Introduction
Mesenchymal stem cells (MSCs) are a group of cells
with the capacity of self-renewal and multilineage differentiation.
Recent studies showed that MSCs could be used as cellular vectors
for targeted delivery of therapeutic agents to tumors (1). MSCs can be derived from a variety of
tissue sources such as bone marrow, umbilical cord and adipose
tissue, however, these adult stem cell sources were still
inadequate for clinical use due to their limited supply and
expansion, and further more, inevitable invasion when collected
(2). Fortunately, a new source of
mesenchymal stem cells emerged at a historic moment - human
embryonic stem cell-derived mesenchymal cells (hESC-MSCs).
hESC-MSCs are easier to be standardized, non-invasive and greatly
proliferate in vitro, which might be an alternative for
traditional adult stem cell therapy (3).
Microvesicles (MVs) are small, spherical membrane
fragments, 50–1,000 nm in diameter and believed to be involved in
multiple aspects of cancer progression (4,5). It
was reported that MVs could deliver antitumor microRNAs, thus
inhibiting tumor growth and stimulating apoptosis (6). Recent studies demonstrated that human
umbilical cord blood-derived mesenchymal stem cells (UC-MSCs)
inhibited the growth of K562 (7).
Particularly, stem cells are a rich source of soluble factors and
MVs. Based on these findings, we assumed that hESC-MSCs might also
suppress the growth of leukemia cells, and in which MVs probably
played an important role.
Materials and methods
Cell culture
The human embryonic stem cell line (NJ15) was kindly
provided by Dr Lianju Qin, from the Nanjing Maternity and Child
Health Care Hospital, Nanjing, China under a Materials Transfer
Agreement. Human umbilical cord-derived mesenchymal stem cell
(UC-MSCs) and bone marrow-derived mesenchymal stem cells (BM-MSCs)
were provided by Dr Wei Zhu, School of Medicine, Jiangsu
University. K562 and HL60 cells were provided by Dr Jun Qian,
Affiliated People's Hospital of Jiangsu University. The hESCs were
cultured on mitomycin C (Roche, USA)-pretreated mouse embryonic
fibroblast (MEF) and maintained in the hESCs medium: DMEM/F12
(Gibco, Grand Island, NY, USA) 80 ml, knockout serum replacement
(Gibco) 20 ml, 1% non-essential amino acid (Gibco), 1 mM
L-glutamine (Gibco), 0.1 mM β-mercaptoethanol (Sigma Chemical Co.,
St. Louis, MO, USA), 4 ng/ml basic fibroblast growth factor
(Gibco).
We harvested hESC-MSCs based our previous study
(8). Briefly, hESCs were directly
seeded onto a plate coated with 0.1% gelatin and cultured in the
MSC growth medium: DMEM low-glucose medium supplemented with 10%
fetal bovine serum (ExCell Bio, FND500, Australia). One week later,
cells were passaged and cultured in normal subculture (Named P1).
After serial passages, further purification was achieved.
Human UC-MSCs, BM-MSCs and leukemia cell lines K562
and HL60 were maintained in DMEM medium (Gibco) supplemented with
10% FBS.
Cell growth and cell viability
assays
To evaluate the effects of hESC-MSCs on the
proliferation of K562 and HL60, 1×105 leukemia
cells/well were cultured solely (as control) or incubated with
hESC-MSCs in DMEM medium without serum (hESC-MSCs were pre-treated
by mitomycin C and cultured overnight prior to coculture with
leukemia cells). After 48-h coculture, leukemia cells were
collected and counted under a microscope. For Transwell cocultures
(Transwell, 3-µm pore size; Corning, USA), hESC-MSCs were plated at
a concentration of 1×105 cells/well in the upper
compartment and an equal number of K562 and HL60 cells were plated
in the lower compartment 24 h later. In direct contact groups,
1×105 leukemia cells were in coculture with hESC-MSCs at
ratios of 50:1 and 0.5:1 of hESC-MSCs. After 24-h coculture,
leukemia cells were collected and assessed with a Cell Counting
Kit-8 (CCK-8) assay. To evaluate the effects of MVs on the growth
of leukemia cells, K562 and HL60 were incubated with different dose
of MVs in DMEM medium without serum. Forty-eight hours later, cell
viability was analyzed by CCK-8 method. Into each well, 100 µl cell
suspension was added with 10 µl CCK-8 reagent (Dojindo, Japan) and
incubated for 2 h at 37°C. Finally, the optical density (OD) values
were read on an enzyme-labeled ELx800 (Bio-Tek, Winooski, VT, USA)
at 450 nm. We calculated the proliferation inhibition rate (IR) by
the following formula: IR = [1 - (mean OD value of test groups) /
(mean OD value of control groups)] × 100%.
CCK-8 assay was used to determine the number of
living cells in cell proliferation assays by utilizing
[2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,
4-disulfophenyl)-2H-tetrazolium, monosodium salt] (WST-8). WST-8
decreased by dehydrogenases in cells to give an orange colored
product (formazan), which was directly proportional to the number
of viable cells.
MVs isolation and characterization by
electron microscopy
To collect MVs, hESC-MSCs were cultured in DMEM
medium depleted of serum. Forty-eight hours later hESC-MSC-MVs were
purified following the standardized procedure reported by Gadkari
et al and Bruno et al (9,10). In
brief, the supernatants (or conditioned medium) of hESC-MSCs were
centrifuged at 2,000 × g for 5 min and then 12,000 × g for 15 min
to remove cell fragments and impurities. Then, conditioned medium
was centrifuged (Beckman Coulter Optima L-90K, Beckman Coulter,
Fullerton, CA, USA) at 100,000 × g for at least 1 h, washed in the
DMEM containing 25 mM HEPES (Gibco) and followed by a second
ultracentrifugation in the same way. We removed the excess fluid
and added 200 µl PBS to resuspend the pellets. The protein content
of hESC-MSC-MVs was analyzed by BCA method (Sigma).
To ensure that the isolated pellets were
hESC-MSC-MVs, we identified them under a scanning electron
microscope (SEM; Hitachi, Japan) and a transmission electron
microscope (TEM; Philips, FEI, Eindhoven, The Netherlands)
respectively.
Characterization of autophagosomes by
TEM
Leukemia cells were first fixed by 3% glutaraldehyde
plus 2% formaldehyde in 0.1 M sodium cacodylate buffer solution.
