Introduction
Immune suppression in cancer patients is one of
major issues that promotes tumor progression and inhibits the
efficiency of anticancer treatment. DNA damage response in cancer
cells following radiotherapy or chemotherapy engages host immune
effectors that may contribute to eradication of cancer.
Nevertheless, in many cases, it is likely that the stimulation in
response to DNA damage is insufficient to activate the immune
system in order to overcome the cancer. Therefore, enhancement of
immune activity with additional therapeutic drugs may be promising
to increase the antitumor effect in combination with radiotherapy
or chemotherapy.
To improve the efficacy of immunotherapy, numerous
studies have sought to develop novel strategies including
immune-checkpoint inhibitors that can overcome the
immunosuppressive environment (1–3). Among
the several immune systems, one of the major activating
immunoreceptors, natural-killer group 2 member D (NKG2D), plays a
central role in the antitumor effect. NKG2D is expressed in several
types of immune cells, including NK, NKT, γδ T and CD8+
αβ T cells that exert cytotoxicity against cancer cells (4,5).
Therefore, reactivation of the NKG2D-mediated immune response is
important for invoking an initial response in terms of cancer
immunotherapy. Major histocompatibility complex class I-related
chains A and B (MICA/B), ligands for the NKG2D immune receptor, in
human cells are often constitutively upregulated in cancer cells,
but are not effectively recognized in an immunosuppressive
environment (6). Regardless of the
adapted environment, further activation of MICA/B is able to
promote an antitumor effect. For example, DNA damage is one of the
activators of MICA/B in cancer cells. Consistent with the evidence
of the upregulation of MICA/B expression by DNA damage, ionizing
radiation (IR) and chemotherapeutic agents enhance NK cell-mediated
killing of cancer cells (7–9). DNA double-strand breaks (DSBs) are the
most critical DNA lesions induced by IR. Gene expression following
DNA damage is regulated by kinases that are also involved in repair
and cell-cycle checkpoint arrest (10). Ataxia telangiectasia, mutated (ATM),
which activates the repair and checkpoint machinery, is a ‘first
responder’ to DSBs (11). During
homologous recombination (HR), a major DSB repair pathway, cells
activate ATM- and Rad3-related (ATR) protein kinase, followed by
Chk1 (11,12). Previous studies have revealed that
the ATR/Chk1 pathway plays an important role in the expression of
MICA/B after DNA damage. However, although the requirement of DNA
damage signaling in MICA/B expression has been reported, variations
in the responsiveness of MICA/B among cancer cell lines after DNA
damage have not been thoroughly investigated (7).
In the present study, we first investigated DNA
damage-dependent MICA/B expression on the surface of cancer cells
in relation to tissue type and tumor-suppressor gene status.
Neither tissue type nor Rb/p53 status influenced MICA/B expression.
Notably, however, various cancer cell lines did not express MICA/B
in response to IR or aphidicolin, a DNA polymerase inhibitor which
induces DNA damage by blocking DNA replication. These insensitive
cell lines did not respond even after carbon-ion irradiation, which
induces robust DNA damage signaling. Next, we screened compounds
that affect chromatin relaxation since we hypothesized that MICA/B
gene expression in insensitive cells may be suppressed by chromatin
disorganization in the tumor environment. We found that treatment
with a histone deacetylase inhibitor (HDACi; vorinostat) restored
DNA damage-induced MICA/B expression in insensitive cells. In
addition, inhibition of Suv39 or G9a histone methyltransferase
activity, both of which promote chromatin relaxation via the HDAC
axis, restored MICA/B expression in insensitive cells after IR
(13–15). The restored MICA/B expression by
HDACi was prevented by inhibition of ATR or depletion of E2F1.
Furthermore, our titration analysis revealed that a low-dose of
HDACi was sufficient to restore DNA damage-dependent MICA/B
expression in insensitive cells. Finally, inhibition of HDAC
restored IR-dependent cytotoxic NK activity in insensitive cells.
Thus, MICA/B expression in insensitive cells can be restored by
therapeutic HDACi or inhibition of Suv39/G9a activity.
Collectively, our results demonstrate that inhibition of the
HDAC/Suv39/G9a pathway may potentiate the therapeutic efficacy of
radiotherapy and DNA-damaging chemotherapy by reactivating
MICA/B-mediated killing of cancer cells.
Materials and methods
Cell lines, reagents and
irradiation
The cancer cell lines were cultured in Dulbecco's
modified Eagles medium, RPMI-1640 medium (both from Wako, Tokyo,
Japan) or McCoy's 5A medium (Gibco, Carlsbad, CA, USA) supplemented
with 10% fetal calf serum (FCS) at 37°C in a humidified mixture of
95% air and 5% CO2. X-rays were generated at 200 kVp and
20 mA using copper (0.5 mm)-aluminum (1.0 mm) filters. Exposure to
carbon-ion irradiation (290 MeV/n, LET ~70 keV/µm) was performed at
the Heavy Ion Medical Accelerator (HIMAC) facility of the National
Institute of Quantum and Radiological Science and Technology
(Chiba, Japan), or at the Gunma University Heavy Ion Medical Center
(GHMC; Gunma, Japan) (290 MeV/n, LET ~70 keV/µm). Before IR, 10 µM
of ATR inhibitor (ATRi), VE821 (Axon), was added at 30 min. At this
concentration, the drug specifically inhibited the target. The DNA
polymerase inhibitor aphidicolin (Wako) was added at a final
concentration of 4 µM, and the cultures were then incubated for 24
h.
Analysis of MICA/B surface expression
by fluorescence-activated cell sorting (FACS)
Cancer cells were incubated for 24 h after exposure
to 10 Gy X-rays, carbon ions, or aphidicolin treatment, and then
harvested for FACS analysis. Adherent cells were harvested by
shake-off in 2 mM EDTA/phosphate-buffered saline (PBS) without
trypsinization according to the method described by Clayton et
al (16). Harvested cells were
washed with FACS solution (ice-cold PBS containing 2% newborn calf
serum, 1 mM EDTA, 0.01% w/v NaN3), and then stained with
MICA/B antibodies for 20 min at 4°C. Cells undergoing apoptosis
were detected using Annexin V. MICA/B expression was analyzed in
cells doubly negative for propidium iodide (PI) (Sigma-Aldrich, St.
Louis, MO, USA) and Annexin V (BioLegend, San Diego, CA, USA). FACS
was performed on a FACSCalibur instrument using the CellQuest
software. FACS data were analyzed using the FlowJo v. 9.3 software
(Tree Star, Inc., Ashland, OR, USA). Expression levels of surface
MICA/B were determined as mean fluorescence intensity (MFI) of
anti-MICA/B normalized against the MFI of an isotype control
antibody. The IR-induced fold increase in expression level was
calculated by dividing the MFI of irradiated cells (IR-MFI) by the
MFI of non-irradiated cells (non-IR-MFI). Reproducible results in
all FACS experiments were obtained from two or more independent
experiments. A representative FACS histogram is shown for each
analysis.
Drug screening focusing on factors
that influence chromatin remodeling
The T98G cell line, an insensitive cell line, was
used in the screening analysis. Each inhibitor was added 2 h before
cells were exposed to X-rays, and the cells were harvested 24 h
post-IR. MICA/B expression was analyzed by FACS. Drug information,
including concentration in the media, is listed in Table I.
 | Table I.Inhibitors used in the present
study. |
Table I.
Inhibitors used in the present
study.
| Name | Effect | Supplier | Cat. no. | Dose |
|---|
| Vorinostat
(SAHA) | HDAC inhibitor | Focus
Biomolecules | 10-1067 |
1 µM |
| TSA | HDAC inhibitor | Wako Pure Chemical
Industries | 203-17561 | 100 nM |
| Chaetocin | Suv39 histone
methyltransferase inhibitor | Abcam | ab144534 | 50
nM |
| Bix-01294 | G9a histone
methyltransferease inhibitor | Cayman Chemical
Company | 13124 |
5 µM |
| 5-Aza-cytidine | DNA
methyltransferase inhibitor | TCI Chemicals | A2033 |
1 µM |
| Zebularine | DNA
methyltransferase inhibitor | Tocris
Bioscience | 2293 | 10
µM |
| NU9056 | HAT inhibitor KAT5,
p300, pCAF and GCN5 | Tocris
Bioscience | 4903 |
5 µM |
siRNA transfection
siControl and siE2F1 were obtained from Ambion
Silencer® Select siRNA (Life Technologies, Carlsbad, CA,
USA). siRNA transfection was performed using HiPerFect (Qiagen,
Valencia, CA, USA) as previously described (17). Briefly, siRNA transfection was
carried out in suspended cells following trypsinization. After 24
h, cells were re-transfected with siRNA in suspension following
trypsinization. Prior to analysis, cells were incubated for 48 h
after the second transfection.
