Introduction
Colorectal cancer (CRC) is one of the leading causes
of cancer-related death in the world. Phytochemicals are promising
anticancer agents given their remarkable chemical structure and
diverse biological activities (1)
and the prevention and treatment of cancer by dietary
phytochemicals that inhibit cell growth is an exciting aspect.
Subsequently, results from epidemiological studies have shown that
the consumption of cruciferous vegetables could contribute to
reduce the risk of CRC and other cancers, and the chemoprotective
effects of cruciferous vegetables have been reported in
carcinogen-induced colon cancer animal models (2).
Indole-3-carbinol (I3C) is a major bioactive
component of cruciferous vegetables, such as broccoli, cabbage,
brussels sprouts and cauliflower, which has been paid more
attention as a cancer preventive or chemotherapeutic agent
(3). As a major acid condensation
product of I3C, 3,3-Diindolylmethane (DIM) is readily detected in
the liver and feces of rodents fed I3C, whereas the parent I3C was
not detected in these animals, suggesting DIM may contribute to the
observed physiological effects of dietary I3C. Indeed, DIM has been
documented to inhibit cell proliferation and induce apoptosis in
colorectal cancer cells (4), and
other types of cancer cells including prostate (5), pancreas (6), breast (7), bladder (8) and hepatoma cancer (9). Mechanistically, DIM suppressed
proliferation via activating peroxisome proliferator-activated
receptor γ (PPARγ) and Nur77 activity in CRC cells, as well as
inducing apoptosis through inactivating AKT and NF-κB activity in
breast cancer cells (10).
Moreover, DIM and its derivatives induce endoplasmic reticulum (ER)
stress-mediated upregulation of death receptor 5 (DR5), causing
pancreatic cancer cell apoptosis (6). We have previously reported that DIM
stimulates ATF3 expression by ATF4-mediated pathway (4) which mediates apoptosis of colorectal
cancer cells (2). Given both ATF3
and ATF4 are closely associated with ER stress response, DIM could
trigger ER stress and subsequently induce growth inhibition and
apoptosis in CRC.
Cyclin D1, a well-identified oncogenic protein, is
often overexpressed in various types of cancer cells and tumors. It
plays crucial roles in cell cycle machinery by activating
cyclin-dependent kinase (CDK) 4/6, which subsequently
phosphorylates and inactivates retinoblastoma protein (pRb),
resulting in the progression from G1 to S phase of the cell cycle
(reviewed in ref. 11). Cyclin D1
functions as a critical regulator in DNA repair, suggesting
targeting cyclin D1 may be beneficial in both pRb-negative and
-positive cancer cells (12). A
large number of chemicals have been shown to downregulate cyclin D1
expression in different cancer cells by triggering multiple
signaling pathways (13). It has
been shown that DIM downregulates cyclin D1 in breast cancer cells,
which can be blocked by proteasome inhibitor (14). However, the effect of DIM on cyclin
D1 expression and the underlying mechanism(s) in colorectal cancer
cells remains to be investigated.
In the present study, we examined the effect of DIM
on cyclin D1 expression in CRC cells, and found that DIM caused
cyclin D1 downregulation independent of PPARγ and protease
activity. Furthermore, we revealed that DIM-triggered ER stress
mediated cyclin D1 translation inhibition.
Materials and methods
Reagents
DIM and cycloheximide were purchased from
Sigma-Aldrich (St. Louis, MO, USA). MG132 and epoxomicin was
obtained from Merck Millipore (Billerica, MA, USA). Antibodies for
cyclin D1, cyclin D3, cyclin E, Ubiquitin, Actin, ATF4 were from
Santa Cruz Biotechnology (Santa Cruz, CA, USA); Antibodies against
CHOP, PERK and total eIF2α were obtained from Cell Signaling
Technology (Danvers, MA, USA). Control siRNA (#6201) and specific
siRNA against PERK (#9024) were purchased from Cell Signaling
Technology. Cell culture media were purchased from (BioWhittaker,
Rockland, ME, USA). All other reagents were purchased from Thermo
Fisher Scientific Inc., (Pittsburgh, PA, USA), unless others
specified.
Cell culture
Human colorectal adenocarcinoma HCT-116, SW480,
HT-29, LoVo and Caco-2 cells were purchased from the American Type
Culture Collection (ATCC; Manassas, VA, USA). HCT-116 cells were
cultured in McCoys 5A; SW480 and LoVo were cultured in RPMI-1640
and Ham's F-12, respectively; HT-29 and Caco-2 cells were cultured
in Dulbecco's modified Eagles medium (DMEM). All media were
supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin
and 100 µg/ml streptomycin. Cells were kept at 37°C under a
humidified atmosphere of 5% CO2.
Plasmid and transient
transfection
Wild-type and S52A mutation of eIF2 alpha construct
(pcDNA.CD2/WT-eIF2A and pcDNA.CD2/S52A-eIF2A) were kind gifts from
Dr David Ron. Transient transfection was performed using PolyJet
reagent (SignaGen Laboratories, Rockville, MD, USA) according to
the manufacturers instruction.
RNA interference
HCT-116 cells were seeded on 6-well plates at a
density of 3.0×105 cells/well overnight. Control siRNA
and siPERK (Cell Signaling Technology) was transfected at a final
concentration of 100 nM using PepMute transfection reagent
(SignaGen Laboratories) according to the manufacturers
instruction.
