Introduction
Hepatocellular carcinoma (HCC) is one of the most
frequently diagnosed cancer across the globe, and the most common
primary liver tumor with increasing incidence worldwide. Targeted
therapy is one of the effectively therapeutic options for advanced
HCC during the past few years (1).
Sorafenib is a multitargeted tyrosine kinase inhibitor for the
treatment of HCC that blocks the Ras, VEGFR, PDGFR, FLT3 and KIT
kinases, which increases the rate of apoptosis and inhibits cell
proliferation, migration and tumor angiogenesis (2). However, acquired resistance to
sorafenib has been found in HCC patients, which results in poor
prognosis. The limited survival benefit from these clinical trials
suggests the existence of primary and acquired sorafenib resistance
mechanisms in HCC cells. Recently, some studies report novel
molecular mechanism of sorafenib resistance in HCC cells. A
low-molecular weight and cysteine-rich proteins, metallothionein
(MT)-1G is reported as a critical regulator and promising
therapeutic target of sorafenib resistance in human HCC cells
(3). Using in vivo RNAi
screening, Rudalska et al suggest that Mapk14 blockade is a
promising approach to overcoming therapy resistance of human HCC
(4). Tumor-associated neutrophils
(TANs) mediated the intratumoral infiltration of macrophages and
Treg cells by secreting CCL2 and CCL17, which stimulated
neovascularization, enhanced HCC growth and metastasis, and
contributed to sorafenib resistance, suggesting that TAN depletion
could enhance the efficacy of sorafenib as an anti-HCC therapeutic
(5). However, the resistance
mechanism remains poorly understood.
Tumor cells often overexpress immune checkpoint
proteins to allow them to evade the host immune system by
inhibiting T-cell attack. One of these immune checkpoint proteins
is programmed death-ligand-1 (PD-L1), which binds to programmed
death-1 (PD-1) expressed on T-cells, B-cells, dendritic cells and
natural killer T-cells to suppress anticancer immunity (6). Therefore, anti-PD-L1 and anti-PD-1
antibodies have been used for the treatment of cancer, showing
promising outcomes (7,8). Moreover, PD-L1 also overexpressed
drug-resistant cancer cells, such as enzalutamide-resistant
prostate cancer (9),
cisplatin-resistant small cell lung cancer cells (10). Despite the importance of PD-L1 in
tumor immunity and drug resistance, the regulation of PD-L1
expression remains poorly understood. Zhu et al found PDL1
is a direct target of BRD4-mediated gene transcription. BET
inhibitors suppress PD-L1 expression in both immune cells and
ovarian cancer cells (11). Hypoxia
upregulates PD-L1 on mouse and human tumor cell lines and on
macrophages and DCs from naive C57BL/6 mice (12). Transcriptional factor MYC directly
regulates CD47 and PD-L1 at the transcriptional level by binding to
their promoters in human melanoma (13). Lo et al showed that
inflammation increases PD-L1 expression in tumors through
TNF-α-mediated activation of NFκB, leading to transactivation of
CSN5 (14).
Epigenetic changes such as DNA methylation act to
regulate gene expression in normal mammalian development. However,
promoter hypermethylation also plays a major role in cancer through
transcriptional silencing of critical growth regulators such as
tumor suppressor genes (15). DNA
methylation is controlled at several different levels in normal and
tumor cells. The addition of methyl groups is carried out by a
family of enzymes, DNA methyltransferases (DNMTs). DNMTs are
enzymes that catalyze the addition of methyl groups to cytosine
residues in DNA. DNMTs found in mammalian cells include DNMT1,
DNMT3a, and DNMT3b (16,17).
In this study, we modeled sorafenib resistance in
HCC cell lines, and explored the molecular and functional
characteristics of resistant cells. We demonstrate
NFκB/PDL1/STAT3/DNMT1 axis as a mechanism by which HCC cells
develop sorafenib-resistance phenotypes and established its
potential as a new therapeutic target for preventing or overcoming
the acquired resistance to sorafenib.
Materials and methods
Plasmids, cell lines and
chemicals
The shRNA and control vectors for PD-L1, DNMT1,
STAT3 and NFκB were obtained from BMGC RNAi (University of
Minnesota). Cell lines were obtained from American Type Culture
Collection (Manassas, VA, USA). HepG2 and Huh7 cell lines were
grown in DMEM with 10% fetal bovine serum (FBS) (Life Technologies,
Grand Island, NY, USA) at 37°C under 5% CO2. For the
drug treatment, cells were treated with the following reagents used
at concentrations, times and schedules indicated in Results.
Sorafenib, decitabine (5-aza-2′-deoxycytidine or Dacogen) and Bay
11–7082 were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Generation of sorafenib-resistant
cells
HepG2 and Huh7 sorafenib-resistant cells
(HepG2SR and Huh7SR) were cultured
continuously with a step-wise increase of sorafenib concentrations
for 8 weeks (0–20 µM). HepG2 and Huh7 parental cells
(HepG2C and Huh7C) were cultured in parallel
without sorafenib and served as control.
Transfections
Approximately 1×106 cells were seeded
into 6-well plates overnight before transfection. The shRNA or
plasmids were introduced into cells using Lipofectamine™ RNAiMAX or
Lipofectamine™ 2000 reagent (Life Technologies), respectively,
according to the manufacturer's instructions.
Chromatin immunoprecipitation
(ChIP)
ChIP assays were performed as described previously
using EZ-ChIP assay kit (Millipore, Billerica, MA, USA). Briefly,
~2×106 transfected cells were cross-linked with 1%
formaldehyde (Sigma-Aldrich), washed and resuspended in 1% SDS
lysis buffer for sonication, in order to yield DNA fragments with
an average size of 300–500 bp. The lysates were immunoprecipitated
by 5 µg of antibody. Aliquots (1%) were reserved for the negative
control (input DNA). ChIP DNA was quantified by qPCR with Power
SYBR® Green PCR Master Mix. Fold change in binding was
compared using the corresponding input DNA. The primers specific
for DNMT1 or PDL1 gene promoter were: hDNMT1-ChIP1 forward,
AATAGATGGAGGTTGGAT; reverse, AGGCATTCATTCATTCAT. hDNMT1-ChIP2
forward, CTATACACTGTGAGATTCTTG; reverse CTGGC TATACGACCTTAG. The
anti-STAT3, anti-phospho-STAT3 (Tyr705), anti-NFκB and
anti-phospho-NFκB (Cell Signaling Technology) were used.
Clonogenic assays
Methylcellulose colony formation assays were
performed in MethoCult® medium (Stem Cell Technologies,
Canada) according to the manufacturer's instructions. Briefly, at 6
h after transfection or exposure to drugs, 500 cells were harvested
and diluted in 0.3 ml of IMDM + 2% FBS (Stem Cell Technologies),
then mixed diluted cells in MethoCult® medium.