Immunofluorescence staining and
microscopy
Immunofluorescence (IF) staining was performed as
previously described (18). Images
were captured on an Olympus BX51 microscope with identical exposure
times.
Antibodies
Antibodies against the following proteins were used
for FACS, immunoblotting and IF staining: MICA/B (6D4; BioLegend),
mouse IgG2a isotype Ctrl (MOPC-173), Annexin V (BioLegend), pChk1
S345, Chk1, Ku80 (all from Cell Signaling Technology, Inc.,
Beverly, MA, USA) and E2F1 (C-20; Santa Cruz Biotechnology, Inc.,
Santa Cruz, CA, USA).
NK-mediated cytotoxicity assay
The NK-92 cell line which was used as an effector
was obtained from the American Type Culture Collection (ATCC;
Manassas, VA, USA) and maintained in Alpha-MEM (Gibco) containing
12.5% (v/v) horse serum, 12.5% (v/v) fetal bovine serum and human
recombinant IL-2. The cytotoxicity of NK-92 against T98G cells was
assessed using the DELFIA EuTDA Cytotoxicity Assay Reagent kit
AD0116 (PerkinElmer, Inc., Waltham, MA, USA). The experiment was
performed following the manufacturer's instructions. Briefly, NK-92
cells used as effector cells were cultured with anti-CD314 (1D11)
or mouse IgG1 (MOPC-21) (both from BioLegend) for 30 min before
mixing the target cells. T98G cells used as target cells were
cultured with BATDA for 30 min at RT, then the cells were washed 3
times with PBS containing 2% newborn calf serum. BATDA-labeled T98G
cells were plated onto round-bottom 96 well plates at a density of
5,000 cells/well and mixed with effector cells at various
effector/target ratios. Spontaneous release was assessed in T98G
cells incubated with culture medium only, and maximum release was
measured in T98G cells incubated for 30 min with 2% Triton X-100.
After incubation for 4 h, 20 ml of supernatant of each well were
harvested for assessment of released TDA. The time-resolved
fluorescence was assessed by EnVision (PerkinElmer, Inc.). The
percentage of cytotoxicity was calculated by (Experimental release
- Spontaneous release)/(Maximum release - Spontaneous release) ×
100.
Cell proliferation assay
Cell viability following vorinostat treatment was
determined using Cell Proliferation kit I (Roche, Mannheim,
Germany).
Statistical analysis
Statistical significance in the FACS, cell
proliferation and NK-mediated cytotoxicity assays were determined
using Student's two-tailed t-test or the Mann-Whitney U test, both
in SigmaPlot 12.0.
Results
Cancer cell lines exhibit a wide range