Semi-quantitative reverse
transcription PCR
Total RNA of cells was isolated by E.Z.N.A Total RNA
kit (Omega Bio-Tek, Inc., Norcross, GA, USA) according to the
manufacturers protocol. Then RNA (1 µg) was reverse transcribed
using Verso cDNA synthesis kit (Thermo Fisher Scientific). PCR was
performed using GoTaq Green Master Mix PCR Mixture (Promega,
Madison, WI, USA) with primers for human cyclin D1, XBP1, ATF3 and
GAPDH as follows: cyclin D1, forward, 5-ATGGAACACCAGCTCCTGTGCTGC-3
and reverse, 5-TCAGATGTCCACGTCCCGCACGT-3; XBP1, forward,
5-CCTTGTAGTTGAGAACCAGG-3 and reverse, 5-GGGGCTTGGTATATATGTGG-3;
ATF3: forward, 5-GTTTGAGGATTTTGCTAACCTGAC-3 and reverse,
5-AGCTGCAATCTTATTTCTTTCTCGT-3; GAPDH, forward,
5-GGGCTGCTTTTAACTCTGGT-3 and reverse, 5-TGGCAGGTTTTTCTAGACGG-3.
Thermal cycling conditions were as follows: 95°C for 2 min,
followed by 25 cycles of 95°C for 30 sec, 60°C for 30 sec and 72°C
for 30 sec, and final extension at 72°C for 5 min. Each PCR product
was electrophoresed on agarose gel and viewed using ethidium
bromide staining under ultraviolet light. The intensity of bands
was analyzed by densitometry using the GAPDH band as a relative
control.
Western blot analysis
Cells were washed with phosphate-buffered saline
(PBS) and lysed using radio immunoprecipitation assay buffer (50
mmol/l Tris-HCl pH 7.4, 150 mmol/l NaCl, 1 mmol/l EDTA, 1% Triton
X-100, 1% sodium deoxycholate, 0.1% SDS) supplemented with 1X
protease inhibitor cocktail solution (Calbiochem, San Diego, CA,
USA) and phosphatase inhibitor (1 mmol/l
Na3VO4, 1 mmol/l NaF) and centrifuged at
13,000 × g for 10 min at 4°C. The supernatants were collected to
determine protein concentration by the BCA protein assay (Pierce,
Rockford, IL, USA) using bovine serum albumin (BSA) as the
standard. Then protein samples (30 mg) were mixed with an equal
amount of 2x Laemmli buffer and boiled for 5 min, subsequently
subjected to SDS-PAGE. The proteins were transferred to
