Introduction
Colorectal cancer (CRC) is one of the most serious
types of cancer worldwide, in terms of both morbidity and
mortality. Despite improvements in medical therapies, the efficacy
of chemotherapy has reached a plateau and the 5-year survival rate
of patients with untreated metastatic CRC (mCRC) is still below
10%, and the underlying molecular basis remains to be clearly
defined (1). Cancer cells often
exhibit intrinsic resistance to chemotherapeutic agents, or develop
resistance over time with treatment. The lack of responsiveness to
chemotherapy is an important problem that needs to be resolved.
Cetuximab, a chimeric monoclonal antibody targeting
the epidermal growth factor receptor (EGFR), has markedly improved
the prognosis in patients with metastatic CRC (mCRC) who harbor
wild-type KRAS and BRAF in their tumors (1–4).
Furthermore, new findings regarding RAS (NRAS)
mutations can further assist in predicting the therapeutic effects
of anti-EGFR antibody therapy (5–7).
CPT-11, one of the major cytotoxic agents in mCRC, is converted
into an active metabolite, SN38, which then acts as a topoisomerase
I inhibitor. Several studies have shown synergism between cetuximab
and DNA-damaging agents, such as CPT-11, in vitro and in
vivo (8,9). Although cetuximab may enhance the
tumor response to some chemotherapeutic agents, including CPT-11
(10–12), the mechanism underlying this
synergism remains unclear.
Mammalian heat shock proteins (HSPs) are known to be
molecular chaperones in protein-protein interactions, acting as
anti-apoptotic proteins and contribute to cell survival (13). HSPs have been classified into four
major families, based on their molecular weights: HSP90, HSP70,
HSP60 and ‘small’ HSPs (15–30 kDa), including HSP27 (14). Their expression can contribute to
the malignant properties of cancer cells, including tumorigenicity,
treatment resistance and apoptosis inhibition (14–18).
Recently, HSP27 has been identified as a treatment target for
several cancers, and clinical trials using an antisense
oligonucleotide, OGX427, which inhibits HSP27 expression, have been
performed in patients with prostate, bladder, ovarian, breast and
non-small cell lung cancer, but not CRC (19). The therapy has been reported to be
feasible and effective. In previous studies, we showed that HSP27
might contribute to resistance to 5-fluorouracil in CRC, in
vitro and in vivo (20–22).
It has also been reported that HSP27 expression is involved in
resistance to CPT-11 and doxorubicin in vitro (23,24).
HSP27 is also believed to be involved in multidrug resistance
(25–27). Thus, we considered that cetuximab
might promote sensitivity to CPT-11 and SN38 via suppression of
HSP27.
The Janus kinase (JAK)/signal transducer and
activator of transcription (STAT) signaling pathway and, in
particular, STAT3, which is a transcription factor, are known to
have oncogenic potential (28,29).
In CRC cells, activated STAT3 causes resistance to CPT-11 and
inhibition of STAT3 strongly enhances the cytotoxic action of
CPT-11 (30,31). STAT3 activation in CRC patients is
also associated with adverse clinical outcomes, supporting its
potential roles as a prognostic biomarker and/or a therapeutic
target (32). Furthermore, STAT3
has been reported to regulate HSP27 in breast epithelial cells
(33).
We hypothesized that cetuximab might promote
sensitivity to CPT-11 and SN38, via suppression of HSP27 through
blocking of the JAK/STAT signaling pathway. The aim of the present
study was to assess whether cetuximab promoted SN38 sensitivity,
and to elucidate the molecular mechanism of the change in SN38
sensitivity caused by cetuximab in CRC cells with various
mutational statuses.
Materials and methods
Drugs, colorectal cancer cell lines
and cell culture conditions
Cetuximab (2 mg/ml) and SN38, the active metabolite
of CPT-11, were kindly provided in powder form by Merck
Laboratories (Darmstadt, Germany) and Yakult Honsha Co., Ltd.
(Tokyo, Japan), respectively. SN38 was dissolved in dimethyl
sulfoxide (DMSO) to a final concentration of 10 mM. For in
vitro experiments, stock solutions of drugs were diluted in
phosphate-buffered saline (PBS).
We used four human CRC cell lines: Caco2, WiDR,
SW480 and SW620. They were cultured in Dulbeccos modified Eagles
medium ((DMEM; Gibco, Charlestown, MA, USA), supplemented with 10%
fetal bovine serum (FBS) and 1% penicillin/streptomycin. Cells were
cultured at 37°C in 5% CO2. Authentication of all cell
lines was confirmed by investigating the RAS mutation status
of each cell line using a polymerase chain reaction (PCR)-based
method (conducted by SRL, Co., Tokyo, Japan). The mutational status
of BRAF for cell lines was obtained from previous reports
(34,35). The results are summarized in
Table I.
 | Table I.Mutation status of colon cancer cell
lines. |
Table I.
Mutation status of colon cancer cell
lines.
|
| Caco2 | WiDR | SW480 | SW620 |
|---|
| RAS | Wild-type | Wild-type | KRAS
mutation (Codon G13D) | KRAS
mutation (Codon G12V) |
| BRAF | Wild-type | Exon 15
(V600E) | Wild-type | Wild-type |
Proliferation and cytotoxicity
assays
We conducted two experiments. One was to determine
cetuximab sensitivity, while the other investigated the combined
effects of SN38 and cetuximab or SN38 and AG490, an inhibitor of
JAK2, instead of cetuximab. Here, 5×103 cells/well were
seeded in 96-well plates. Then, tumor cells were treated with 1, 10
and 100 µg/ml cetuximab or a fixed cetuximab concentration of 10
µg/ml with SN38 or a fixed AG490 concentration of 40 µM with SN38
concentrations ranging from 0.01 to 30 µg/ml for 48 h, followed by
overnight incubation in serum-free medium. For cetuximab/SN38 or
AG490/SN38 co-treatment experiments, cells were pretreated with
cetuximab or AG490 for 15 min before the SN38 addition. Cell
viability was evaluated by the reduction of methylthiazol
tetrazolium to formazan (0.5 mg/ml). The absorbance of each well
was measured at 540 and 600 nm using a microplate spectrophotometer
(Immuno Reader; Nalgen Nunc International, Rochester, NY, USA).
Viability was assessed as the percentage of viable cells compared
with untreated cells (100% viable). The assessment was based on
three independent experiments.
