Introduction
Malignant glioma is a type of tumor that is derived
from the glial cells in the nervous system. Patients commonly
succumb to this deadly disease within 5 years upon being diagnosed
(1). Glioma is classified into 4
pathological grades according to the World Health Organization
(WHO). Among them, grade IV, also called glioblastoma multiforme
(GBM), is recognized clinically as the most frequent and malignant
category (2). Currently,
therapeutic strategies involve 3 approaches, which consist of
maximal tolerable surgical resection paired with radiation and
chemotherapy. The combination of these therapies is capable of
adding only months of additional survival. The diffuse invasion of
tumor cells into the surrounding brain tissues imparts the major
challenge for therapy. Although early radical surgical
interventions attempt to remove the entire affected brain
hemisphere, patients are usually subjugated to cancer cells that
have crossed into the other hemisphere (3). Even now, with advanced microsurgical
techniques, recurrence is still often inevitable. Glioma typically
reoccurs within 1–2 cm of the primary tumor (4). Hence, a primary challenge is to
prevent tumor cells from uncontrolled delamination and infiltration
into other brain regions.
Cell migration is a finely tuned biological process
that requires an elaborately assembled leading edge and dynamic
interaction between the migrating cell and the extracellular matrix
(ECM) (5). Actin-myosin molecular
motors exert the main contractile stress and in particular, myosin
II plays an important role in glioma invasion (6). Normally, sturdy cell attachments
prevent cells from detaching and emigrating. These cell attachments
are mediated by cell-cell and cell-matrix receptors, such as
integrins, cadherins and neural cell adhesion molecules. Detachment
of those cells entails the activation of proteases, such as matrix
metalloproteinases, which dissociate tumor cells from the
neighboring environment (cells and ECM) (7–9).
Consequently, this promotes dynamic alterations of actin and
adhesion at the cell membrane, and activation of respective
downstream signaling pathways that govern adhesion and
migration.
Nicotinic acid (NA), a member of the vitamin B
family, is well known for its functions in the treatment and
prevention of atherosclerosis. NA is one of the most effective
agents capable of offering protection against cardiovascular risk
factors by increasing high density lipoprotein (HDL) levels, while
simultaneously decreasing very low density lipoprotein (VLDL) and
low density lipoprotein (LDL) levels (10). Moreover, NA has been ascertained to
function by downregulating intracellular cyclic adenosine
monophosphate (cAMP), the major intracellular mediator of
prolipolytic stimuli, and subsequently decreasing cellular levels
of free fatty acids (11). Notably,
another study recently indicated that NA is also able to boost
intracellular calcium compartmentalization. Moreover, it further
disclosed that a high concentration of NA (50 mM) is able to
co-activate TRPV1-4 transducing calcium. Concomitantly, we
demonstrated that NA over 30 mM is able to markedly disassemble the
cytoskeleton and lead to the depigmentation of the Xenopus
embryo (12–14).
Given the crucial function of cytoskeletal stress
fibers in cell migration, our discovery of the de novo
biological function of NA warrants a further investigation into
daunting GBM. We initially examined distinct gradients of NA on the
malignant glioblastoma cell line U251. Expectedly, we determined
that both 3.5 and 7.0 mM of NA were able to detach U251 from
culture dishes. Next we investigated F-actin and β-tubulin cellular
patterns and found that 7.0 mM of NA treatment specifically
abrogated the stress fiber distribution of the cytoskeleton.
Supplementation with extra ECM molecules such as collagen, gelatin,
poly-L-ornithine and laminin in 7.0 mM were unable to restore
cellular stress in comparison to supplementation in
phosphate-buffered saline (PBS)-treated U251 cells. This implies
that 7 mM of NA probably modulates cytoskeletal stress via a
specific biological process instead of non-specific biological
toxicity. Based on these data, we propose that NA treatment may be
developed into an effective therapeutic method for malignant
glioma.
Materials and methods
Sources and maintenance of cells
U251 glioblastoma cell were provided by the Cell
Bank of Type Culture Collection of Chinese Academy of Sciences
(Shanghai, China). U251 cells were cultured in Dulbecco's modified
Eagle's medium (DMEM) supplemented with 10% fetal calf serum (both
from HyClone, Logan, UT, USA). Primary mouse neurons and glia were
isolated from the brains of 13–15 day-old fetal ICR mice (Jackson
Laboratory, Bar Harbor, ME, USA).
Primary mouse neuron/glia culture
Primary mouse neurons and glia cells were isolated
from the brain of fetal ICR mice 13–15 days old. The primary mouse
neurons were cultured in neurobasal media supplemented with 2% B27
(both from Invitrogen), 0.5 mM glutamine and 25 µM glutamate (both
from Sigma). The primary glia cells were cultured in the DMEM
supplemented with 10% FBS. All cell lines were incubated at 37.5°C
with 5% CO2.
Cell viability assays
The MTT assays were carried out as described to
determine the viability of U251 cells as well as normal glia and
neurons (15). To perform this,
cells were cultured in 96-well plates with 1 mg/ml of MTT (Amresco,
LLC, Solon, OH, USA) for 4 h. The medium was then carefully
aspirated, and 100 µl of dimethyl sulfoxide (DMSO) was added to
each well to dissolve the MTT formazan crystals. The optical
density was assessed at 570 nm with an iMark microplate absorbance
reader (Bio-Rad, Hercules, CA, USA).
Immunocytochemistry
U251 cells were cultured on Lab-Tek chamber slides
(Sigma-Aldrich). After treatment with PBS or NA, the cells were
fixed with 4% paraformaldehyde and permeabilized with 0.4% Triton
X-100 at room temperature. The cells were then blocked with bovine
serum albumin (5%; Amresco) and incubated with a primary antibody
at 4°C overnight. Primary antibodies used were: β-tubulin (1:400;
556321; BD Transduction Laboratories™, San Jose, CA, USA),
p-paxillin (1:200; 44-720G), paxillin (1:200; AHO0492), p-cortactin
(1:400; 44-856) (all from Invitrogen), myosin IIA (M8064; 1:100;
Sigma-Aldrich). The cells were subsequently incubated with PE or
FITC-conjugated secondary antibodies (Santa Cruz Biotechnology,
Santa Cruz, CA, USA) at room temperature, and labeled with DAPI
(Sigma-Aldrich) to identify cell nuclei (16). F-actin stress fibers were labeled
with rhodamine phalloidin (5 U/ml; R415; Invitrogen Life
Technologies) in PBS for 15 min at room temperature as previously
described (12). Fluorescence was
detected using an Olympus IX81S1F-3 laser confocal scanning
microscope (Olympus, Tokyo, Japan).