nitrocellulose membranes (Osmonics, Minnetonka, MN, USA), which
were blocked with TBS containing 0.1 % Tween-20 (TBST) and 5%
non-fat milk for 1 h at room temperature, followed by incubation
with a specific primary antibody (1:1,000) in at 4°C overnight.
After three washes with TBST, the blots were incubated with
peroxidase-conjugated IgG for 1 h at room temperature, visualized
using ECL (Amersham Biosciences, Piscataway, NJ, USA) and
quantified by Scion Image Software (Scion Corp., Frederick, MD,
USA).
Results
Effect of DIM on cyclin D1 expression
in colorectal cancer cells
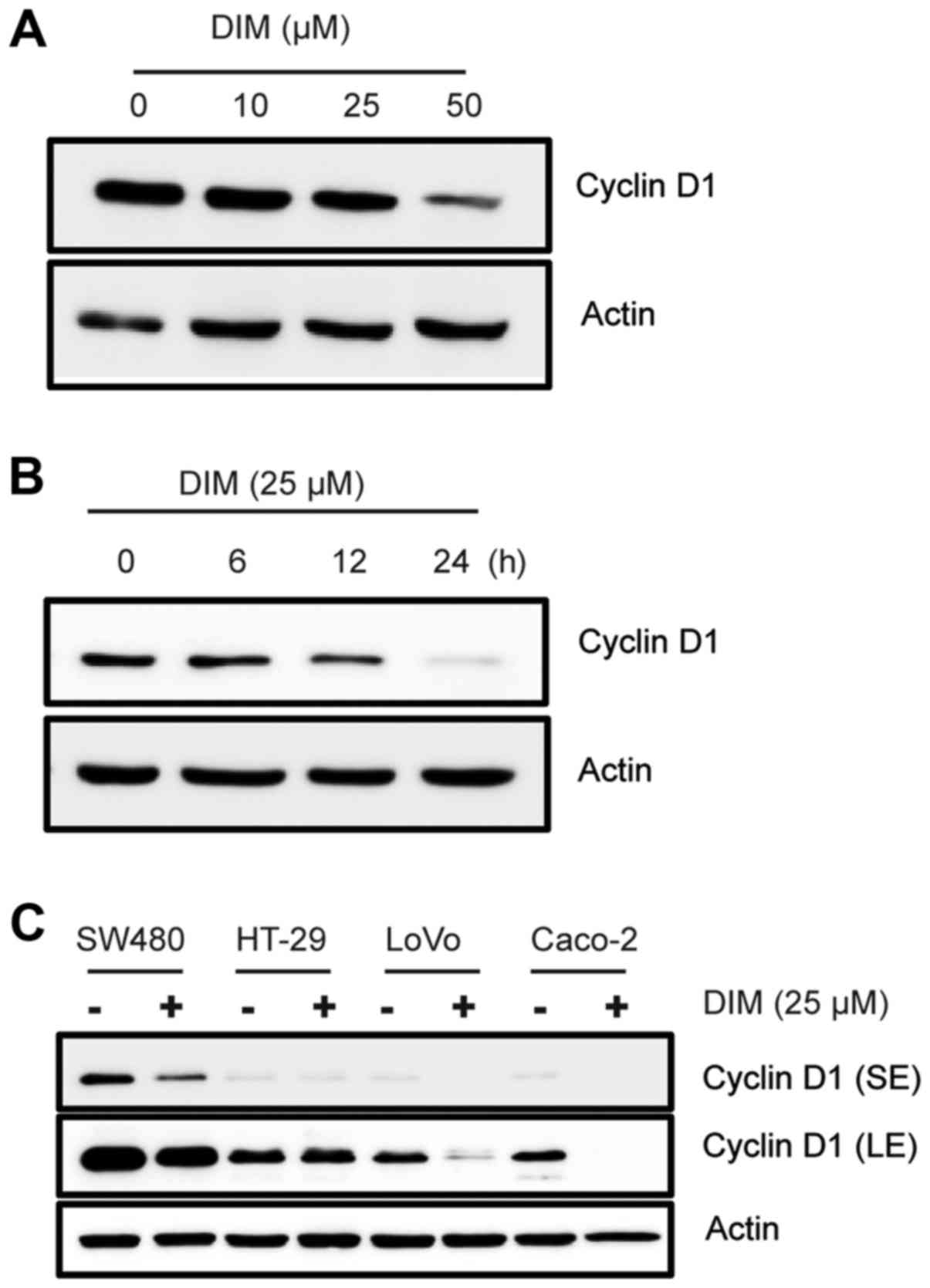
To investigate the effect of DIM on cyclin
expression in colorectal cancer cells, we treated HCT-116 cells
with DIM at different dose- and time-dependent manner. As a result,
DIM downregulated cyclin D1 and D3 expression in a dose- and
time-dependent manner (Fig. 1A and
B), but not cyclin E. Since DIM treatment exhibited the
strongest decrease in cyclin D1 expression, we further investigated
the alteration of cyclin D1 expression in other colorectal cancer
cells. Various cancer cells were treated with 25 µM DIM for 24 h
and cyclin D1 expression was measured. The result indicated that
DIM consistently decreased cyclin D1 in SW480, LoVo and CaCo-2
cells (Fig. 1C), suggesting that
cyclin D1 downregulation could be a potential mechanism for the
anti-proliferative effect of DIM.
Effect of DIM on cyclin D1 mRNA
expression and protein stability
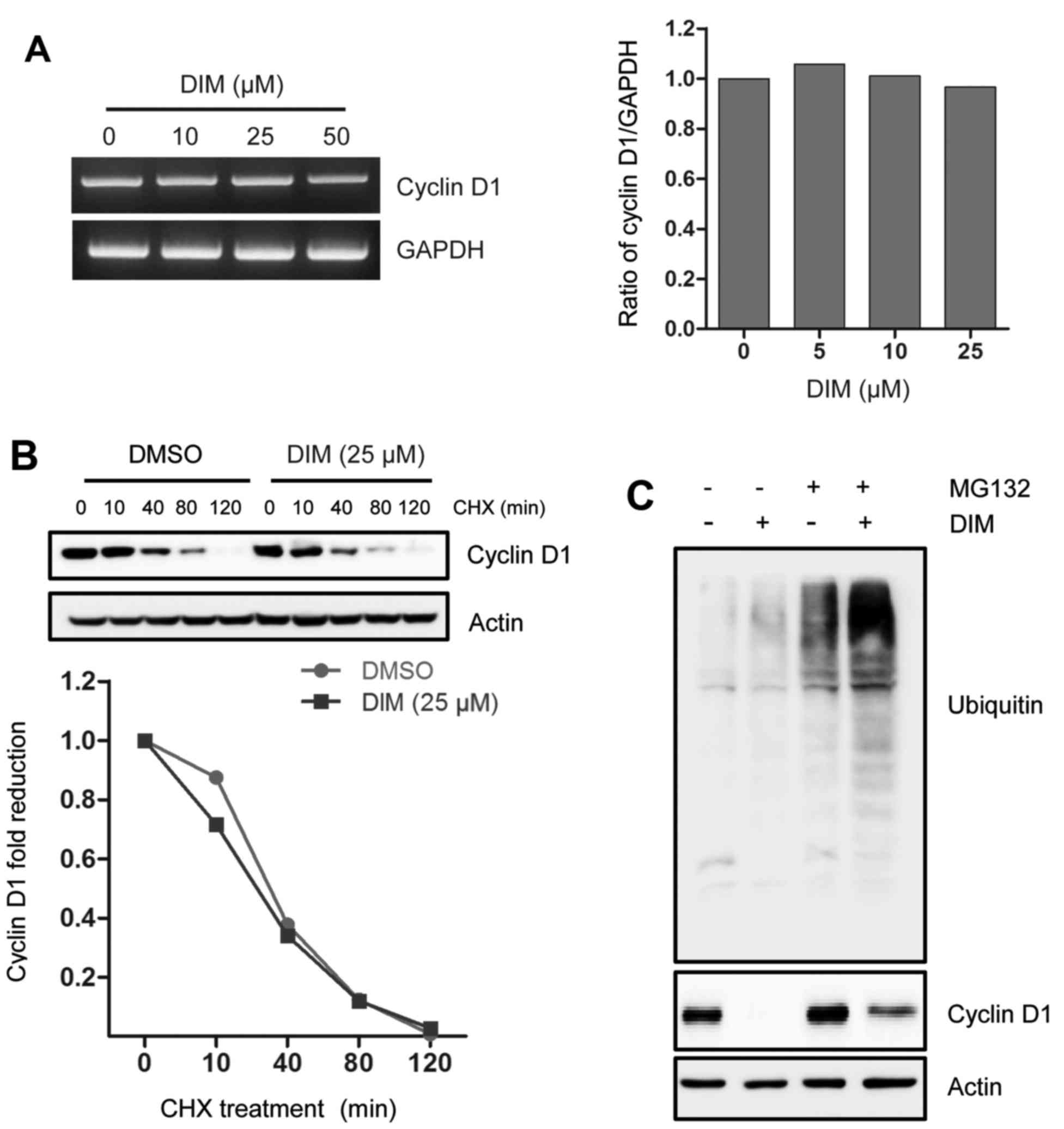
To better understand the underlying mechanism by
which DIM reduced cyclin D1 expression, we assessed the effect of
DIM on cyclin D1 mRNA expression and protein stability. As seen in
Fig. 2A, the level of cyclin D1
mRNA was not altered in the presence of DIM. Moreover, we examined
cyclin D1 protein stability, showing that DIM did not affect its
turnover rate when blocking protein synthesis (Fig. 2B). This result was further examined