Western blot analysis
Whole-cell extracts (20 µg/lane) were
electrophoresed through 7.5% sodium dodecyl sulfate polyacrylamide
gels (Bio-Rad Laboratories, Hercules, CA, USA) and were transferred
to an Immuno-Blot polyvinylidene fluoride membrane (Bio-Rad
Laboratories). The membranes were blocked for 1 h in PBS
(Gibco-BRL) with 0.5% Tween-20 (PBS-T) and 5% non-fat dry milk at
room temperature, then incubated at 4°C overnight with anti-human
HSP27 mouse monoclonal antibody (1:2,000; G3.1; Lab Vision Corp.,
Fremont, CA, USA), anti-human β-actin mouse monoclonal antibody
(1:5,000; AC74; Sigma-Aldrich, St. Louis, MO, USA), and anti-human
STAT3 mouse monoclonal antibody (1:200; C-20; Santa Cruz
Biotechnology, Santa Cruz, CA, USA). The membranes were incubated
for 30 min with horseradish peroxidase-conjugated anti-mouse
immunoglobulin G (IgG) (1:2,500; Promega Corp., Madison, WI, USA).
Proteins were detected using the ECL-Plus reagent (GE Healthcare
Life Sciences, Little Chalfont, UK) according to the manufacturers
protocol. Each experiment was performed in triplicate. AG490
(Abcam, Cambridge, UK), JAK/STAT and a JAK2 inhibitor were
reconstituted in DMSO and stored at −20°C until used.
Statistical analysis
Data are reported as means ± standard deviation.
Statistical analyses were performed using the Students t-test or
the Mann-Whitney U test. P<0.05 were considered to indicate a
statistically significant difference.
Results
RAS and BRAF mutation status
The mutation status of each CRC cell line is shown
in Table I. KRAS and
NRAS mutations, which predict the efficacy of cetuximab
treatment, were not detected in Caco2 or WiDR. However, the G13D
and G12V KRAS mutations were detected in SW480 and SW620,
respectively. The exon 15 mutation in BRAF was detected only
in WiDR. From these results, Caco2 was selected as the ‘positive’
control because it had no mutation in KRAS, NRAS, or
BRAF. WiDR, SW480 and SW620 were used as ‘negative’ controls
because of KRAS or BRAF mutations.
Expression of HSP27 in CRC cell
lines
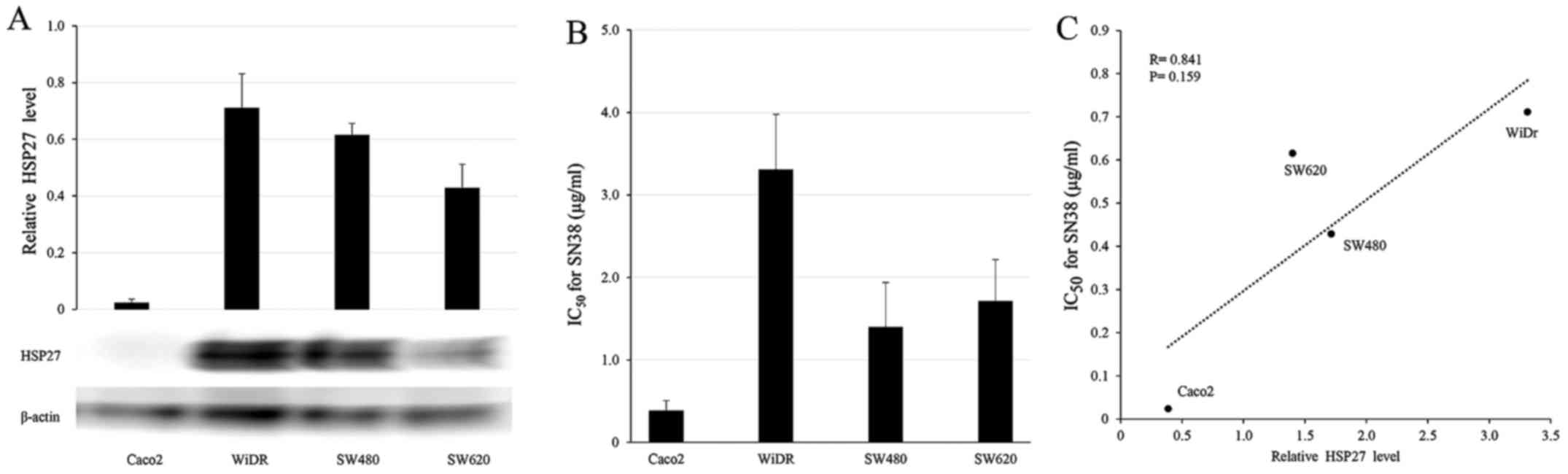
Western blot analysis was conducted to determine
HSP27 protein levels in the four CRC cell lines (Fig. 1A). WiDR, SW480 and SW620 cells
showed high HSP27 protein levels and relative resistance to SN38,
whereas Caco2 showed a low level of HSP27 protein and high
sensitivity to SN38 (Fig. 1B). The
HSP27 protein levels tended to correlate with the concentration of
SN38, resulting in a 50% growth inhibition, but the results were
not significant (IC50; Fig.
1C; R=0.841, P=0.159).
