Introduction
Hepatocellular carcinoma (HCC) is ranked as the
fifth most common type of cancer worldwide and is considered as the
third most common cause of cancer-related deaths (1). The first-line therapeutic protocol for
HCC patients includes surgical treatment, such as liver resection
and liver transplantation, but only for early-stage patients.
Unfortunately, most HCC patients are often diagnosed at a late
stage at which chemotherapy presents a unique advantage in the
treatment for these patients (2).
Due to the development of drug resistance in the treatment of HCC,
it is necessary to develop novel chemotherapy drugs for the
treatment of HCC.
The molecular mechanism underlying the development
of HCC may be the disruption of a number of genes that function in
different regulatory pathways, producing several molecular variants
of HCC (3). Deregulation of E2F1
transcriptional activity is observed in a variety of cancers,
including HCC, suggestive of the vital role of E2F1 in HCC
pathogenesis (4). E2F1 is a
transcription factor that controls cell fate including apoptosis. A
recent study (5) reported that the
downregulation of the transcription factor E2F1 was a key event
contributing to the efficient induction and execution of ER
stress-mediated cell apoptosis, and indicated that disruption of
endoplasmic reticulum (ER) stress homeostasis, coupled with E2F1
gene expression modulation, may represent a new valuable target for
the development of novel therapeutic strategies against
chemoresistant tumor malignancies. Therefore, compounds that both
negatively regulate the expression of E2F1 and trigger an ER
stress-mediated apoptotic process may exhibit potential antitumor
activity on chemoresistant cancers, such as HCC.
Celastrol, a natural compound extracted from
Tripterygium wilfordii, has demonstrated promising antitumor
activities in various cancer including HCC (6). Nonetheless, the exact mechanisms of
action of celastrol in HCC have not been fully elucidated. It was
reported that celastrol induces apoptosis evoked by ER stress
(7,8). However, the effect of celastrol on
E2F1 expression has never been reported. In the present study, we
aimed to investigate the action of celastrol in HCC, with an
emphasis on E2F1 modulation.
Materials and methods
Chemicals and reagents
Celastrol was purchased from Pie & Pie
Technologies (Shenzhen, China). Proteasome inhibitor MG132 was
obtained from Calbiochem (San Diego, CA, USA), PS-341 was obtained
from Millennium Pharmaceuticals (Cambridge, MA, USA) and epoxomycin
was purchased from the Peptide Institute (Osaka, Japan). Z-VAD-FMK
was obtained from Beyotime Institute of Biotechnology (Shanghai,
China). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT) was purchased from Amresco (Solon, OH, USA). A stock
solution of celastrol was prepared at 50 mM in dimethyl sulfoxide
(DMSO) and stored at −20°C. The following antibodies were used:
PARP, caspase-9 and cleaved caspase-3 (Cell Signaling Technology,
Beverly, MA, USA), E2F1 (Santa Cruz Biotechnology, Santa Cruz, CA,
USA) and actin (Sigma, St. Louis, MO, USA).
Cell culture
The HCC cell lines HepG2 and BEL-7402 were obtained
from the Cell Bank of the Chinese Academy of Sciences (Shanghai,
China). The lung adenocarcinoma cell line A549, large cell lung
cancer line NCI-H460 and the breast cancer cell line MDA-MB231 were
obtained from the American Tissue Culture Collection (ATCC;
Manassas, VA, USA). Cells were maintained in Dulbeccos modified
Eagles medium (DMEM) supplemented with 10% fetal bovine serum (FBS)
in a humidified incubator under 5% CO2 at 37°C as
previously described (9).
Cell morphology
HepG2 cells were grown in culture flasks to 80–90%
confluency. Then, the cells were treated with the indicated
concentrations of celastrol for 18 h. Morphological changes in the
cells were observed using an inverted microscope and
photographed.