of DNA damage-induced MICA/B expression, independent of tissue type
and Rb/p53 status
Cell surface expression of MICA/B is upregulated
after DNA damage (7). Previous
studies have reported that spontaneous MICA/B expression varies
widely among cancer cell lines (19–21).
In the present study, to investigate variety of DNA damage-induced
MICA/B expression among cancer cells, in relation to tissue type
and tumor-suppressor gene status, MICA/B surface expression was
examined in several cancer cell lines following exposure to IR. To
assess human MICA/B expression, levels of MICA/B on the cell
surface were analyzed by FACS. To ascertain that we were analyzing
MICA/B expression in living cells, we assessed only PI and Annexin
V double-negative cells (data not shown). Consistent with previous
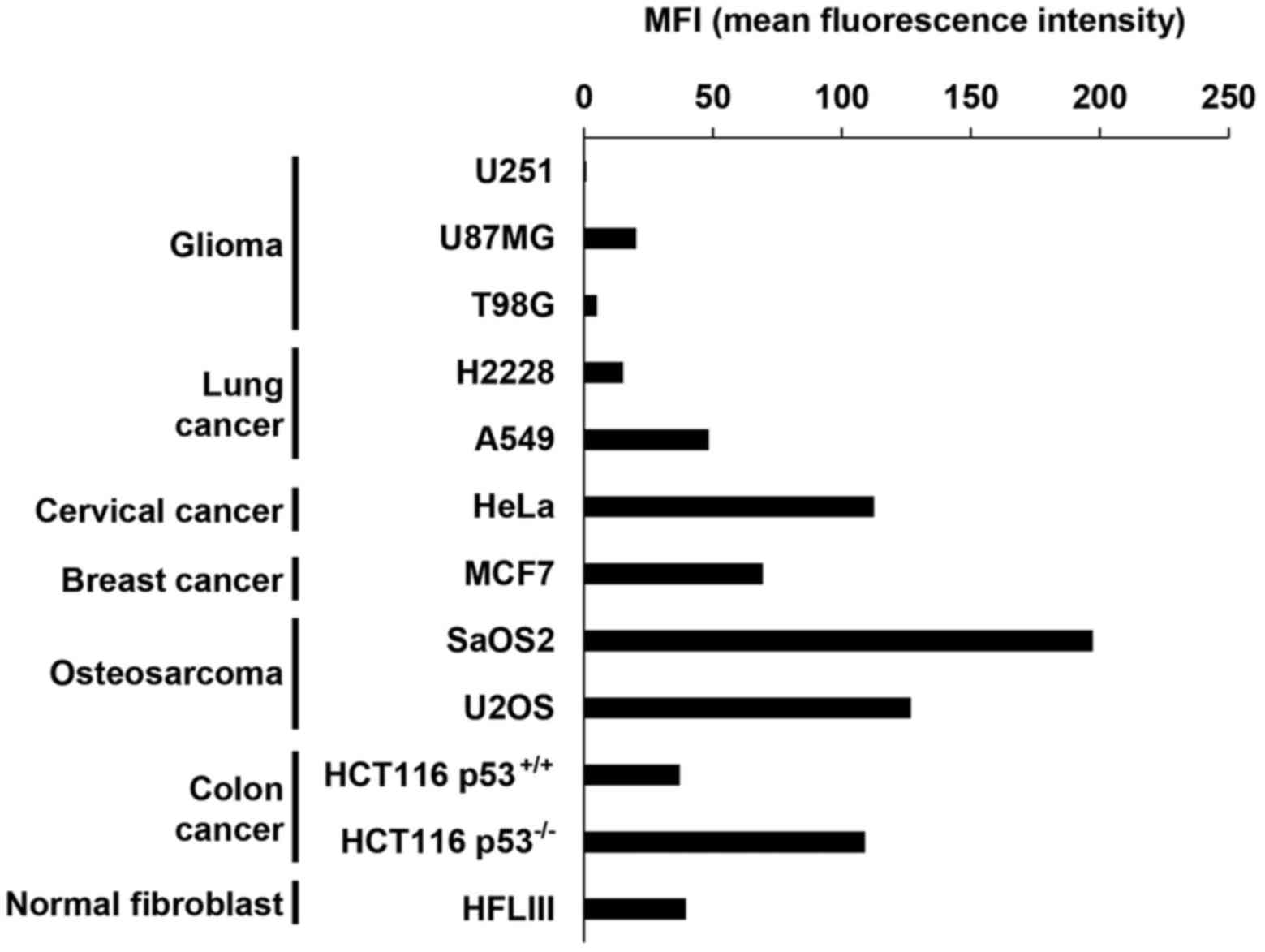
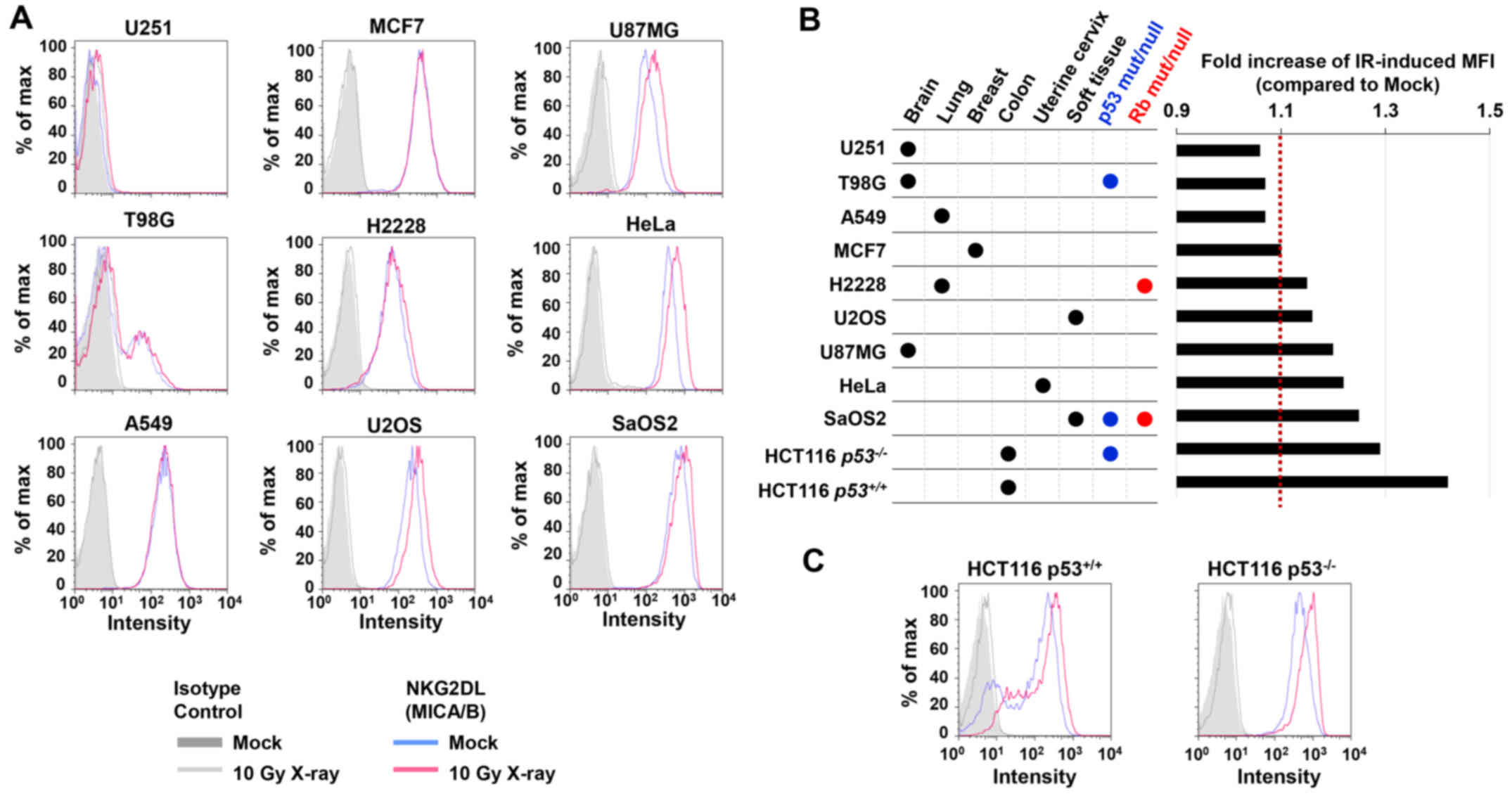
studies, the basal level of MICA/B expression in the absence of DNA
damage varied among the cell types (Fig. 1). At 24 h after 10 Gy X-rays, we
found that H2228, U2OS, U87MG, HeLa and SaOS2 cells exhibited high
levels of MICA/B induction, whereas lower levels of induction were
observed in U251, T98G, A549 and MCF7 cells (Fig. 2A and B). Notably, X-ray-induced
MICA/B expression was not obviously correlated with tissue types
(Fig. 2A and B). We also
investigated MICA/B expression in relation to the status of
tumor-suppressor genes, Rb and p53. However, we did not observe any
correlation between mutation status and MICA/B expression (Fig. 2B and C).
X-ray-induced DSBs are mostly repaired by either HR
or non-homologous end joining (NHEJ) within a few hours after
X-rays (10). Therefore, we
wondered whether DNA damage signaling following X-irradiation was
insufficient to induce MICA/B in insensitive cancer cells since the
DNA damage is rapidly repaired, and the signal is consequently not
maintained for a long period of time (17,22).
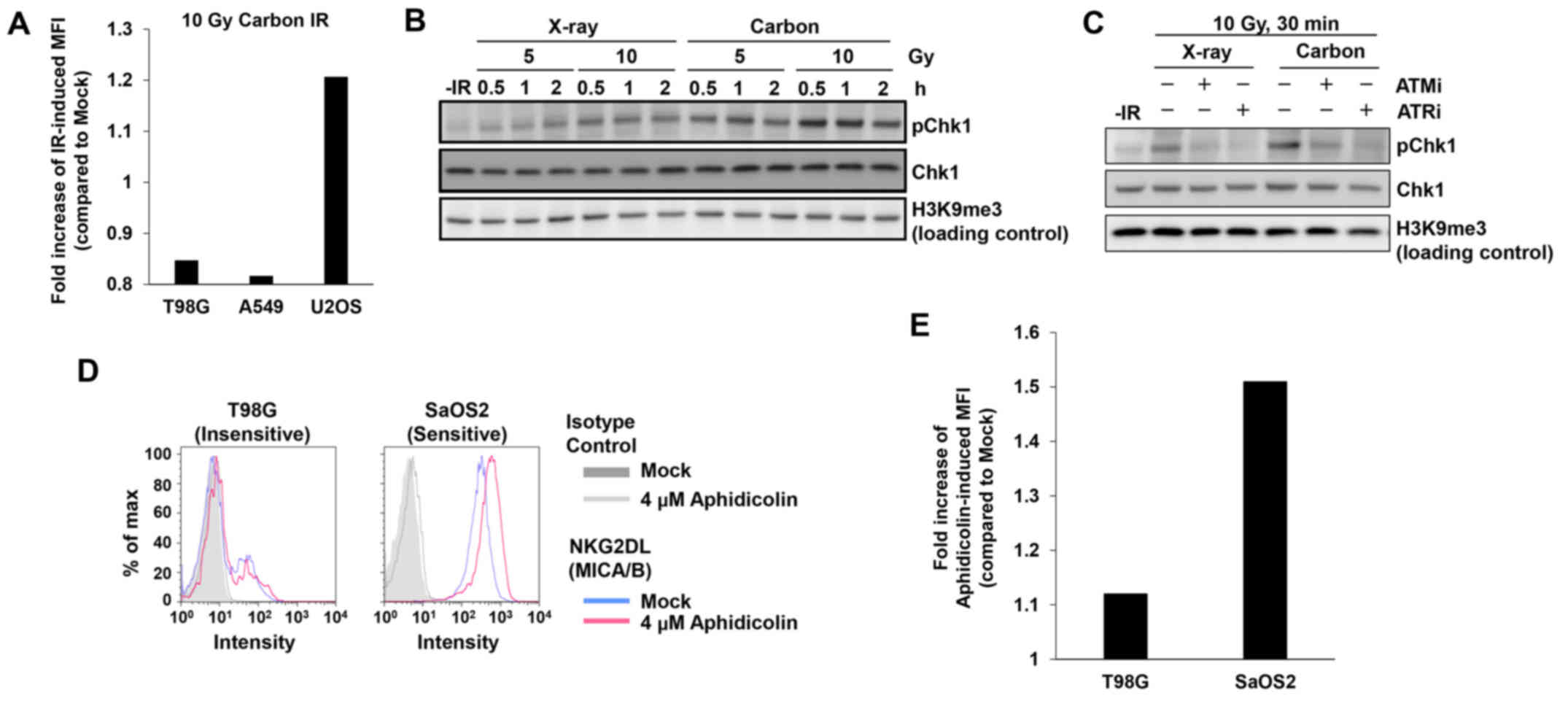
To address this issue, we tested whether heavy ion irradiation
could activate MICA/B in insensitive T98G or A549 cell lines. In
contrast to X-rays, heavy ion irradiation induces complex DNA
lesions that delay the speed of DSB repair (23,24).
However, the insensitive cell lines did not express MICA/B even
after carbon-ion irradiation (Fig.
3A). High levels of DNA damage signaling after carbon-ion
irradiation were confirmed by Chk1 S345 phosphorylation (pChk1),
since Chk1 has a central role in the upregulation of MICA/B
expression after DNA damage (Fig.