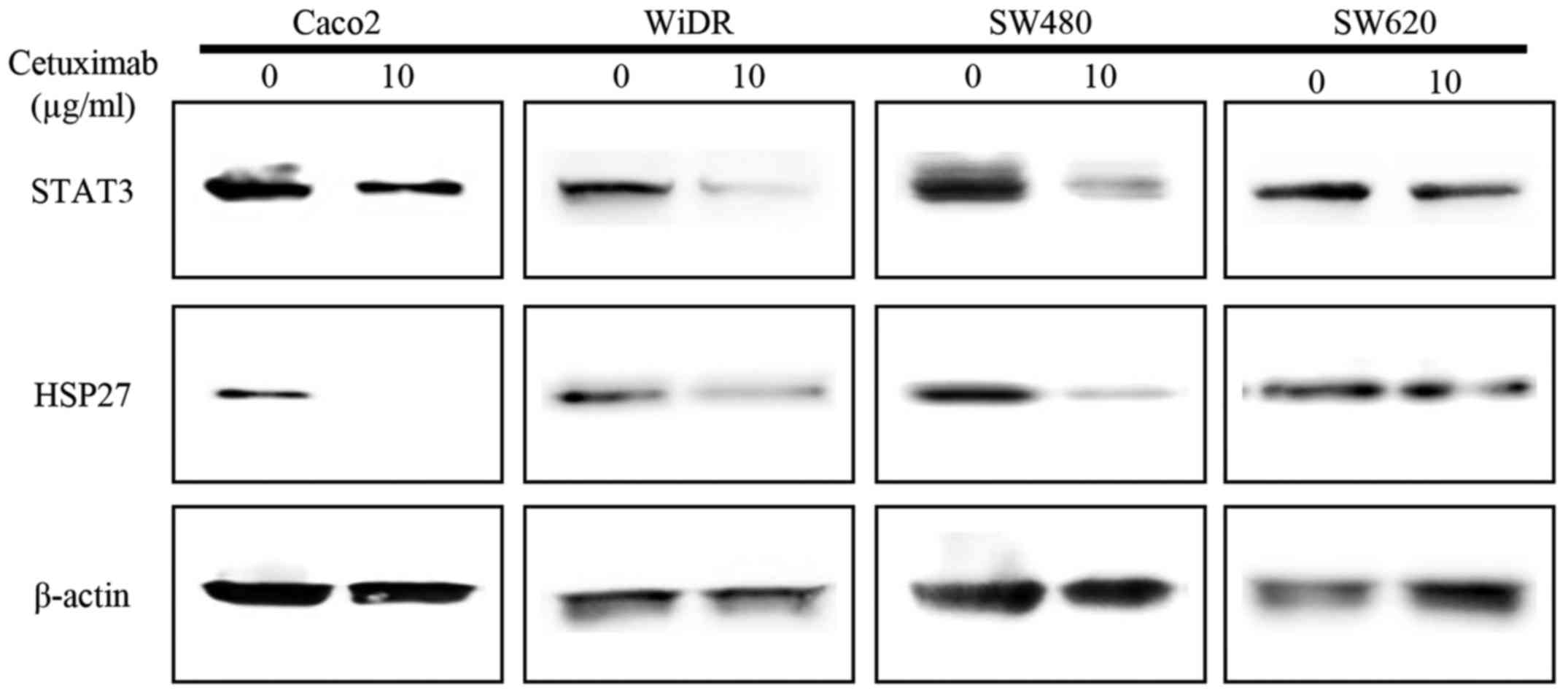
HSP27 downregulation by exposure to
cetuximab
HSP27 and STAT3 levels with exposure to cetuximab in
the four CRC cells were evaluated by western blotting. STAT3
protein was expressed constitutively in the four CRC cell lines
with no exposure to cetuximab. Levels of HSP27 and STAT3 were
greatly downregulated in Caco2, WiDR and SW480 cells with exposure
to 10 µg/ml cetuximab for 24 h vs. untreated cells. However, SW620
cells showed no significant difference in the expression of HSP27
or STAT3 with cetuximab exposure (Fig.
2).
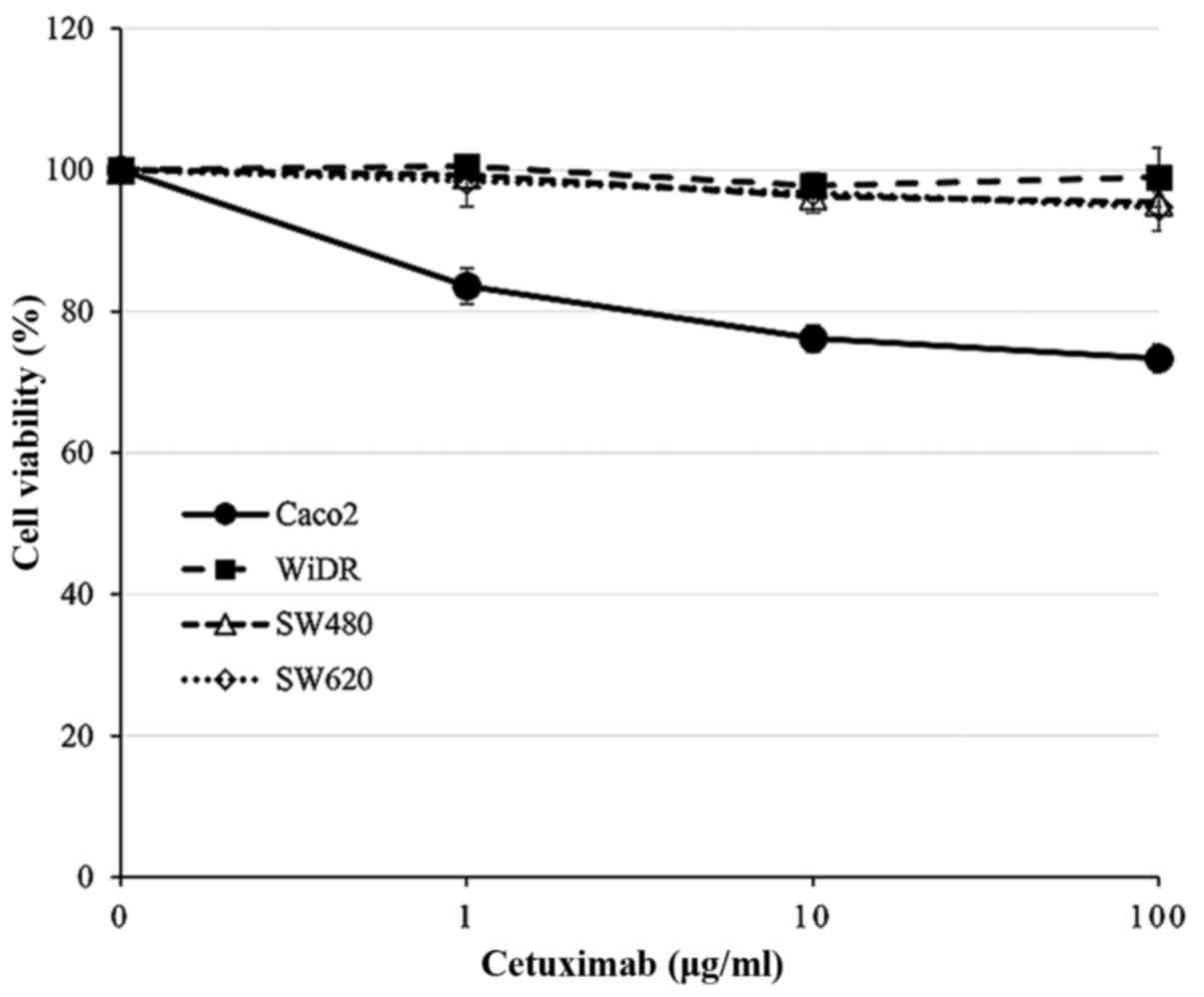
Proliferation assay using cetuximab in
CRC cell lines
Cell proliferation was assessed 48 h after exposure
to cetuximab alone using the MTT assay. As shown in Fig. 3, little cytotoxic effect of
cetuximab treatment (concentrations from 1 to 100 µg/ml) was
observed in WiDR, SW480 or SW620 cells. However, the proliferation
of Caco2 cells was suppressed according to cetuximab
concentration.
