Introduction
Hepatocellular carcinoma (HCC) is the fifth most
common malignant tumor in the world, especially in Southeast Asia
and Africa (1). Hepatitis B and C
viral infections, chronic alcohol consumption and aflatoxin B1
exposure are predominant risk factors for HCC development (2). Although understanding of HCC
pathogenesis and clinical therapies for HCC treatment have been
achieved, HCC incidence and survival of HCC patients are still
unsatisfactory (3). Therefore,
effective prognostic indicators and therapeutic strategy for HCC
are needed.
Antidepressants are clinically prescribed to cancer
patients for the treatment of various disorders, including
depression, psychiatric disorders, and chronic pain (4,5). In
addition, antidepressant drugs reduce hot flashes in patients
treated with chemotherapy and protect normal cells from radiation
and chemotherapy (6). Antitumor
effects of selective serotonin (SSRIs) and serotonin and
norepinephrine reuptake inhibitors (SNRIs) have been reported in
several cancer cell lines. Fluoxetine, an SSRI antidepressant, was
shown to inhibit the growth of tumor-derived cell lines by
promoting apoptosis through activation of MAPK and caspase-3
pathways in human carcinoma, osteosarcoma, glioma, and
hepatocellular carcinoma cells (7–9).
Recently, SNRIs, duloxetine and milnacipran, were shown to damage
HepG2 cells (10).
Tricyclic antidepressants (TCAs), including
desipramine are an alternative to SSRIs. They are first-line drugs
for pharmacological treatment of neuropathic pain (11). Therapeutic effect of desipramine is
attributed to inhibition of norepinephrine reuptake (12). TCAs have been reported to reduce the
viability of various cell lines (13,14)
and induce neurotoxicity associated with Parkinson's disease
(15). However, potential antitumor
effects of desipramine on hepatocellular carcinoma cells have not
been explored and are the focus of this study.
Materials and methods
Cell culture and reagents
Hep3B cells were obtained from the Korea Cell Line
Bank (KCLB, Seoul, Korea) and grown in Dulbecco's modified Eagle's
medium nutrient mixture F-12 HAM (DMEM F-12 HAM) supplemented with
10% fetal bovine serum, 1% antibiotics, 5 mM L-glutamine.
Desipramine, 4′,6-diamidino-2-phenylindole (DAPI) and
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl
benzimidazolyl-carbocyanine iodide (JC-1) was purchased from Enzo
Life Sciences (Plymouth Meeting, PA, USA).
2′,7′-Dichlorodihydrofluorescin diacetate (DCFH-DA) was purchased
from Thermo Fisher Scientific (Waltham, MA, USA).
Cell viability assay
Cell viability assay was measured by using Cell
Counting Kit-8 (CCK-8; Enzo Life Sciences). Briefly, Hep3B cells
were seeded into 96-well plates (5000 cells/well) and cultured for
24 h in DMEM F-12 HAM containing 10% fatal bovine serum. After
treatment of desipramine (30–500 µM) for 24 h, the CCK-8 reagent
was added to each well, and the cells were incubated at 37°C for an
additional 2 h. Absorbance was measured at 450 nm using a
spectrophotometer (Spectra Max M5; Molecular Devices, Sunnyvale,
CA, USA). For the treatment of N-acetylcysteine (NAC), cells were
treated with desipramine (3–100 µM) with or without 10 mM NAC for
24 h. Cells were then applied to CCK-8 assay for cell viability
assay.
LDH (lactic dehydrogenase) activity
measurement
LDH activity was measured by a chemical colorimetric
method using cytotoxic detection kit (Takara Bio Inc., Shinga,
Japan). Briefly, after 48 h incubation of the Hep3B cells in
12-well plates (1×104 cells/well), the cells were
treated with desipramine (10–500 µM). After 24 h incubation,
culture media were collected and centrifuged at 10,000 × g for 10
min. Optical density values were measured at 490 nm using a
spectrophotometer (Spectra Max M5; Molecular Devices).
Measurement of intracellular reactive
oxygen species (ROS) production
Intracellular ROS can oxidize the non-fluorescent
DCF to the highly fluorescent DCF. Thus, intracellular ROS can be
measured by fluorescence intensity of DCF by a fluorometer.
Briefly, after 48 h incubation of Hep3B cells in 12-well plates
(1×104 cells/well), cells were treated with desipramine
(3–100 µM) for 24 h. Hep3B cells were then treated with 10 µM
DCFH-DA for 30 min at 37°C. Cells were observed under a
fluorescence microscope (IX-81; Olympus Corp.). Fluorescence
intensity was calculated using a spectrophotometer at excitation
and emission wavelengths of 488 and 515 nm, respectively.
MMP (mitochondrial transmembrane
potential) assessment by JC-1 staining
After 48 h incubation of Hep3B cells in 12-well
plates (1×104 cells/well), cells were treated with
desipramine (3–100 µM) for 24 h. Cells were then incubated with 10
µg/ml JC-1 for 10 min. JC-1-labelled cells were observed under a
fluorescence microscope (IX-81; Olympus Corp.). JC-1 fluorescence
was measured using spectrophotometer with 550 nm excitation/600 nm
emission for red fluorescence and 485 nm excitation/535 nm emission
wavelengths for green fluorescence.