Western blotting
Western blot analyses for whole-cell lysates were
performed using the following primary antibodies: paxillin
(1:4,000), p-paxillin (1:5,000). Detection was carried out using
HRP-conjugated secondary antibodies and an enhanced
chemiluminescence substrate (GE Healthcare, Piscataway, NJ, USA).
Membranes were stripped and reblotted for β-actin (1:1,000; A5316;
Sigma-Aldrich) as a loading control.
RT-PCR
The mRNA levels of target genes were analyzed using
semi-quantitative RT-PCR. Total RNA was extracted from U251 cells
with an RNA Simple Total RNA kit (Tiangen, Beijing, China), and
reverse transcription was performed using the M-MLV First Strand
kit (Invitrogen). Reverse transcription products were then
amplified by PCR using the HotStart Taq Master Mix kit (Tiangen).
Following a ‘hot start’ at 95°C for 3 min, the samples were cycled
at 95°C for 30 sec, 55°C for 30 sec, and 72°C for 20 sec. The total
number of cycles used were: 21 cycles for paxillin and 20
cycles for GAPDH. RT-qPCR was carried out with the
SYBR-Green Master Mix (Thermo Fisher Scientific, Inc., Waltham, MA,
USA), and samples were analyzed using a QuantStudio™ 7 Flex
Real-Time PCR System. Following a ‘hot start’ at 50°C for 2 min and
95°C for 10 min, the samples were then cycled at 95°C for 10 min,
60°C for 30 sec, and 72°C for 30 sec for 40 cycles. The samples
were then given a final 5 min extension at 72°C. Primers used for
PCR were: 5′-CTGCTGGAACTGAACGCTGTA-3′ (forward) and
5′-GGGGCTGTTAGTCTCTGGGA-3′ (reverse) for paxillin, and
5′-TGTGGGCATCAATGGATTTGG-3′ (forward) and
5′-ACACCATGTATTCCGGGTCAAT-3′ (reverse) for GAPDH.
Sample collection and ethics
statement
All paraffin GBM samples were collected from the
Affiliated Hospital of KMUST, Medical Faculty of Kunming University
of Science and Technology (Kunming, China). All specimens had
confirmed pathological diagnosis and were classified according to
the WHO criteria.
Since these clinical materials were for research
purposes only, we obtained written informed consent forms from all
patients, directly. Prior consent from all patients and approval
from the Ethics Committee at the Affiliated Hospital of Kunming
University of Science and Technology were obtained.
Methods involving live animals were carried out in
accordance with the guidelines and regulations enacted and enforced
by the Chinese National Ministry of Science and Technology as well
as the National Ministry of Health. All experimental protocols were
approved by the Institutional Laboratory Animal Ethics Committee at
Kunming University of Science and Technology.
Results
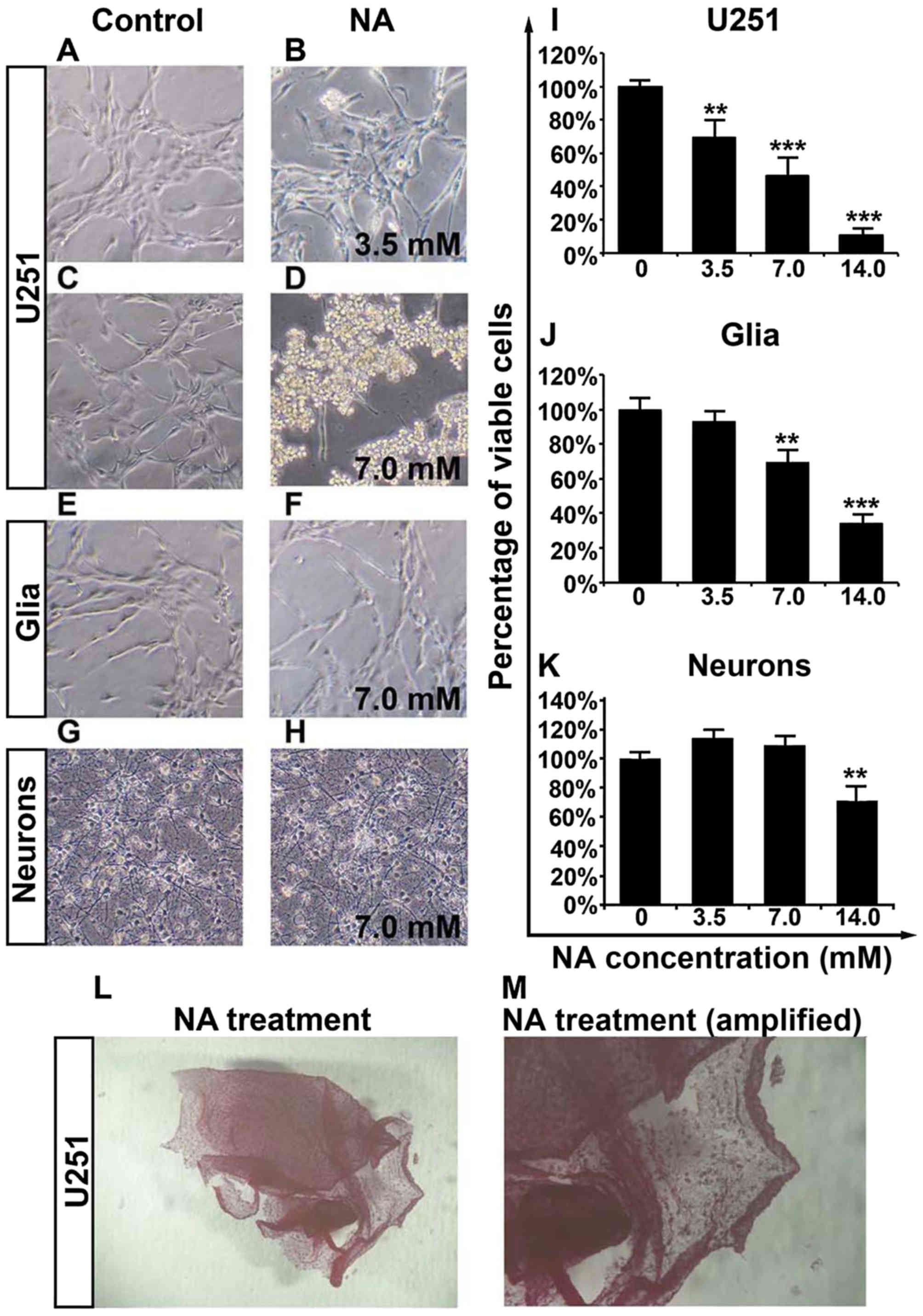
Overnight treatment of U251 cells with
NA causes detachment and decreased viability
We have previously reported that NA is able to
modify cytoskeletal structures in 3T3 cells (12). To assess whether NA has any
potential effects on malignant glioma, we carried out overnight
(12-h) treatment of cultured U251 GBM cells with various
concentrations of NA. As shown in Fig.