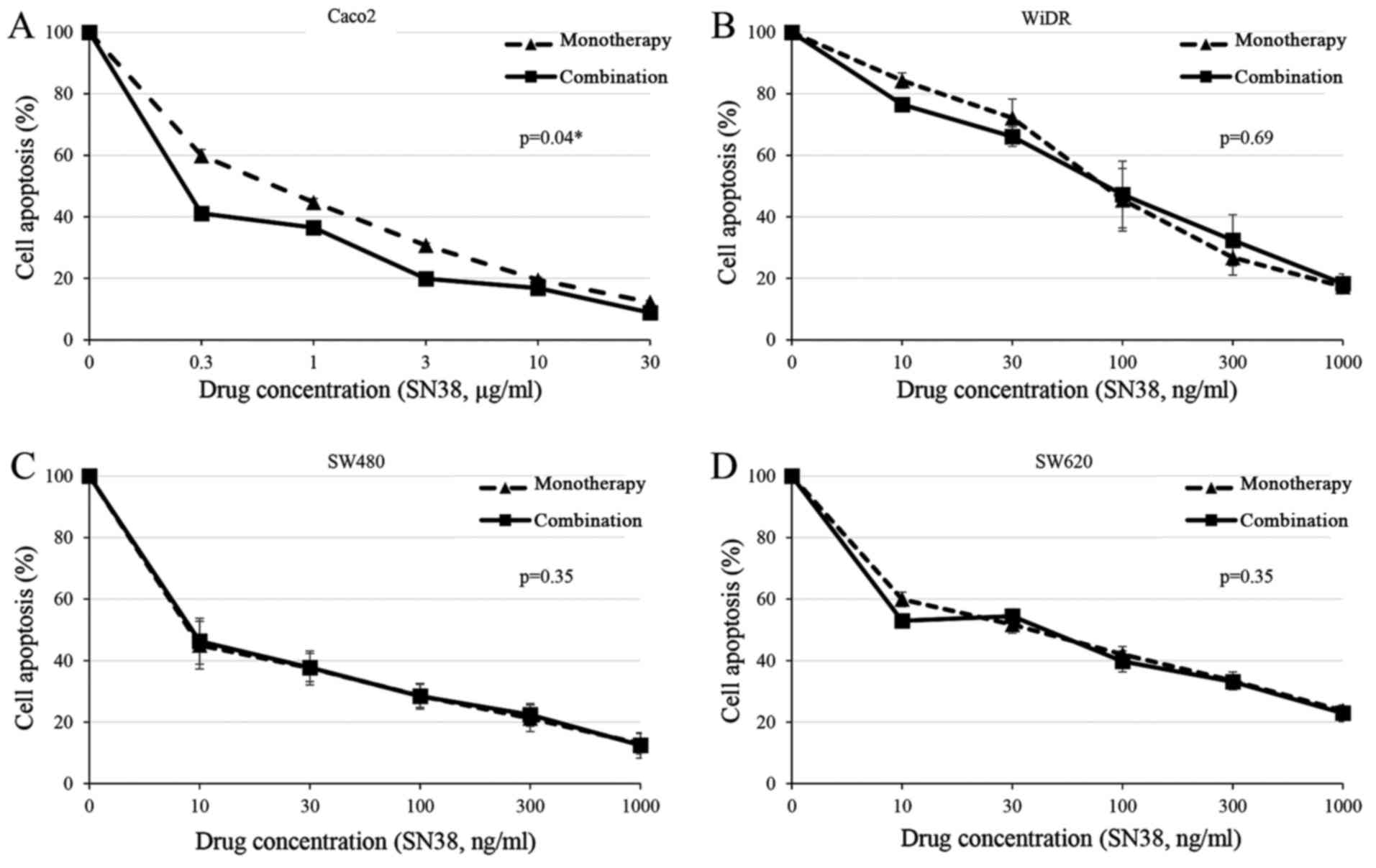
Cytotoxic effect of cetuximab in
combination with SN38 in CRC cell lines
The cytotoxic effects of cetuximab in combination
with SN38 were examined. CRC cells were treated with serial
concentrations of SN38 (from 0.01 to 30 µg/ml) in the presence or
absence of cetuximab. Combined drug effects were evaluated in the
four CRC cell lines. Fig. 4 shows
representative effects of the cetuximab and SN38 combination.
Treatment of Caco2 cells with 10 µg/ml cetuximab plus various
concentrations of SN38 enhanced the cytotoxic effects
significantly, depending on the SN38 concentration, compared with
treatment with SN38 alone (Fig. 4A;
P=0.04). However, there was no significant combination effect in
the three other cell lines (Fig.
4B-D). The addition of cetuximab (at a concentration causing
~20% inhibition) reduced the IC50 value of SN38 from
0.52 to 0.31 µg/ml (40% reduction) in Caco2. These results suggest
a synergistic effect of cetuximab and SN38 in Caco2 cells.