Subsequently 1.1 ml of the MethoCult mixture was dispensed into a
35-mm dish. Colonies were scored in 7–10 days.
Western blotting
After the various treatments, the whole cellular
lysates were prepared by harvesting the cells in 1X cell lysis
buffer [20 mM HEPES (pH 7.6), 150 mM NaCl and 0.1% NP40]
supplemented with 1X phosphatase inhibitor Cocktail 2 and 3
(Sigma-Aldrich), 1 mM PMSF (Sigma-Aldrich) and 1X protease
inhibitors (protease inhibitor cocktail set III,
Calbiochem-Novabiochem, San Diego, CA, USA). Protein was resolved
by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis
and transferred onto PVDF membranes (Amersham, Piscataway, NJ,
USA). The antibodies used were β-actin (Santa Cruz Biotechnology);
DNMT1 (New England Biolabs, Ipswich, MA, USA), the anti-STAT3,
anti-phospho-STAT3 (Tyr705), anti-NFκB and anti-phospho-NFκB,
anti-PD-L1 (Cell Signaling Technology).
RNA isolation, cDNA preparation and
quantitative PCR
RNA was isolated using miRNAeasy kit (Qiagen)
according to the manufacturer's instructions. Reverse transcription
for obtaining cDNA was performed according to the manufacturer's
instructions (Invitrogen). The expression of DNMT1, PD-L1,
CDH1 and GAPDH gene was evaluated by SYBR Green
Quantitative PCR. Expression of the target genes was measured using
the ∆CT approach. The primers were: DNMT1 forward,
5′-CCAGATGAGGACAATGAG-3′; reverse, 5′-AGCAAGACAACCATAATCA-3′.
PD-L1 forward, 5′-TCCACTCAATGCCTCAAT-3′; reverse,
5′-GAAGACCTCACAGACTCAA-3′. GAPDH forward,
5′-ACAGGATTGACAGATTGA-3′; reverse, 5′-TATCGGAATTAACCAGACA-3′.
CDH1 forward, 5′-AGAACGCATTGCCACATACAC-3′; reverse,
GAGGATGGTGTAAGCGATGG-3′.
Bisulfite sequencing
Total DNA sample (2 µg) was converted and purified
using EpiTect Bisulfite kit (Qiagen) according to the
manufacturer's instructions. The −251 to +139 region within the
CDH1 CpG was amplified from bisulfite-treated DNA sample by PCR
using the following primers: forward,
5′-TTTTTTTTGATTTTAGGTTTTAGTGAG-3′; reverse,
5′-ACTCCAAAAACCCATAACTAACC-3′. The PCR products were subcloned
using the TA Cloning® kit (Invitrogen), and sequenced by
Genewiz Company.
Statistical analysis
The qPCR and colony assay were analyzed using the
Student's t-test. Correlation data were performed with Pearson
correlation coefficients. The statistical analysis were carried out
using GraphPad Prism 5.0. Differences were considered statistically
significant at P<0.05. All P-values were two-tailed.
Results
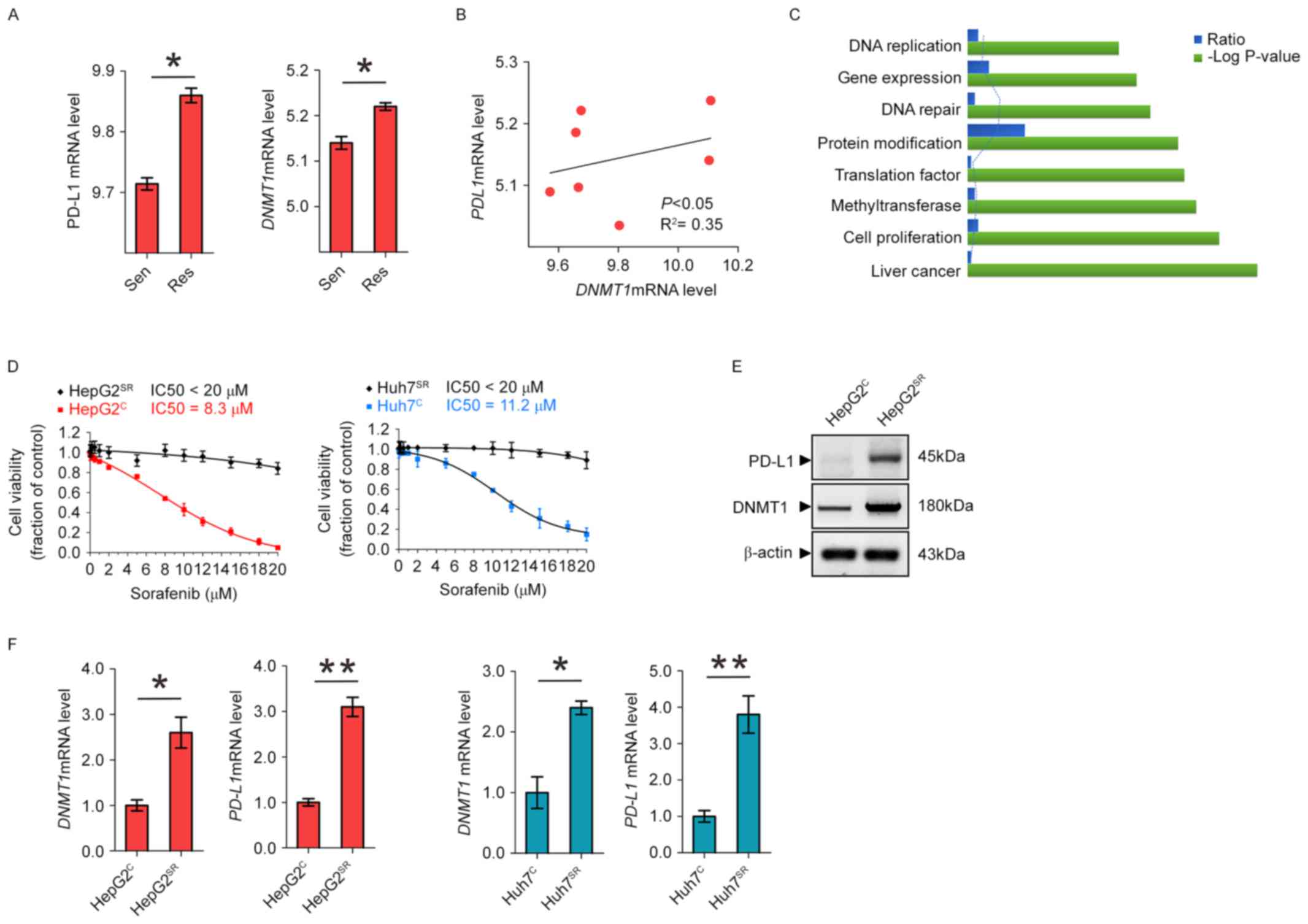
Highly upregulated DNMT1 is positively
correlated with PD-L1 overexpression in sorafenib-resistant HCC
cells
To determine whether PD-L1 and DNMT1 expression is
upregulated in sorafenib-resistant HCC cells, we analyzed the GEO
data discovered a significant PD-L1 and DNMT1 upregulation in
sorafenib-resistant HCC mice (Fig.