using proteasomal inhibitor MG-132. As shown in Fig. 2C, a proteasome inhibitor MG-132
marginally restored cyclin D1 expression in the presence of DIM. It
is suggested that DIM marginally affects proteasomal pathway for
cyclin D1 degradation, although the level of ubiquitinated proteins
is increased by blocking ubiquitin-proteasome pathway.
Taking together, DIM deceased cyclin D1 independent
of degradation pathway and these data implicated that DIM-induced
cyclin D1 downregulation could occur at the translational
level.
Effects of signaling pathways on
DIM-mediated cyclin D1 downregulation
DIM has been reported to inhibit proliferation of
colorectal cancer cells through activating PPARγ activity (15). Thus, PPARγ antagonist was treated in
HCT-116 cells. As shown in Fig. 3A,
cyclin D1 was not affected by a PPARγ antagonist GW9662 in the
presence of DIM, indicating that its reduction was not associated
with PPARγ activation. To further examine other possible signaling
pathways mediated by the downregulation of cyclin D1 by DIM, we
treated cells with various inhibitors, including proteasome
inhibitor epoxomicin, cysteine protease inhibitor leupeptin, serine
protease inhibitor PMSF, pan-caspase inhibitor z-VAD-fmk, NF-κB
inhibitor Bay 11–7082 (BAY), PKC inhibitor Rottlerin (RO), p38MAPK
inhibitor SB203580 (SB), ERK inhibitor PD98059 (PD), and PI3K
inhibitor LY294002 (LY). As shown in Fig. 3B, epoxomicin apparently increased
basal cyclin D1 expression, suggesting basal cyclin D1 was mainly
regulated by the proteasome pathway. However, compared to treatment
with different inhibitors alone, co-treated with DIM consistently
downregulated cyclin D1 expression, except of RO and PD compound
because they decreased basal cyclin D1 expression as well (Fig. 3B and C). Collectively, PPARγ and
several kinase pathways seemed not to affect DIM-mediated cyclin D1
downregulation.