1A-D, while PBS had some effect, incubation in NA lifted U251
cells from the culture dishes, and cells treated with 7.0 mM of NA
detached more markedly than cells treated with 3.5 mM of NA. The
detached cells remained adhered to each other to form sheet-like
structures (Fig. 1L and M).
Notably, 7.0 mM of NA had less impact on primary mouse glia and
neurons as compared with the U251 cells (Fig. 1E-H), suggesting that the molecular
target(s) of NA are abundant in GBM cells but not in primary glia
or neurons.
We next employed an MTT assay and found that
increasing concentrations (3.5, 7.0 and 14.0 mM) of NA decreased
the viability of U251 cells in a dose-dependent manner (Fig. 1I). Likewise, a dose-dependent
decrease in the viability of primary glia was observed for NA, but
the effect was weaker than on U251 cells and was insignificant at
3.5 mM (Fig. 1J). The similar
response to NA between U251 cells and normal glia could be
explained by the fact that glioma cells are derived from glia and
therefore share some common biological background (17). Conversely, the viability of primary
neurons was only decreased by 14.0 mM, but not by lower dosages
(3.5 and 7.0 mM) of NA (Fig. 1K).
Hence, by carefully titrating the concentration of NA, it is
possible to obtain an optimal dosage at which NA selectively
targets glioma cells while sparing most normal glia and
neurons.
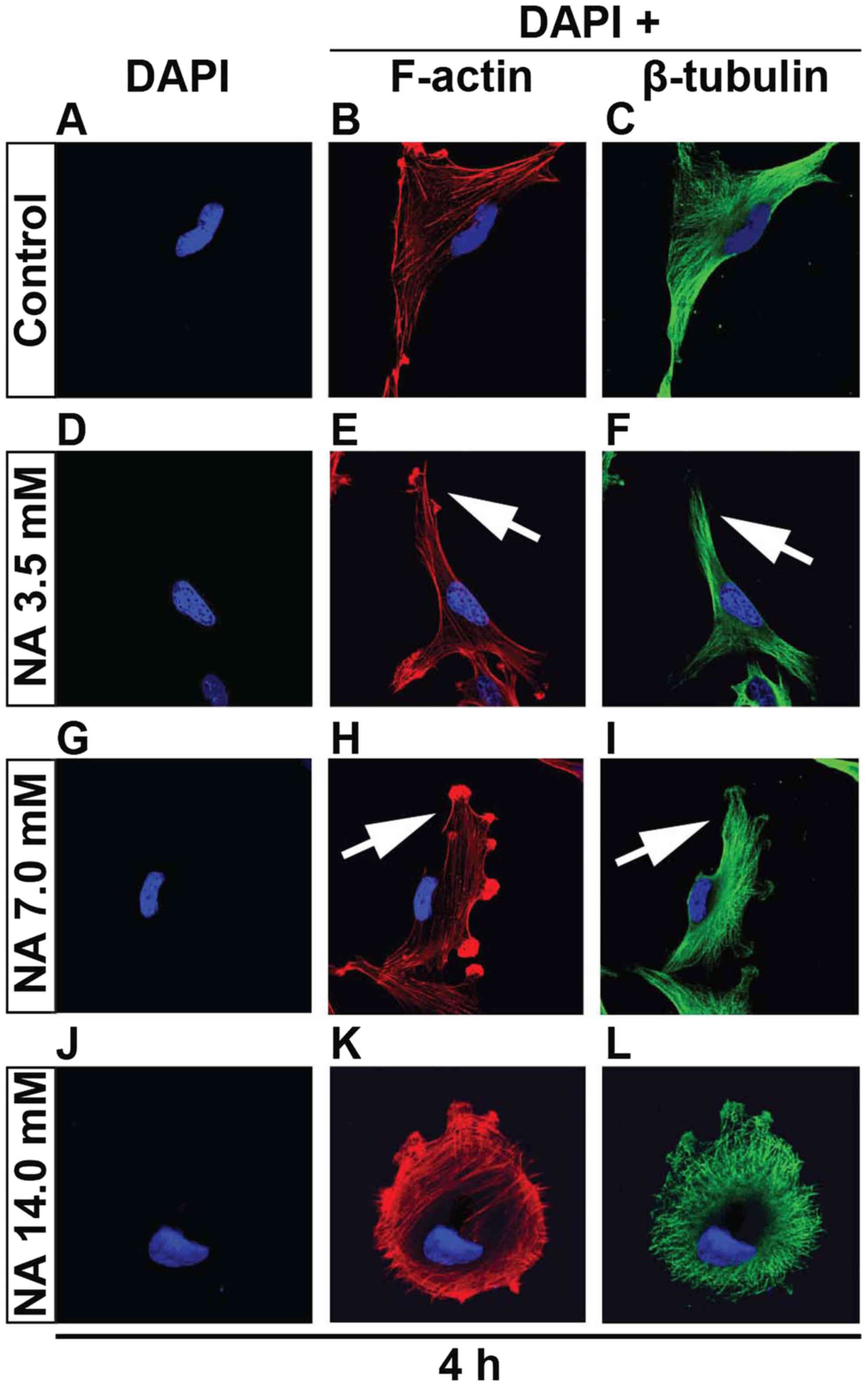
Short-term treatment of U251 cells
with NA alters cytoskeletal structures
Given that overnight incubation of NA completely
detaches U251 cells from the petri dish, it was necessary to cut
down the time frame in order to capture the initial event. Thus we
shortened the culturing time from 12 to 4 h since the U251 cells
were still attached. That said, 14.0 mM of NA greatly decreased the
attachment of U251 cells, and tethered cells assembled (Fig. 2J-L). With gentle shakes the tethered
cells detached. Again utilizing the 4-h culture time, F-actin and
β-tubulin staining revealed, particularly in 7.0 mM of NA
treatment, that F-actin fibers were disoriented and entangled close
to the plasma membrane (Fig. 2G-I).
Likewise, β-tubulin fibers lack tension and are loosely arranged.
Although 3.5 mM of NA has a relatively minor effect on cytoskeletal
stress, there were still discernable kinks in the F-actin fibers
close to the plasma membrane (Fig.