Western blot analysis of MAPKs
Hep3B cells were treated with desipramine (100 µM)
at each time point (1, 2, 4, and 8 h, respectively). In addition,
cells were treated with desipramine (3–10 µM) with/without
pre-incubation of MAPK inhibitors (PD98059, SB203580, and SP600125)
for 24 h. Cells were harvested and lysed in RIPA buffer containing
protease inhibitor cocktail (Roche, Indianapolis, IN, USA). Protein
homogenates were separated on SDS-PAGE gels and transferred to PVDF
membranes (Bio-Rad, Hercules, CA, USA). After blocking for 1 h with
5% non-fat dry milk, the membranes were incubated overnight at 4°C
with antibodies against total or phosphorylated extracellular
signal-regulated kinase 1/2 (ERK1/2), c-JUN N-terminal kinase
(JNK), p38, or β-actin (Cell Signaling Technology, Danvers, MA,
USA). Next, the membranes were incubated with the appropriate
HRP-conjugated secondary antibodies (Cell Signaling Technology) for
1 h and the bands were detected using enhanced chemiluminescence.
The blots were scanned by a Bio-Rad ChemiDoc XRS and the intensity
of each protein was quantified by Quantity One 4.5.0 software
(Bio-Rad).
Ca2+ measurement
Intracellular Ca2+ concentration was
measured by using the fluorescent dye, Fura-2/AM (Thermo Fisher
Scientific). Hep3B cells were incubated in 6-well plates
(1×104 cells/well) on laminin-coated coverglass. Cells
were then washed with the HEPES-TRIS buffer, and loaded with 5 µM
Fura-2/AM, together with 0.025% pluronic F127 (Thermo Fisher
Scientific) for 30 min. The fluorescence was monitored under a
fluorescence microscope (IX-81; Olympus Corp.) with 340 and 380 nm
excitation/510 nm emission wavelengths. The equation
[Ca2+] = (R - Rmin) / (Rmax - R)Sf
× Kd was used to convert the Fura-2/AM ratios to intracellular
concentrations. R is the Fluo-3/AM 340/380 ratio.
Statistical analysis
The data are reported as the mean ± SEM. Statistical
significance was analyzed by using one-way analysis of variance
(ANOVA) with Bonferroni's post-hoc test (Prism 5.0.3, GraphPad
Software Inc., San Diego, CA, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
Effects of desipramine on cell
viability of Hep3B cells
In order to determine the anti-proliferative
potential of desipramine in hepatocellular carcinoma cells,
viability of Hep3B cells was investigated using the CCK-8 assay
after treatment with 1, 3, 10, 30, 100, 300, and 500 µM desipramine
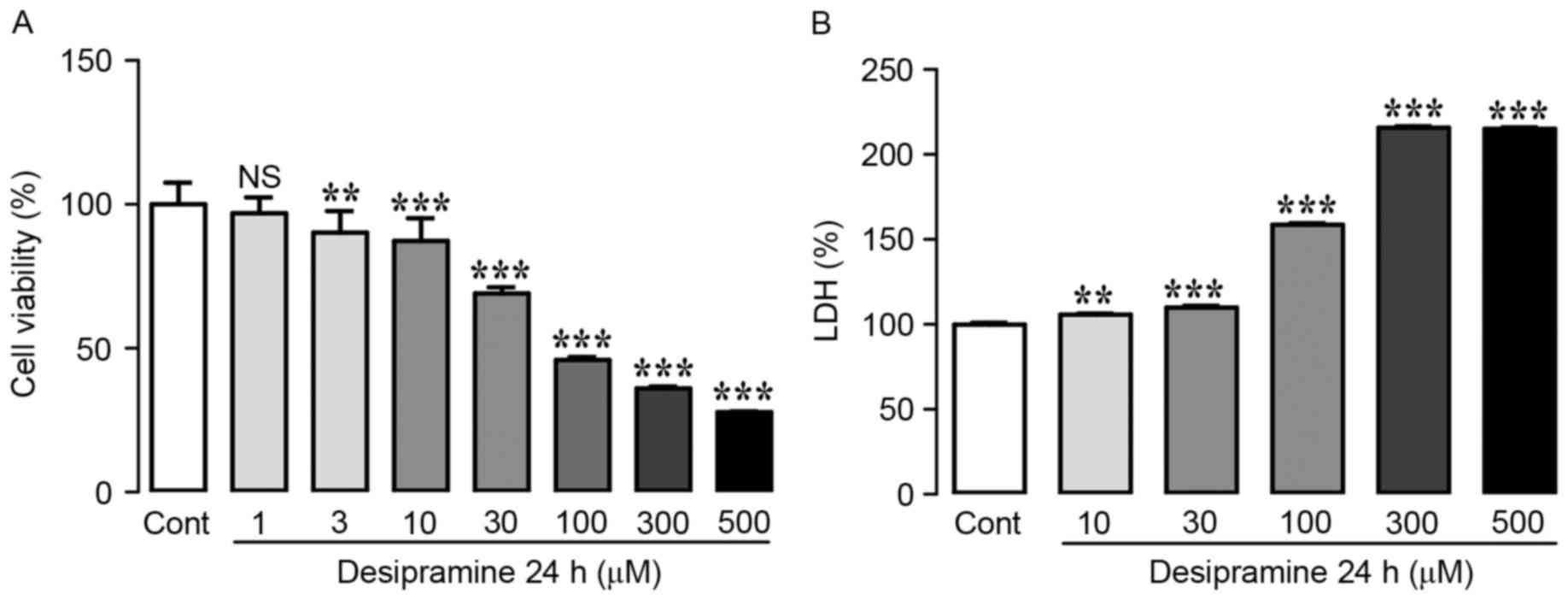
for 24 h. As shown in Fig. 1A,
desipramine induced cell death in a dose-dependent manner.