Cell viability
Cells were treated with celastrol at the indicated
concentrations and time points. Cell viability was estimated by MTT
assay, as previously described (10). Cells were cultured into a 96-well
plate (1×104/well), and incubated with the indicated
concentrations of celastrol (solvent of DMSO as the control group)
when the cells were 80–90% confluent. After 44 h of treatment with
celastol, 10 µl stock MTT solution was added to the culture medium
(0.5 mg/ml final concentration) for incubation for an additional 4
h. Then, the medium was removed, and 150 µl DMSO was added to
dissolve the solid formazan. The absorbance of each well was read
at 490 nm using a microplate reader (Thermo Fisher Scientific,
Inc., Waltham, MA, USA). The cell viability was calculated as a
percent ratio of the absorbance of the sample cells to the
absorbance of the control cells. Log (inhibitor) vs. response
non-linear fit was used to estimate the IC50 value
(GraphPad Prism 6.0; GraphPad Software, Inc., La Jolla, CA,
USA).
Apoptosis assay
Cell apoptosis was detected using the Annexin V
apoptosis detection kit (BD Biosciences, San Jose, CA, USA)
according to the manufacturer's instructions. Briefly, cells were
treated with celastrol at the indicated concentrations for 18 h,
trypsinized and washed twice with cold phosphate-buffered saline
(PBS). Then, the washed cell pellet was resuspended in 1.0 ml
binding buffer at a concentration of 106 cells/ml and
stained with 10 µl of Annexin V along with 20 µl 7-AAD for 15 min
on ice in the dark. The samples were analyzed by flow cytometry (BD
FACSCallbur) within 1 h.
RNA preparation and RT-PCR
Total RNA was extracted using the TRIzol reagent
(Invitrogen, Carlsbad, CA, USA) and the phenol-chloroform
extraction method according to the manufacturer's protocol. Total
RNA (2 µg) was annealed with Oligo(dT) primers at 65°C for 5 min.
The cDNA was synthesized using a First Strand cDNA Synthesis kit
(Fermentas, Hanover, MD, USA). Primers for E2F1 detection were as
follows: sense primer, 5′-ACCAGGGTTTCCAGAGATGC-3′ and antisense
primer: 5′-CACCACACAGACTCCTTCCC-3′ (11). The reaction mix contained: 2.5 µl
10X Ex Taq buffer, 2 µl dNTP mixture, 200 nM forward and reverse
primers, 100 ng cDNA template, 0.25 µl Takara Ex Taq and
ddH2O up to 25 µl volume. The PCR cycling conditions
consisted of the following: 98°C for 10 sec for denaturation, 57°C
for 30 sec for annealing and 72°C for 30 sec for extension, for a
total of 30 cycles. Products of RT-PCR were separated by 1.5%
agarose gel electrophoresis and detected using a gel imaging
system.
siRNA assays and cell
transfection
E2F1-specific RNA was designed and synthesized by
Shanghai GenePharma Co. (Shanghai, China). Using Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol, HepG2 cells were transfected with 100 nM
double-stranded siRNA oligonucleotides. After 48 h transfection,
cells were treated with celastrol for additional 18 h, followed by
western blotting and MTT assay, as previously decribed (12). The siRNA sequences of E2F1 were as
follows: 5′-GAGGAGUUCAUCAGCCUUU-3′.
Western blot analysis
Cells were lysed in radioimmunoprecipitation assay
lysis buffer [50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.1% sodium
dodecyl sulfate (SDS), 1% Triton X-100, 1% deoxycholate, 1 mM
ethylenediaminetetraacetic acid, 1 mM phenylmethanesulfonyl
fluoride, 1 mM NaF, 1 mM sodium vanadate and protease inhibitors
cocktail (Sigma)] and cleared by centrifugation to obtain
whole-cell lysates. Then, equal amounts of proteins were subjected
to SDS-polyacrylamide gel electrophoresis, and transferred to
nitrocellulose membranes (Pall Corporation, Ann Arbor, MI, USA).