1A). In addition, as shown in Fig.
1B, we found that in HCC mice, higher levels of DNMT1 are
accompanied by PD-L1 overexpression, while lower expression of
DNMT1 is observed in mice carrying lower PDL1 mRNA level,
indicating of a positive correlation between these variables
(P<0.05). To better understand which biological functions are
affected by long-term sorafenib treatment, we conducted functional
annotations using DAVID bioinformatics resources 6.7. Enrichment
scores for Gene Ontology (GO) categories in overlapped genes
(Fig. 1C). To further understand
the sorafenib resistance mechanism, we established two HCC
sorafenib-resistant cell lines, HepG2SR and
Huh7SR, by the stepwise increase of drug dosages and
continuous culture in drug-containing medium for 2 months. The
final concentrations were 20 µM of sorafenib, which exerted
sufficient inhibitory action and were in the range of clinically
achievable levels (18).
HepG2SR and Huh7SR exhibited significantly
higher IC50 value than their parental control
HepG2C and Huh7C (Fig. 1D). Notably, comparing to the
parental cells, the protein expression and RNA level of DNMT1 and
PD-L1 were all increased in HepG2SR and
Huh7SR (Fig. 1E and F).
These results support the highly upregulated DNMT1 is positively
correlated with PD-L1 overexpression in sorafenib-resistant HCC
cells.
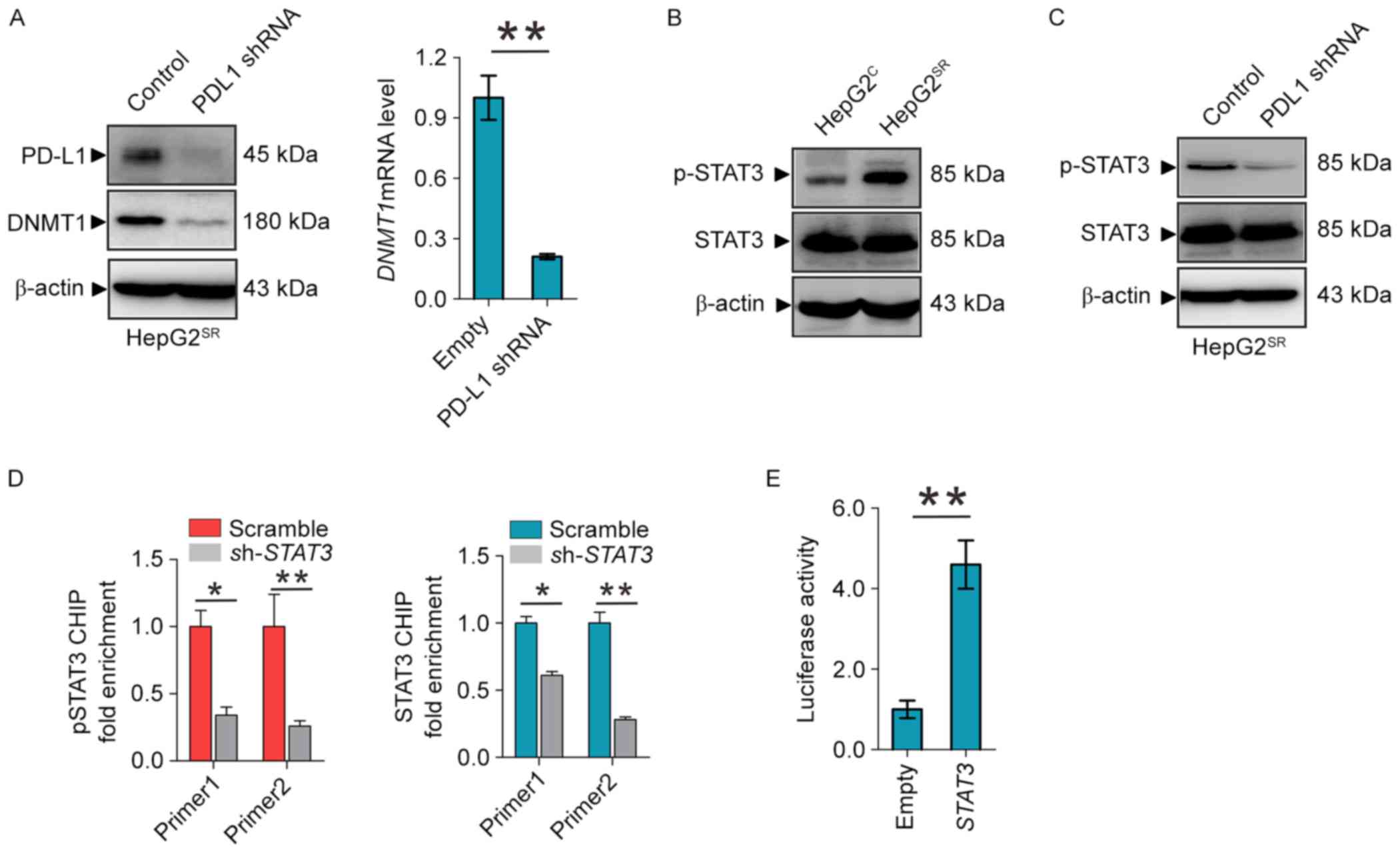
PD-L1 regulates DNMT1 through STAT3
signaling pathway
Given the positive correlations of DNMT1 and PD-L1
expression in sorafenib-resistant HCC, we proposed the existence of
a regulatory loop between DNMT1 signaling and PD-L1 machinery, in
which PD-L1 regulates the expression of DNMT1. As shown in Fig. 2A, PD-L1 depletion by shRNA in
HepG2SR and Huh7SR, as expected, decrease
PD-L1 and DNMT1 protein and mRNA levels. To elucidate how PD-L1
regulates the DNMT1 gene, we focused on the STAT3/DNMT1 network,
since STAT3 has been shown to regulate DNMT1 transcription in
cancer and play a key role in drug-resistant (19). First, we found that phosphor-STAT3
was overexpressed in HepG2SR compared with
HepG2C (Fig. 2B).