3B) (7). The ATM/ATR dependency
of this effect was confirmed by the decrease in pChk1 in the
presence of an ATM- or ATR-specific inhibitor (Fig. 3C) (25). To further confirm the poor
responsiveness in insensitive cells, we treated insensitive T98G
cells with aphidicolin, a DNA polymerase inhibitor, which induces
DNA damage by blocking DNA replication (26). Similarly to IR, aphidicolin
treatment did not activate MICA/B in insensitive T98G cells
(Fig. 3D and E). These data reveal
that the limited response of MICA/B expression in insensitive cell
lines is due to neither IR-induced damage complexity nor the type
of DNA damage. Instead, other factors are likely involved in the
suppression of DNA damage-dependent MICA/B activation.
Inhibition of histone deacetylase
activity restores DNA damage-induced MICA/B expression in
insensitive cancer cells
Next, we speculated that the insensitivity may be
dependent on the suppression of the MICA/B gene expression due to
an alteration in chromatin structure in cancer cells (27–29).
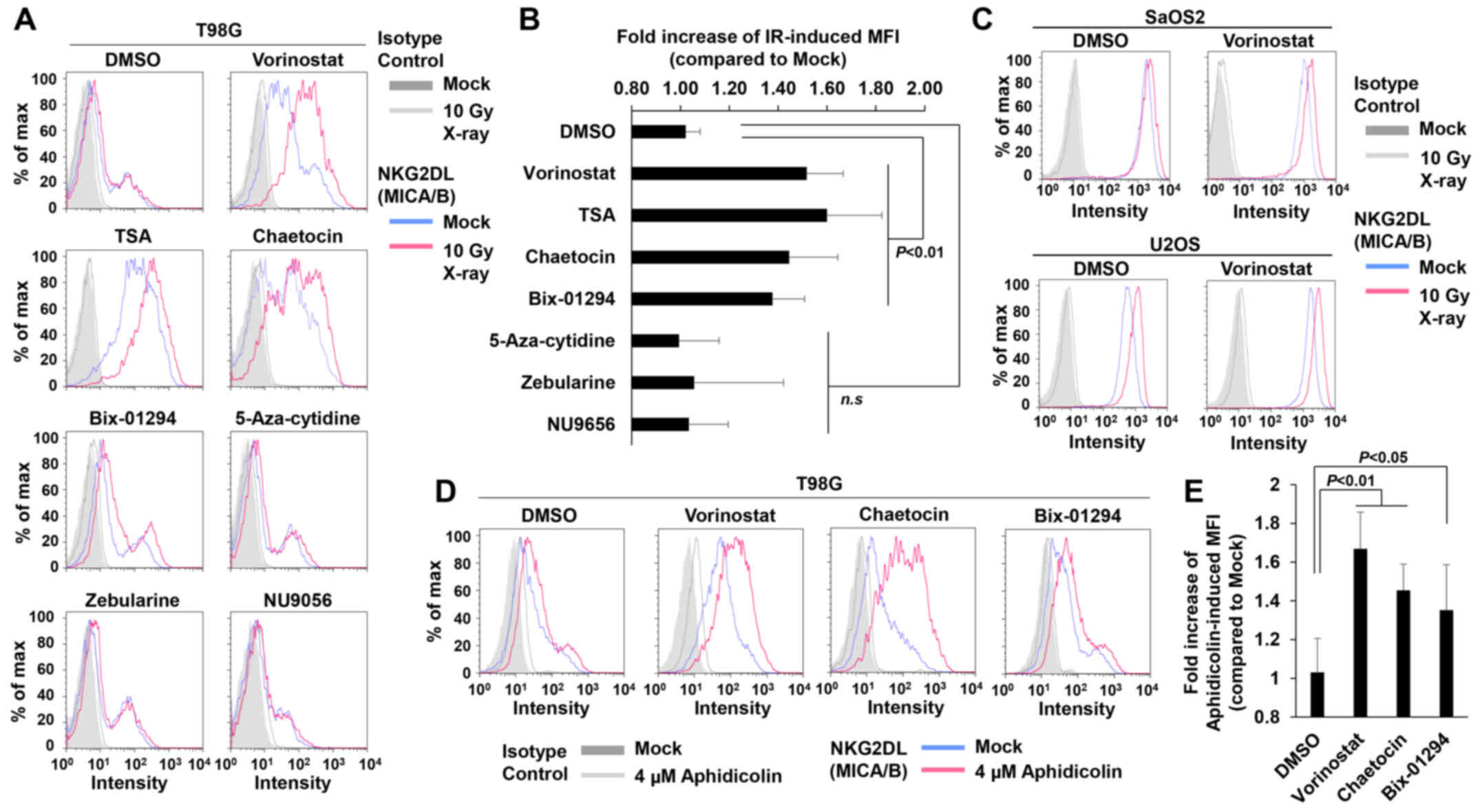
To test this idea, we conducted a screening for compounds that
restored MICA/B expression by targeting chromatin remodeling
factors in insensitive T98G cells (Fig.
4A and Table I). The results
revealed that vorinostat, an HDACi, substantially restored
IR-dependent MICA/B induction in insensitive T98G (Fig. 4A and B) and U251 cells (data not
shown), although the drug exerted little effect on other
insensitive A549 and MCF7 cell lines (data not shown). In contrast,
in sensitive U2OS and SaOS2 cells, IR also increased MICA/B
expression even further in the presence of vorinostat (Fig. 4C). Consistent with previous
findings, vorinostat alone increased MICA/B expression in
insensitive as well as sensitive cells (Fig. 4 and Table II) (30–32).
Similarly to vorinostat, trichostatin A (TSA; HDAC inhibitor) and
the histone methyltransferase inhibitors; chaetocin (Suv39
inhibitor) and Bix-01294 (G9a inhibitor), which also relieve
chromatin compaction, increased MICA/B expression on their own, and
more importantly, treatment with these inhibitors restored
IR-dependent MICA/B induction in insensitive cells (Fig. 4A and B, and Tables I and II). By contrast, neither DNA
methyltransferase nor histone acetyltransferase (HAT) inhibitors
affected MICA/B expression (Fig. 4A and
B, and Tables I and II). Although the effects of these
inhibitors may be influenced by the conditions of the drug
treatment, e.g., concentration or duration, our screening analysis
revealed that inhibition of HDAC/Suv39/G9a was able to restore and
enhance DNA damage-induced MICA/B expression. To further
consolidate the notion that HDAC/Suv39/G9a inhibition restores
MICA/B expression in insensitive cells, we analyzed the MICA/B
surface expression +/− HDAC, Suv39 or G9a inhibitor after
aphidicolin treatment (Fig. 4D and
E). Similar to the result after IR, aphidicolin-induced MICA/B
expression was restored by HDAC/Suv39/G9a inhibition.
| Table II.MFI in T98G, U2OS and SaOS2 cells in
the presence of inhibitors +/− X-ray. |
Table II.
MFI in T98G, U2OS and SaOS2 cells in
the presence of inhibitors +/− X-ray.