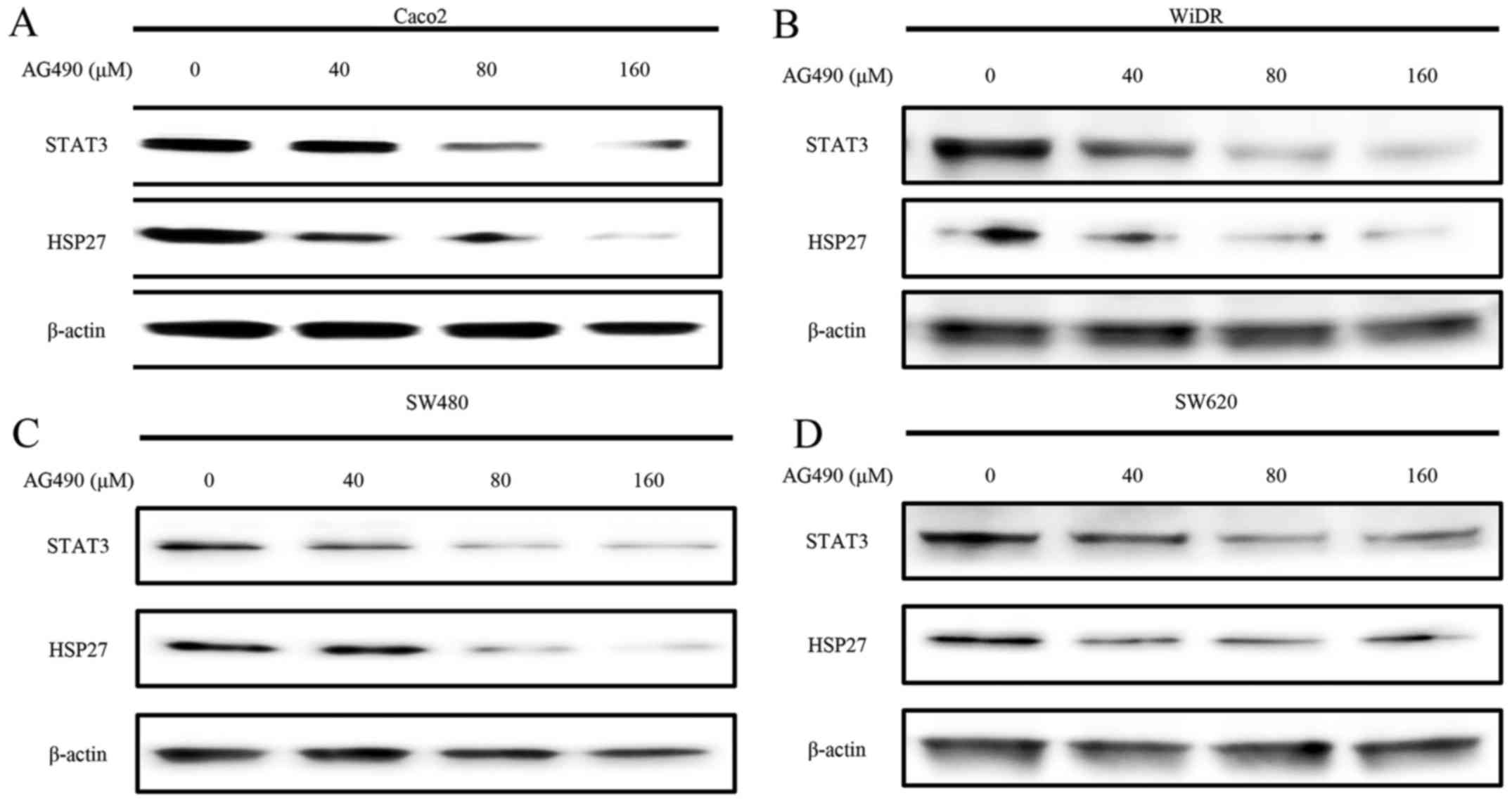
HSP27 downregulation by exposure to
AG490
Western blot analyses showed a
concentration-dependent decrease in the levels of STAT3 and HSP27
at 24 h after AG490 (JAK2 inhibitor) exposure in Caco2, SW480 and
WiDR cells. STAT3 and HSP27 were almost undetectable with 160 µM
AG490 in these three cell lines. However, no significant decrease
in the levels of STAT3 or HSP27 was observed in SW620 cells
(Fig. 5).
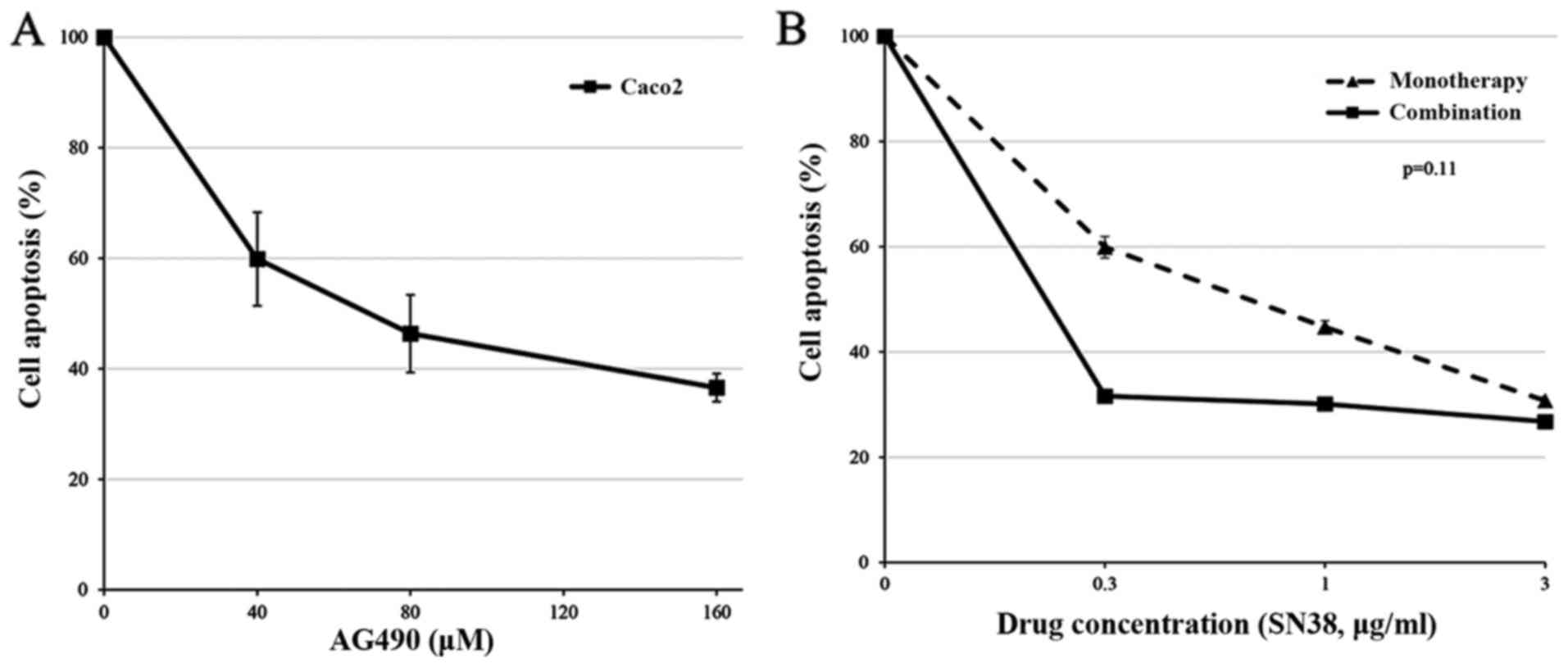
Cytotoxic effect of AG490 in
combination with SN38 in Caco2 cells
The cytotoxic effects of AG490, in combination with
SN38, were examined. Cell proliferation was assessed 48 h after
exposure to AG490 alone using the MTT assay. Fig. 6A shows that the proliferation of
Caco2 cells was suppressed based upon the AG490 concentration.