After blocking with 5% skim milk, the membranes were incubated with
primary antibodies at 4°C overnight, and the membrane-bound
antibodies were visualized using goat anti-rabbit or anti-mouse
horseradish peroxidase-conjugated secondary antibodies (1:5,000
dilution; 1–2 h) and a chemiluminescent substrate (Thermo Fisher
Scientific, Rockford, IL, USA). Equal loading of protein was
confirmed by measuring total actin. The quantification of protein
was analyzed using ImageJ software (National Institutes of Health,
Bethesda, MD, USA). The dilution rates of primary antibodies used
were: PARP, caspase-9 and cleaved caspase-3 (1:1,000); E2F1
(1:500); actin (1:100,00).
Statistical analysis
All experiments were repeated at least three times
and the data are presented as the mean ± standard deviation unless
noted otherwise. Differences between data groups were evaluated for
significance using the Students t-test of unpaired data by SPSS
version 17.0 for Windows (SPSS, Inc., Chicago, IL, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
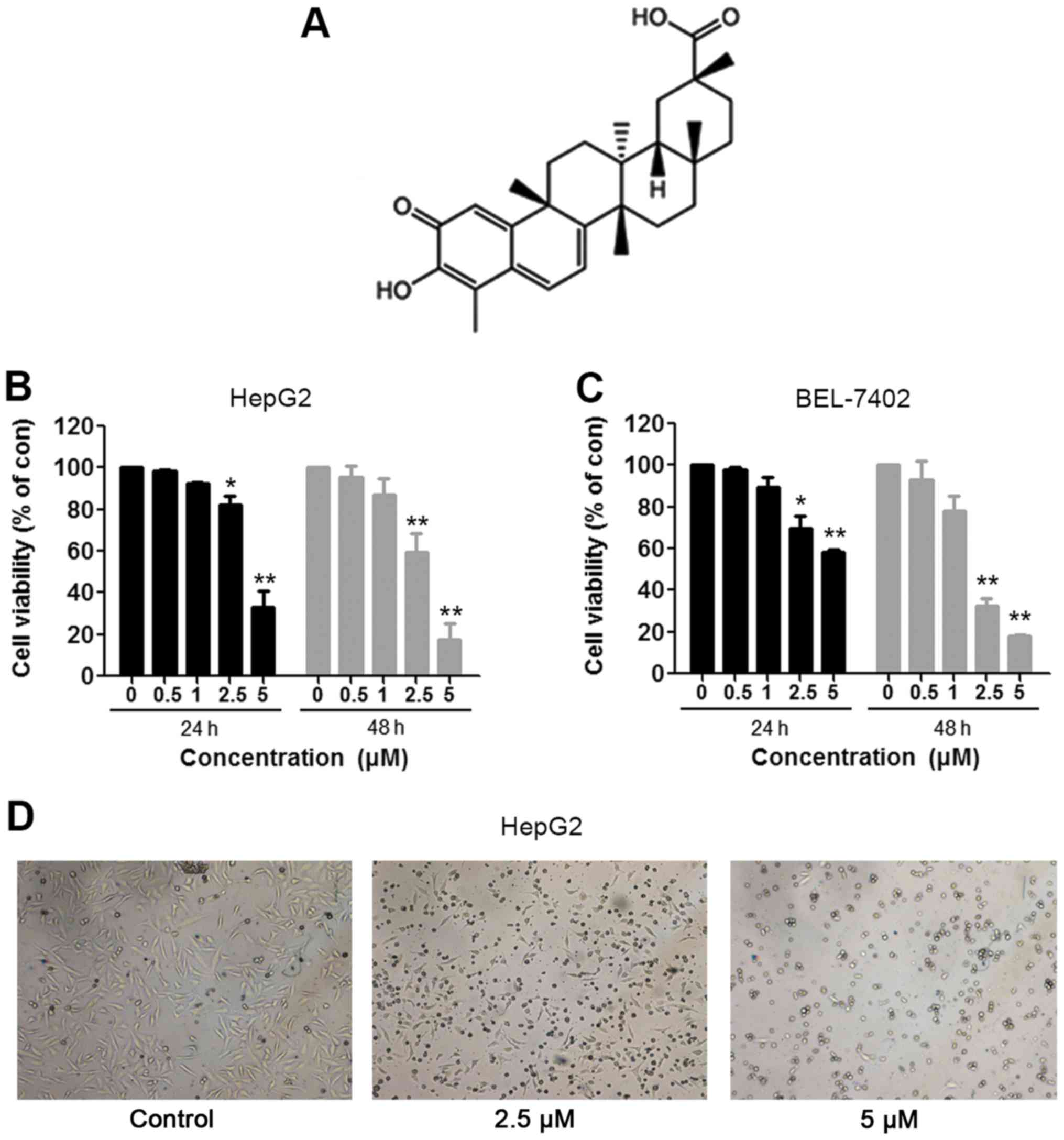
Celastrol inhibits the growth of HCC
cells
To evaluate the effect of celastrol on the growth of
HCC cells, the MTT assay was used. HepG2 and Bel-7402 cells were
treated with the indicated concentrations (0.5, 1, 2.5 and 5 µM) of
celastrol (Fig. 1A) for 24 and 48
h. The assay results showed that compared to the control cells,
HepG2 cells incubated for 24 h exhibited viabilities of 98.5, 92.5,