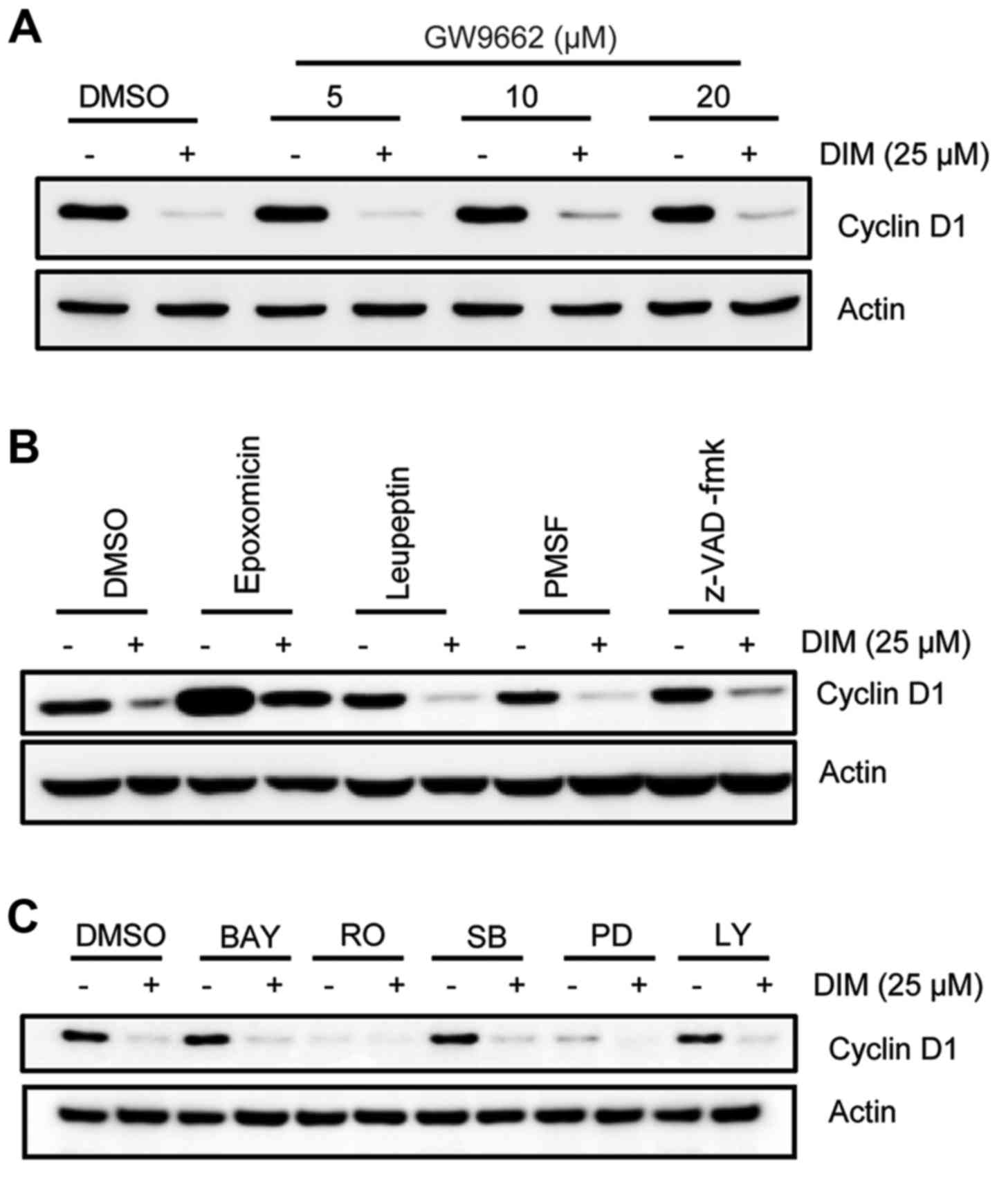 | Figure 3.Effect of signal pathways on
DIM-mediated cyclin D1 downregulation. (A) HCT-116 cells were
pretreated with GW9662 dose-dependently for 1 h, followed by
incubation with 25 µM DIM for another 24 h. Cell lysates were
analyzed using cyclin D1 antibody. Actin was as a loading control.
(B and C) HCT-116 cells were pretreated with 1 µM epoxomicin, 100
µM leupeptin, 1 mM PMSF, 50 µM z-VAD-fmk, 10 µM BAY11-7082 (BAY),
10 µM rottlerin (RO), 10 µM SB203580(SB), 40 µM PD98059 (PD), 10 µM
LY294002 (LY) for 1 h, then incubated with 25 µM DIM for 24 h. The
lysates were analyzed by western blot analysis. |
Effect of ER stress response on cyclin
D1 downregulation by DIM
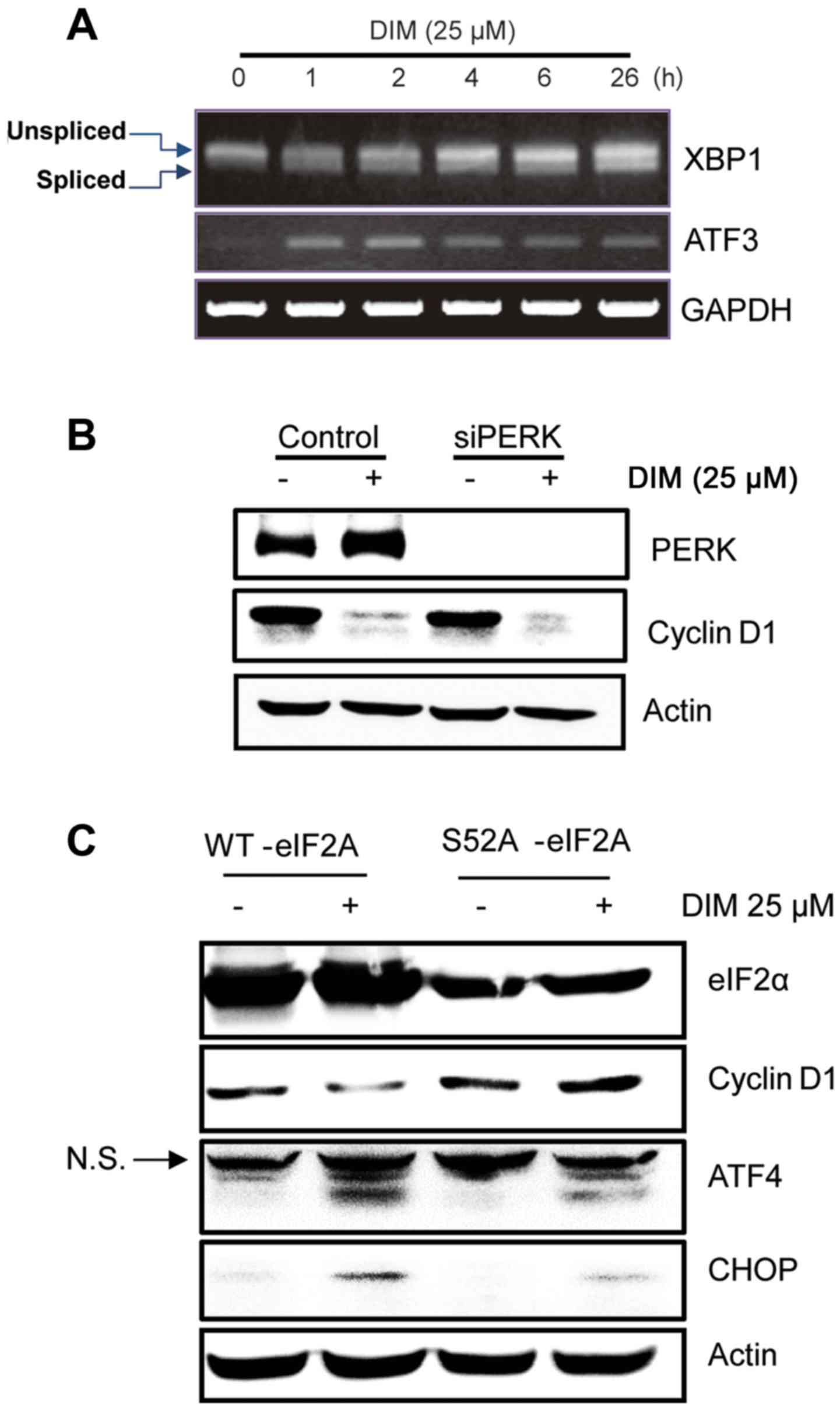
To further elucidate the signaling pathways involved
in DIM-induced cyclin D1 translation suppression, we examined
whether DIM triggered ER stress response of colorectal cancer
cells. It is known that ER stress inhibits the translation of
cyclin D1, mediated by the phosphorylation of eIF2α. As expected,
DIM induced x-box-binding-protein-1 (XBP-1) mRNA splicing and ATF3
expression in a time-dependent manner (Fig. 4A). These are the markers for ER
stress. Next, we measured whether PERK, one of upstream component
of elF2α, contributes to DIM-mediated cyclin D1 downregulation.
Blocking PERK did not seem to affect DIM-mediated cyclin D1
downregulation (Fig. 4B),
indicating that DIM may affect elF2α by other kinases. Given the
critical role of eIF2α phosphorylation in ER stress-mediated
protein translation inhibition, we then asked whether eIF2α protein
phosphorylation mediated cyclin D1 downregulation by DIM. As shown
in Fig. 4C, transfecting eIF2α
construct harboring an S52A mutation attenuated ER stress response
as assessed by CHOP and ATF4 upregulation, indeed restoring cyclin
D1 expression in the presence of DIM. It suggested that ER
stress-mediated protein synthesis inhibition played key roles in
cyclin D1 downregulation by DIM in CRC cells.
Discussion
There is a large number of evidence showing that the
high intake of cruciferous vegetables is inversely associated with
the risk of CRC in humans (16). As