2D-F). NA treatment is detrimental to cell-matrix adhesion
probably due to either abrogation of components within the ECM or
integrin adhesome disruption or both, since the tension exerted on
adhesions requires the integrity of both ECM and integrin adhesome
(18,19).
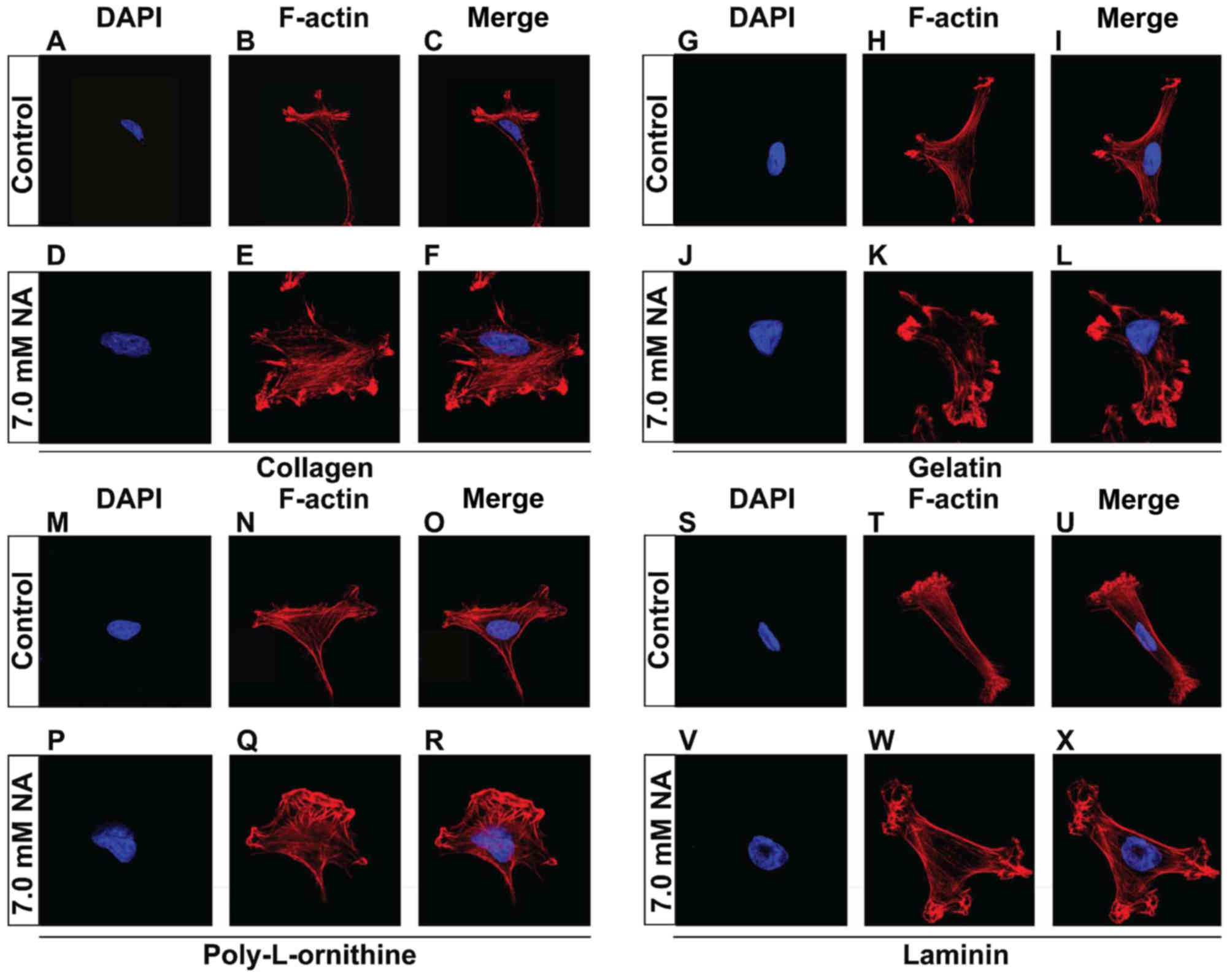
Compensation of ECM components failed
to restore the tension of actin fibers
The short-term effects on cytoskeletal structures
and cell detachment caused by prolonged treatment, suggest that NA
inhibits the adhesion of U251 cells to the matrix. In vivo,
cell-matrix adhesion depends on ECM components, in particular
fibrous proteins (such as collagens, fibronectins and laminins) and
proteoglycans (such as chondroitin sulphate, heparan sulphate,
keratan sulphate and hyaluronic acid) that are synthesized locally
and meshed into an organized structure to form the fundamental
framework for many tissues (20).
In the context of cancer biology, increased deposition of ECM has
been associated with higher mortality in cancer patients due to
increased invasion and metastasis (21). It is therefore important to test
whether NA also affects cell adhesion on ECM components. To do
this, U251 cells were cultured on petri dishes that were pre-coated
with additional collagen (Fig.
3A-F), gelatin (Fig. 3G-L),
poly-L-ornithine (Fig. 3M-R) or
laminin (Fig. 3S-X), allowed to
attach, and subsequently treated with 7.0 mM of NA for 4 h. Similar
to what we observed previously with the unsupplemented plates, U251
cells that were cultured on plates coated with various ECM
components also exhibited disoriented F-actin fibers that entangled
close to plasma membrane (Fig.
3A-X). These results suggest that NA interferes with the
ability of U251 cells to adhere to the ECM from an intracellular
aspect, rather than directly targeting the ECM.