Second, PD-L1 knockdown dephosphorylates STAT3 at Tyr705 in
HepG2SR and Huh7SR (Fig. 2C). In addition, ChIP showed that
STAT3 knockdown diminished the binding of total and phospho-STAT3
in DNMT1 promoter (Fig. 2D).
Reporter assays revealed that STAT3 inactivation by shRNA disrupted
the luciferase activities driven by DNMT1 promoter region
containing STAT3 binding elements (Fig.
2E). These results confirmed STAT3 transcript regulates the
DNMT1 gene, and suggest that PD-L1 specifically regulate DNMT1
through STAT3 signaling in sorafenib resistance to HCC.
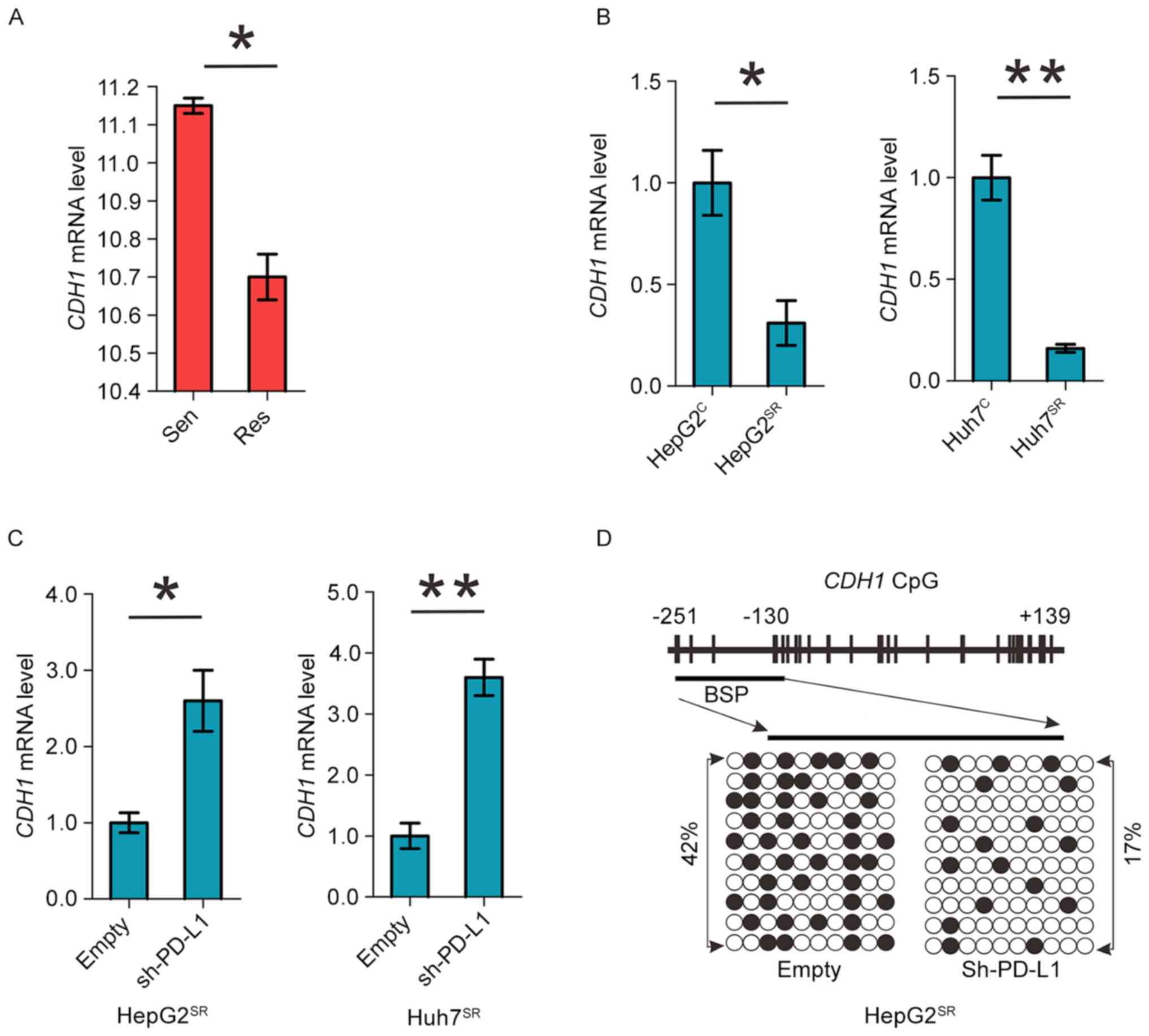
PD-L1 induces DNMT1-dependent DNA
hypomethylation and restores the expression of methylation-silenced
CDH1
Cadherin 1 (CDH1) is a cell-cell adhesion molecule
and functions as a metastasis suppressor in HCC. It is
epigenetically silenced and its downregulation associates with poor
prognosis in HCC (20,21). As shown in Fig. 3A and B we found significant CDH1
downregulation in sorafenib-resistant HCC mice and cell model.
Important, knockdown of PDL1 can increase CDH1 mRNA level in
HepG2SR and Huh7SR cells (Fig. 3C). As PD-L1 regulates DNMT1, we
proposed that PD-L1 downregulation might mediate CDH1 restoration
via promoter DNA hypermethylation. We analyzed CDH1 promoter
methylation status using bisulfite sequencing in PD-L1
shRNA-transfected HepG2SR and Huh7SR cells,
and found a >25% change (42% in empty versus 17% in PDL1 shRNA)
from hyper- to unmethylated in CDH1 promoter (Fig. 3D). Thus, PD-L1 induces
DNMT1-dependent DNA hypomethylation and restores the expression of
methylation-silenced CDH1.
