Atn treatment. Cell viability and western blot assays were used to
detect variations in cell growth and protein expression. Notably,
CIP2A silencing enhanced Atn-induced growth inhibition (Fig. 4C and D) and promoted Atn-induced
apoptosis effects (Fig. 4E and
F).
To further confirm the role of the CIP2A/PP2A/pAkt
axis in mediating induction of apoptosis of TNBC cells by Atn, we
generated MDA-MB-231 cells with ectopic overexpression of CIP2A by
transient transfection (Fig. 4G).
Compared with wild-type cells, the expression level of pAkt was
upregulated in the CIP2A-overexpressing cells. Cell viability was
used to detect variations in cell growth. Notably, CIP2A
overexpression abolished Atn-induced growth inhibition (Fig. 4H) and inhibited Atn-induced
apoptosis effects (Fig. 4I). These
findings demonstrated that CIP2A, at least in part, plays a
critical role in Atn-triggered TNBC proliferation inhibition. Atn
also enhanced growth inhibition and apoptosis in CIP2A-silencing
TNBC cells. Thus, these results confirmed that Atn induced cell
proliferation inhibition and apoptosis, at least in part, by
downregulating CIP2A.
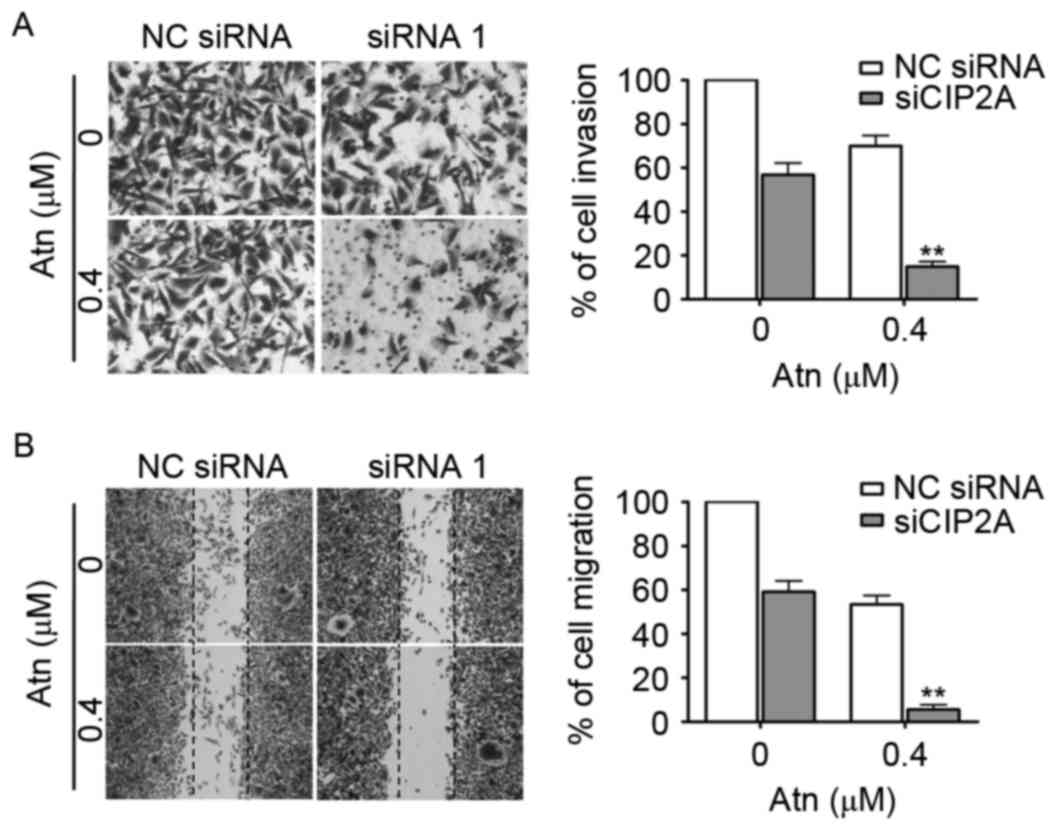
Silencing CIP2A enhances Atn-inhibited
invasive behavior of TNBC cells
Previous studies, including ours reported that CIP2A
promoted the aggressiveness of breast cancer (23,24).