Treatment of Hep3B cells with 1 µM desipramine did not affect cell
viability, whereas 3–500 µM desipramine induced a significant
decrease in cell viability (90.2±2.9, 87.4±3.0, 69.1±2.1, 45.9±1.1,
36.1±0.7, and 27.8±0.2% at 3, 10, 30, 100, 300, and 500 µM, vs.
control cells, respectively) (Fig.
1A). As LDH is used as an indicator of cytotoxicity because of
LDH release upon loss of cell membrane integrity, we also
determined LDH activity after desipramine treatment (10–500 µM).
Desipramine treatment markedly increased LDH activity in a
dose-dependent manner (105.7±0.8, 109.8±1.1, 150.3±8.4, 227.7±12.1,
and 255.7±40.7% at 10, 30, 100, 300, and 500 µM, vs. control cells,
respectively). Collectively, these results demonstrate that
desipramine inhibits the proliferation of Hep3B cells (Fig. 1B).
Effects of desipramine on ROS
production in Hep3B cells
As ROS production activates pro-apoptotic signaling
pathways, we analyzed the involvement of ROS in desipramine-induced
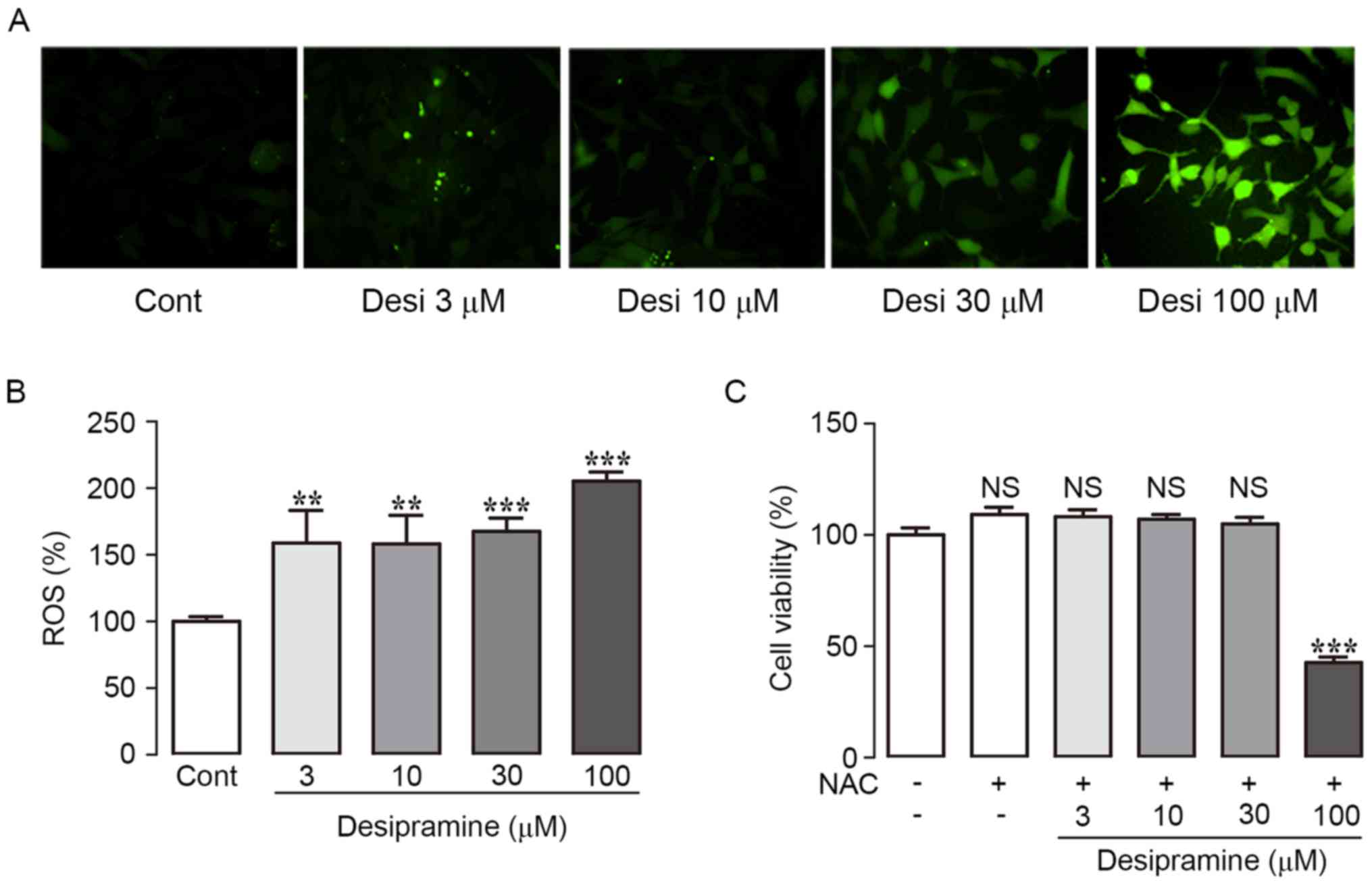
apoptosis in Hep3B cells. ROS levels were assessed using DCFH-DA.