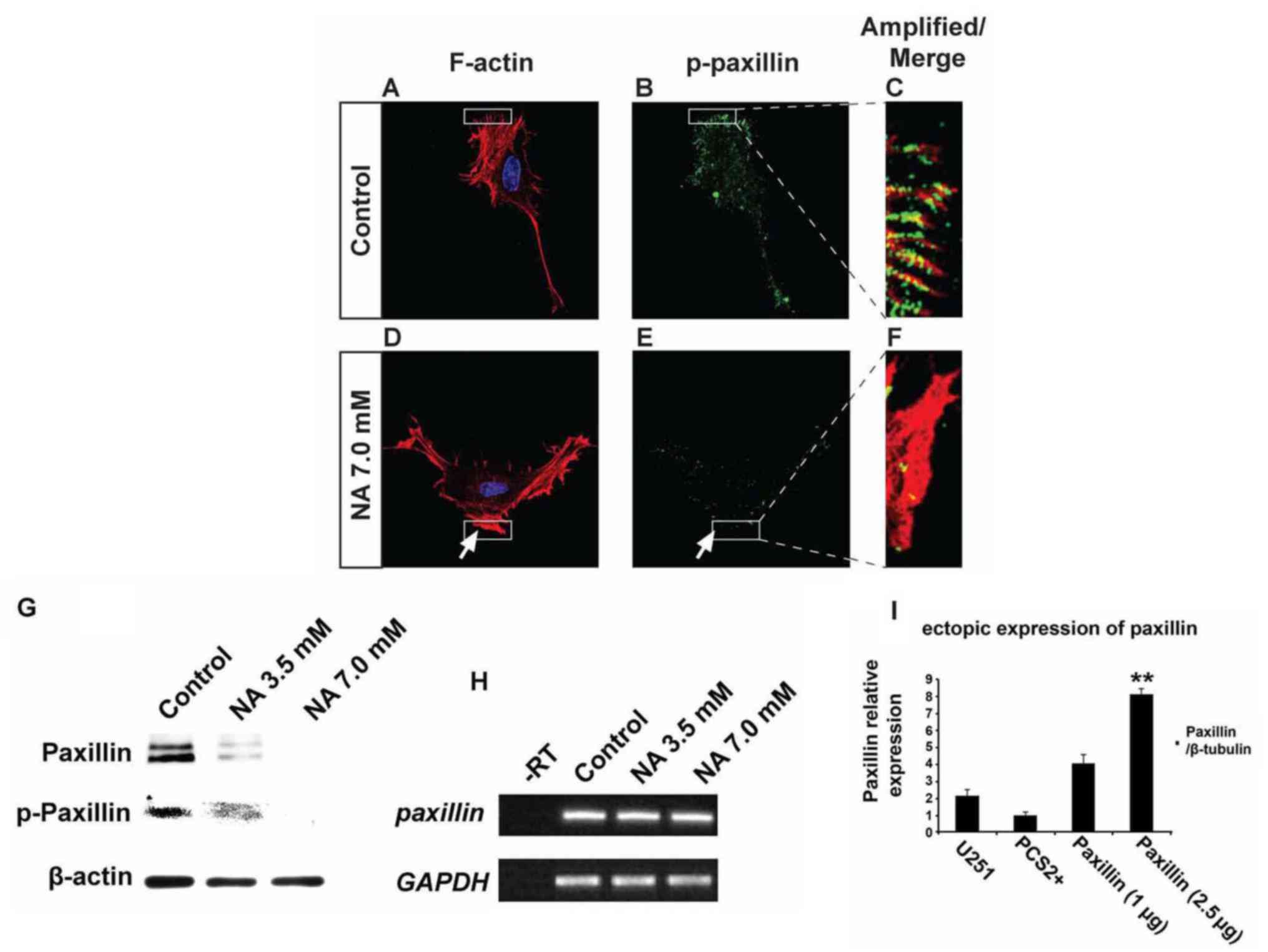
NA impedes the assembly of leading
edge
Phosphorylation of paxillin is known to play pivotal
roles in the rearrangement of the actin cytoskeletal network at
focal adhesion sites, and can serve as an important indicator for
active cell migration (22). Our
immunocytochemistry and western blot results revealed that U251
cells treated with 7.0 mM NA for 4 h had significantly decreased
levels of phosphorylated paxillin (p-paxillin) and total paxillin
when compared with untreated cells (Fig. 4A-G). In agreement with these
findings, western blot analyses revealed a dose-dependent decrease
of both total and phosphorylated paxillin levels upon treatment
with 3.5 and 7.0 mM of NA (Fig.
4G). However, mRNA levels of paxillin were unaffected
(Fig. 4H), suggesting that NA
downregulates paxillin post-transcriptionally. Phosphorylation of
tyrosine residues in cortactin has also been shown to regulate
focal adhesion turnover and cell migration (23,24),
while myosin IIA generates the major contractile forces by pulling
on actin stress fibers at the trailing end of migrating cells
(25–27). We found that in U251 cells incubated
in 7.0 mM of NA for 4 h, phosphorylated cortactin (p-cortactin)
markedly decreased (Fig. 5A-F), and
myosin IIA dissociated from F-actin stress fibers (Fig. 5G-L). Based on these data, we
conclude that short-term NA treatment can disrupt focal adhesion