Next, we examined the effect of CIP2A depletion on Atn-inhibited
invasive behavior. The invasion assay was performed in highly
invasive MDA-MB-231 cells using Matrigel-coated 24-well
microchemotaxis chambers. As shown in Fig. 5A, MDA-MB-231 cells were treated with
CIP2A siRNA (100 nM) then Atn (0.4 µM), and cell invasion was
determined after 20 h. CIP2A depletion markedly suppressed the
invasion and enhanced the effect of Atn. In addition, we explored
the effect of CIP2A depletion on migration, MDA-MB-231 cells were
treated with CIP2A siRNA (100 nM) then Atn (0.4 µM), and cell
migration was determined after 48 h. As shown in Fig. 5B, CIP2A depletion significantly
decreased MDA-MB-231 cell migration and enhanced the effect of
Atn.
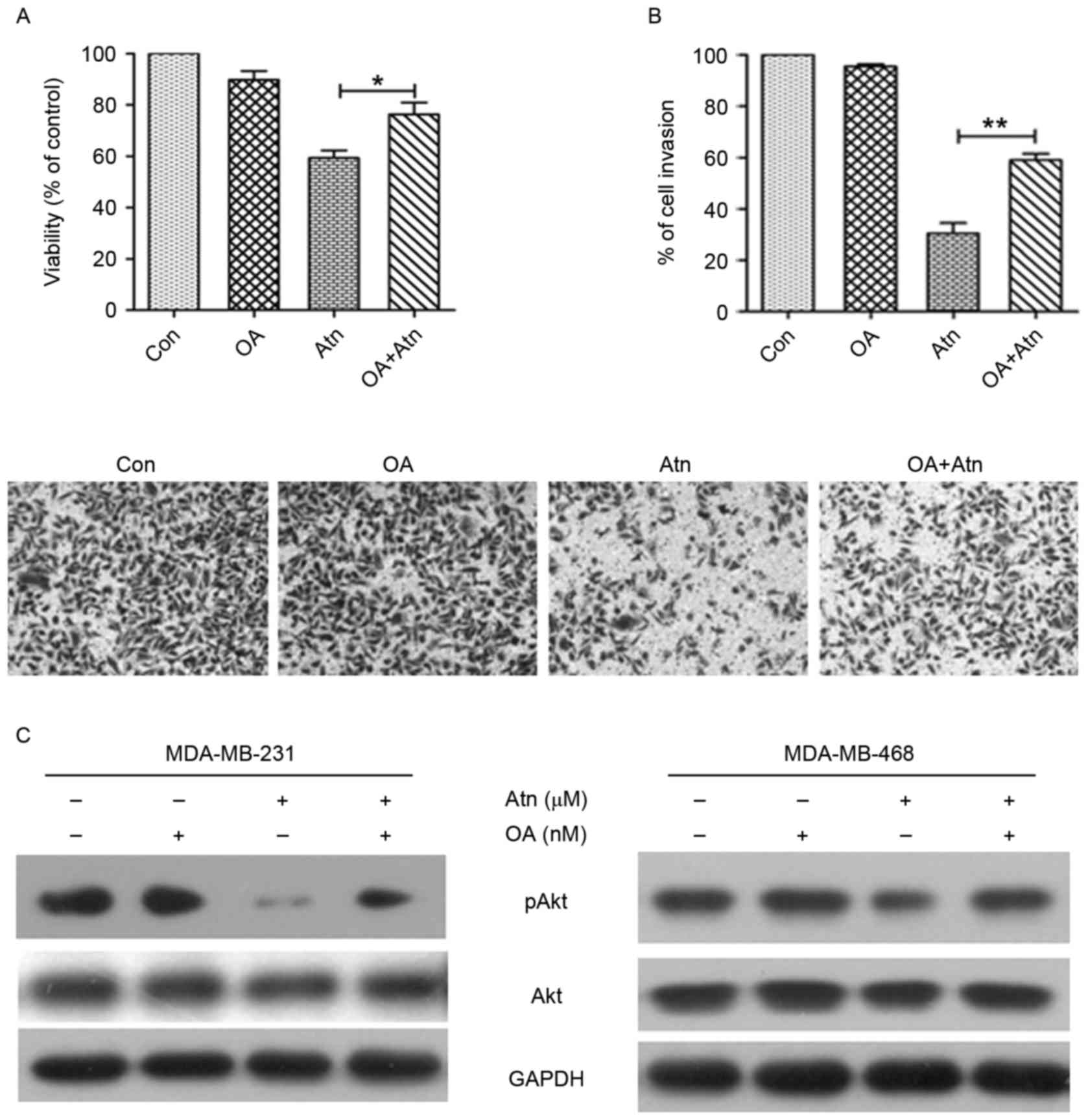
Reactivation of PP2A is essential for
Atn-inhibited proliferation and invasion
To evaluate whether Atn inhibited cell proliferation
and invasion is due to reactivation of PP2A activity, we compared
cell proliferation and invasion in Atn-treated cells in the
presence and absence of PP2A inhibitor okadaic acid (OA). Our
results showed that OA significantly reversed the cell
proliferation and invasion inhibited by Atn (Fig. 6A and B). We next examined whether
the inhibition of PP2A had any effect in the phosphorylation status
of Akt, the target of PP2A. Consistent with our expectation,
treatment of OA rescued Akt phosphorylation in Atn-treated cells.