| Cell line | Reagent | MFI (−IR) | MFI (10 Gy) | Fold increase of
IR-induced MFI |
|---|
| T98G | DMSO | 3.65 | 3.79 | 1.02 |
|
| Vorinostat | 18.94 | 32.13 | 1.76 |
|
| TSA | 67.66 | 108.0 | 1.60 |
|
| Chaetocin | 8.59 | 13.13 | 1.44 |
|
| Bix-01294 | 4.37 | 5.72 | 1.38 |
|
| 5-Aza-cytidine | 4.76 | 4.54 | 0.99 |
|
| Zebularine | 5.48 | 5.94 | 1.06 |
|
| NU9056 | 4.92 | 4.95 | 1.03 |
| U2OS | DMSO | 84.70 | 105.50 | 1.25 |
|
| Vorinostat | 190.89 | 251.0 | 1.31 |
| SaOS2 | DMSO | 168.91 | 207.61 | 1.23 |
|
| Vorinostat | 389.26 | 493.79 | 1.27 |
Restoration of MICA/B expression by
HDACi is dependent on ATR and E2F1, a transcription factor that
responds to DNA damage
DNA damage-dependent NKG2DL activation requires
ATM/ATR (7). Therefore, we
investigated whether the restoration of MICA/B expression by HDAC
inhibition is dependent on these kinases. As expected, treatment
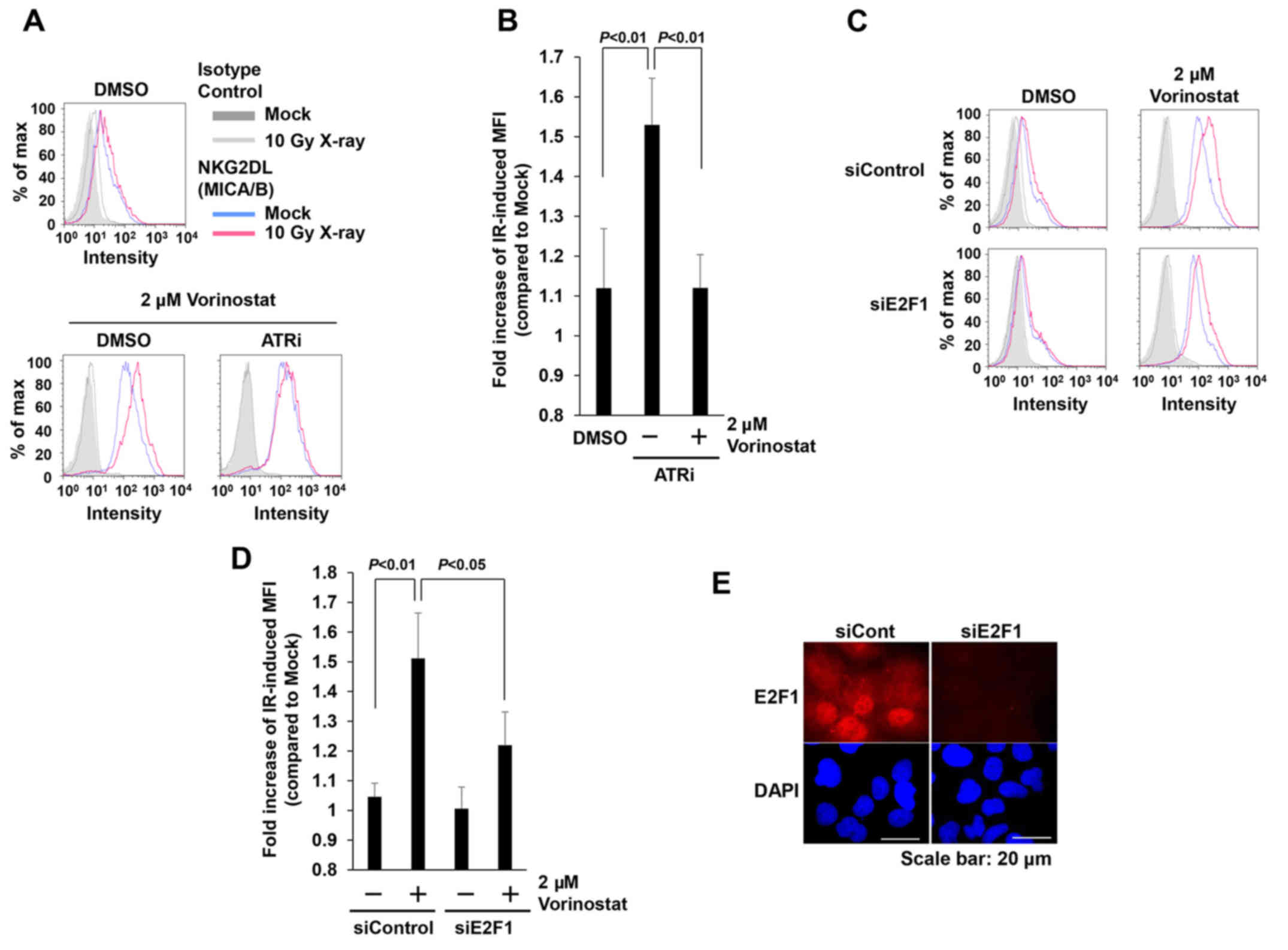
with the ATR inhibitor (ATRi) decreased IR-dependent MICA/B
expression restored by HDACi (Fig. 5A
and B). In mice, NKG2DL expression is controlled by the
transcription factor E2F (33). To
assess the involvement of E2F1 in the restoration of IR-induced
MICA/B expression by vorinostat, we evaluated MICA/B expression
following depletion of E2F1 by siRNA. Depletion of E2F1 in
insensitive cells decreased IR-dependent MICA/B expression in the
presence of HDACi (Fig. 5C-E).
Thus, our results indicate that restoration of MICA/B expression by
vorinostat is mediated by ATR-dependent DNA damage signaling, and
that transcription is regulated by the E2F1 pathway.
Low-dose HDACi restores DNA
damage-dependent MICA/B expression
MICA/B expression in cancer cells is increased
following treatment with HDACi alone (30–32).
Inhibition of HDAC alters global chromatin structure, resulting in
dysregulation of gene expression (34). Such dynamic genome alterations
sometimes cause severe side-effects in normal cells. Therefore,
high concentrations of HDACi should be avoided in order to decrease
side-effects in normal tissues (35). One advantage of radiotherapy is that
it can directly target tumors; consequently, identification of the
minimal dose of HDACi that can restore/enhance MICA/B expression
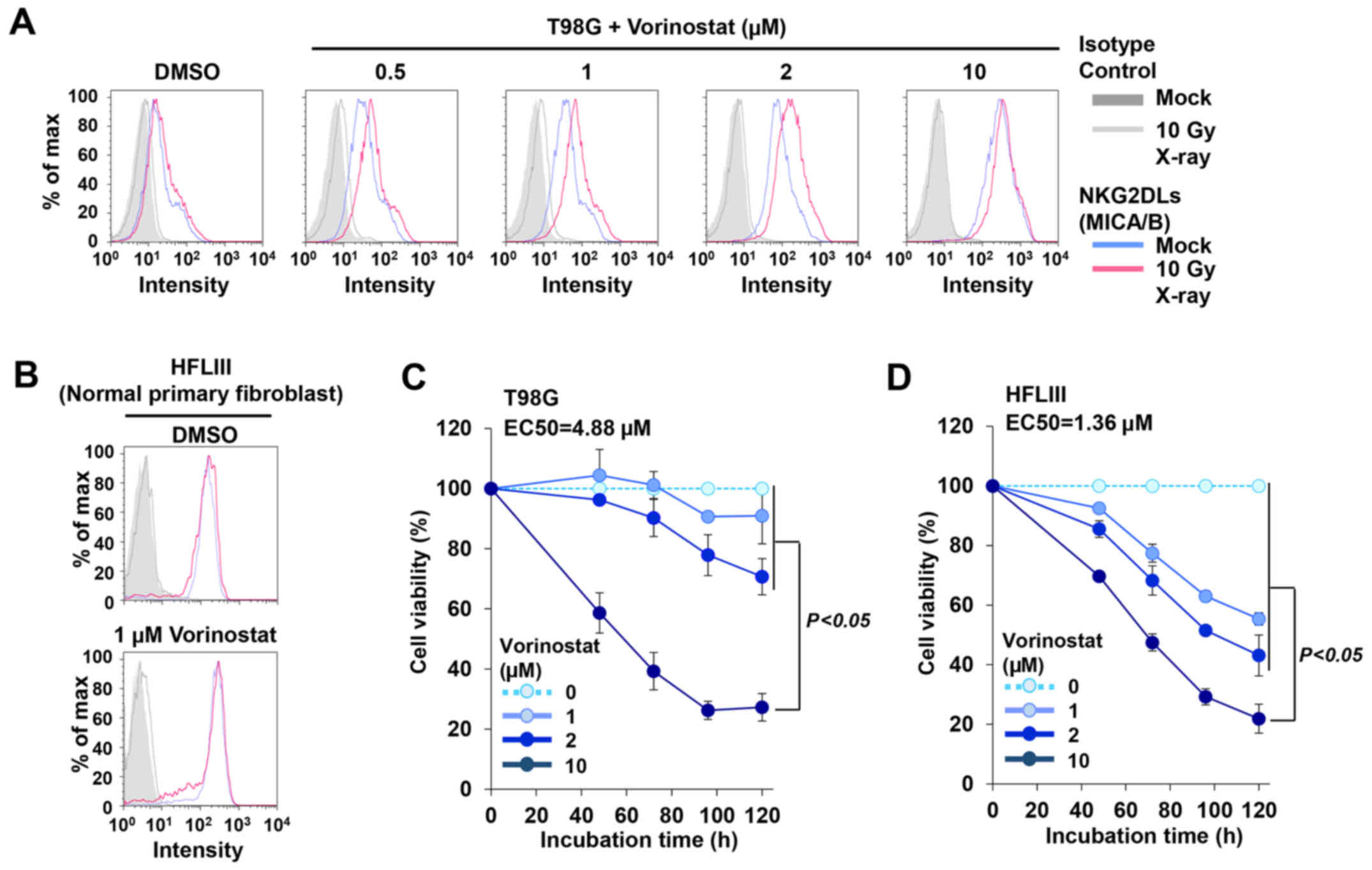
following IR may facilitate clinical applications (36). Thus, we titrated vorinostat against
insensitive cells to determine the lowest concentration of HDACi
that can restore IR-induced MICA/B expression. We selected
vorinostat for the titration analysis since treatment with TSA,
chaetocin and Bix-01294 caused severe cell toxicity compared with
vorinostat (data not shown). In addition, vorinostat has been used
in clinical practice (37). Similar
to the data in Fig. 4A and B,
vorinostat alone increased MICA/B expression in insensitive cells
in a dose-dependent manner (Table
III). Notably, the titration analysis revealed that 0.5–2 µM
vorinostat restored DNA damage-dependent MICA/B expression in
insensitive T98G cells (Fig. 6A and
Table III). IR-dependent
enhancement was not observed in the presence of 10 µM vorinostat,
possibly due to saturation of transcriptional activity (Fig. 6A and Table III). In contrast, 1 µM vorinostat
did not induce MICA/B expression in normal human fibroblast cells
after IR (Fig. 6B). Next, we
investigated whether treatment with the lower concentration of
vorinostat causes toxicity in cancer cells or normal fibroblast
cells. A concentration of 10 µM vorinostat markedly curtailed cell
growth in both cancer and normal cells (Fig. 6C and D). Although 1–2 µM vorinostat
decreased the growth rate of normal fibroblast cells by ~50%, we
confirmed that the growth inhibition was significantly lower than
that induced by the 10-µM vorinostat treatment (Fig. 6D).