Desipramine treatment significantly increased intracellular ROS
generation in a dose-dependent manner (158.8±10.5, 158.1±15.7,
167.6±3.8, and 205.3±7.6% at 3, 10, 30, and 100 µM vs. control
cells, respectively) (Fig. 2A and
B). In addition, treatment with the ROS scavenger NAC (10 mM)
significantly attenuated desipramine-induced cell death, according
to the CCK-8 assay (Fig. 2C).
Effects of desipramine on MMP in Hep3B
cells
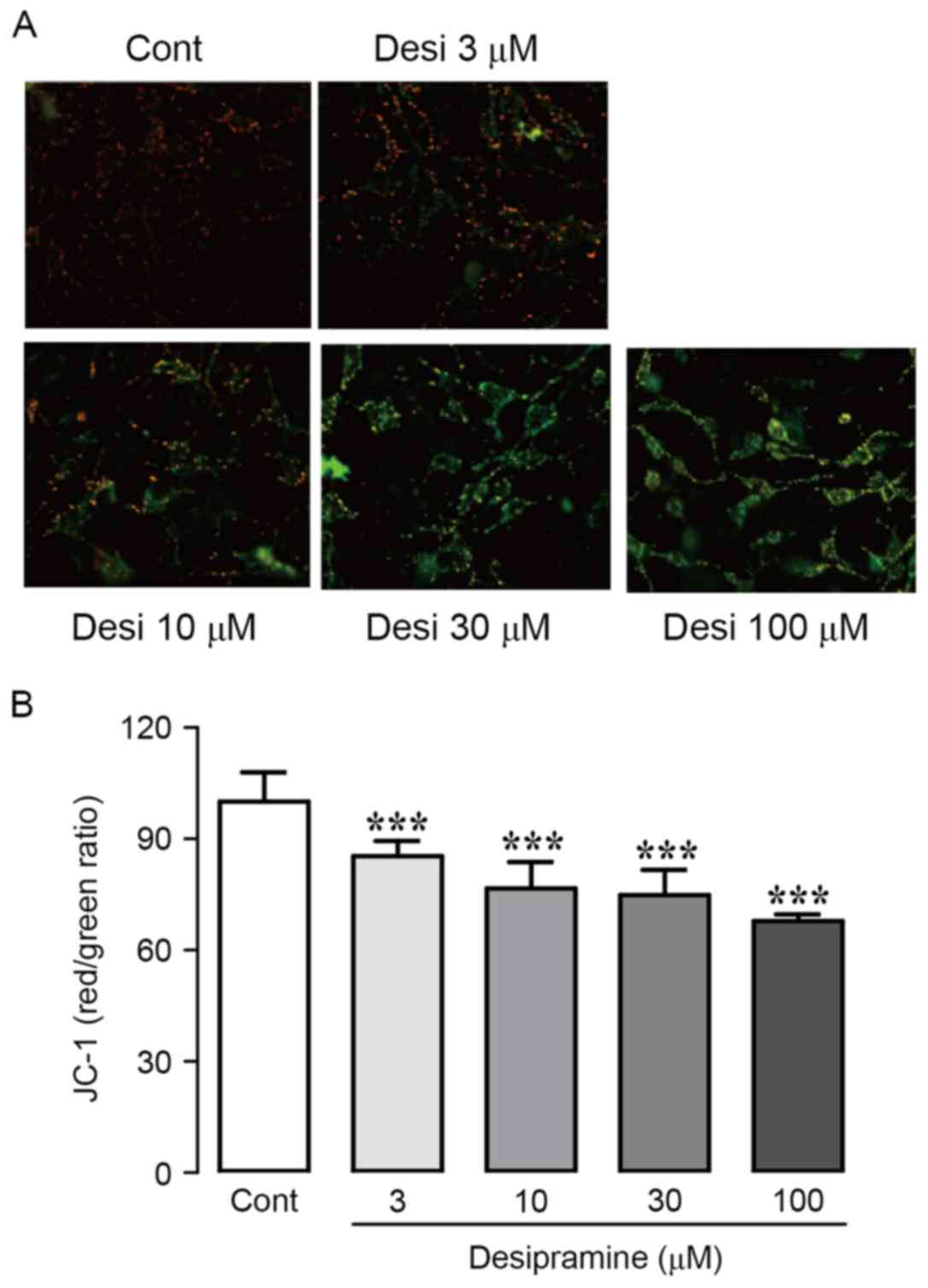
In order to analyze mitochondrial activity in the
presence of desipramine, we observed the effect of desipramine on
MMP using JC-1, an MMP-sensing dye. During apoptosis, mitochondrial
depolarization occurs, altering the fluorescence of JC-1 from red
(aggregates) to green (monomers). Desipramine significantly reduced
MMP in Hep3B cells in a dose-dependent manner, indicated by a shift
from red to green JC-1 fluorescence (Fig. 3A). To corroborate these results, we
monitored the fluorescence intensity of JC-1 using
spectrophotometry. As concentration of desipramine increased, the
ratio of red/green fluorescence intensity markedly decreased
(85.4±1.2, 76.6±2.9, 74.8±2.0, and 67.9±1.7% at 3, 10, 30, and 100
µM vs. control cells, respectively) (Fig. 3B).