Similar results were also observed in MDA-MB-468 cells (Fig. 6C). Taken together, these findings
suggested that reactivation of PP2A is essential for Atn-inhibited
proliferation and invasion via CIP2A inhibition.
Discussion
TNBC is a heterogeneous disease for which there is
no targeted therapy as they do not express the respective drug
targets (5). Moreover, TNBC is a
highly aggressive form of breast cancer that is associated with
early peak of recurrence, advanced stage at diagnosis and poorer
outcome in comparison with non-TNBCs; in the advanced setting,
responses to radiation therapy and chemotherapy lack durability.
Finally, women who are diagnosed with TNBC tend to be younger than
non-TNBC patients (25). Thus,
there is an urgent need to develop a targeted therapy to inhibit
the progression and metastasis of TNBC.
Our previous observations found that Atn inhibits
proliferation and induces apoptosis in TNBCs cells by the
inhibition of STAT3 activation. Atn was identified to inhibit STAT3
binding to genomic DNA by disrupting hydrogen bond binding between
DNA and STAT3. In addition, Atn augmented
Taxotere®-induced TNBC cell cytotoxicity (18). Here, we further showed that Atn was
also able to cause a significant dose- and time-dependent decrease
of CIP2A at both mRNA and protein levels in TNBCs (Fig. 2).
We tested the expression of CIP2A downstream
molecule Akt and found that Atn treatment downregulated pAkt (but
not total Akt) in both MDA-MB-231 (Fig.
3A) and MDA-MB-468 (Fig. 3B)
cells. CIP2A is an endogenous inhibitor of PP2A and the
dephosphorylation of Akt is widely regulated by PP2A. Next, we
detected PP2A phosphatase activity and found that Atn significantly
upregulated PP2A activity in both MDA-MB-231 and MDA-MB-468 cells
(Fig. 3D). PP2A is a
serine/threonine phosphatase that has a critical role in regulating
various cellular processes, including signaling transduction,
protein synthesis, cell cycle determination, metabolism, apoptosis
and stress response (26). Because
loss of PP2A function has been identified in various malignant
diseases such as cancer of the lung, liver, colon, and breast, it
has been reported that enhancing PP2A activity could be an
effective approach for cancer treatment (27). Thus, we showed that by Atn-induced
inhibition of CIP2A, activity of PP2A was significantly enhanced
and expression of pAkt was downregulated in TNBCs cells.
Moreover, we demonstrated that knockdown of CIP2A
enhanced the proliferation inhibition and apoptosis effect of Atn
in TNBCs and activated PP2A (Fig.
4C-F). Furthermore, ectopic expression of CIP2A upregulated the
expression of pAkt (Fig. 4G),
indicating that PP2A was inactivated and then Akt was activated.
Then, we examined whether ectopic expressed CIP2A affected the
treatment of Atn. Atn can downregulate CIP2A expression in
MDA-MB-231 cell overexpressed CIP2A (Fig. 4G). We also detected proliferation
and apoptosis of MDA-MB-231 cell overexpressed CIP2A (Fig. 4H and I), and found that CIP2A
overexpression overcame the proliferation inhibition and apoptosis
effect of Atn. These data validated the mechanism by which
Atn-induced cancer cell apoptosis in TNBC cells, that is, induction
of cancer cell apoptosis, at least in part, by inhibiting CIP2A and
enhancing pAkt downregulation.
Migration and invasion are two important
prerequisites of TNBC cancer progression and metastasis. Our
previous research showed that CIP2A depletion markedly inhibited
the invasive and migratory abilities of MDA-MB-231 cells. We
investigated whether CIP2A depletion could enhance the effects of
Atn in MDA-MB-231 cell line and found that silencing CIP2A
significantly enhanced Atn-induced migration and invasion
inhibition in MDA-MB-231 cells (Fig.