Then cells were fixed with 2% osmium tetroxide and dehydrated with
propylene oxide and ethanol. After that, cells were embedded, and
ultrathin sections were cut and stained in uranyl acetate as well
as lead citrate, and observed under a TEM (Philips).
Western blot analysis
Leukemia cells (1×105 cells/well) were
cultured solely (as control) or incubated with hESC-MSC-MVs (30 and
60 µg/ml) in DMEM medium without serum. Forty-eight hours later,
total protein of leukemia cells was extracted and 100 µg protein
was subjected to 12% SDS-PAGE and subsequently electroblotted onto
PVDF membranes. These reactive bands were analyzed with enhanced
chemiluminescence (ECL, Millipore, USA) reagents. Signals were
captured using an Image Quant™ LAS-4000 Mini Imager (GE Healthcare,
Piscataway, NJ, USA). All primary antibodies including rabbit
anti-GAPDH (cat. no. 5174), anti-Beclin-1 (cat. no. 3738),
anti-LC3B (cat. no. 2775), anti-Bax (cat. no.2772), anti-Bcl-2
(cat. no. 2876) were purchased from Cell Signaling Technology (CST
Beverly, MA, USA). Primary antibodies were detected using goat
anti-rabbit secondary antibody (Thermo Fisher Scientific, Rockford,
IL, USA). The protein density of each band was detected using
Quantity One v4.62 software (Bio-Rad, Inc, Berkeley, CA, USA) for
semiquantitative evaluation.
Flow cytometer analysis
Apoptosis of tumor cells was measured using Annexin
V-FITC/PI apoptosis kit (BD Biosciences, San Jose, CA, USA). In
total, 1×105 leukemia cells were collected and stained
with 5 µl Annexin V-FITC and PI successively under normal
temperature for 15 min away from light, and analyzed within an hour
on flow cytometer (FACSCalibur; BD Biosciences).
Statistical analysis
The one-way ANOVA with multiple comparisons was
adopted to evaluate the difference among three or more groups. Each
experiment was independently carried out at least three times. Data
are shown as the mean ± standard deviation (SD) and p-value ≤0.05
was statistically significant.
Results
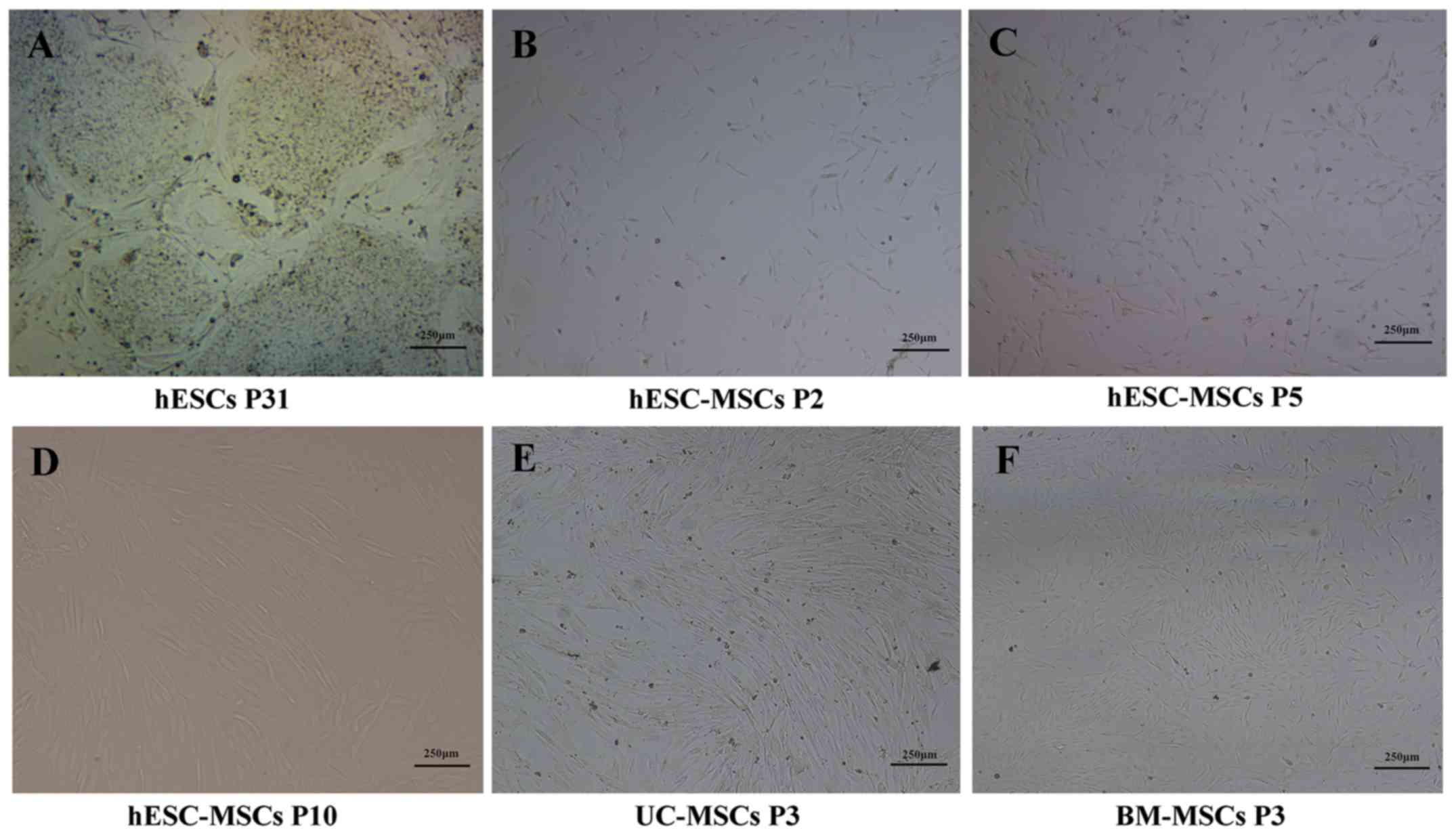
Generation of human embryonic stem
cell derived-mesenchymal stem cells (hESC-MSCs)
hESCs (P31) were originally cultured under
feeder-dependent conditions (Fig.
1A). Approximately 3 days after cultured in MSC medium, long
spindle cells began appearing around the colonies. When these long
spindle cells reached >75% confluence (~7 days later), cells
were digested and seeded onto new gelatin-coated cultures. With the
removal of colony structures, heterogeneous cells became
homogeneous and gradually began to be more slender and confluent
(Fig. 1B and C). At passage 10,
some shuttle fibroblast-like cells were in the majority and
arranged regularly similar to UC-MSCs and BM-MSCs (Fig. 1D-F).