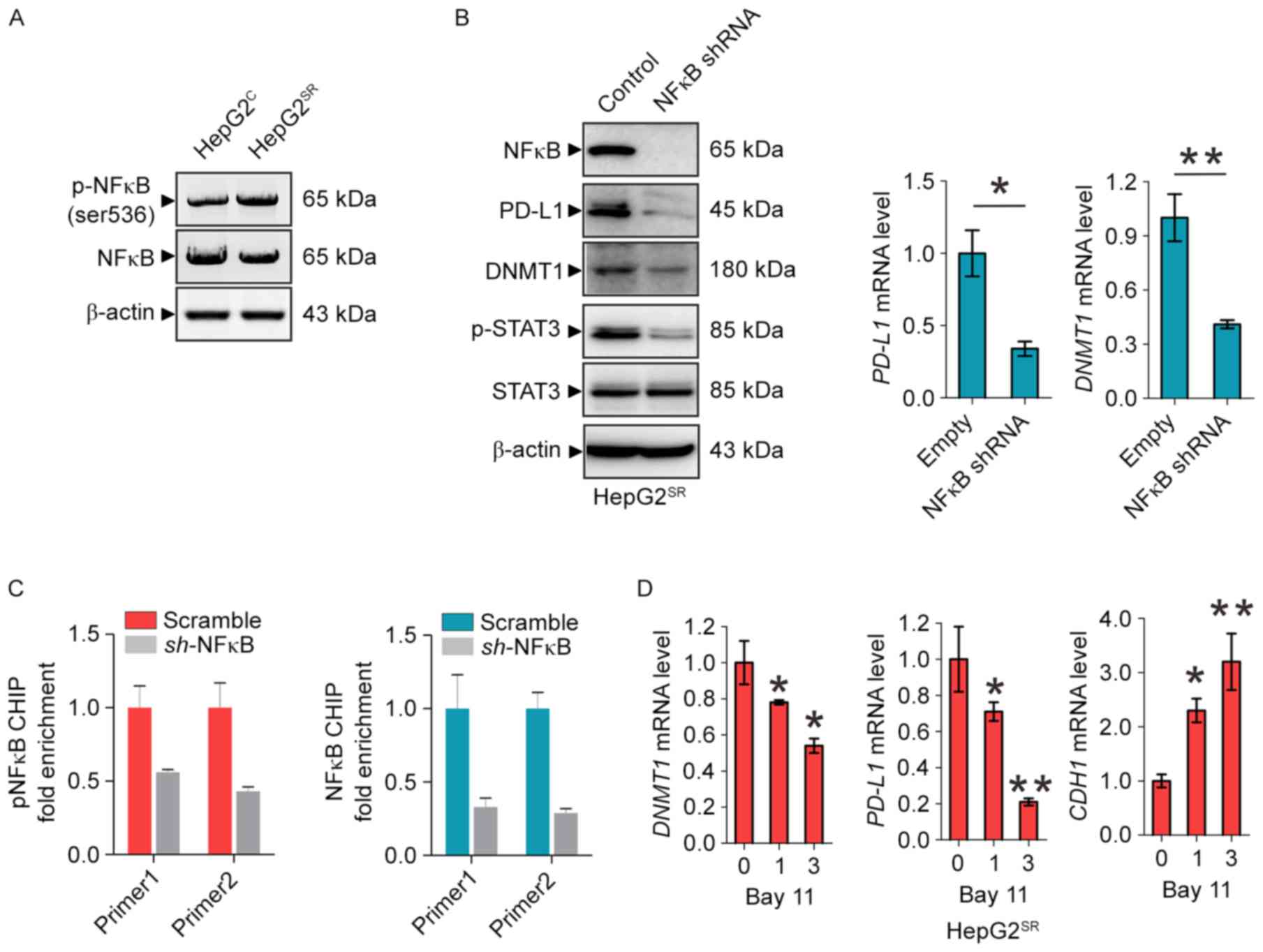
Inactivation of NFκB blocks
PD-L1/Stat3/DNMT1 pathway in sorafenib-resistant HCC cells
To provide further insight into the mechanism
underlying PD-L1/STAT3/DNMT1 signaling in sorafenib-resistant HCC,
we considered the transcriptional factor NFκB may play a key role.
Indeed, as shown in Fig. 4A, NFκB
was activated in HepG2SR cells. NFκB knockdown by shRNA
in HepG2SR cells significantly decreased PDL1 protein
and mRNA levels followed by the dephosphorylation of STAT3 and
downregulated DNMT1 expression (Fig.
4B). To elucidate how NFκB regulates PD-L1, we investigated
whether NFκB can directly transcriptionally regulate PDL1. Firstly,
it was predicted that PDL1 promoter area has an NFκB binding site.
Secondly, ChIP demonstrated ~1.5-fold of enrichment reduction of
NFκB on PD-L1 promoter in HepG2SR cells after NFκB shRNA
transfection supporting that PD-L1 is one of the NFκB binding
targets (Fig. 4C). Of note, as
shown in Fig. 4D, when
HepG2SR cells were treated at 1 and 3 M of Bay 11, a
specific NFκB inhibitor (22), the
mRNA level of PD-L1 and DNMT1 were significantly reduced, and the
CDH1 was markedly increased. These data suggest that inactivation
of NFκB blocks the PD-L1/Stat3/DNMT1 pathway in
sorafenib-resistance HCC cells.
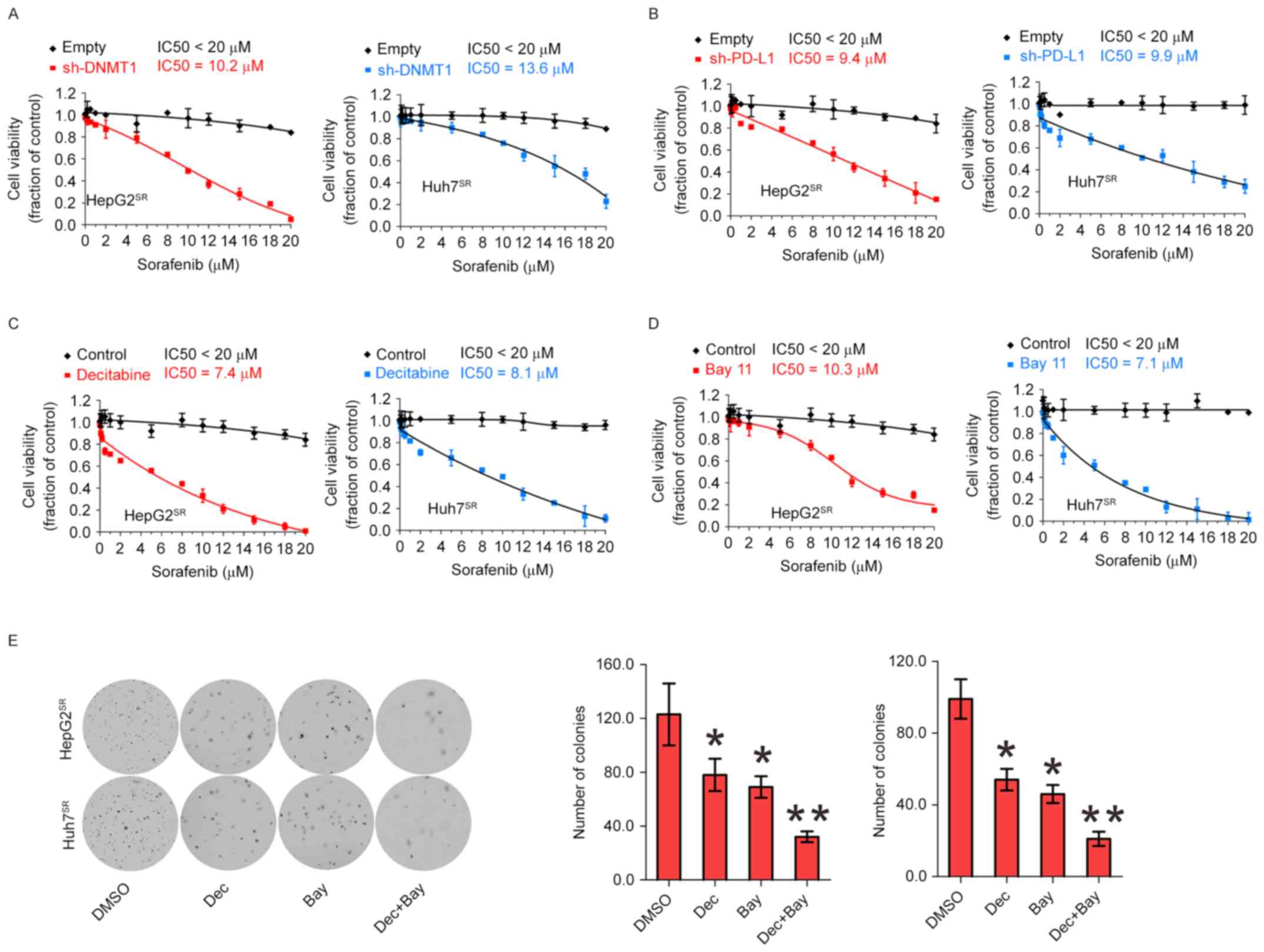
Genetic or pharmacological disruption
of PD-L1 or DNMT1 sensitizes HCC resistance to sorafenib
Because of activation of PD-L1 and DNMT1 signaling
in sorafenib resistance to HCC, we speculated that genetic or
pharmacological disruption of PD-L1 and DNMT1 function could be an
alternative strategy to impair sorafenib-resistant cell
proliferation. When PD-L1 or DNMT1 was depleted by their specific
shRNA in sorafenib-resistant cells, then exposing the cells to 0–20
µM sorafenib for 72 h, cell growth IC50 value for PD-L1
or DNMT1 knockdown was significantly decreased, respectively,
compared with their empty control, suggesting that abrogation of
PDL1 and DNMT1 restores sorafenib sensitivity (Fig. 5A and B). Next, we considered
pharmacological inhibition using their inhibitor to sensitize
resistant cells to sorafenib treatment. As shown in Fig. 5C, treatment with decitabine, a DNMT1
inhibitor, induced more pronounced inhibition on cell proliferation
rate in HepG2SR and Huh7SR. Because
anti-PD-L1 can not inhibit PD-L1 expression but activity.
Therefore, we consider Bay 11 can be used as PD-L1 inhibitor
(Fig. 4D). Indeed, 3 M of Bay 11
induced sensitized sorafenib-resistant cells (Fig. 5D). Decitabine and Bay 11 sensitized
resistant cells to sorafenib treatment alone, and in a potential
cooperation of NFκB/PDL1/DNMT1 in controlling sorafenib-resistant
cell growth. We treated with decitabine and Bay 11 alone or both,
and colony assays showed combination of decitabine and Bay 11
significantly disrupted the colony forming capability of
HepG2SR and Huh7SR cells (Fig. 5E). Collectively, these data support
the potential targeted combination therapies to enhance current and
emerging sorafenib therapies for HCC.
Discussion
Hepatocellular cancer (HCC) is a leading cause of
cancer-related death worldwide, and the incidence of HCC is