5). To further investigate the effect of PP2A activation in Atn
induced TNBC proliferation and invasion inhibition, we co-incubated
OA, the inhibitor of PP2A, with Atn in TNBC cells. The results
showed that loss of PP2A function can antagonize the inhibition
effect of Atn in TNBC cell proliferation and invasion (Fig. 6A and B) through Akt pathway
(Fig. 6C). These results confirmed
that PP2A reactivation plays a critical role in Atn induced CIP2A
inhibition of downregulation mediated TNBC proliferation and
invasion.
In conclusion, we demonstrated the Atn novel
therapeutic mechanism in TNBC, that is, enhancement of
PP2A-dependent pAkt downregulation by inhibition of CIP2A. Our
results showed that designing agents to reactivate PP2A may be a
feasible strategy for the development of novel TNBC therapies.
Further studies on the detailed molecular modification of the
CIP2A/PP2A/Akt signaling axis by Atn and exploring its possible
application in other cancers are warranted.
Acknowledgements
This study was supported by grants from the National
Natural Sciences Foundation of China (grant no. 81400157); the
Natural Science Foundation of Hubei Provincial Department of
Education (grant nos. Q20152106 and Q20162114); the Natural Science
Foundation of Hubei Province of China (grant nos. 2016CFB528 and
2015CFB212); the Foundation of Health and Family planning
Commission of Hubei Province (grant nos. WJ2015MB291 and
WJ2017F065, 067); the Faculty Development Grants from Hubei
University of Medicine (grant nos. 2014QDJZR06, 2014QDJZR08 and
2015QDJZR16); the Foundation of Hubei University of Medicine (grant
nos. FDFR201605 and FDFR201609); and the Key Discipline Project of
Hubei University of Medicine.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang ZT, Chen ZJ, Jiang GM, Wu YM, Liu T,
Yi YM, Zeng J, Du J and Wang HS: Histone deacetylase inhibitors
suppress mutant p53 transcription via HDAC8/YY1 signals in triple
negative breast cancer cells. Cell Signal. 28:506–515. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Deng XS, Wang S, Deng A, Liu B, Edgerton
SM, Lind SE, Wahdan-Alaswad R and Thor AD: Metformin targets Stat3
to inhibit cell growth and induce apoptosis in triple-negative
breast cancers. Cell Cycle. 11:367–376. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shu S, Lin CY, He HH, Witwicki RM,
Tabassum DP, Roberts JM, Janiszewska M, Huh SJ, Liang Y, Ryan J, et
al: Response and resistance to BET bromodomain inhibitors in
triple-negative breast cancer. Nature. 529:413–417. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Junttila MR, Puustinen P, Niemelä M, Ahola
R, Arnold H, Böttzauw T, Ala-aho R, Nielsen C, Ivaska J, Taya Y, et
al: CIP2A inhibits PP2A in human malignancies. Cell. 130:51–62.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rincón R, Cristóbal I, Zazo S, Arpí O,
Menéndez S, Manso R, Lluch A, Eroles P, Rovira A, Albanell J, et
al: PP2A inhibition determines poor outcome and doxorubicin
resistance in early breast cancer and its activation shows
promising therapeutic effects. Oncotarget. 6:4299–4314. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tsukamoto S, Huang Y, Umeda D, Yamada S,
Yamashita S, Kumazoe M, Kim Y, Murata M, Yamada K and Tachibana H:
67-kDa laminin receptor-dependent protein phosphatase 2A (PP2A)
activation elicits melanoma-specific antitumor activity overcoming
drug resistance. J Biol Chem. 289:32671–32681. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li W, Ge Z, Liu C, Liu Z, Björkholm M, Jia
J and Xu D: CIP2A is overexpressed in gastric cancer and its
depletion leads to impaired clonogenicity, senescence, or
differentiation of tumor cells. Clin Cancer Res. 14:3722–3728.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ren J, Li W, Yan L, Jiao W, Tian S, Li D,
Tang Y, Gu G, Liu H and Xu Z: Expression of CIP2A in renal cell
carcinomas correlates with tumour invasion, metastasis and
patients' survival. Br J Cancer. 105:1905–1911. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu CY, Shiau CW, Kuo HY, Huang HP, Chen
MH, Tzeng CH and Chen KF: Cancerous inhibitor of protein
phosphatase 2A determines bortezomib-induced apoptosis in leukemia
cells. Haematologica. 98:729–738. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xiao X, He Z, Cao W, Cai F, Zhang L, Huang
Q, Fan C, Duan C, Wang X, Wang J, et al: Oridonin inhibits
gefitinib-resistant lung cancer cells by suppressing
EGFR/ERK/MMP-12 and CIP2A/Akt signaling pathways. Int J Oncol.