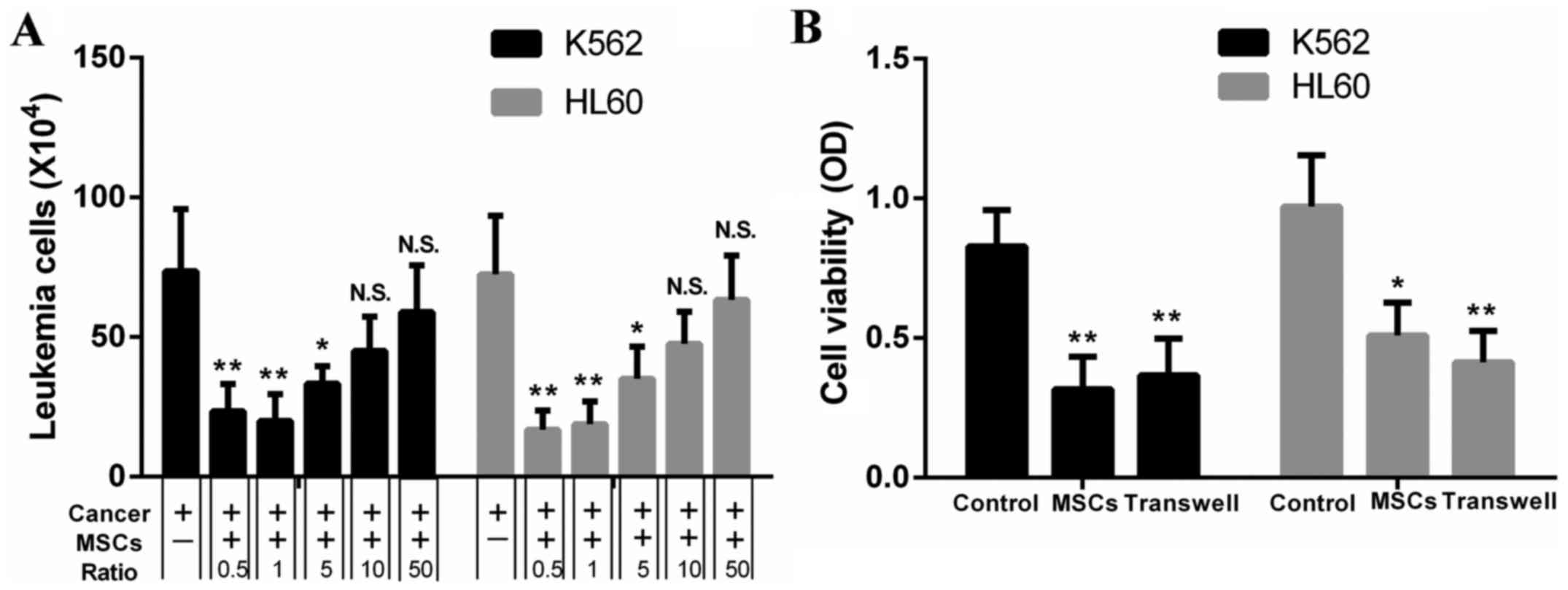
hESC-MSCs inhibit the proliferation of
leukemia cells
We observed that hESC-MSCs showed an inhibitory
effect on proliferation of leukemia cells which was
concentration-dependent. The number of K562 and HL60 cells alone,
cocultured with hESC-MSCs at ratios of 50:1 and 0.5:1 as shown in
Fig. 2, the amount of K562 and HL60
cells rose with the increasing ratio of leukemia cells to hESC-MSCs
over the range of 0.5:1-50:1 (Fig.
2A). To determine whether this effect was realized through
paracrine or cell-to-cell contact, the Transwell chamber technology
was used and CCK8-assay was adopted to evaluate cell viability. The
inhibition rate of K562 for direct contact and Transwell coculture
groups was 61.3±15.7 versus 51.3±4.6%. In HL60, the rate was
46.6±13.2 versus 57.2±8.8% and there was no significant difference
between these groups. Results indicated that cell-to-cell contact
might not be necessary, since this effect still existed in
Transwell cocultures (Fig. 2B).
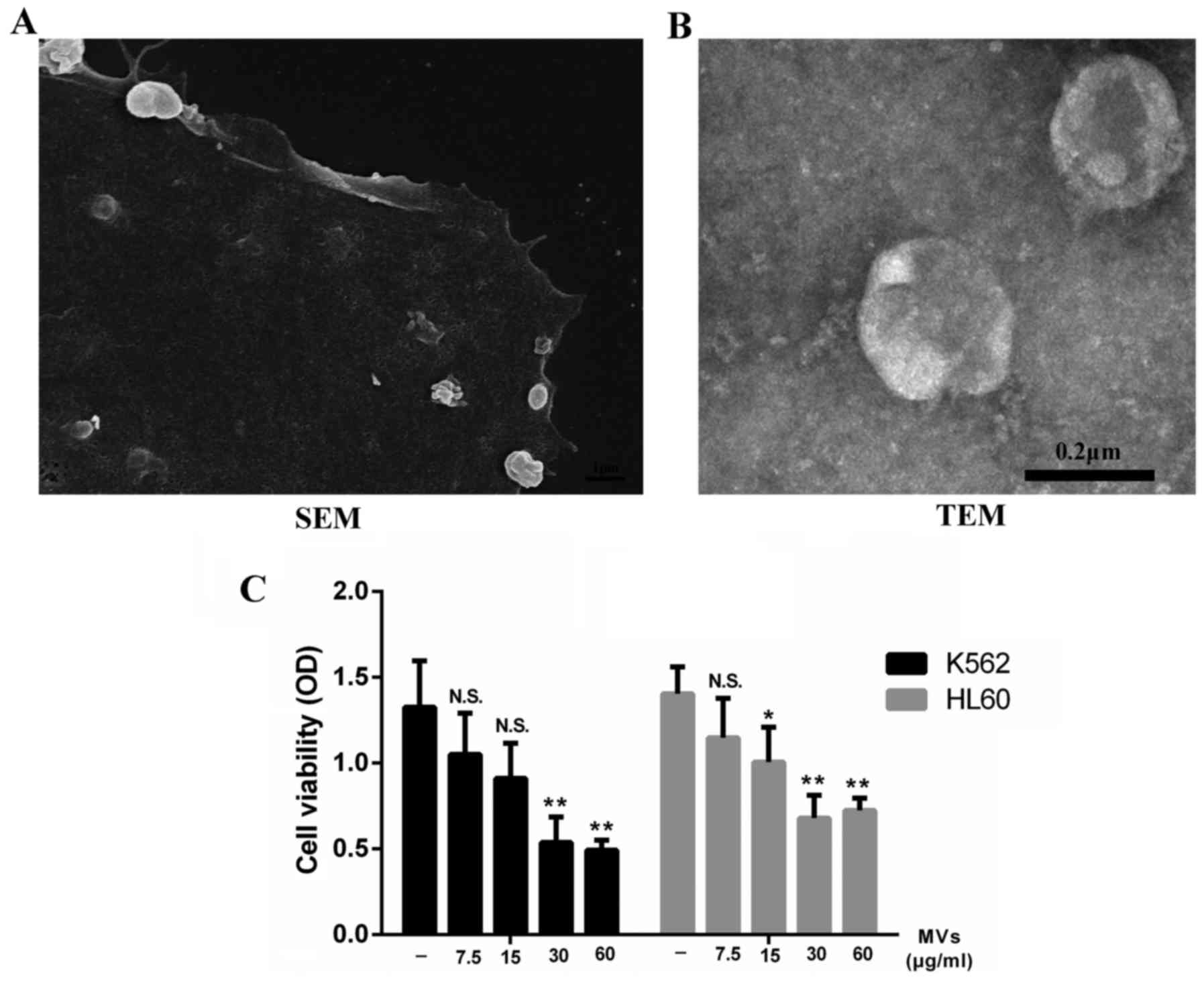
hESC-MSCs-MVs inhibit the
proliferation of leukemia cells
Supernatants of hESC-MSCs were ultracentrifuged and
the collected pellets were captured under a SEM and TEM separately.