increasing in many parts of the world (23). Sorafenib is one of multiple targeted
agents utilized by oncologists, and is FDA approved for treatment
of a wide range of human cancers, including kidney, melanoma,
prostate, ovarian, pancreatic, lung cancer and HCC. However, only
rarely do sorafenib treated tumors regress completely, and the
therapeutic effects of the drug are often temporary, most patients
ultimately develop drug resistance and suffer relapses (24). Therefore, more effective therapeutic
strategies and precision medicines to improve the prognosis of HCC
patients with sorafenib resistance are urgently needed. Recent
studies have shown that some mechanism was found in
sorafenib-resistant cell lines. A tumor suppressor gene
angiopoietin-like protein 1 (ANGPTL1) positively correlates with
sorafenib sensitivity in HCC cells and human HCC tissues. ANGPTL1
directly interacts with and inactivates the MET receptor, which
contributes to Slug suppression through inhibition of the
extracellular receptor kinase/protein kinase B (ERK/AKT)-dependent
early growth response protein 1 (Egr-1) pathway. ANGPTL1 may serve
as a novel MET receptor inhibitor for advanced HCC therapy
(25). Another study found PROX1
positively correlates with sorafenib resistance in HCC cells and
may be a new potential strategy for improving sorafenib efficacy
(26).
PD-L1, a ligand of PD1, is highly upregulated on
many kinds of tumor cells, including melanoma, ovarian, and lung
cancers. Because programmed death-1 (PD-1) and PD-L1 blockade have
yielded promising clinical effects, understanding the regulatory
mechanism of PD-L1 may identify biomarkers and/or develop
combinatorial strategies for clinical use. Recently, PD-L1
overexpression also was reported in resistant cells. PD-L1 was
expressed at a high level in SCLC cells (H69 and H82) resistant to
cisplatin versus parental cells (10). Bishop et al showed that ENZ
resistance is associated with high frequency of PD-1/L1 therapy
targets, both in the tumor and circulating immune cells (9). Cancer epigenetics involves the causes
of CpG island hypermethylation in tumor suppressor genes leading to
transcriptional silencing. DNMT1 is a member of DNA
methyltransferase family which includes DNMT1, DNMT3a and DNMT3b.
Methylation occurs predominantly at cytosine-C5 in the context of
CpG dinucleotides, and is established and maintained by three DNA
methyltransferases, DNMT3a, DNMT3b, and DNMT1. DNMT3a and DNMT3b
have mostly de novo DNA methylation activity, whereas DNMT1
plays a central role in preserving the patterns of DNA methylation
through cell division (27). DNMT1
and DNA methylation is often considered as a key epigenetic
regulatory mechanism and potential target for resistance to drugs
(28).
Although PD-L1 and DNMT1 has been shown to be
overexpressed in human cancer and drug resistance, respectively,
whether and how PD-L1/DNMT1 axis affect sorafenib resistance to HCC
remains largely unexplored. In this study, we mainly focused on
DNMT1, because GEO data analysis showed DNMT1 overexpression in
sorafenib resistance HCC cells, but not DNMT3a and 3b (data not
shown). In vitro study found DNMT1 was upregulated in
sorafenib-resistant HCC cells, which is consistent to GEO assay.