| Table III.MFI in T98G cells using
vorinostat. |
Table III.
MFI in T98G cells using
vorinostat.
| Cell line | Reagent | Dose (µM) | MFI (−IR) | MFI (10 Gy) | Fold increase of
IR-induced MFI |
|---|
| T98G | Vorinostat | 0 | 3.88 | 4.38 | 1.13 |
|
|
| 0.5 | 7.76 | 9.61 | 1.24 |
|
|
| 1.0 | 10.56 | 13.63 | 1.29 |
|
|
| 2.0 | 19.93 | 32.45 | 1.63 |
|
|
| 10.0 | 50.45 | 61.39 | 1.22 |
HDACi restores IR-induced NK-mediated
cytotoxicity in insensitive cells
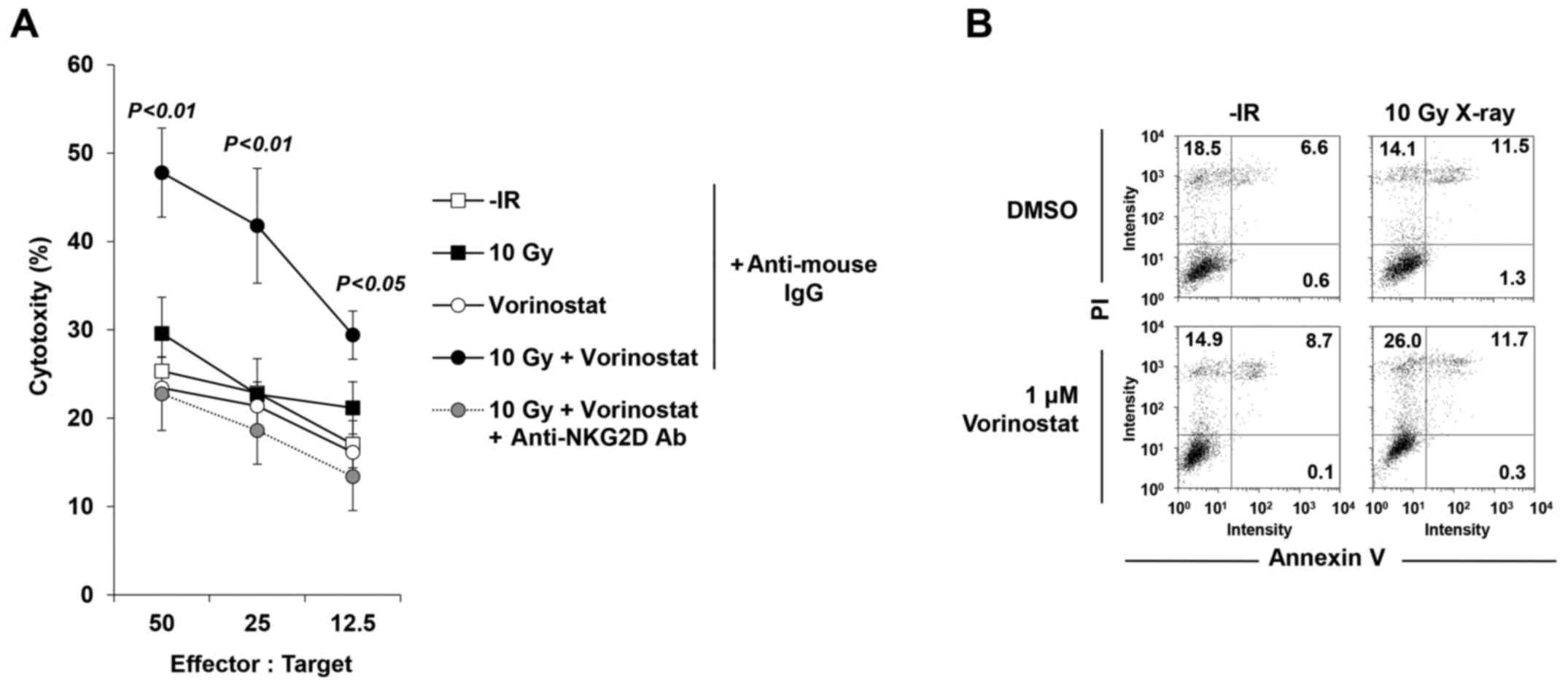
Next, to confirm whether the restored MICA/B
expression affects NK cell-mediated cytotoxicity, an NK cell
cytotoxicity assay was performed with/without vorinostat in
insensitive T98G cells. Treatment with vorinostat substantially
restored the activity of T98G lysis, suggesting the restoration of
T98G cell susceptibility to NK-92-mediated killing (Fig. 7A). Notably, the increase in
cytotoxicity was critically dependent on the NKG2D-MICA/B
interaction, since it was completely abolished by addition of a
blocking anti-NKG2D Ab (Fig. 7A),
whereas addition of an isotype control mAb did not significantly
affect NK-92-mediated lysis (Fig.
7A). We examined levels of PI and Annexin V to confirm that 10
Gy +/− 1 µM vorinostat in the absence of NK-92 cells did not cause
cell death at 24 h after IR, suggesting that neither IR nor HDACi
(or both) affected cell viability in this time range of analysis
(Fig. 7B).
Collectively, these data strongly demonstrate that
DNA damage-dependent MICA/B expression and NK cell cytotoxicity in
insensitive cancer cells can be restored by inhibition of the HDAC
pathway.
Discussion
NKG2DL is constitutively expressed in various tumor
cells (19,21). In addition, ATM/ATR signaling
contributes to NKG2DL expression induced by DNA-damaging agents
both in vivo and in vitro (7). In the present study, we investigated
whether cancer cell lines showed distinct responsiveness of MICA/B
expression after DNA damage. Our data provide the first
demonstration that there is considerable variation in MICA/B
expression among cancer cells in response to DNA damage. Notably,
neither the complexity of IR-induced damage nor the type of DNA
damage influenced the restoration of DNA damage-dependent MICA/B
expression in insensitive cancer cells. This observation supports
the important notion that stimulation by DNA damage alone cannot
effectively overcome the suppressive phenotype in insensitive
cancer cells.