assembly in U251 cells. In vivo, such disruption could lead
to the inhibition of glioma cell migration by preventing firm
attachment to the ECM.
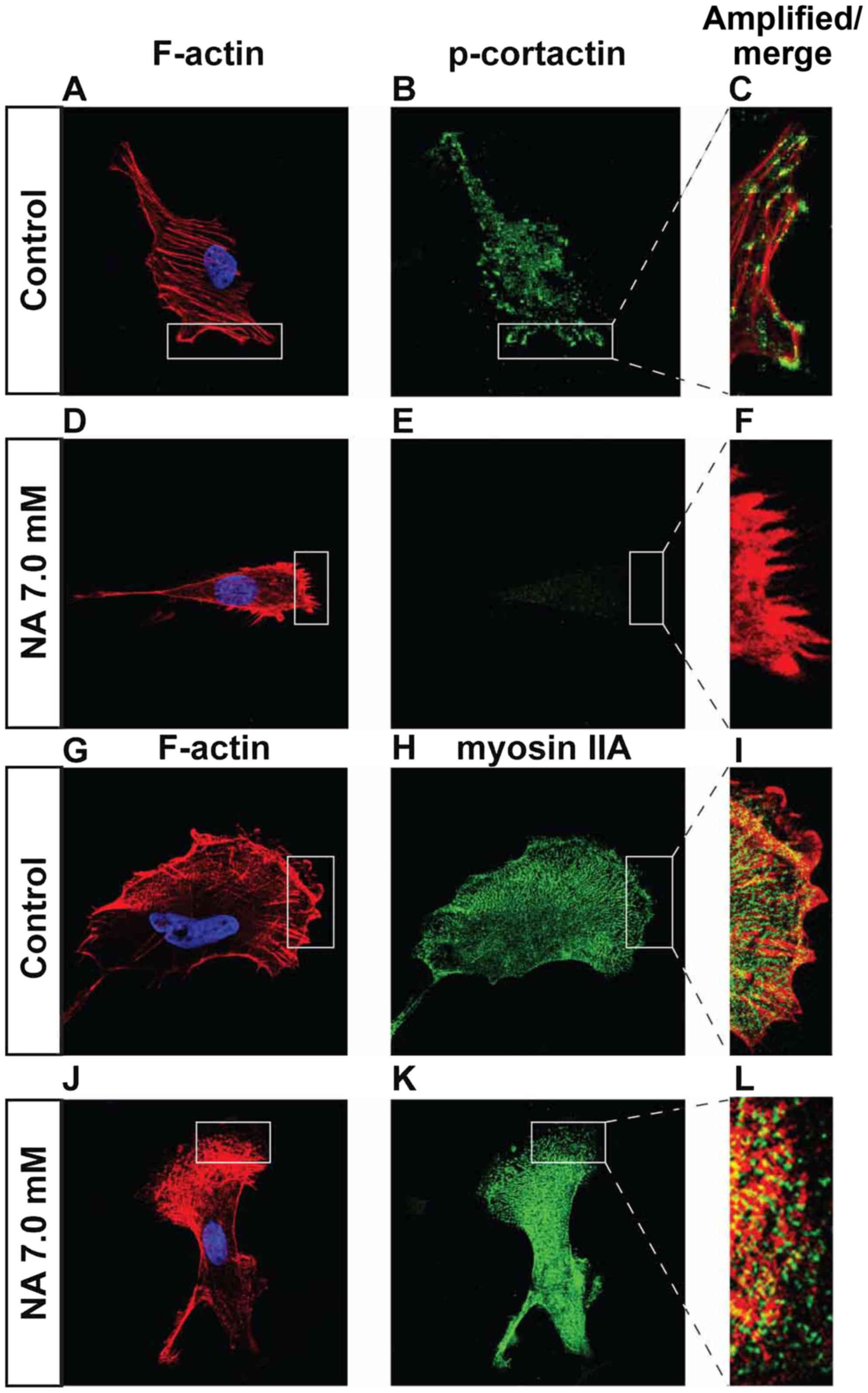 | Figure 5.Effects of short-term nicotinic acid
(NA) treatment on p-cortactin and myosin IIA. U251 cells were
treated with the indicated concentrations of NA for 4 h. DAPI
staining for nuclei (blue) as well as rhodamine phalloidin labeling
for F-actin (red in A, D, C, F and G, J, I, L), immunocytochemistry
for p-cortactin (green in B, C, E and F) and myosin IIA (green in
H, I, K and L) were carried out as described in Materials and
methods. |
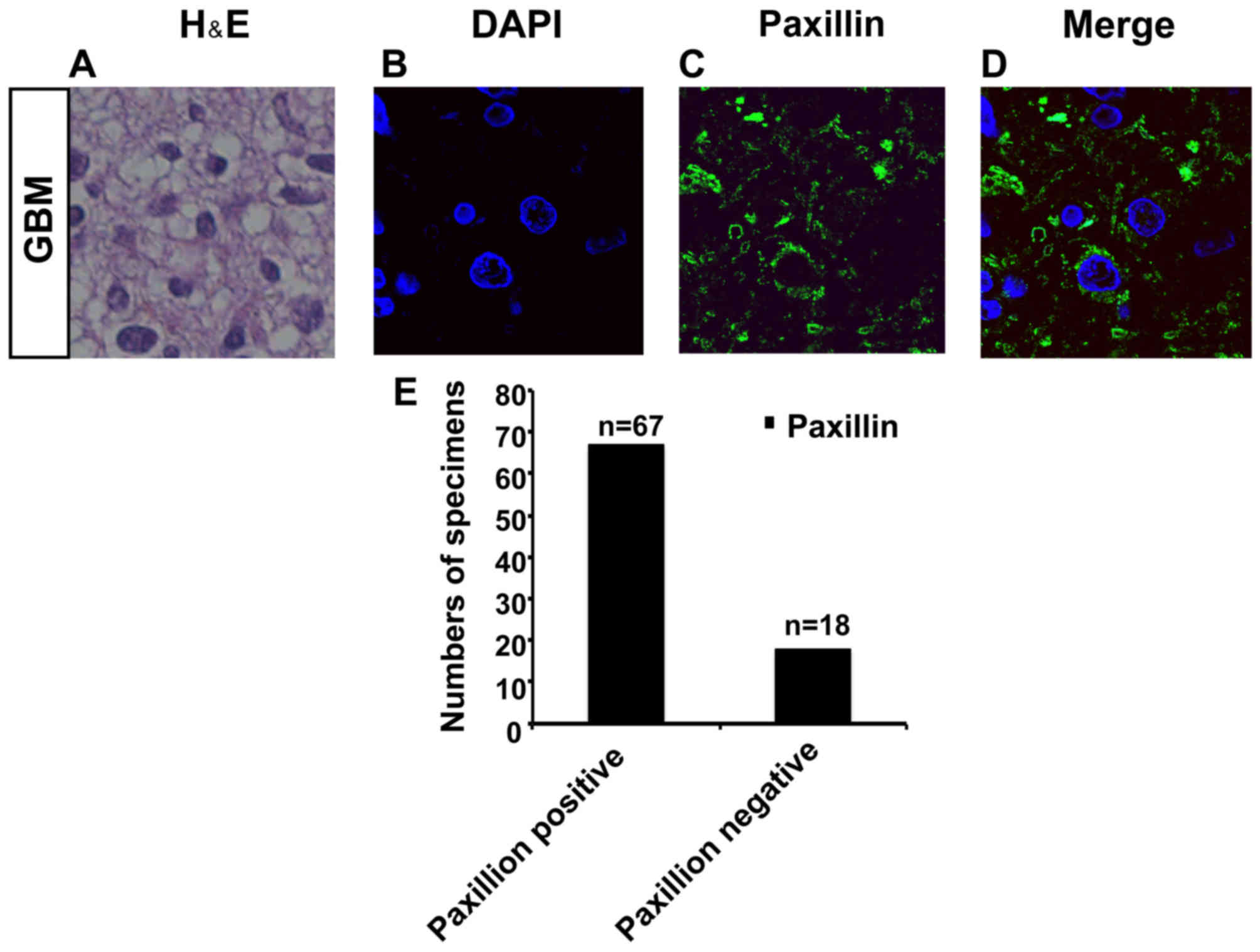
Paxillin is expressed in specimens of
GBM
Οur immunocytochemistry and western blot analysis
results showed that 4 h of treatment with 7.0 mM NA significantly
decreased the levels of phosphorylated paxillin (p-paxillin) and
total paxillin in U251 cells in comparison to untreated U251 cells
(Fig. 4A-G). This indicates that
paxillin is expressed in U251 cells and is a target of NA. We
therefore investigated the expression pattern of paxillin in
specimens of GBM.
We collected specimens from 85 GBM patients
(Fig. 6A). Interestingly, by
immunostaining, we found that paxillin was expressed in 67 samples
out of a total of 85 specimens (Fig.
6B-E). This corroborates that paxillin is expressed in GBM, and
the findings support further investigations into NA as a potential
therapy for malignant glioma.