Given both DNMT1 and PDL1 were significantly upregulated in
sorafenib-resistant HCC cells and positively correlated by GEO
assay, we proposed PD-L1 augments DNMT1 expression. We demonstrated
that downregulation of PD-L1 in sorafenib-resistant cells might
result from deregulation of DNMT1 at both protein and mRNA levels,
leading to the upregulation of tumor suppressor CDH1 and the
reduction of CDH1 promoter methylation. Moreover, depletion of
PD-L1 or DNMT1 enhanced cell growth arrest, suggesting that
PD-L1/DNMT1 axis plays a key role in survival and proliferation of
sorafenib-resistant HCC cells. Accordingly, inhibition of
PD-L1/DNMT1 axis may contribute to the resistance of
molecular-targeted therapy. DNMT1 inhibitor caused a gradual
decrease in self-renewal and tumorigenicity, and upregulation of
apoptosis- and differentiation-related genes in HCC (29,30).
Decitabine is a useful demethylation agent in clinic. It can
decrease DNMT1 expression and cell cycle and is commonly used as a
single agent to treat patients with MDS and elderly patients with
AML (30,31). PDL1 antibodies, such as durvalumab
and avelumab, are selective, high-affinity, human antibodies that
block PD-L1 binding to PD-1, allowing T cells to recognize and kill
tumor cells (7,32). However, these antibodies can only
inhibit PD-L1 activity but not expression, which limits the use to
overcome HCC resistance to sorafenib. Some transcranial factors
were reported to directly regulate PD-L1. MYC regulates CD47 and
PD-L1 at the transcriptional level by binding to their promoters in
human melanoma (13). Moreover,
PD-L1 is a direct target of HIF1-α and blockade of PD-L1 under
hypoxia enhanced myeloid-derived suppressor cell-mediated T cell
activation. Lo et al showed that inflammation increases
PD-L1 expression in tumors through TNF-α-mediated activation of
NFκB, leading to transactivation of CSN5 (14). In this study, upregulation of
phosphorylation of NFκB-p65 (Ser536) was positively associated with
PD-L1 expression in sorafenib resistance HCC cells. Because NFκB
knockdown significantly decreased both protein and mRNA levels of
PD-L1 and DNMT1, especially, diminishing the binding of PD-L1
promoter, it demonstrated that PD-L1 is one of a direct target of
NFκB in sorafenib-resistant cells. Notably, NFκB inhibitor Bay-11
can decrease PD-L1 expression, which can be used to treat resistant
cells to a PD-L1 inhibitor. Cell proliferation results showed that
both decitabine and Bay 11 sensitize resistant cells to sorafenib
treatment, further supporting that cellular function of PD-L1/DNMT1
signaling is required to sustain survival and proliferation of
sorafenib-resistant cells. Importantly, combination of decitabine
and Bay 11 synergistically inhibited colony formation of HCC
resistant cells to sorafenib indicating that dual inactivation of
PD-L1 and DNMT1 synergistically disrupts the cell growth and
proliferation of sorafenib-resistant HCC cells.
For mechanism study, first we elucidated how PD-L1
regulates the DNMT1 gene. As phosphorylated STAT3 can bind specific
DNA elements resulting in transcriptional activation. Previously it
was reported that STAT3 binds DNMT1 promoter and positively
regulates DNMT1 transcription (33), we proposed that PD-L1 augments DNMT1
expression by STAT3 in sorafenib-resistant HCC cells. We found
depletion of PD-L1 significantly downregulated phosphorylation of
STAT3 followed by the DNMT1 expression. Then CHIP and promoter
reporter assay confirmed that STAT3 transcriptionally regulate
DNMT1. Of note, it is likely that STAT3 is downstream of PD-L1. One
report suggests that STAT3 as an upstream target regulates PD-L1
expression (34). There may be a
feedback loop between STAT3 and PD-L1 which need further
investigation.
Taken together, our results identified a mechanistic
and functional link between PD-L1 and DNMT1-dependent DNA
methylation in HCC cells resistant to sorafenib. These results also
established the clinical potential strategy for targeting
NFκB/PD-L1/STAT3/DNMT1 axis aimed to improve sorafenib efficacy and
overcoming sorafenib resistance.
Acknowledgments
The authors would like to thank Dr Ningling Kang at
the Hormel Institute, University of Minnesota for providing HCC
cell lines, and Lin Yang at the Mayo Clinic, Rochester, MN, USA for
assistance in English editing. This study was supported by grants
from Foundation of Jilin Provincial Development and Reform
Commission (KY20160002; 3J115AJ73428, Yahui Liu), Jilin University
Fund for Excellent Young Teacher (no. 419080500355, Bai Ji), and
Youth Fund from the Department of Science and Technology, Jilin
Province, China (no. 20140520026JH, Bai Ji).
References
|
1
|
Di Maio M, Daniele B and Perrone F:
Targeted therapies: Role of sorafenib in HCC patients with
compromised liver function. Nat Rev Clin Oncol. 6:505–506. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wilhelm S, Carter C, Lynch M, Lowinger T,
Dumas J, Smith RA, Schwartz B, Simantov R and Kelley S: Discovery
and development of sorafenib: A multikinase inhibitor for treating
cancer. Nat Rev Drug Discov. 5:835–844. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sun X, Niu X, Chen R, He W, Chen D, Kang R
and Tang D: Metallothionein-1G facilitates sorafenib resistance
through inhibition of ferroptosis. Hepatology. 64:488–500. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rudalska R, Dauch D, Longerich T, McJunkin
K, Wuestefeld T, Kang TW, Hohmeyer A, Pesic M, Leibold J, von Thun
A, et al: In vivo RNAi screening identifies a mechanism of
sorafenib resistance in liver cancer. Nat Med. 20:1138–1146. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z,
Chen EB, Fan J, Cao Y, Dai Z and Zhou J: Tumor-associated
neutrophils recruit macrophages and T-regulatory cells to promote
progression of hepatocellular carcinoma and resistance to
sorafenib. Gastroenterology. 150:1646–1658.e17. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Goodman A, Patel SP and Kurzrock R:
PD-1-PD-L1 immune-checkpoint blockade in B-cell lymphomas. Nat Rev
Clin Oncol. 14:203–220. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Massard C, Gordon MS, Sharma S, Rafii S,
Wainberg ZA, Luke J, Curiel TJ, Colon-Otero G, Hamid O, Sanborn RE,
et al: Safety and efficacy of durvalumab (MEDI4736), an
anti-programmed cell death ligand-1 immune checkpoint inhibitor, in
patients with advanced urothelial bladder cancer. J Clin Oncol.
34:3119–3125. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Reck M, Rodríguez-Abreu D, Robinson AG,
Hui R, Csőszi T, Fülöp A, Gottfried M, Peled N, Tafreshi A, Cuffe
S, et al: KEYNOTE-024 Investigators: Pembrolizumab versus
chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl
J Med. 375:1823–1833. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bishop JL, Sio A, Angeles A, Roberts ME,
Azad AA, Chi KN and Zoubeidi A: PD-L1 is highly expressed in
Enzalutamide-resistant prostate cancer. Oncotarget. 6:234–242.