Our drug screening analysis demonstrated that
histone H3K9 modification is a key process involved in the
restoration and enhancement of MICA/B expression, even in the
absence of DNA damage. HDACs deacetylate multiple lysine residues
of histones, resulting in chromatin compaction (38). Therefore, inhibition of HDAC
activity leads to chromatin relaxation. HDAC inhibition also
affects the responsiveness of gene expression. Since gene silencing
is caused by the chromatin condensation in the promoter region,
forced genome-wide relaxation by HDACi treatment can restore gene
expression even when the DNA at the promoter region is highly
methylated (28,39). Similar to the role of HDAC,
Suv39/G9a, a methyltransferase of histone H3K9, promotes chromatin
compaction. HDACs and Suv39/G9a function in the same axis, and the
balance of their activities controls chromatin structure. In the
drug screening analysis, we found that inhibition of Suv39/G9a
activity also enhanced MICA/B expression. In the present study, we
revealed that inhibition of HDAC, Suv39 or G9a activity increased
MICA/B expression in both insensitive and sensitive cell lines in
the absence of DNA damage (30–32).
Recently, Baragaño Raneros et al demonstrated that MICA/B is
highly methylated in several acute myeloid leukemia cell lines
(28). From these observations, we
proposed the idea that MICA/B is frequently downregulated by gene
silencing as a consequence of tumor development, however, by the
inhibition of HDAC/Suv39/G9a activity, MICA/B gene expression may
be restored by the reactivation of MICA/B transcription at the
relaxed promoter region.
In the present study, we found that low-dose HDACi
sufficiently restored DNA damage-dependent MICA/B expression, which
was dependent on DNA damage signaling via the ATR pathway. ATR
activates Chk1, which transduces downstream signals to control gene
expression in response to DNA damage. In addition, we found that
IR-induced MICA/B expression requires E2F1. Collectively, these
data reveal that the ATR/Chk1 signal promotes E2F1-dependent
transcriptional activity, which is required for MICA/B expression.
However, future studies may be required to determine the mechanism
of the signaling cascade from ATR/Chk1 to E2F1. Notably, DNA damage
alone did not induce MICA/B expression in insensitive cells;
rather, it was restored in the presence of HDACi. Moreover, we
wondered whether the restoration of MICA/B expression is dependent
on IR or HDACi when it is combined. Although the current data may
not fully answer the question, we believe that the results suggest
that the MICA/B gene reacquires IR-responsiveness when MICA/B gene
silencing is restored by vorinostat. In addition, we found that the
surface expression of MICA/B following DNA damage was not restored
by HDAC inhibition in H2228, A549 or MCF7 cell lines (data not
shown). These data may suggest that the non-responsiveness of
MICA/B expression in these cell lines is caused by other
mechanisms, e.g., mutations in the MICA/B gene or dysfunction of
the MICA/B protein; this issue may be addressed in a future study.
Previous studies demonstrated that HDAC inhibition prevents DSB
repair (40). Therefore, we
assessed whether inhibition of NHEJ affects the expression of
MICA/B in cancer cells, however, we did not see an obvious change
in expression (data not shown). Further studies are required to
assess precisely how DNA repair influences MICA/B expression
following DNA damage.
Vorinostat and panobinostat are approved in the US
for the treatment of cutaneous T cell lymphoma and multiple
myeloma, respectively. Thus, single-agent administration of an
HDACi has already been shown as useful cancer treatment (39). Conversely, the biggest advantage of
radiotherapy is the ability to target the tumor without systemic
side-effects. However, despite the improvement of therapeutic
efficacy due to development of novel radiotherapy technologies,
metastases can occur, since it is technically unfeasible to
irradiate all the disseminated micro-metastatic cancer cells. It is
therefore important to find the best cohort for radiotherapy. The
therapeutic benefit of radiotherapy in combination with HDACi has
already been shown in vivo and in vitro (41,42).
Our findings support the idea that HDACi may be a good cohort for
radiotherapy, since the combination therapy may treat not only a
gross tumor, but also micro-metastatic cancer cells by immune
response. Alternatively, upregulation of MICA/B can be a productive
marker for the efficacy of therapeutic agents. For instance, by
combining HDACi treatment with radioimmunoconjugate therapy,
another potential application in preclinical and clinical settings
(43), the exposed ligands may be
used as a target for elimination of cancer cells.
In summary, this is the first study revealing the
wide variation in MICA/B surface expression in cancer cell lines
following DNA damage. Notably, inhibition of HDAC activity was able
to restore DNA damage-dependent MICA/B expression in insensitive
cells. Our analysis revealed that the restored MICA/B expression
was mediated by ATR and E2F1 signaling. Previous studies revealed
that HDACi treatment exerts its anticancer effects via multiple
mechanisms, including prevention of DNA repair and promotion of the
adaptive immune response (44,45).
In addition to these effects, our data demonstrate that HDACs may
contribute to MICA/B (NKG2D)-mediated antitumor effects after IR.
Thus, combination therapy using HDACi and radiotherapy or DNA
damage-inducing chemotherapy represent valid and feasible
approaches to cancer therapy.
Acknowledgements
We would like to thank Michie Akaishizawa, Yoshimi
Omi and Shiho Nakanishi for their technical and administrative
assistance in the present study. The present study was supported by
the Astellas Foundation for Research on Metabolic Disorders, the
Terumo Life Science Foundation, and the JSPS KAKENHI (grant no.
26701005). The present study was carried out as part of the
Research Project with Heavy Ions at NIRS-HIMAC and GHMC. We would
like to thank the NIRS-HIMAC and GHMC engineering staff for
providing support for our heavy ion experiments.
References
|
1
|
Navarro Gras A, Björklund AT and Chekenya
M: Therapeutic potential and challenges of natural killer cells in
treatment of solid tumors. Front Immunol. 6:2022015.PubMed/NCBI
|
|
2
|
Robert C, Long GV, Brady B, Dutriaux C,
Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C,
Kalinka-Warzocha E, et al: Nivolumab in previously untreated
melanoma without BRAF mutation. N Engl J Med. 372:320–330.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brahmer J, Reckamp KL, Baas P, Crinò L,
Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE,
Holgado E, et al: Nivolumab versus docetaxel in advanced
squamous-cell non-small-cell lung cancer. N Engl J Med.
373:123–135. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bauer S, Groh V, Wu J, Steinle A, Phillips
JH, Lanier LL and Spies T: Activation of NK cells and T cells by
NKG2D, a receptor for stress-inducible MICA. Science. 285:727–729.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Raulet DH and Guerra N: Oncogenic stress
sensed by the immune system: Role of natural killer cell receptors.
Nat Rev Immunol. 9:568–580. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lanier LL: NKG2D receptor and its ligands
in host defense. Cancer Immunol Res. 3:575–582. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gasser S, Orsulic S, Brown EJ and Raulet
DH: The DNA damage pathway regulates innate immune system ligands
of the NKG2D receptor. Nature. 436:1186–1190. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fine JH, Chen P, Mesci A, Allan DS, Gasser
S, Raulet DH and Carlyle JR: Chemotherapy-induced genotoxic stress
promotes sensitivity to natural killer cell cytotoxicity by
enabling missing-self recognition. Cancer Res. 70:7102–7113. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Soriani A, Zingoni A, Cerboni C, Iannitto
ML, Ricciardi MR, Di Gialleonardo V, Cippitelli M, Fionda C,
Petrucci MT, Guarini A, et al: ATM-ATR-dependent up-regulation of
DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic
agents results in enhanced NK-cell susceptibility and is associated
with a senescent phenotype. Blood. 113:3503–3511. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shibata A and Jeggo PA: DNA double-strand
break repair in a cellular context. Clin Oncol. 26:243–249. 2014.
View Article : Google Scholar
|
|
11
|
Shiloh Y: ATM and related protein kinases:
Safeguarding genome integrity. Nat Rev Cancer. 3:155–168. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rhind N: Changing of the guard: How ATM
hands off DNA double-strand break signaling to ATR. Mol Cell.
33:672–674. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shinkai Y and Tachibana M: H3K9
methyltransferase G9a and the related molecule GLP. Genes Dev.