Effect of desipramine on the MAPK
signaling in Hep3B cells
In order to identify molecular mechanisms underlying
anti-proliferative effects of desipramine in Hep3B cells, we
determined protein expression patterns of MAPKs (ERK1/2, JNK, and
p38) involved in pro-apoptotic signaling pathways. Results showed
that phospho-ERK levels were significantly increased after 100 µM
desipramine treatment for 1, 2, and 4 h compared with control cells
(185.2±10.1, 151.5±9.8, and 138.5±6.1% at 1, 2, and 4 h vs. control
cells, respectively). In addition, phospho-p38 and JNK expression
levels were significantly increased after 100 µM desipramine
treatment for 1 and 2 h compared with control cells (135.1±4.8 and
116.4±2.3% increase in p-38, 118.9±2.3 and 113.9±2.8% increase in
p-JNK vs. control cells, respectively) (Fig. 4A and B). Pre-incubation with 20 µM
PD98059 (ERK1/2 inhibitor), SB203580 (p38 inhibitor), or SP600125
(JNK inhibitor) followed by exposure to desipramine (3–100 µM)
revealed that all three inhibitors abolished the anti-proliferative
effect of desipramine in Hep3B cells (Fig. 4C).
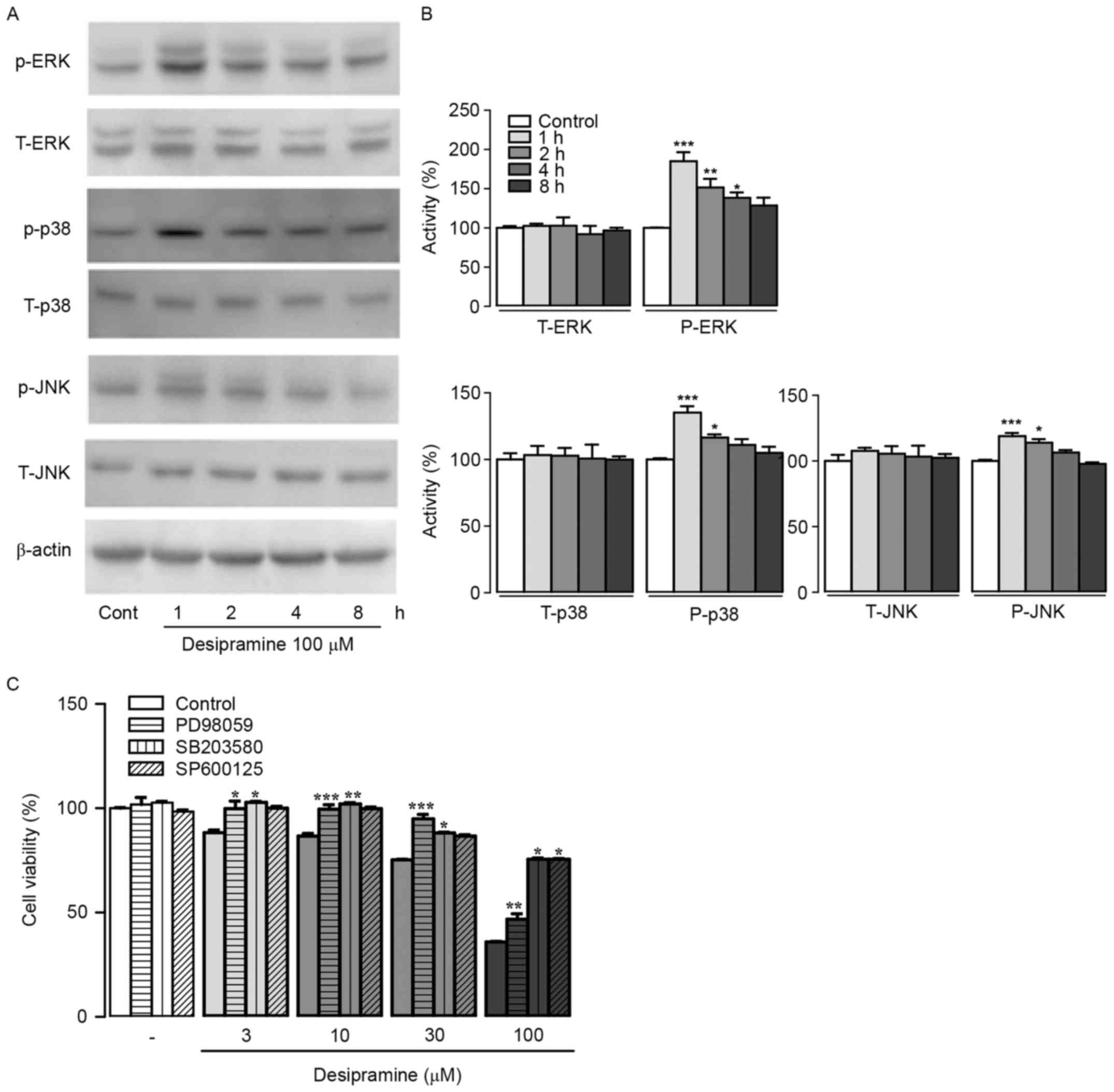 | Figure 4.Effects of desipramine on MAPK protein
expression in Hep3B cells. (A) Cell extracts from
desipramine-treated Hep3B cells at different time points (1, 2, 4,
and 8 h, respectively) were analyzed using western blotting. (B)
Total and phosphorylated forms of ERK1/2, p38, and JNK were
quantified with scanning densitometry. β-actin was used as the
loading control. (C) The cells were pre-incubated with MAPK
inhibitors (PD98059, SB203580, and SP600125) followed by
desipramine (3–100 µM) treatments. Cell viability was determined
using the CCK-8 assay. Data are expressed as % changes ± SEM vs.