We observed that the hESC-MSC-MVs were small, spherical membrane
fragments, several hundreds nanometers in diameter (Fig. 3B) and released from cell surface or
by exocytose (Fig. 3A). We tested
the content of MVs through a BCA protein assay kit resulting in
93.7 µg/100 µl.
Considering that paracrine might play an important
part in the inhibitory effect, we incubated K562 and HL60 cells
with different levels of MVs ranging from 0 to 60 µg/ml. Data
showed that MVs were able to inhibit the growth of leukemia cells
as well as hESC-MSCs. This effect was enhanced by increasing the
dose of MVs. However, this inhibitory rate did not increase
obviously when the concentration of MVs reached 30 µg/ml (Fig. 3C).
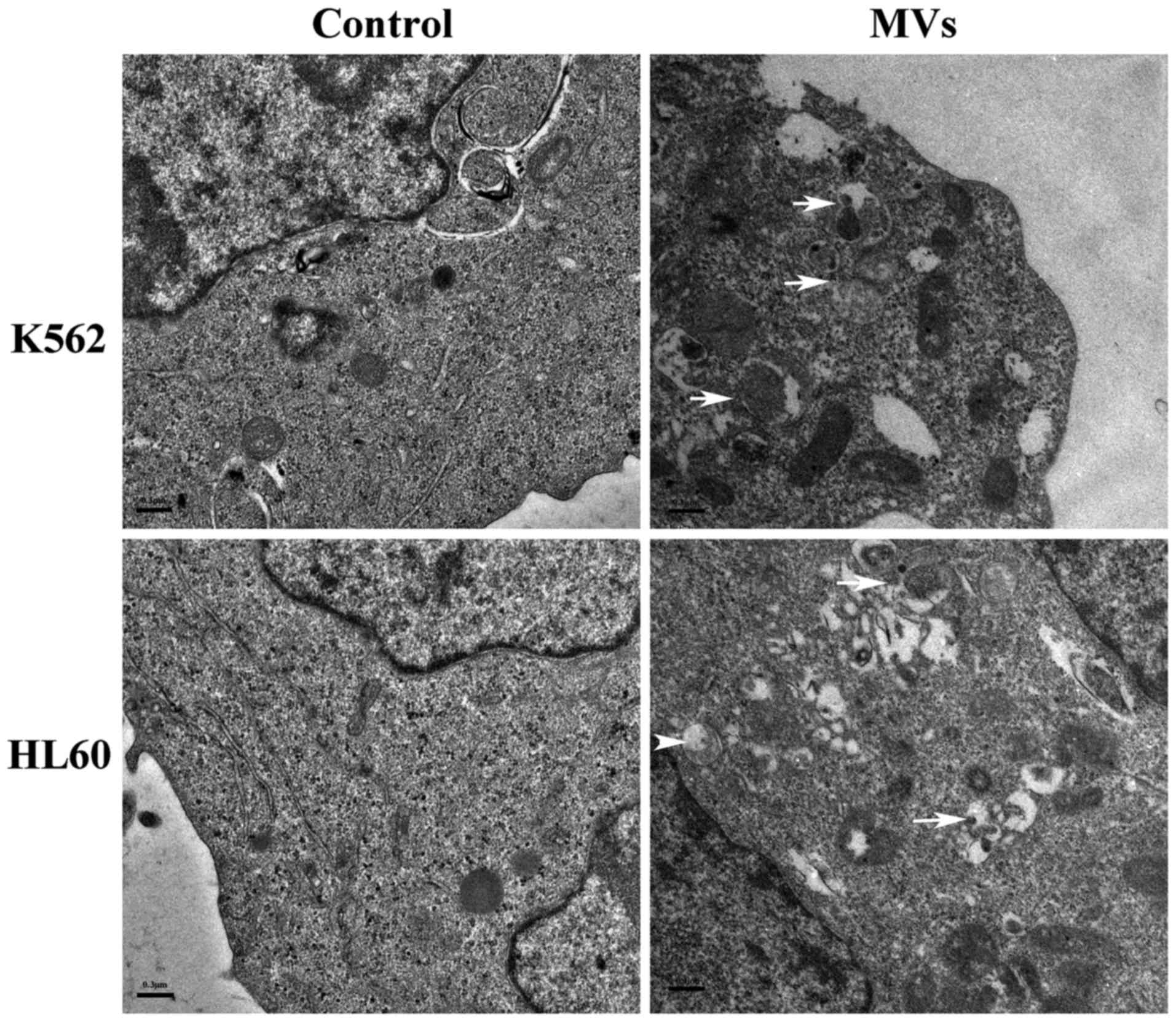
hESC-MSC-MVs upregulate autophagic
activity in K562 and HL60 cells
After 48-h incubation with 0 and 30 µg/ml MVs, K562
and HL60 cells were identified under TEM. While treated groups
showed many autophagosomes, electron-dense vacuoles, which
contained degraded organelles such as mitochondria and endoplasmic
reticulum, such vacuoles were absent in control groups (Fig. 4).