82.0 and 33.2%, respectively, and the HepG2 cells treated for 48 h
presented viabilities of 95.6, 87.0, 59.4 and 17.4%, respectively
(Fig. 1B). The IC50
values of HepG2 cells for 24 and 48 h of celastrol treatment were
4.016 and 2.766 µM, respectively. In addition, the similar
inhibitory effect also occurred in the BEL-7402 cells. As shown in
Fig. 1C, relative to the control
cells, BEL-7402 cells treated for 24 h exhibited viabilities of
97.9, 89.7, 69.6 and 58.6%, respectively, and the BEL-7402 cells
treated for 48 h presented viabilities of 92.9, 78.0, 32.4 and
18.1%, respectively. The IC50 values of BEL-7402 cells
for 24 and 48 h treatment of celastrol were 6.079 and 1.856 µM,
respectively. In addition, obvious morphological changes were
observed following treatment of celastrol in the HepG2 cells
(Fig. 1D). Our results indicated
that celastrol inhibited the viability of the HCC cells in a dose-
and time-dependent manner.
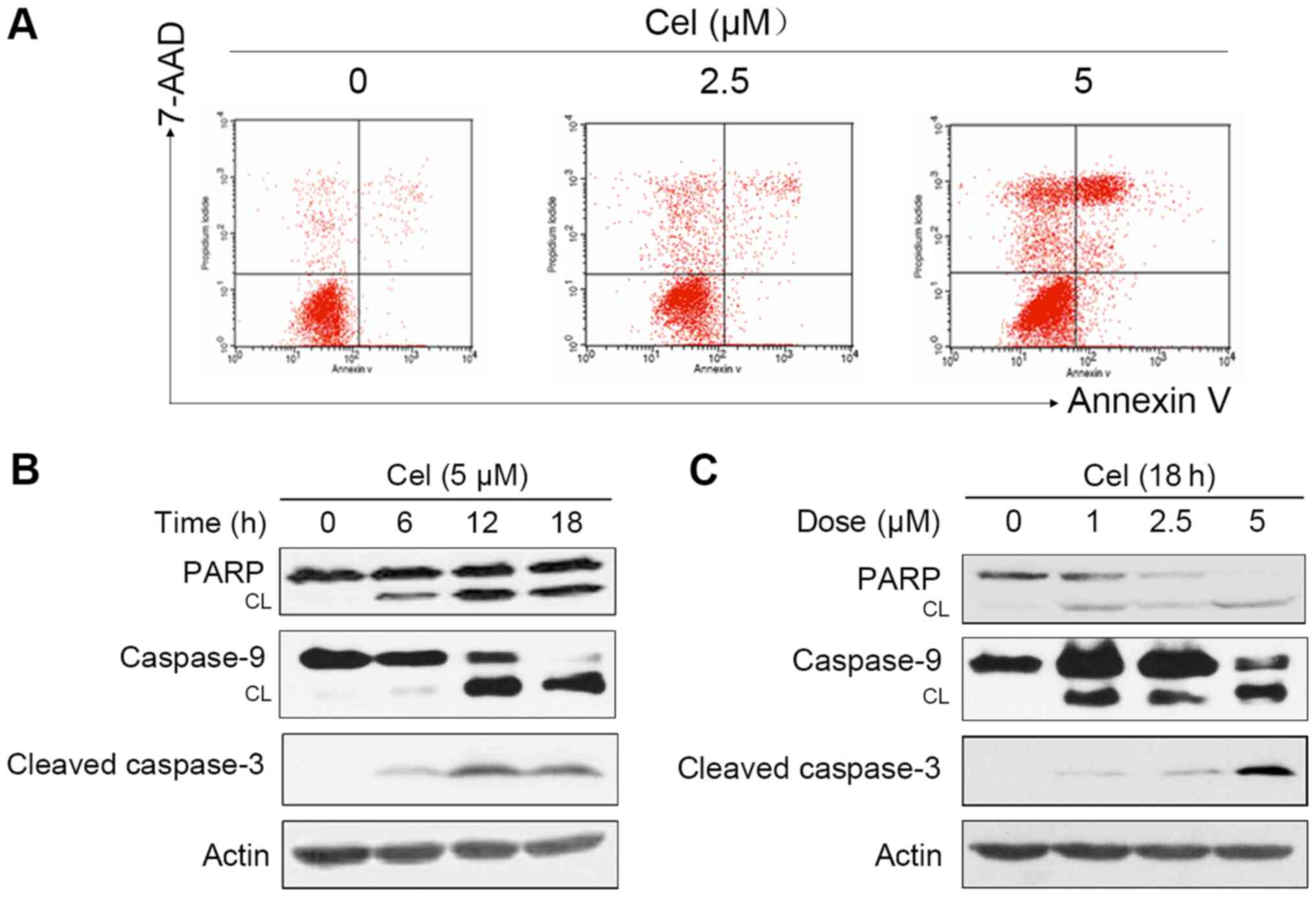
Celastrol induces apoptosis in HepG2
cells
We investigated whether the cytotoxic effect of
celastrol was associated with apoptosis in the HepG2 cells. Annexin
V/7-AAD-staining and flow cytometric assay showed that treatment of
the HepG2 cells with celastrol for 24 h caused apoptosis in a
proportion of the cells (Fig. 2A).
To further dissect the molecular mechanism underlying
celastrol-induced apoptosis, the effects of celastrol on caspase
activation which is a pivotal step in apoptosis were examined.