the control group. Differences between the groups were analyzed
using a one-way ANOVA followed by Bonferroni's post-hoc test.
*P<0.05, **P<0.01, ***P<0.001 vs. control. Cont,
control. |
Effects of desipramine on
[Ca2+]i in Hep3B cells
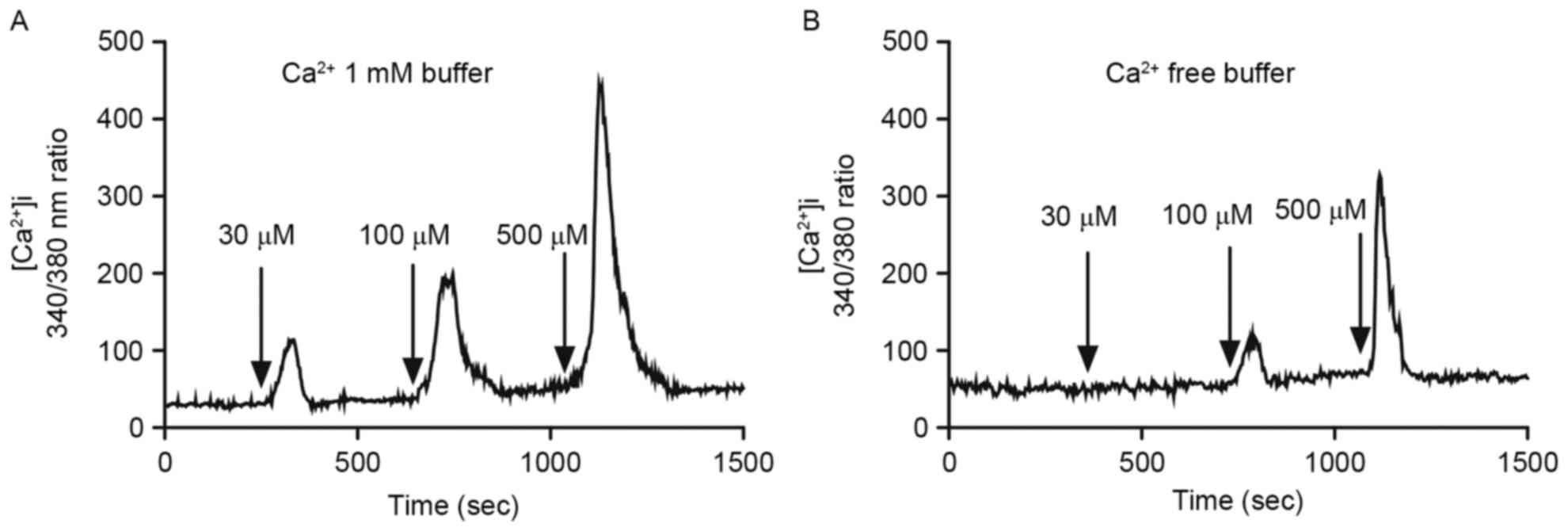
Since abnormal [Ca2+]i rise may lead to
interruption of ion flux, protein dysfunctions, or apoptosis
(16), we analyzed
[Ca2+]i after desipramine treatment (30, 100, and 500
µM) in Ca2+-free or 1 mM Ca2+-buffer using
Fura-2/AM, a fluorescent Ca2+-sensitive dye. Desipramine
induced [Ca2+]i increase in a concentration-dependent
manner in 1 mM Ca2+-containing buffer (Fig. 5A). Similarly, [Ca2+]i
increase occurred after 100 and 500 µM desipramine treatment in
Ca2+-free buffer (Fig.
5B).