a major component of cruciferous vegetables, I3C and its
condensation product DIM exhibited anti-tumorigenic effect in
different types of cancer cells and in animal models (17). Given the important role of I3C/DIM
in cancer chemoprevention, the multiple mechanisms responsible for
the anti-proliferative effect of DIM have been proposed, including
the regulation of cell cycle regulators such as p21, p27, cyclin D1
or E as well as CKDs 2, 4, 6, which in part was attributed to the
effect of DIM on Sp1 transcriptional activity (18). Herein, we further identified that
cyclin D1 was downregulated by DIM via ER-stress-mediated protein
synthesis inhibition, which provided new mechanism for the
potential chemo-preventive or therapeutic function of DIM in
CRC.
It has been documented that I3C induced G1 cell
cycle arrest in prostate and breast cancer cells, which is
accompanied with cyclin D1 downregulation (19,20).
Thus, it is not surprising that we found DIM decreased cyclin D1 in
CRC cells. However, cyclin D1 in HT-29 cells was not substantially
altered in the presence of DIM (Fig.
1C), which was in agreement with a previous report (15) that DIM analogues like DIM-C-pPhCF3
and DIM-C-pPhtBu did not affect the expression of cyclin D1 in
HCT-15 or HT-29 cells, suggesting that DIM-induced cyclin D1
downregulation was dependent on cell content.
Several PPARγ agonists have been reported to
downregulate cyclin D1 expression at transcriptional or
post-transcriptional level as part of the mechanism for causing G1
cell cycle arrest or growth inhibition through receptor-dependent
and -independent pathways. We have previously observed that PPARα/γ
dual ligand MCC-555 decreased cyclin D1 in pancreatic cancer cells
in a PPARγ-dependent manner (21).
In contrast, our finding in the present study demonstrated that DIM
altered cyclin D1 expression independent of PPARγ activity. In
addition, a DIM derivate did not affect cyclin D1 expression
although it activated PPARγ in CRC cells (15). The evidence suggested that DIM could
exhibit multiple growth-inhibitory mechanisms which varies in
different types of cancer cells, and is dependent on cell
content.