Discussion
Although NA and its derivatives have been
intensively studied for decades, new functions and mechanisms of
action for them continue to emerge. For example, nicotinamide, the
amide of NA that has the same vitamin functions, was shown to block
proliferation and induce apoptosis of chronic lymphocytic leukemia
(CLL) cells (28). Recently, we
reported that treatment of 3T3 cells with NA leads to dynamic
changes in intracellular calcium concentration and disassembly of
the cytoskeletal structures (12).
These observations prompted us to test whether NA has any effects
on malignant glioma cells, which are known for their high invasive
activity. We found that NA had marked effects on assembly of
leading edge and cell-matrix adhesion, which suggests roles in the
inhibition of glioma cell invasion. These results revealed a novel
function of NA in the regulation of tumor cell migration, and set
the stage for testing NA as a potential therapy for malignant
glioma.
When applied overnight, NA causes detachment and
decreased viability of the U251 GBM cells, effects that appear to
be selective for U251 cells over normal neurons and glia (Fig. 1). Although the molecular basis for
this selectivity remains unknown, our subsequent study revealed
that cell death caused by NA is due to activation of the apoptotic
pathway (Yang et al, unpublished data). Notably,
nicotinamide has been shown to induce apoptosis in CLL cells by
regulation of the sirtuin SIRT1 (28). Since SIRT1 also plays important
roles in the apoptosis of malignant glioma cells (29), it may be of interest to investigate
whether and how NA affects sirtuins and their downstream targets
such as p53 (30,31).
Unlike overnight treatment, short-term incubation of
U251 cells in NA does not cause any detectable cell death, but
instead impairs assembly of leading edge and inhibits cell-ECM
interactions. The decrease in cell-matrix interaction is manifested
by disrupting cytoskeletal structures and focal adhesion assembly.
Similar to what we observed previously in 3T3 cells, NA treatment
modifies F-actin and β-tubulin filaments in a dose-dependent
response that correlates well with an increase in the intracellular
calcium level. However, direct treatment of 3T3 cells with
CaCl2 alters the structures of F-actin stress fibers but
not microtubules (12), suggesting
that the effect of NA on microtubules is independent of or less
sensitive to the increased calcium levels. In line with the latter
possibility, 3.5 mM of NA induces subtle modifications of F-actin
stress fibers, but has no detectable effect on microtubules,
whereas 7.0 mM of NA is able to remodel both structures (Fig. 2D-I). By contrast, 3.5 mM of NA is
sufficient to cause a marked decrease in the protein level of
paxillin (Fig. 4G). Therefore, the
disrupted focal adhesion assembly likely precedes and probably
causes the alterations in the cytoskeletal structure, consistent
with previous studies showing that integrin-mediated cell-matrix
adhesion is important for the maintenance of both F-actin and
microtubule filaments (19,32).
Short-term treatment with 7 mM of NA results in
extensive disruption of focal adhesion assembly, as indicated by
markedly diminished paxillin and p-cortactin as well as
mislocalized myosin IIA. In particular, paxillin protein is almost
completely lost (Fig. 4E-G).
However, paxillin mRNA remains essentially unchanged
(Fig. 4H), suggesting an efficient
post-transcriptional regulation by NA. One possible explanation for
these phenomena is the cleavage of paxillin by calpains, the
calcium-activated proteases, which have been demonstrated to
interfere with focal adhesion assembly and cell migration (19,33).
Indeed, we observed a dose-dependent increase of calpain activity
in U251 cells treated with 3.5 and 7.0 mM of NA (Yang et al,
unpublished data). Thus, paxillin is possibly an important effector
that mediates NA-induced inhibition of cell-matrix interaction. To
test this hypothesis, we attempted to rescue the effects caused by
NA by transfecting U251 cells with a plasmid that encodes paxillin.
However, although the mRNA of exogenous paxillin was
expressed at high levels (Fig. 4I),
we were unable to detect any paxillin protein in transfected cells
upon NA treatment (data not shown). This is in line with our
observation that paxillin is effectively and post-transcriptionally
downregulated by NA. Future efforts may be focused on targeting the
turnover of paxillin protein, such as by blocking calpain activity.
Additionally, we collected specimens from 85 GBM patients and
evaluated the expression pattern of paxillin. Notably, we found
that paxillin was expressed in 67 samples out of a total of 85
specimens (Fig. 6A-E).
Although this result is preliminary, it is an
encouraging first step towards testing NA as a potential therapy
for malignant glioma in an animal model. Since NA is widely used as
an antidyslipidemic drug, the abundant clinical data that are
available may greatly facilitate the trials of this vitamin in
other diseases such as cancer. It should be noted, though, that we
are still facing some challenges when exploring the clinical
applications of NA. First of all, the mechanisms of action for NA
have not been fully elucidated. In particular, it is not clear how
NA is metabolized and how signals in the cells trigger the
intracellular calcium spike. To this end we are investigating
whether nicotinamide, which has the same vitamin function as NA but
no lipid-regulating activity (34),
can mimic NA in affecting cell-cell and cell-matrix adhesion. It
also remains to be determined whether the cell-surface NA receptor
GPR109A, which mediates the NA lipid-regulating activity, as well
as the TRPV channels that have been reported to be downstream
targets of NA (10,13), are involved in the cellular
processes that we observed for NA in the present study. Secondly, a
high dosage of NA is known to cause adverse effects (34). Some of these effects, such as
flushing, are mediated by GPR109A and TRPV channels, which may be
involved in the antitumor activity of NA that we reported in the
present study. Fortunately, these side-effects are not
life-threatening and may be acceptable for the treatment of highly
deadly diseases such as malignant glioma. In conclusion, our data
supports the potential application of NA as a therapy for malignant
glioma, and further investigation is warranted into the effects of
NA on other invasive tumors.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (81200878 to J.L.), and the
Recruited Talent Program of KMUST (KKZ3201560014 to J.L.).
References
|
1
|
Cuddapah VA, Robel S, Watkins S and
Sontheimer H: A neurocentric perspective on glioma invasion. Nat
Rev Neurosci. 15:455–465. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dandy WE: Removal of right cerebral
hemisphere for certain tumors with hemiplegia: Preliminary report.
J Am Med Assoc. 90:823–825. 1928. View Article : Google Scholar
|
|
4
|
Hou LC, Veeravagu A, Hsu AR and Tse VC:
Recurrent glioblastoma multiforme: A review of natural history and
management options. Neurosurg Focus. 20:E52006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ridley AJ, Schwartz MA, Burridge K, Firtel
RA, Ginsberg MH, Borisy G, Parsons JT and Horwitz AR: Cell
migration: Integrating signals from front to back. Science.