48:2608–2618. 2016.PubMed/NCBI
|
|
13
|
Seshacharyulu P, Pandey P, Datta K and
Batra SK: Phosphatase: PP2A structural importance, regulation and
its aberrant expression in cancer. Cancer Lett. 335:9–18. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schönthal AH: Role of serine/threonine
protein phosphatase 2A in cancer. Cancer Lett. 170:1–13. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lv P, Wang Y, Ma J, Wang Z, Li JL, Hong
CS, Zhuang Z and Zeng YX: Inhibition of protein phosphatase 2A with
a small molecule LB100 radiosensitizes nasopharyngeal carcinoma
xenografts by inducing mitotic catastrophe and blocking DNA damage
repair. Oncotarget. 5:7512–7524. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cho MK, Park JW, Jang YP, Kim YC and Kim
SG: Potent inhibition of lipopolysaccharide-inducible nitric oxide
synthase expression by dibenzylbutyrolactone lignans through
inhibition of I-kappaBalpha phosphorylation and of p65 nuclear
translocation in macrophages. Int Immunopharmacol. 2:105–116. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yao X, Zhu F, Zhao Z, Liu C, Luo L and Yin
Z: Arctigenin enhances chemosensitivity of cancer cells to
cisplatin through inhibition of the STAT3 signaling pathway. J Cell
Biochem. 112:2837–2849. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Feng T, Cao W, Shen W, Zhang L, Gu X, Guo
Y, Tsai HI, Liu X, Li J, Zhang J, et al: Arctigenin inhibits STAT3
and exhibits anticancer potential in human triple-negative breast
cancer therapy. Oncotarget. 8:329–344. 2017.PubMed/NCBI
|
|
19
|
Cao W, Liu Y, Zhang R, Zhang B, Wang T,
Zhu X, Mei L, Chen H, Zhang H, Ming P, et al: Homoharringtonine
induces apoptosis and inhibits STAT3 via IL-6/JAK1/STAT3 signal
pathway in Gefitinib-resistant lung cancer cells. Sci Rep.
5:84772015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu H, Gu Y, Wang H, Yin J, Zheng G, Zhang
Z, Lu M, Wang C and He Z: Overexpression of PP2A inhibitor SET
oncoprotein is associated with tumor progression and poor prognosis
in human non-small cell lung cancer. Oncotarget. 6:14913–14925.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu Y, Dong Y, Zhang B and Cheng YX: Small
compound 6-O-angeloylplenolin induces caspase-dependent apoptosis
in human multiple myeloma cells. Oncol Lett. 6:556–558.
2013.PubMed/NCBI
|
|
23
|
Li S, Feng TT, Guo Y, Yu X, Huang Q, Zhang
L, Tang W and Liu Y: Expression of cancerous inhibitor of protein
phosphatase 2A in human triple negative breast cancer correlates
with tumor survival, invasion and autophagy. Oncol Lett.
12:5370–5376. 2016.PubMed/NCBI
|
|
24
|
Come C, Laine A, Chanrion M, Edgren H,
Mattila E, Liu X, Jonkers J, Ivaska J, Isola J, Darbon JM, et al:
CIP2A is associated with human breast cancer aggressivity. Clin
Cancer Res. 15:5092–5100. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wright HJ, Arulmoli J, Motazedi M, Nelson
LJ, Heinemann FS, Flanagan LA and Razorenova OV: CDCP1 cleavage is
necessary for homodimerization-induced migration of triple-negative
breast cancer. Oncogene. 35:4762–4772. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cristobal I, Rincon R, Manso R, Caramés
C1, Zazo S2, Madoz-Gúrpide J2, Rojo F3 and García-Foncillas J:
Deregulation of the PP2A inhibitor SET shows promising therapeutic
implications and determines poor clinical outcome in patients with
metastatic colorectal cancer. Clin Cancer Res. 21:347–356. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu HC, Hung MH, Chen YL, Chu PY, Wang CY,
Chao TT, Liu CY, Shiau CW and Chen KF: Erlotinib derivative
inhibits hepatocellular carcinoma by targeting CIP2A to reactivate
protein phosphatase 2A. Cell Death Dis. 5:e13592014. View Article : Google Scholar : PubMed/NCBI
|