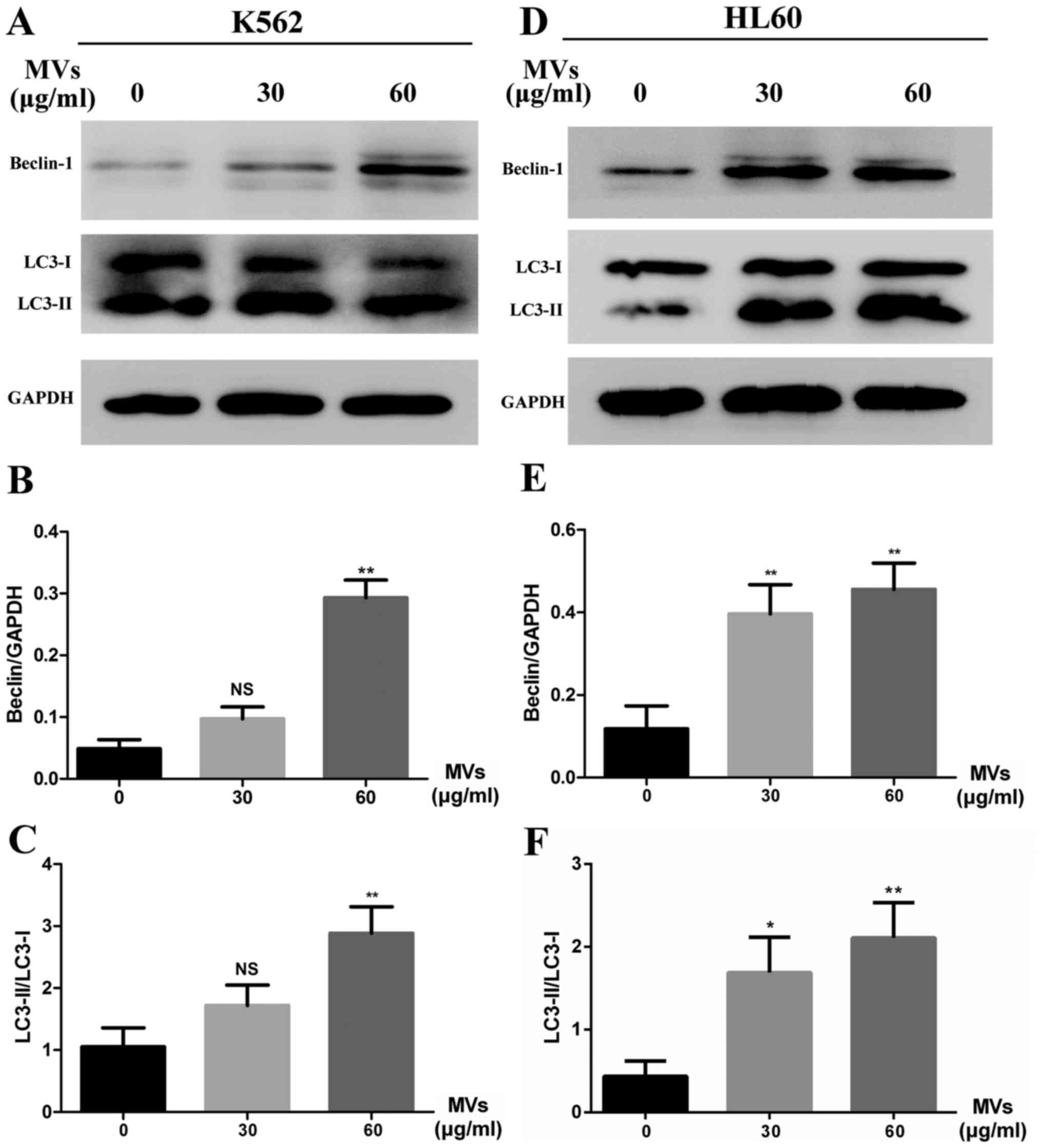
To confirm MVs influence autophagic activity in
leukemia cells, western blot analysis of Beclin-1 elevation and LC3
conversion was performed. In the experiment groups, K562 and HL60
cells were incubated with 30 and 60 µg/ml of MVs for 48 h. Results
confirmed increasing level of Beclin-1 and LC3-II formation, which
indicated that MVs could apparently upregulate the autophagic
activity compared with untreated groups (Fig. 5A and D). Semiquantitative analysis
are shown of Beclin-1 levels (Fig. 5B
and E) and LC3 conversion (Fig. 5C
and F).