302:1704–1709. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Beadle C, Assanah MC, Monzo P, Vallee R,
Rosenfeld SS and Canoll P: The role of myosin II in glioma invasion
of the brain. Mol Biol Cell. 19:3357–3368. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Demuth T and Berens ME: Molecular
mechanisms of glioma cell migration and invasion. J Neurooncol.
70:217–228. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kwiatkowska A and Symons M: Signaling
determinants of glioma cell invasion. Glioma Signaling Springer.
121–141. 2013.https://doi.org/10.1007/978-94-007-4719-7_7
View Article : Google Scholar
|
|
9
|
Wolfenson H, Lavelin I and Geiger B:
Dynamic regulation of the structure and functions of integrin
adhesions. Dev Cell. 24:447–458. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Offermanns S: The nicotinic acid receptor
GPR109A (HM74A or PUMA-G) as a new therapeutic target. Trends
Pharmacol Sci. 27:384–390. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gille A, Bodor ET, Ahmed K and Offermanns
S: Nicotinic acid: Pharmacological effects and mechanisms of
action. Annu Rev Pharmacol Toxicol. 48:79–106. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li J, Li Y, Zhang P, Niu H and Shi Y:
Nicotinic acid modulates intracellular calcium concentration and
disassembles the cytoskeleton. Mol Med Rep. 10:2805–2810.
2014.PubMed/NCBI
|
|
13
|
Ma L, Lee BH, Clifton H, Schaefer S and
Zheng J: Nicotinic acid is a common regulator of heat-sensing
TRPV1-4 ion channels. Sci Rep. 5:89062015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ma L, Lee BH, Mao R, Cai A, Jia Y, Clifton
H, Schaefer S, Xu L and Zheng J: Nicotinic acid activates the
capsaicin receptor TRPV1: Potential mechanism for cutaneous
flushing. Arterioscler Thromb Vasc Biol. 34:1272–1280. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sobottka SB and Berger MR: Assessment of
antineoplastic agents by MTT assay: Partial underestimation of
antiproliferative properties. Cancer Chemother Pharmacol.
30:385–393. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Qu J, Rizak JD, Li X, Li J and Ma Y:
Melatonin treatment increases the transcription of cell
proliferation-related genes prior to inducing cell death in C6
glioma cells in vitro. Oncol Lett. 6:347–352.
2013.PubMed/NCBI
|
|
17
|
Camand E, Peglion F, Osmani N, Sanson M
and Etienne-Manneville S: N-cadherin expression level modulates
integrin-mediated polarity and strongly impacts on the speed and
directionality of glial cell migration. J Cell Sci. 125:844–857.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vicente-Manzanares M, Choi CK and Horwitz
AR: Integrins in cell migration - the actin connection. J Cell Sci.
122:199–206. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Winograd-Katz SE, Fässler R, Geiger B and
Legate KR: The integrin adhesome: From genes and proteins to human
disease. Nat Rev Mol Cell Biol. 15:273–288. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Frantz C, Stewart KM and Weaver VM: The
extracellular matrix at a glance. J Cell Sci. 123:4195–4200. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gilkes DM, Semenza GL and Wirtz D: Hypoxia
and the extracellular matrix: Drivers of tumour metastasis. Nat Rev
Cancer. 14:430–439. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Deakin NO and Turner CE: Paxillin comes of
age. J Cell Sci. 121:2435–2444. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kruchten AE, Krueger EW, Wang Y and
McNiven MA: Distinct phospho-forms of cortactin differentially
regulate actin polymerization and focal adhesions. Am J Physiol
Cell Physiol. 295:C1113–C1122. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lin Y, Wu Y, Li J, Dong C, Ye X, Chi YI,
Evers BM and Zhou BP: The SNAG domain of Snail1 functions as a
molecular hook for recruiting lysine-specific demethylase 1. EMBO
J. 29:1803–1816. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Aguilar-Cuenca R, Juanes-García A and
Vicente-Manzanares M: Myosin II in mechanotransduction: Master and
commander of cell migration, morphogenesis, and cancer. Cell Mol
Life Sci. 71:479–492. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kuo JC, Han X, Hsiao CT, Yates JR III and
Waterman CM: Analysis of the myosin-II-responsive focal adhesion
proteome reveals a role for β-Pix in negative regulation of focal
adhesion maturation. Nat Cell Biol. 13:383–393. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
van den Dries K, Meddens MB, de Keijzer S,
Shekhar S, Subramaniam V, Figdor CG and Cambi A: Interplay between
myosin IIA-mediated contractility and actin network integrity
orchestrates podosome composition and oscillations. Nat Commun.
4:14122013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Audrito V, Vaisitti T, Rossi D, Gottardi
D, D'Arena G, Laurenti L, Gaidano G, Malavasi F and Deaglio S:
Nicotinamide blocks proliferation and induces apoptosis of chronic
lymphocytic leukemia cells through activation of the
p53/miR-34a/SIRT1 tumor suppressor network. Cancer Res.
71:4473–4483. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Qu Y, Zhang J, Wu S, Li B, Liu S and Cheng
J: SIRT1 promotes proliferation and inhibits apoptosis of human
malignant glioma cell lines. Neurosci Lett. 525:168–172. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sasca D, Hähnel PS, Szybinski J, Khawaja
K, Kriege O, Pante SV, Bullinger L, Strand S, Strand D, Theobald M,
et al: SIRT1 prevents genotoxic stress-induced p53 activation in
acute myeloid leukemia. Blood. 124:121–133. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li L, Wang L, Li L, Wang Z, Ho Y, McDonald
T, Holyoake TL, Chen W and Bhatia R: Activation of p53 by SIRT1
inhibition enhances elimination of CML leukemia stem cells in
combination with imatinib. Cancer Cell. 21:266–281. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Byron A, Askari JA, Humphries JD,
Jacquemet G, Koper EJ, Warwood S, Choi CK, Stroud MJ, Chen CS,
Knight D, et al: A proteomic approach reveals integrin activation
state-dependent control of microtubule cortical targeting. Nat
Commun. 6:61352015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cortesio CL, Boateng LR, Piazza TM, Bennin
DA and Huttenlocher A: Calpain-mediated proteolysis of paxillin
negatively regulates focal adhesion dynamics and cell migration. J
Biol Chem. 286:9998–10006. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Offermanns S: Activation of platelet
function through G protein-coupled receptors. Circ Res.
99:1293–1304. 2006. View Article : Google Scholar : PubMed/NCBI
|