Discussion
In the present study, we demonstrated that
desipramine inhibits proliferation of Hep3B cells by inducing
apoptosis, which suggests the antitumor potential of this drug in
hepatocellular carcinoma. Some antidepressants (e.g., paroxetine,
fluvoxamine and sertraline) have potent anticancer properties
against various cancer cell lines (17,18).
In contrast, these drugs were also reported to stimulate or not
affect proliferation of tumor cells (19,20).
Cytotoxic effects of desipramine were demonstrated in several
cancer cell lines, including prostate cancer, colon, renal tubular,
and glioma cells (21–23). Consistent with previous
observations, we found that viability of Hep3B cells was decreased
after desipramine treatment in a dose-dependent manner (Fig. 1A). Furthermore, increased LDH
release confirmed the cell damage caused by desipramine (Fig. 1B).
ROS play an important role in oxidative stress,
which is primarily generated in mitochondria. In tumor cells,
increased ROS generation promotes proliferation, altered
metabolism, and angiogenesis, and is controlled by the
oxidant/anti-oxidant balance system (24). When this system is impaired,
excessive amounts of ROS eventually lead to tumor cell death. Our
results demonstrated that desipramine treatment markedly induced
ROS production in Hep3B cells, an effect prevented by treatment
with NAC, a ROS scavenger (Fig. 2).
In addition, desipramine decreased MMP in Hep3B cells (Fig. 3). Overproduction of ROS causes
mitochondria damage, loss of MMP, and eventually, apoptosis via
mitochondria-mediated cell death pathway (25).
MAPK signaling pathways are known regulators of cell
survival, proliferation, and stress response and are responsible
for the apoptotic cascade in a number of cancer cell lines. Many
anticancer agents activate MAPK pathways in various cell types
(26). We examined the effects of
desipramine on the expression of three major MAPK proteins (ERK1/2,
JNK, and p38) in Hep3B cells. Our results suggest that desipramine
significantly inhibits the phosphorylation of ERK1/2, JNK, and p38.
Furthermore, exposure to MAPK inhibitors suppressed
desipramine-induced cell death (Fig.
4). These results indicate that MAPK signaling pathways play an
important role in desipramine-induced cell death in Hep3B
cells.
Intracellular Ca2+ is closely linked to
ROS production with accumulation leading to apoptotic cell death
(27). Ca2+ overload
triggers the opening of the permeability transition pore, which is
associated with mitochondrial cell death pathways of apoptosis
(28). Increase in intracellular
Ca2+ has been associated with apoptosis in tumor cells
(9,29). In this study, [Ca2+]i
increased in response to desipramine treatment in
Ca2+-free and 1 mM Ca2+ buffer (Fig. 5). These results indicate that
desipramine causes Ca2+ influx or release of
Ca2+ from endoplasmic reticulum. Consequently, increase
in intracellular Ca2+ participates in
desipramine-induced apoptosis of Hep3B cells.
In conclusion, the current study provides new
evidence that desipramine induces apoptosis of hepatocellular
carcinoma cells by increasing ROS production, reducing MMP,
promoting accumulation of intracellular Ca2+, and
increasing the activity of MAPK proteins (ERK1/2, JNK, and p38). We
further propose desipramine as a potential anticancer agent against
HCC.
Acknowledgements
This study was supported by the research funds of
Korean Ministry of Science (2011–0013872) and the National Research
Foundation of Korea (NRF) grant funded by the Korea government
(MSIP) (2016R1A2B1010904).
References
|
1
|
Paul SB, Shalimar Sreenivas V, Gamanagatti
SR, Sharma H, Dhamija E and Acharya SK: Incidence and risk factors
of hepatocellular carcinoma in patients with hepatic venous outflow
tract obstruction. Aliment Pharmacol Ther. 41:961–971. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: Epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kang TW and Rhim H: Recent advances in
tumor ablation for hepatocellular carcinoma. Liver Cancer.
4:176–187. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Riblet N, Larson R, Watts BV and
Holtzheimer P: Reevaluating the role of antidepressants in
cancer-related depression: A systematic review and meta-analysis.
Gen Hosp Psychiatry. 36:466–473. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Smith EM, Pang H, Cirrincione C, Fleishman
S, Paskett ED, Ahles T, Bressler LR, Fadul CE, Knox C,
Le-Lindqwister N, et al: Alliance for Clinical Trials in Oncology:
Effect of duloxetine on pain, function, and quality of life among
patients with chemotherapy-induced painful peripheral neuropathy: A
randomized clinical trial. JAMA. 309:1359–1367. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lieb J: Antidepressants, prostaglandins
and the prevention and treatment of cancer. Med Hypotheses.