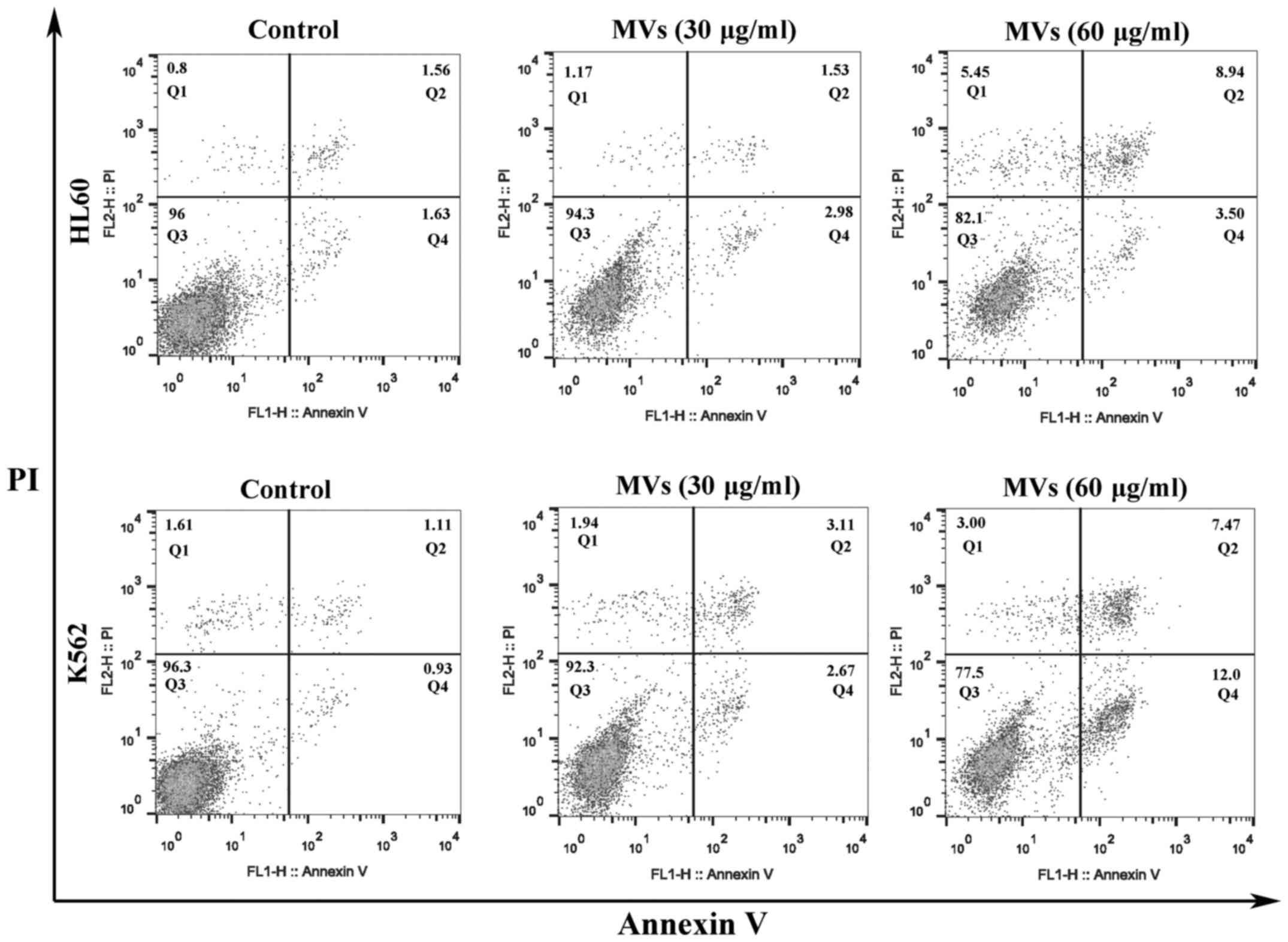
hESC-MSC-MVs promote apoptosis in
leukemia cells
K562 and HL60 cells were stained with Annexin V/PI
kit and flow cytometric analysis indicated that the apoptosis ratio
of leukemia cells increased with the higher concentration of MVs
(Fig. 6).
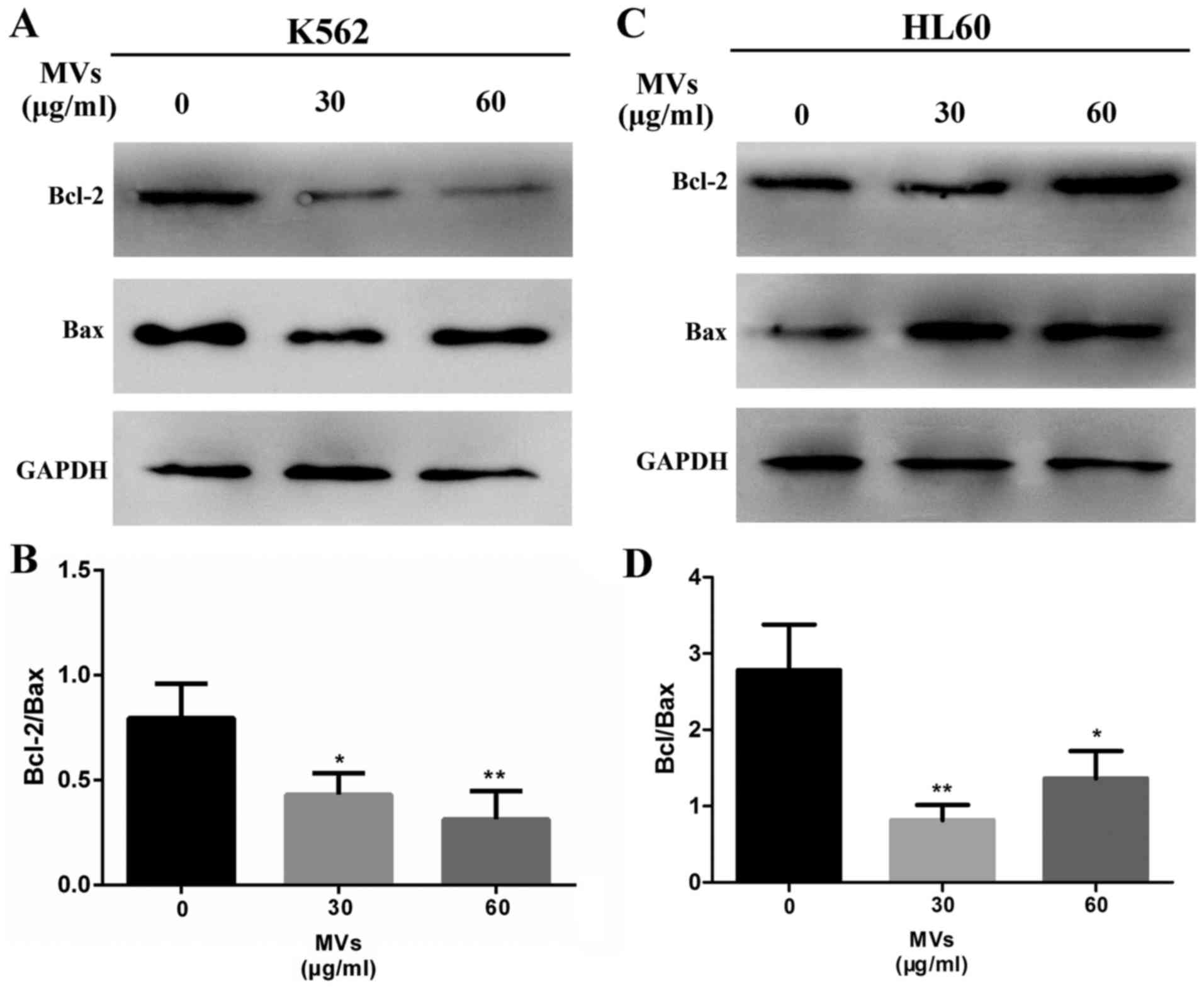
Western blotting was conducted to detect the ratio
of Bcl-2 to Bax, which is a common way to assess the level of
apoptosis. Results showed that the ratio was significantly
decreased in MV-treated groups (Fig.