2015.PubMed/NCBI
|
|
10
|
Yan F, Pang J, Peng Y, Molina JR, Yang P
and Liu S: Elevated cellular PD1/PD-L1 expression confers acquired
resistance to cisplatin in small cell lung cancer cells. PLoS One.
11:e01629252016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhu H, Bengsch F, Svoronos N, Rutkowski
MR, Bitler BG, Allegrezza MJ, Yokoyama Y, Kossenkov AV, Bradner JE,
Conejo-Garcia JR, et al: BET Bromodomain inhibition promotes
anti-tumor immunity by suppressing PD-L1 expression. Cell Rep.
16:2829–2837. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Noman MZ, Desantis G, Janji B, Hasmim M,
Karray S, Dessen P, Bronte V and Chouaib S: PD-L1 is a novel direct
target of HIF-1α, and its blockade under hypoxia enhanced
MDSC-mediated T cell activation. J Exp Med. 211:781–790. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Casey SC, Tong L, Li Y, Do R, Walz S,
Fitzgerald KN, Gouw AM, Baylot V, Gütgemann I, Eilers M, et al: MYC
regulates the antitumor immune response through CD47 and PD-L1.
Science. 352:227–231. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lo J, Lau EY, Ching RH, Cheng BY, Ma MK,
Ng IO and Lee TK: Nuclear factor kappa B-mediated CD47
up-regulation promotes sorafenib resistance and its blockade
synergizes the effect of sorafenib in hepatocellular carcinoma in
mice. Hepatology. 62:534–545. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Baylin SB: DNA methylation and gene
silencing in cancer. Nat Clin Pract Oncol. 2:(Suppl 1). S4–S11.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Esteller M: Epigenetics in cancer. N Engl
J Med. 358:1148–1159. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Esteller M, Fraga MF, Paz MF, Campo E,
Colomer D, Novo FJ, Calasanz MJ, Galm O, Guo M, Benitez J, et al:
Cancer epigenetics and methylation. Science. 297:1807–1808. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Spinzi G and Paggi S: Sorafenib in
advanced hepatocellular carcinoma. N Engl J Med. 359:2497–2498;
author reply 2498–2499. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ and
Settleman J: Drug resistance via feedback activation of Stat3 in
oncogene-addicted cancer cells. Cancer Cell. 26:207–221. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang C, Li J, Huang T, Duan S, Dai D,
Jiang D, Sui X, Li D, Chen Y, Ding F, et al: Meta-analysis of DNA
methylation biomarkers in hepatocellular carcinoma. Oncotarget.
7:81255–81267. 2016.PubMed/NCBI
|
|
21
|
Huang J, Wang Y, Guo Y and Sun S:
Down-regulated microRNA-152 induces aberrant DNA methylation in
hepatitis B virus-related hepatocellular carcinoma by targeting DNA
methyltransferase 1. Hepatology. 52:60–70. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen L, Ruan Y, Wang X, Min L, Shen Z, Sun
Y and Qin X: BAY 11–7082, a nuclear factor-κB inhibitor, induces
apoptosis and S phase arrest in gastric cancer cells. J
Gastroenterol. 49:864–874. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Llovet JM, Villanueva A, Lachenmayer A and
Finn RS: Advances in targeted therapies for hepatocellular
carcinoma in the genomic era. Nat Rev Clin Oncol. 12:4362015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen J, Jin R, Zhao J, Liu J, Ying H, Yan
H, Zhou S, Liang Y, Huang D, Liang X, et al: Potential molecular,
cellular and microenvironmental mechanism of sorafenib resistance
in hepatocellular carcinoma. Cancer Lett. 367:1–11. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen HA, Kuo TC, Tseng CF, Ma JT, Yang ST,
Yen CJ, Yang CY, Sung SY and Su JL: Angiopoietin-like protein 1
antagonizes MET receptor activity to repress sorafenib resistance
and cancer stemness in hepatocellular carcinoma. Hepatology.
64:1637–1651. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu Y, Ye X, Zhang JB, Ouyang H, Shen Z,
Wu Y, Wang W, Wu J, Tao S, Yang X, et al: PROX1 promotes
hepatocellular carcinoma proliferation and sorafenib resistance by
enhancing β-catenin expression and nuclear translocation. Oncogene.
34:5524–5535. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jin B and Robertson KD: DNA
methyltransferases, DNA damage repair, and cancer. Adv Exp Med
Biol. 754:3–29. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Min HY, Lee SC, Woo JK, Jung HJ, Park KH,
Jeong HM, Hyun SY, Cho J, Lee W, Park JE, et al: Essential role of
DNA methyltransferase 1-mediated transcription of insulin-like
growth factor 2 in resistance to histone deacetylase inhibitors.
Clin Cancer Res. 23:1299–1311. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jiang C and Gong F: DNA methyltransferase
1: A potential gene therapy target for hepatocellular carcinoma?
Oncol Res Treat. 39:448–452. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Welch JS, Petti AA, Miller CA, Fronick CC,
O'Laughlin M, Fulton RS, Wilson RK, Baty JD, Duncavage EJ, Tandon
B, et al: TP53 and decitabine in acute myeloid leukemia and
myelodysplastic syndromes. N Engl J Med. 375:2023–2036. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Duncavage EJ, Uy GL, Petti AA, Miller CA,
Lee YS, Tandon B, Gao F, Fronick CC, O'Laughlin M, Fulton RS, et
al: Mutational landscape and response are conserved in peripheral
blood of AML and MDS patients during decitabine therapy. Blood.
129:1397–1401. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nagaya T, Nakamura Y, Sato K, Harada T,
Choyke PL, Hodge JW, Schlom J and Kobayashi H: Near infrared
photoimmunotherapy with avelumab, an anti-programmed death-ligand 1
(PD-L1) antibody. Oncotarget. 8:8807–8817. 2017.PubMed/NCBI
|
|
33
|
Zhang Q, Wang HY, Woetmann A, Raghunath
PN, Odum N and Wasik MA: STAT3 induces transcription of the DNA
methyltransferase 1 gene (DNMT1) in malignant T lymphocytes. Blood.
108:1058–1064. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang WB, Yen ML, Liu KJ, Hsu PJ, Lin MH,
Chen PM, Sudhir PR, Chen CH, Chen CH, Sytwu HK, et al:
Interleukin-25 mediates transcriptional control of PD-L1 via STAT3
in multipotent human mesenchymal stromal cells (hMSCs) to suppress
Th17 responses. Stem Cell Rep. 5:392–404. 2015. View Article : Google Scholar
|