Caco2 cells were treated with serial concentrations of SN38 (from
0.3 to 3.0 µg/ml) in the presence or absence of a fixed AG490
concentration (40 µM) for 48 h. Fig.
6B shows the representative effects of the AG490 and SN38
combination. Treatment of Caco2 cells with 40 µM AG490 plus the
various concentrations of SN38 enhanced the cytotoxic effects
comparable to that the effect with cetuximab plus SN38; however,
the result was not significantly better than that obtained with
treatment with SN38 alone (Fig. 6B;
P=0.11).
Discussion
Cetuximab is a human-mouse chimeric immunoglobulin
(IgG1) monoclonal antibody for EGFR that is approved for use in
patients with mCRC and an absence of mutations in KRAS,
NRAS and BRAF (4–7). Some
studies have shown synergistic effects of cetuximab and CPT-11 in
wild-type RAS and BRAF mCRC patients (4,8,9). It is
believed that cetuximab may enhance the tumor response to CPT-11
and its active metabolite, SN38, in addition to its ‘original’
antitumor effects. However, the molecular mechanism of this
synergistic effect has not yet been fully investigated (10–12).
HSP27 is known to be a stress-activated, adenosine
triphosphate-independent cytoprotective chaperone with many notable
properties, including resistance to chemotherapy in several
cancers. In CRC cells, HSP27 has been reported to be a resistance
factor for CPT-11, as well as a prognostic factor (24–27).
The JAK/STAT signaling pathway, and especially the
transcription factor STAT3, have been implicated in the regulation
of drug sensitivity (28,29). In CRC cells, activated STAT3 causes
resistance to CPT-11 and inhibition of STAT3 strongly enhances the
cytotoxic action of CPT-11 (30,31).
STAT3 activation in CRC patients is associated with adverse
clinical outcomes, consistent with its potential roles as a
prognostic biomarker and a chemoprevention and/or therapeutic
target (32). Furthermore, STAT3
has been reported to regulate HSP27 in breast epithelial cells
(33). Thus, we considered that
cetuximab might promote sensitivity to CPT-11 and its active
metabolite, SN38, via suppression of the HSP27 through blocking the
JAK/STAT signaling pathway.
Consistent with previous reports, HSP27 protein
levels and SN38 sensitivity had a tendency to be correlated in the
CRC cell lines (Fig. 1C; R=0.841,
P=0.159) (24). Exposure to 10
µg/ml cetuximab and various concentrations of AG490, an inhibitor
of JAK2, showed suppression of STAT3 and HSP27 protein
levels except in the KRAS G12V mutant cell line, SW620.
Synergistic effects of cetuximab in combination with SN38 have been
confirmed only in RAS wild-type and BRAF wild-type
cells (Caco2), and not in the three other RAS- or
BRAF-mutated cells. Furthermore, the combined effects of
AG490 plus SN38 exhibited a similar tendency to that of cetuximab
plus SN38, but these results were not significant in Caco2 cells.
These results indicate that cetuximab may promote sensitivity to
SN38 via suppression of HSP27, through blocking of the JAK/STAT
signaling pathway in Caco2 cells. A synergistic effect was only
seen at lower concentrations of SN38. The reason is uncertain;
however, higher concentrations of SN38 have a strong cytotoxicity
as a single agent in Caco2 cells. Therefore, the synergism with
cetuximab may be masked by the strong effect of SN38.
The present study revealed two novel and important
findings. First, cetuximab may cause suppression of HSP27 through
blocking the JAK/STAT signaling pathway in CRC cells, because AG490
caused a concentration-dependent decrease in the level of HSP27,
similar to cetuximab. This suggests that HSP27 may be a downstream
mediator of the JAK/STAT signaling pathway in CRC cells. Second,
suppression of HSP27 caused by cetuximab was observed even in
RAS- or BRAF-mutated cells (WiDR and SW480). Based on
this result, it is expected that cetuximab promotes sensitivity to
SN38, even in RAS- or BRAF-mutated cells. However, no
combination effect of cetuximab and SN38 was observed in RAS- or
BRAF-mutated cells. The reason for this may be that suppression of
HSP27 alone, through blocking of the JAK/STAT signaling pathway,
did not overcome the power of the RAS/RAF signaling pathway (also
known as the MAPK pathway), accelerated by the presence of
RAS or BRAF mutations. To the best of our knowledge,
there are no reports focused on the power balance of the RAS/RAF
and JAK/STAT signaling pathway in CRC. We speculate that the
RAS/RAF signaling pathway has a stronger potential than the
JAK/STAT signaling pathway because the RAS mutation is the only
proven predictive marker for anti-EGFR antibody treatments. There
are no reports on similar mutations in the JAK/STAT signaling
pathway in CRC (5–7,36).
Furthermore, no suppression of HSP27 and STAT3 by cetuximab or
AG490 was observed in SW620 cells with the KRAS G12V
mutation. The reason for this is uncertain; however, a previous
study reported that mutant KRAS could activate STAT3 in
SW620 cells (37). Thus, SW620 may
be a unique CRC cell line that does not respond to stimulation by
cetuximab or AG490 in the JAK/STAT signaling pathway.
We acknowledge several limitations of the present
study. First, there is the absence of in vivo data.
Secondly, we used only wild-type CRC cell lines and not
CPT-11-refractory CRC cell lines. However, we used four CRC cell
lines with different RAS and BRAF mutational statuses, which could
affect the combination effect with cetuximab and SN38. Therefore,
we believe that our data is clinically relevant. Further research
is required to strengthen our hypothesis. Third, the changes in the
protein levels of HSP27 and STAT3 by exposure to cetuximab or AG490
suggest that phosphorylation of HSP27 and STAT3 are also likely to
gather a positive response. However, after 48 h of exposure to
cetuximab, no changes in the phosphorylation of HSP27 and STAT3
were observed (data not shown). The reason could be that peak STAT3
phosphorylation occurs within 15–60 min in response to various
stimuli, and even in the presence of a continuous cytokine, STAT3
activation decreases over several hours (38). Furthermore, HSP27 phosphorylation
does not last more than several hours (39). Therefore, suppression of
phosphorylated HSP27 and STAT3 was not confirmed after a 48-h
exposure to cetuximab. Molecular mechanisms of the synergism
between cetuximab and SN38 may be related not only to HSP27
suppression but also other factors, such as immunological effects.
Recently, STAT3 inhibition can mediate anticancer effects by
multiple mechanisms, including cell-autonomous effects and
immunological effects (40).