7).
Discussion
Previous studies showed that UC-MSCs could inhibit
the proliferation of K562 (7) after
coculture. BM-MSCs were found to inhibit the proliferation of
malignant cells of hematopoietic origin (11,12).
However, these MSCs had many limitations such as inadequate supply
and inevitable invasion when collected. Our previous study focused
on the acquisition and characterization of hESC-MSCs and found that
they shared many characteristics with traditional MSCs, including
morphology, self-renewal and differentiation (8). Particularly, our hESC-MSCs could be
cultured for at least 26 passages (data not shown), which might
become a better source of MSCs. On the basis of this analysis and
previous work, we replaced traditional UC-MSCs or BM-MSCs with
hESC-MSCs and received the expected results that they could also
inhibit leukemia cells hESC-MSCs. However, it was notable that
Ramasamy et al and Goldstein et al reported MSCs
could form a cancer stem cell niche in which tumor cells could
preserve the potential of proliferating and sustaining the
malignant process (12,13). According to the studies of Fonseka
et al and Psaila et al the anti-proliferative effect
of UC-MSCs contributed to arrest the growth of K562 cells in rest
stage (G0/G1 phase), which might confer tumor cells a better
survival and self-renewal ability (7,14). The
transplantation of human MSCs to mice showed tumorigenicity,
attracting attention to this new therapy (15,16).
Considering these problems, we focused on the active components in
the MSCs culture supernatants.
We observed that the inhibitory effect still existed
in Transwell coculture and there was no significant difference
compared with the direct contact group, which provided further
evidence that paracrine might play an important role. However,
Fonseka et al pointed out that the anti-proliferative effect
was reduced in Transwell and supernatant groups, and direct
cell-to-cell contact might play a significant role (7). This variation might be contributed to
different types and pretreatment of MSCs and, more importantly, the
culture conditions. In addition, the effect might be not obvious if
tumor cells were cocultured with the supernatants of MSCs directly
because the concentration of active components within would be too
low. Considering the above, we concentrated on the MSC culture
supernatants and obtained MVs. Under TEM, MVs were electron-dense
vacuoles, several hundred nanometers in diameter and released from
the cell membrane surface or by exocytosis (17,18).
As expected, MVs inhibited the growth of K562 and HL60 cells in a
concentration-dependent manner, which further strengthened the
evidence that paracrine or MVs did play an important role. Similar
findings were provided by Yang et al. In rheir study,
exosomes from ACHN cells inhibited Jurkat T cell proliferation,
which also showed a concentration-dependent manner (19). MVs were expected to become new
material of biotherapy for cancer. Under electron microscope, we
found many electron-dense vacuoles containing extensively degraded
organelles such as mitochondria and endoplasmic reticulum in
MV-treated groups. These vacuoles were similar to
autophagosomes.
Autophagy, or ‘self-eating’, performs a very
important function in many physiological and pathological
processes. For example, it provides cells with metabolic precursors
as well as energy under starvation and at the same time alleviates
stress by removing long lived proteins and damaged organelles
(20). However, dysregulated or
excessive autophagy certainly can cause autophagic cell death, also
called ‘the type II programmed cell death’ (21,22).
In view of this, some anti-neoplastic therapies have focused on
inducing autophagy in human cancer cell lines (23,24).
TEM was widely perceived as a ‘gold standard’ test for autophagy,
the mark of autophagy under TEM was the presence of autophagosome
accumulation. We observed many autophagosomes in MV-treated groups
and suspected that MVs might upregulate autophagy in leukemia
cells. Then, western blotting was performed to measure the
expression level of autophagy-related proteins. In our test,
another important finding was that MVs upregulated the Beclin-1
protein level in leukemia cells. Notably, Beclin-1 was the first
gene that had been proved to be related with mammalian autophagy
(25). It was reported that
Beclin-1 could inhibit cancer initiation and showed low levels in
human breast carcinoma (26). It
should be clearly noted that autophagy is much more than just a
promoter or inhibitor of cancers and the relationship between them
is intricate and complex (27).
Another important problem is the relationship between autophagy and
apoptosis. These two programmed cell death pathways were confirmed
to coregulate the survival and death of cells (28,29).
In some cases, autophagy inhibited apoptosis to adapt to metabolic
and environmental stress; in others, it coordinated with apoptosis
to result in cell death (30).
Cancer cells have various ways to avoid apoptosis, the important
means by which organisms deal with defective cells (31). Inducing apoptosis in tumor cells is
believed to be a common strategy to fight against cancer (32). Other laboratories have provided
evidence that exosomes or MVs could promote apoptosis in tumor
cells (33,34). Our data showed that MVs induced
autophagy and excessive autophagy might induce apoptosis, which
explained in part the inhibitory effect of MVs on leukemia cells.
However, the molecular mechanism of leukemia cell autophagy and
apoptosis induced by MVs still remains unclear and need further
study.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81571221) and the Natural
Science Foundation of Jiangsu Province (grant no. BK20151346).
References
|
1
|
Hu YL, Fu YH, Tabata Y and Gao JQ:
Mesenchymal stem cells: A promising targeted-delivery vehicle in
cancer gene therapy. J Control Release. 147:154–162. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Drissi H, Gibson JD, Guzzo RM and Xu RH:
Derivation and chondrogenic commitment of human embryonic stem
cell-derived mesenchymal progenitors. Methods Mol Biol. 1340:65–78.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brown PT, Squire MW and Li WJ:
Characterization and evaluation of mesenchymal stem cells derived
from human embryonic stem cells and bone marrow. Cell Tissue Res.
358:149–164. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang H, Bai M, Deng T, Liu R, Wang X, Qu
Y, Duan J, Zhang L, Ning T, Ge S, et al: Cell-derived microvesicles
mediate the delivery of miR-29a/c to suppress angiogenesis in
gastric carcinoma. Cancer Lett. 375:331–339. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wu S, Ju GQ, Du T, Zhu YJ and Liu GH:
Microvesicles derived from human umbilical cord Wharton's jelly
mesenchymal stem cells attenuate bladder tumor cell growth in vitro
and in vivo. PLoS One. 8:e613662013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fonsato V, Collino F, Herrera MB,
Cavallari C, Deregibus MC, Cisterna B, Bruno S, Romagnoli R,
Salizzoni M, Tetta C, et al: Human liver stem cell-derived
microvesicles inhibit hepatoma growth in SCID mice by delivering
antitumor microRNAs. Stem Cells. 30:1985–1998. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fonseka M, Ramasamy R, Tan BC and Seow HF:
Human umbilical cord blood-derived mesenchymal stem cells
(hUCB-MSC) inhibit the proliferation of K562 (human
erythromyeloblastoid leukaemic cell line). Cell Biol Int.