25:781–788. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Melcher M, Schmid M, Aagaard L, Selenko P,
Laible G and Jenuwein T: Structure-function analysis of SUV39H1
reveals a dominant role in heterochromatin organization, chromosome
segregation, and mitotic progression. Mol Cell Biol. 20:3728–3741.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tachibana M, Sugimoto K, Fukushima T and
Shinkai Y: Set domain-containing protein, G9a, is a novel
lysine-preferring mammalian histone methyltransferase with
hyperactivity and specific selectivity to lysines 9 and 27 of
histone H3. J Biol Chem. 276:25309–25317. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Clayton A, Mitchell JP, Court J, Linnane
S, Mason MD and Tabi Z: Human tumor-derived exosomes down-modulate
NKG2D expression. J Immunol. 180:7249–7258. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shibata A, Moiani D, Arvai AS, Perry J,
Harding SM, Genois MM, Maity R, van Rossum-Fikkert S, Kertokalio A,
Romoli F, et al: DNA double-strand break repair pathway choice is
directed by distinct MRE11 nuclease activities. Mol Cell. 53:7–18.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nakajima NI, Hagiwara Y, Oike T, Okayasu
R, Murakami T, Nakano T and Shibata A: Pre-exposure to ionizing
radiation stimulates DNA double strand break end resection,
promoting the use of homologous recombination repair. PLoS One.
10:e01225822015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Groh V, Rhinehart R, Secrist H, Bauer S,
Grabstein KH and Spies T: Broad tumor-associated expression and
recognition by tumor-derived gamma delta T cells of MICA and MICB.
Proc Natl Acad Sci USA. 96:6879–6884. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Diefenbach A and Raulet DH: Strategies for
target cell recognition by natural killer cells. Immunol Rev.
181:170–184. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pende D, Rivera P, Marcenaro S, Chang CC,
Biassoni R, Conte R, Kubin M, Cosman D, Ferrone S, Moretta L, et
al: Major histocompatibility complex class I-related chain A and
UL16-binding protein expression on tumor cell lines of different
histotypes: Analysis of tumor susceptibility to NKG2D-dependent
natural killer cell cytotoxicity. Cancer Res. 62:6178–6186.
2002.PubMed/NCBI
|
|
22
|
Deckbar D, Jeggo PA and Löbrich M:
Understanding the limitations of radiation-induced cell cycle
checkpoints. Crit Rev Biochem Mol Biol. 46:271–283. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shibata A, Conrad S, Birraux J, Geuting V,
Barton O, Ismail A, Kakarougkas A, Meek K, Taucher-Scholz G,
Löbrich M, et al: Factors determining DNA double-strand break
repair pathway choice in G2 phase. EMBO J. 30:1079–1092. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schmid TE, Dollinger G, Beisker W, Hable
V, Greubel C, Auer S, Mittag A, Tarnok A, Friedl AA, Molls M, et
al: Differences in the kinetics of gamma-H2AX fluorescence decay
after exposure to low and high LET radiation. Int J Radiat Biol.
86:682–691. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jazayeri A, Falck J, Lukas C, Bartek J,
Smith GC, Lukas J and Jackson SP: ATM- and cell cycle-dependent
regulation of ATR in response to DNA double-strand breaks. Nat Cell
Biol. 8:37–45. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rothkamm K, Krüger I, Thompson LH and
Löbrich M: Pathways of DNA double-strand break repair during the
mammalian cell cycle. Mol Cell Biol. 23:5706–5715. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kato N, Tanaka J, Sugita J, Toubai T,
Miura Y, Ibata M, Syono Y, Ota S, Kondo T, Asaka M, et al:
Regulation of the expression of MHC class I-related chain A, B
(MICA, MICB) via chromatin remodeling and its impact on the
susceptibility of leukemic cells to the cytotoxicity of
NKG2D-expressing cells. Leukemia. 21:2103–2108. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Raneros Baragaño A, Martín-Palanco V,
Fernandez AF, Rodriguez RM, Fraga MF, Lopez-Larrea C and
Suarez-Alvarez B: Methylation of NKG2D ligands contributes to
immune system evasion in acute myeloid leukemia. Genes Immun.
16:71–82. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Baylin SB: DNA methylation and gene
silencing in cancer. Nat Clin Pract Oncol. 2:(Suppl 1). S4–S11.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Skov S, Pedersen MT, Andresen L, Straten
PT, Woetmann A and Odum N: Cancer cells become susceptible to
natural killer cell killing after exposure to histone deacetylase
inhibitors due to glycogen synthase kinase-3-dependent expression
of MHC class I-related chain A and B. Cancer Res. 65:11136–11145.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Armeanu S, Bitzer M, Lauer UM, Venturelli
S, Pathil A, Krusch M, Kaiser S, Jobst J, Smirnow I, Wagner A, et
al: Natural killer cell-mediated lysis of hepatoma cells via
specific induction of NKG2D ligands by the histone deacetylase
inhibitor sodium valproate. Cancer Res. 65:6321–6329. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Diermayr S, Himmelreich H, Durovic B,
Mathys-Schneeberger A, Siegler U, Langenkamp U, Hofsteenge J,
Gratwohl A, Tichelli A, Paluszewska M, et al: NKG2D ligand
expression in AML increases in response to HDAC inhibitor valproic
acid and contributes to allorecognition by NK-cell lines with
single KIR-HLA class I specificities. Blood. 111:1428–1436. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jung H, Hsiung B, Pestal K, Procyk E and
Raulet DH: RAE-1 ligands for the NKG2D receptor are regulated by
E2F transcription factors, which control cell cycle entry. J Exp
Med. 209:2409–2422. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dokmanovic M, Clarke C and Marks PA:
Histone deacetylase inhibitors: Overview and perspectives. Mol
Cancer Res. 5:981–989. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Su H, Altucci L and You Q: Competitive or
noncompetitive, that's the question: Research toward histone
deacetylase inhibitors. Mol Cancer Ther. 7:1007–1012. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Avallone A, Piccirillo MC, Delrio P,
Pecori B, Di Gennaro E, Aloj L, Tatangelo F, D'Angelo V, Granata C,
Cavalcanti E, et al: Phase 1/2 study of valproic acid and
short-course radiotherapy plus capecitabine as preoperative
treatment in low-moderate risk rectal cancer-V-shoRT-R3 (Valproic
acid - short Radiotherapy - rectum 3rd trial). BMC Cancer.
14:8752014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Krug LM, Kindler HL, Calvert H, Manegold
C, Tsao AS, Fennell D, Öhman R, Plummer R, Eberhardt WE, Fukuoka K,
et al: Vorinostat in patients with advanced malignant pleural
mesothelioma who have progressed on previous chemotherapy
(VANTAGE-014): A phase 3, double-blind, randomised,
placebo-controlled trial. Lancet Oncol. 16:447–456. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gaszner M and Felsenfeld G: Insulators:
Exploiting transcriptional and epigenetic mechanisms. Nat Rev
Genet. 7:703–713. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
West AC and Johnstone RW: New and emerging
HDAC inhibitors for cancer treatment. J Clin Invest. 124:30–39.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Miller KM, Tjeertes JV, Coates J, Legube
G, Polo SE, Britton S and Jackson SP: Human HDAC1 and HDAC2
function in the DNA-damage response to promote DNA nonhomologous
end-joining. Nat Struct Mol Biol. 17:1144–1151. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Blattmann C, Oertel S, Thiemann M, Dittmar
A, Roth E, Kulozik AE, Ehemann V, Weichert W, Huber PE, Stenzinger
A, et al: Histone deacetylase inhibition sensitizes osteosarcoma to
heavy ion radiotherapy. Radiat Oncol. 10:1462015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kim JH, Shin JH and Kim IH: Susceptibility
and radiosensitization of human glioblastoma cells to trichostatin
A, a histone deacetylase inhibitor. Int J Radiat Oncol Biol Phys.
59:1174–1180. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cornelissen B, Kersemans V, Darbar S,
Thompson J, Shah K, Sleeth K, Hill MA and Vallis KA: Imaging DNA
damage in vivo using gammaH2AX-targeted immunoconjugates. Cancer
Res. 71:4539–4549. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Munshi A, Kurland JF, Nishikawa T, Tanaka
T, Hobbs ML, Tucker SL, Ismail S, Stevens C and Meyn RE: Histone
deacetylase inhibitors radiosensitize human melanoma cells by
suppressing DNA repair activity. Clin Cancer Res. 11:4912–4922.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
West AC, Smyth MJ and Johnstone RW: The
anticancer effects of HDAC inhibitors require the immune system.
Oncoimmunology. 3:e274142014. View Article : Google Scholar : PubMed/NCBI
|