Cetuximab, when used in combination with FOLFIRI [one of standard
chemotherapy regimens for mCRC that does no induce immunogenic cell
death (ICD)], induces ICD in vitro and in vivo
(41). Therefore, the molecular
mechanisms of the synergism between cetuximab and SN38 potentially
involve multiple mechanisms, including ICD induced by cetuximab via
STAT3 inhibition, in addition to our hypothesis. A better
understanding of these mechanisms will lead to novel treatments for
CRC.
In conclusion, cetuximab may promote sensitivity to
SN38 via suppression of HSP27 through blocking the JAK/STAT
signaling pathway in RAS wild-type CRC cells.
References
|
1
|
Van Cutsem E, Köhne CH, Hitre E, Zaluski
J, Chien Chang CR, Makhson A, DHaens G, Pintér T, Lim R, Bodoky G,
et al: Cetuximab and chemotherapy as initial treatment for
metastatic colorectal cancer. N Engl J Med. 360:1408–1417. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lièvre A, Bachet JB, Le Corre D, Boige V,
Landi B, Emile JF, Côté JF, Tomasic G, Penna C, Ducreux M, et al:
KRAS mutation status is predictive of response to cetuximab therapy
in colorectal cancer. Cancer Res. 66:3992–3995. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Grávalos C, Cassinello J, Fernández-Rañada
I and Holgado E: Role of tyrosine kinase inhibitors in the
treatment of advanced colorectal cancer. Clin Colorectal Cancer.
6:691–699. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pietrantonio F, Petrelli F, Coinu A, Di
Bartolomeo M, Borgonovo K, Maggi C, Cabiddu M, Iacovelli R, Bossi
I, Lonati V, et al: Predictive role of BRAF mutations in patients
with advanced colorectal cancer receiving cetuximab and
panitumumab: A meta-analysis. Eur J Cancer. 51:587–594. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schwartzberg LS, Rivera F, Karthaus M,
Fasola G, Canon JL, Hecht JR, Yu H, Oliner KS and Go WY: PEAK: A
randomized, multicenter phase II study of panitumumab plus modified
fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab
plus mFOLFOX6 in patients with previously untreated, unresectable,
wild-type KRAS exon 2 metastatic colorectal cancer. J Clin Oncol.
32:2240–2247. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Peeters M, Oliner KS, Price TJ, Cervantes
A, Sobrero AF, Ducreux M, Hotko Y, André T, Chan E, Lordick F, et
al: Analysis of KRAS/NRAS mutations in a phase III study of
panitumumab with FOLFIRI compared with FOLFIRI alone as second-line
treatment for metastatic colorectal cancer. Clin Cancer Res.
21:5469–5479. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Heinemann V, von Weikersthal LF, Decker T,
Kiani A, Vehling-Kaiser U, Al-Batran SE, Heintges T, Lerchenmüller
C, Kahl C, Seipelt G, et al: FOLFIRI plus cetuximab versus FOLFIRI
plus bevacizumab as first-line treatment for patients with
metastatic colorectal cancer (FIRE-3): A randomised, open-label,
phase 3 trial. Lancet Oncol. 15:1065–1075. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Meyerhardt JA and Fuchs CS: Epidermal
growth factor receptor inhibitors and colorectal cancer. Oncology
(Williston Park). 18:(Suppl 14). 35–38. 2004.PubMed/NCBI
|
|
9
|
Bokemeyer C, Van Cutsem E, Rougier P,
Ciardiello F, Heeger S, Schlichting M, Celik I and Köhne CH:
Addition of cetuximab to chemotherapy as first-line treatment for
KRAS wild-type metastatic colorectal cancer: Pooled analysis
of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer.
48:1466–1475. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sung FL, Poon TC, Hui EP, Ma BB, Liong E,
To KF, Huang DP and Chan AT: Antitumor effect and enhancement of
cytotoxic drug activity by cetuximab in nasopharyngeal carcinoma
cells. In Vivo. 19:237–245. 2005.PubMed/NCBI
|
|
11
|
Nakata E, Hunter N, Mason K, Fan Z, Ang KK
and Milas L: C225 antiepidermal growth factor receptor antibody
enhances the efficacy of docetaxel chemoradiotherapy. Int J Radiat
Oncol Biol Phys. 59:1163–1173. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cunningham D, Humblet Y, Siena S, Khayat
D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype
C, et al: Cetuximab monotherapy and cetuximab plus irinotecan in
irinotecan-refractory metastatic colorectal cancer. N Engl J Med.
351:337–345. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Concannon CG, Gorman AM and Samali A: On
the role of Hsp27 in regulating apoptosis. Apoptosis. 8:61–70.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Andrieu C, Taieb D, Baylot V, Ettinger S,
Soubeyran P, De-Thonel A, Nelson C, Garrido C, So A, Fazli L, et
al: Heat shock protein 27 confers resistance to androgen ablation
and chemotherapy in prostate cancer cells through eIF4E. Oncogene.
29:1883–1896. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Song TF, Zhang ZF, Liu L, Yang T, Jiang J
and Li P: Small interfering RNA-mediated silencing of heat shock
protein 27 (HSP27) Increases chemosensitivity to paclitaxel by
increasing production of reactive oxygen species in human ovarian
cancer cells (HO8910). J Int Med Res. 37:1375–1388. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sarto C, Valsecchi C, Magni F, Tremolada
L, Arizzi C, Cordani N, Casellato S, Doro G, Favini P, Perego RA,
et al: Expression of heat shock protein 27 in human renal cell
carcinoma. Proteomics. 4:2252–2260. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ciocca DR, Fuqua SA, Lock-Lim S, Toft DO,
Welch WJ and McGuire WL: Response of human breast cancer cells to
heat shock and chemotherapeutic drugs. Cancer Res. 52:3648–3654.
1992.PubMed/NCBI
|
|
18
|
Vargas-Roig LM, Gago FE, Tello O, Aznar JC
and Ciocca DR: Heat shock protein expression and drug resistance in
breast cancer patients treated with induction chemotherapy. Int J
Cancer. 79:468–475. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Matsui Y, Hadaschik BA, Fazli L, Andersen
RJ, Gleave ME and So AI: Intravesical combination treatment with
antisense oligonucleotides targeting heat shock protein-27 and
HTI-286 as a novel strategy for high-grade bladder cancer. Mol
Cancer Ther. 8:2402–2411. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hayashi R, Ishii Y, Ochiai H, Matsunaga A,
Endo T, Hasegawa H and Kitagawa Y: Suppression of heat shock
protein 27 expression promotes 5-fluorouracil sensitivity in colon
cancer cells in a xenograft model. Oncol Rep. 28:1269–1274.