The ubiquitin-proteasome degradation pathway plays a
key role in modulating cell cycle regulators, including cyclin D1,
since they are short-life proteins. Not surprisingly, the 26s
proteasome inhibitors MG-132 and epoximicin both increased basal
cyclin D1 expression of HCT-116 cells in the present study
(Figs. 2C and 3B). A large number of chemicals or drugs
have been documented to trigger cyclin D1 degradation through
proteasome pathway (13). DIM and
its derivate have also been reported to reduce cyclin D1 in MCF-7
and MDA-MB-231 cells, which was blocked by proteasome inhibitor
MG-132 (14). Our finding showed
that proteasome inhibitors failed to completely reverse cyclin D1
downregulation in the presence of DIM, indicating DIM did not
induce proteasome-dependent cyclin D1 degradation (Fig. 2C). However, we did observe that DIM
increased the level of ubiquitinated protein when proteasome
activity was blocked, suggesting DIM could target other proteins
through activating ubiquitin-proteasome pathway. Indeed, Li et
al (22) reported that DIM
selectively induced proteasomal degradation of class I histone
deacetylases in CRC cells. Moreover, various pathway inhibitors
failed to restore cell cyclin D1 expression in the presence of DIM.
Although these inhibitors have previously been verified to block
specific pathways by our group (23,24),
they could harbor other non-identified activities, and induce cell
stress by themselves. Therefore, the signaling pathways involved in
cyclin D1 downregulation by DIM should be further carefully ruled
out.
Accumulating evidence showed that chemicals or drugs
harboring anticancer activity were able to trigger ER stress, which
contributes to cell cycle arrest and apoptosis. DIM induced
apoptosis through ER stress-mediated upregulation of DR5 in
pancreatic cancer cells (6). We
also reported DIM increased ATF3 and ATF4 expression in CRC cells,
both of which can be considered as markers of ER stress. ER
stress-mediated eIF2α phosphorylation leads to nearly global
protein repression by limiting the delivery of initiator Met-tRNAi
to translation machinery, including cyclin D1 (25). In the present study we employed
constitutively active eIF2α construct (S52A mutation) which had
been documented to attenuate ER stress response in HCT-116 cells
(26). As expected, transfection of
mutant eIF2α construct weakened DIM-induced ER stress as evaluated
by examining CHOP and ATF4 expression, and rescued cyclin D1
expression, suggesting DIM halted cyclin D1 protein translation by
triggering ER stress. Moreover, the results were also supported by
the observation that neither cyclin D1 mRNA expression nor protein
stability was affected by DIM. However, the detailed evidence that
DIM inhibited cyclin D1 protein synthesis remains to be further
investigated.
Taken together, we presented here that DIM modulated
cyclin D1 through activating ER stress response, therefore,
providing new insight into its anti-proliferative effect on CRC
cells. Given the multiple signaling pathways induced by ER stress,
it would be meaningful to clarify DIM-induced ER stress pathways
and identify potential anticancer molecules in ongoing
investigations.
Acknowledgements
The present study was supported by the Research
Resettlement Fund for the new faculty of Seoul National University
and partially supported by the Research Institute for Veterinary
Science, Seoul National University. The study was also supported in
part by the Program in Organizational or Personal Cooperation with
Foreign Counterparts (no. 2010630161), the China Scholarship
Council, China.
Glossary
Abbreviations
Abbreviations:
|
DIM
|
3,3-diindolylmethane
|
|
I3C
|
indole-3-carbinol
|
|
ER
|
endoplasmic reticulum
|
|
CRC
|
colorectal cancer
|
References
|
1
|
Lee KW, Bode AM and Dong Z: Molecular
targets of phytochemicals for cancer prevention. Nat Rev Cancer.
11:211–218. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kassie F, Uhl M, Rabot S, Grasl-Kraupp B,
Verkerk R, Kundi M, Chabicovsky M, Schulte-Hermann R and Knasmüller
S: Chemoprevention of 2-amino-3-methylimidazo[4,5-f]quinoline
(IQ)-induced colonic and hepatic preneoplastic lesions in the F344
rat by cruciferous vegetables administered simultaneously with the
carcinogen. Carcinogenesis. 24:255–261. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weng JR, Tsai CH, Kulp SK and Chen CS:
Indole-3-carbinol as a chemopreventive and anti-cancer agent.
Cancer Lett. 262:153–163. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee SH, Min KW, Zhang X and Baek SJ:
3,3-diindolylmethane induces activating transcription factor 3
(ATF3) via ATF4 in human colorectal cancer cells. J Nutr Biochem.
24:664–671. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nachshon-Kedmi M, Yannai S, Haj A and
Fares FA: Indole-3-carbinol and 3,3~-diindolylmethane induce
apoptosis in human prostate cancer cells. Food Chem Toxicol.
41:745–752. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Abdelrahim M, Newman K, Vanderlaag K,
Samudio I and Safe S: 3,3-diindolylmethane (DIM) and its
derivatives induce apoptosis in pancreatic cancer cells through
endoplasmic reticulum stress-dependent upregulation of DR5.
Carcinogenesis. 27:717–728. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rahman KW and Sarkar FH: Inhibition of
nuclear translocation of nuclear factor-{kappa}B contributes to
3,3-diindolylmethane-induced apoptosis in breast cancer cells.