36:793–801. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hu JB, Wang XH, Ma QH, Zhang W, Wang YY
and Wen XM: Preliminary investigation on the differentiation of
human embryonic stem cells into mesenchymal stem cells. Chin J Clin
Lab Sci. 31:276–278. 2013.
|
|
9
|
Gadkari R, Zhao L, Teklemariam T and
Hantash BM: Human embryonic stem cell derived-mesenchymal stem
cells: An alternative mesenchymal stem cell source for regenerative
medicine therapy. Regen Med. 9:453–465. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bruno S, Collino F, Deregibus MC, Grange
C, Tetta C and Camussi G: Microvesicles derived from human bone
marrow mesenchymal stem cells inhibit tumor growth. Stem Cells Dev.
22:758–771. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Song N, Gao L, Qiu H, Huang C, Cheng H,
Zhou H, Lv S, Chen L and Wang J: Mouse bone marrow-derived
mesenchymal stem cells inhibit leukemia/lymphoma cell proliferation
in vitro and in a mouse model of allogeneic bone marrow
transplant. Int J Mol Med. 36:139–149. 2015.PubMed/NCBI
|
|
12
|
Ramasamy R, Lam EW, Soeiro I, Tisato V,
Bonnet D and Dazzi F: Mesenchymal stem cells inhibit proliferation
and apoptosis of tumor cells: Impact on in vivo tumor growth.
Leukemia. 21:304–310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Goldstein RH, Reagan MR, Anderson K,
Kaplan DL and Rosenblatt M: Human bone marrow-derived MSCs can home
to orthotopic breast cancer tumors and promote bone metastasis.
Cancer Res. 70:10044–10050. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Psaila B, Kaplan RN, Port ER and Lyden D:
Priming the ‘soil’ for breast cancer metastasis: The pre-metastatic
niche. Breast Dis. 26:65–74. 2006.2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Luo F, Liu T, Wang J, Li J, Ma P, Ding H,
Feng G, Lin D, Xu Y and Yang K: Bone marrow mesenchymal stem cells
participate in prostate carcinogenesis and promote growth of
prostate cancer by cell fusion in vivo. Oncotarget. 7:30924–30934.
2016.PubMed/NCBI
|
|
16
|
Burns JS, Abdallah BM, Guldberg P, Rygaard
J, Schrøder HD and Kassem M: Tumorigenic heterogeneity in cancer
stem cells evolved from long-term cultures of
telomerase-immortalized human mesenchymal stem cells. Cancer Res.
65:3126–3135. 2005.PubMed/NCBI
|
|
17
|
Raposo G and Stoorvogel W: Extracellular
vesicles: Exosomes, microvesicles, and friends. J Cell Biol.
200:373–383. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gambim MH, do Carmo AO, Marti L,
Veríssimo-Filho S, Lopes LR and Janiszewski M: Platelet-derived
exosomes induce endothelial cell apoptosis through peroxynitrite
generation: Experimental evidence for a novel mechanism of septic
vascular dysfunction. Crit Care. 11:R1072007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang L, Wu X, Wang D, Luo C and Chen L:
Renal carcinoma cell-derived exosomes induce human immortalized
line of Jurkat T lymphocyte apoptosis in vitro. Urol Int.
91:363–369. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fulda S and Kögel D: Cell death by
autophagy: Emerging molecular mechanisms and implications for
cancer therapy. Oncogene. 34:5105–5113. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu L, Alva A, Su H, Dutt P, Freundt E,
Welsh S, Baehrecke EH and Lenardo MJ: Regulation of an ATG7-beclin
1 program of autophagic cell death by caspase-8. Science.
304:1500–1502. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Borthakur G, Duvvuri S, Ruvolo V, Tripathi
DN, Piya S, Burks J, Jacamo R, Kojima K, Ruvolo P, Fueyo-Margareto
J, et al: MDM2 inhibitor, nutlin 3a, induces p53 dependent
autophagy in acute leukemia by AMP kinase activation. PLoS One.
10:e01392542015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Paglin S, Hollister T, Delohery T, Hackett
N, McMahill M, Sphicas E, Domingo D and Yahalom J: A novel response
of cancer cells to radiation involves autophagy and formation of
acidic vesicles. Cancer Res. 61:439–444. 2001.PubMed/NCBI
|
|
25
|
Fu LL, Cheng Y and Liu B: Beclin-1:
Autophagic regulator and therapeutic target in cancer. Int J
Biochem Cell Biol. 45:921–924. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liang XH, Jackson S, Seaman M, Brown K,
Kempkes B, Hibshoosh H and Levine B: Induction of autophagy and
inhibition of tumorigenesis by beclin 1. Nature. 402:672–676. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
White E: The role for autophagy in cancer.
J Clin Invest. 125:42–46. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang C, Kaushal V, Shah SV and Kaushal GP:
Autophagy is associated with apoptosis in cisplatin injury to renal
tubular epithelial cells. Am J Physiol Renal Physiol.
294:F777–F787. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kroemer G and Levine B: Autophagic cell
death: The story of a misnomer. Nat Rev Mol Cell Biol. 9:1004–1010.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gump JM and Thorburn A: Autophagy and
apoptosis: What is the connection? Trends Cell Biol. 21:387–392.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Campisi J: Senescent cells, tumor
suppression, and organismal aging: Good citizens, bad neighbors.
Cell. 120:513–522. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zaharie F, Muresan MS, Petrushev B, Berce
C, Gafencu GA, Selicean S, Jurj A, Cojocneanu-Petric R, Lisencu CI,
Pop LA, et al: Exosome-carried microRNA-375 inhibits cell
progression and dissemination via Bcl-2 blocking in colon cancer. J
Gastrointestin Liver Dis. 24:435–443. 2015.PubMed/NCBI
|
|
34
|
Ristorcelli E, Beraud E, Mathieu S,
Lombardo D and Verine A: Essential role of Notch signaling in
apoptosis of human pancreatic tumoral cells mediated by exosomal
nanoparticles. Int J Cancer. 125:1016–1026. 2009. View Article : Google Scholar : PubMed/NCBI
|