2012.PubMed/NCBI
|
|
21
|
Tsuruta M, Nishibori H, Hasegawa H, Ishii
Y, Endo T, Kubota T, Kitajima M and Kitagawa Y: Heat shock protein
27, a novel regulator of 5-fluorouracil resistance in colon cancer.
Oncol Rep. 20:1165–1172. 2008.PubMed/NCBI
|
|
22
|
Matsunaga A, Ishii Y, Tsuruta M,
Okabayashi K, Hasegawa H and Kitagawa Y: Inhibition of heat shock
protein 27 phosphorylation promotes sensitivity to 5-fluorouracil
in colorectal cancer cells. Oncol Lett. 8:2496–2500.
2014.PubMed/NCBI
|
|
23
|
Garrido C, Mehlen P, Fromentin A, Hammann
A, Assem M, Arrigo AP and Chauffert B: Inconstant association
between 27-kDa heat-shock protein (Hsp27) content and doxorubicin
resistance in human colon cancer cells. The doxorubicin-protecting
effect of Hsp27. Eur J Biochem. 237:653–659. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Choi DH, Ha JS, Lee WH, Song JK, Kim GY,
Park JH, Cha HJ, Lee BJ and Park JW: Heat shock protein 27 is
associated with irinotecan resistance in human colorectal cancer
cells. FEBS Lett. 581:1649–1656. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang F, Zhang P, Shi C, Yang Y and Qin H:
Immunohistochemical detection of HSP27 and hnRNP K as prognostic
and predictive biomarkers for colorectal cancer. Med Oncol.
29:1780–1788. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yu Z, Zhi J, Peng X, Zhong X and Xu A:
Clinical significance of HSP27 expression in colorectal cancer. Mol
Med Rep. 3:953–958. 2010.PubMed/NCBI
|
|
27
|
Tweedle EM, Khattak I, Ang CW, Nedjadi T,
Jenkins R, Park BK, Kalirai H, Dodson A, Azadeh B, Terlizzo M, et
al: Low molecular weight heat shock protein HSP27 is a prognostic
indicator in rectal cancer but not colon cancer. Gut. 59:1501–1510.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bedel R, Thiery-Vuillemin A, Grandclement
C, Balland J, Remy-Martin JP, Kantelip B, Pallandre JR, Pivot X,
Ferrand C, Tiberghien P, et al: Novel role for STAT3 in
transcriptional regulation of NK immune cell targeting receptor
MICA on cancer cells. Cancer Res. 71:1615–1626. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kusaba T, Nakayama T, Yamazumi K, Yakata
Y, Yoshizaki A, Inoue K, Nagayasu T and Sekine I: Activation of
STAT3 is a marker of poor prognosis in human colorectal cancer.
Oncol Rep. 15:1445–1451. 2006.PubMed/NCBI
|
|
30
|
Weber A, Borghouts C, Delis N, Mack L,
Brill B, Bernard AC, Coqueret O and Groner B: Inhibition of Stat3
by peptide aptamer rS3-PA enhances growth suppressive effects of
irinotecan on colorectal cancer cells. Horm Mol Biol Clin Investig.
10:273–279. 2012.PubMed/NCBI
|
|
31
|
Courapied S, Sellier H, de Carné Trécesson
S, Vigneron A, Bernard AC, Gamelin E, Barré B and Coqueret O: The
cdk5 kinase regulates the STAT3 transcription factor to prevent DNA
damage upon topoisomerase I inhibition. J Biol Chem.
285:26765–26778. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Morikawa T, Baba Y, Yamauchi M, Kuchiba A,
Nosho K, Shima K, Tanaka N, Huttenhower C, Frank DA, Fuchs CS, et
al: STAT3 expression, molecular features, inflammation patterns,
and prognosis in a database of 724 colorectal cancers. Clin Cancer
Res. 17:1452–1462. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Song H, Ethier SP, Dziubinski ML and Lin
J: Stat3 modulates heat shock 27kDa protein expression in breast
epithelial cells. Biochem Biophys Res Commun. 314:143–150. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shigeta K, Hayashida T, Hoshino Y,
Okabayashi K, Endo T, Ishii Y, Hasegawa H and Kitagawa Y:
Expression of epidermal growth factor receptor detected by
cetuximab indicates its efficacy to inhibit in vitro and in vivo
proliferation of volorectal cancer cells. PLoS One. 8:e663022013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jhawer M, Goel S, Wilson AJ, Montagna C,
Ling YH, Byun DS, Nasser S, Arango D, Shin J, Klampfer L, et al:
PIK3CA mutation/PTEN expression status predicts response of colon
cancer cells to the epidermal growth factor receptor inhibitor
cetuximab. Cancer Res. 68:1953–1961. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Salazar R and Ciardiello F: Optimizing
Anti-EGFR therapy in colorectal cancer. Clin Cancer Res.
21:5415–5416. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zaanan A, Okamoto K, Kawakami H, Khazaie
K, Huang S and Sinicrope FA: The mutant KRAS gene up-regulates
BCL-XL protein via STAT3 to confer apoptosis resistance that is
reversed by BIM protein induction and BCL-XL antagonism. J Biol
Chem. 290:23838–23849. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Subramaniam A, Shanmugam MK, Perumal E, Li
F, Nachiyappan A, Dai X, Swamy SN, Ahn KS, Kumar AP, Tan BK, et al:
Potential role of signal transducer and activator of transcription
(STAT)3 signaling pathway in inflammation, survival, proliferation
and invasion of hepatocellular carcinoma. Biochim Biophys Acta.
1835:46–60. 2013.PubMed/NCBI
|
|
39
|
Stope MB, Weiss M, Preuss M, Streitbörger
A, Ritter CA, Zimmermann U, Walther R and Burchardt M: Immediate
and transient phosphorylation of the heat shock protein 27
initiates chemoresistance in prostate cancer cells. Oncol Rep.
32:2380–2386. 2014.PubMed/NCBI
|
|
40
|
Yang H, Yamazaki T, Pietrocola F, Zhou H,
Zitvogel L, Ma Y and Kroemer G: Improvement of immunogenic
chemotherapy by STAT3 inhibition. OncoImmunology. 5:e10780612015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Pozzi C, Cuomo A, Spadoni I, Magni E,
Silvola A, Conte A, Sigismund S, Ravenda PS, Bonaldi T, Zampino MG,
et al: The EGFR-specific antibody cetuximab combined with
chemotherapy triggers immunogenic cell death. Nat Med. 22:624–631.
2016. View Article : Google Scholar : PubMed/NCBI
|