Cancer Res. 65:364–371. 2005.PubMed/NCBI
|
|
8
|
Kassouf W, Chintharlapalli S, Abdelrahim
M, Nelkin G, Safe S and Kamat AM: Inhibition of bladder tumor
growth by 1,1-bis(3-indolyl)-1-(p-substituted phenyl)methanes: A
new class of peroxisome proliferator-activated receptor gamma
agonists. Cancer Res. 66:412–418. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gong Y, Firestone GL and Bjeldanes LF:
3,3-diindolylmethane is a novel topoisomerase IIalpha catalytic
inhibitor that induces S-phase retardation and mitotic delay in
human hepatoma HepG2 cells. Mol Pharmacol. 69:1320–1327. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chintharlapalli S, Papineni S, Baek SJ,
Liu S and Safe S: 1,1-Bis(3-indolyl)-1-(p-substituted
phenyl)methanes are peroxisome proliferator-activated receptor
gamma agonists but decrease HCT-116 colon cancer cell survival
through receptor-independent activation of early growth response-1
and nonsteroidal anti-inflammatory drug-activated gene-1. Mol
Pharmacol. 68:1782–1792. 2005.PubMed/NCBI
|
|
11
|
Musgrove EA, Caldon CE, Barraclough J,
Stone A and Sutherland RL: Cyclin D as a therapeutic target in
cancer. Nat Rev Cancer. 11:558–572. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jirawatnotai S, Hu Y, Michowski W, Elias
JE, Becks L, Bienvenu F, Zagozdzon A, Goswami T, Wang YE, Clark AB,
et al: A function for cyclin D1 in DNA repair uncovered by protein
interactome analyses in human cancers. Nature. 474:230–234. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Alao JP: The regulation of cyclin D1
degradation: Roles in cancer development and the potential for
therapeutic invention. Mol Cancer. 6:242007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vanderlaag K, Samudio I, Burghardt R,
Barhoumi R and Safe S: Inhibition of breast cancer cell growth and
induction of cell death by 1,1-bis(3-indolyl)methane (DIM) and
5,5-dibromoDIM. Cancer Lett. 236:198–212. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chintharlapalli S, R III Smith, Samudio I,
Zhang W and Safe S: 1,1-Bis(3-indolyl)-1-(p-substituted
phenyl)methanes induce peroxisome proliferator-activated receptor
gamma-mediated growth inhibition, transactivation, and
differentiation markers in colon cancer cells. Cancer Res.
64:5994–6001. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu QJ, Yang Y, Vogtmann E, Wang J, Han LH,
Li HL and Xiang YB: Cruciferous vegetables intake and the risk of
colorectal cancer: A meta-analysis of observational studies. Ann
Oncol. 24:1079–1087. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Banerjee S, Kong D, Wang Z, Bao B, Hillman
GG and Sarkar FH: Attenuation of multi-targeted
proliferation-linked signaling by 3,3-diindolylmethane (DIM): From
bench to clinic. Mutat Res. 728:47–66. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Firestone GL and Bjeldanes LF:
Indole-3-carbinol and 3–3-diindolylmethane antiproliferative
signaling pathways control cell-cycle gene transcription in human
breast cancer cells by regulating promoter-Sp1 transcription factor
interactions. J Nutr. 133 Suppl:2448S–2455S. 2003.PubMed/NCBI
|
|
19
|
Hong C, Kim HA, Firestone GL and Bjeldanes
LF: 3,3-Diindolylmethane (DIM) induces a G1 cell cycle arrest in
human breast cancer cells that is accompanied by Sp1-mediated
activation of p21WAF1/CIP1 expression. Carcinogenesis.
23:1297–1305. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vivar OI, Lin CL, Firestone GL and
Bjeldanes LF: 3,3-Diindolylmethane induces a G1 arrest in human
prostate cancer cells irrespective of androgen receptor and p53
status. Biochem Pharmacol. 78:469–476. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Min KW, Zhang X, Imchen T and Baek SJ: A
peroxisome proliferator-activated receptor ligand MCC-555 imparts
anti-proliferative response in pancreatic cancer cells by
PPARgamma-independent up-regulation of KLF4. Toxicol Appl
Pharmacol. 263:225–232. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li Y, Li X and Guo B: Chemopreventive
agent 3,3-diindolylmethane selectively induces proteasomal
degradation of class I histone deacetylases. Cancer Res.
70:646–654. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee SH, Bahn JH, Whitlock NC and Baek SJ:
Activating transcription factor 2 (ATF2) controls tolfenamic
acid-induced ATF3 expression via MAP kinase pathways. Oncogene.
29:5182–5192. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang X, Min KW, Wimalasena J and Baek SJ:
Cyclin D1 degradation and p21 induction contribute to growth
inhibition of colorectal cancer cells induced by
epigallocatechin-3-gallate. J Cancer Res Clin Oncol. 138:2051–2060.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sonenberg N and Hinnebusch AG: Regulation
of translation initiation in eukaryotes: Mechanisms and biological
targets. Cell. 136:731–745. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yoon CH, Lee ES, Lim DS and Bae YS: PKR, a
p53 target gene, plays a crucial role in the tumor-suppressor
function of p53. Proc Natl Acad Sci USA. 106:7852–7857. 2009.
View Article : Google Scholar : PubMed/NCBI
|