69:684–689. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chou CT, He S and Jan CR:
Paroxetine-induced apoptosis in human osteosarcoma cells:
Activation of p38 MAP kinase and caspase-3 pathways without
involvement of [Ca2+]i elevation. Toxicol Appl
Pharmacol. 218:265–273. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stepulak A, Rzeski W, Sifringer M, Brocke
K, Gratopp A, Kupisz K, Turski L and Ikonomidou C: Fluoxetine
inhibits the extracellular signal regulated kinase pathway and
suppresses growth of cancer cells. Cancer Biol Ther. 7:1685–1693.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mun AR, Lee SJ, Kim GB, Kang HS, Kim JS
and Kim SJ: Fluoxetine-induced apoptosis in hepatocellular
carcinoma cells. Anticancer Res. 33:3691–3697. 2013.PubMed/NCBI
|
|
10
|
Kuwahara J, Yamada T, Egashira N, Ueda M,
Zukeyama N, Ushio S and Masuda S: Comparison of the anti-tumor
effects of selective serotonin reuptake inhibitors as well as
serotonin and norepinephrine reuptake inhibitors in human
hepatocellular carcinoma cells. Biol Pharm Bull. 38:1410–1414.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Finnerup NB, Attal N, Haroutounian S,
McNicol E, Baron R, Dworkin RH, Gilron I, Haanpää M, Hansson P,
Jensen TS, et al: Pharmacotherapy for neuropathic pain in adults: A
systematic review and meta-analysis. Lancet Neurol. 14:162–173.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dharmshaktu P, Tayal V and Kalra BS:
Efficacy of antidepressants as analgesics: A review. J Clin
Pharmacol. 52:6–17. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Haller I, Lirk P, Keller C, Wang GK,
Gerner P and Klimaschewski L: Differential neurotoxicity of
tricyclic antidepressants and novel derivatives in vitro in a
dorsal root ganglion cell culture model. Eur J Anaesthesiol.
24:702–708. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kitagawa N, Oda M, Nobutaka I, Satoh H,
Totoki T and Morimoto M: A proposed mechanism for amitriptyline
neurotoxicity based on its detergent nature. Toxicol Appl
Pharmacol. 217:100–106. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee MY, Hong S, Kim N, Shin KS and Kang
SJ: Tricyclic antidepressants amitriptyline and desipramine induced
neurotoxicity associated with Parkinson's disease. Mol Cells.
38:734–740. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Clapham DE: Intracellular calcium.
Replenishing the stores. Nature. 375:634–635. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bielecka AM and Obuchowicz E:
Antidepressant drugs as a complementary therapeutic strategy in
cancer. Exp Biol Med (Maywood). 238:849–858. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kubera M, Grygier B, Arteta B, Urbańska K,
Basta-Kaim A, Budziszewska B, Leśkiewicz M, Kołaczkowska E, Maes M,
Szczepanik M, et al: Age-dependent stimulatory effect of
desipramine and fluoxetine pretreatment on metastasis formation by
B16F10 melanoma in male C57BL/6 mice. Pharmacol Rep. 61:1113–1126.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Volpe DA, Ellison CD, Parchment RE,
Grieshaber CK and Faustino PJ: Effects of amitriptyline and
fluoxetine upon the in vitro proliferation of tumor cell lines. J
Exp Ther Oncol. 3:169–184. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li YF and Luo ZP: Desipramine antagonized
corticosterone-induced apoptosis in cultured PC12 cells. Acta
Pharmacol Sin. 23:311–314. 2002.PubMed/NCBI
|
|
21
|
Qi H, Chen HZ and Jin ZJ: Caspase 3 gene
expression and [Ca2+]i homeostasis underlying
desipramine-induced C6 glioma cell apoptosis. Acta Pharmacol Sin.
23:803–807. 2002.PubMed/NCBI
|
|
22
|
Ho CM, Kuo SY, Chen CH, Huang JK and Jan
CR: Effect of desipramine on Ca2+ levels and growth in
renal tubular cells. Cell Signal. 17:837–845. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chang HC, Huang CC, Huang CJ, Cheng JS,
Liu SI, Tsai JY, Chang HT, Huang JK, Chou CT and Jan CR:
Desipramine-induced apoptosis in human PC3 prostate cancer cells:
Activation of JNK kinase and caspase-3 pathways and a protective
role of [Ca2+]i elevation. Toxicology. 250:9–14. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ríos-Arrabal S, Artacho-Cordón F, León J,
Román-Marinetto E, Del Mar Salinas-A, Sensio M, Calvente I and
Núñez MI: Involvement of free radicals in breast cancer.
Springerplus. 2:4042013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Estaquier J, Vallette F, Vayssiere JL and
Mignotte B: The mitochondrial pathways of apoptosis. Adv Exp Med
Biol. 942:157–183. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chang HL, Wu YC, Su JH, Yeh YT and Yuan
SS: Protoapigenone, a novel flavonoid, induces apoptosis in human
prostate cancer cells through activation of p38 mitogen-activated
protein kinase and c-Jun NH2-terminal kinase 1/2. J Pharmacol Exp
Ther. 325:841–849. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Perrone GG, Tan SX and Dawes IW: Reactive
oxygen species and yeast apoptosis. Biochim Biophys Acta.
1783:1354–1368. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Orrenius S, Zhivotovsky B and Nicotera P:
Regulation of cell death: The calcium-apoptosis link. Nat Rev Mol
Cell Biol. 4:552–565. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim TH, Kim JS, Kim ZH, Huang RB, Chae YL
and Wang RS: Induction of apoptosis in MCF-7 human breast cancer
cells by Khz (fusion of Ganoderma lucidum and Polyporus
umbellatus mycelium). Mol Med Rep. 13:1243–1249.
2016.PubMed/NCBI
|