Western blotting revealed that incubation with increasing
concentrations of celastrol led to a significant cleavage of
caspase-9 and −3 in a dose- and time-dependent manner (Fig. 2B and C). We then followed the status
of nuclear enzyme poly(ADP-ribose) polymerase (PARP) that is one of
the main cleavage targets of caspase-3. PARP cleavage was enhanced
in the celastrol-treated HepG2 cells in a dose- and time-dependent
manner (Fig. 2B and C), which is
commonly regarded as an apoptotic marker (13). Collectively, these results showed
that celastrol caused apoptosis and the activation of the caspase
cascade in HepG2 cells.
Celastrol-induced apoptosis in HepG2
cells is caspase-dependent
To further confirm the activation of caspases as an
essential step in the apoptotic pathway induced by celastrol, a
caspase inhibitor benzyloxycarbony (Cbz)-l-Val-Ala-Asp
(OMe)-fluoromethylketone (Z-VAD-FMK) was administered. HepG2 cells
were pretreated with 20 µM Z-VAD-FMK for 1 h, and then incubated
with 5 µM celastrol for an additional 18 h. Morphological results
showed that Z-VAD-FMK partially rescued celastrol-induced cell
death (Fig. 3A). Western blotting
further revealed that celastrol-triggered PARP cleavage was
suppressed by Z-VAD-FMK (Fig. 3B).
Z-VAD-FMK also partly inhibited celastrol-induced cell cytotoxicity
in the HepG2 cells (Fig. 3C). These
data suggested that celastrol induced apoptosis of the HepG2 cells
in a caspase-dependent fashion.
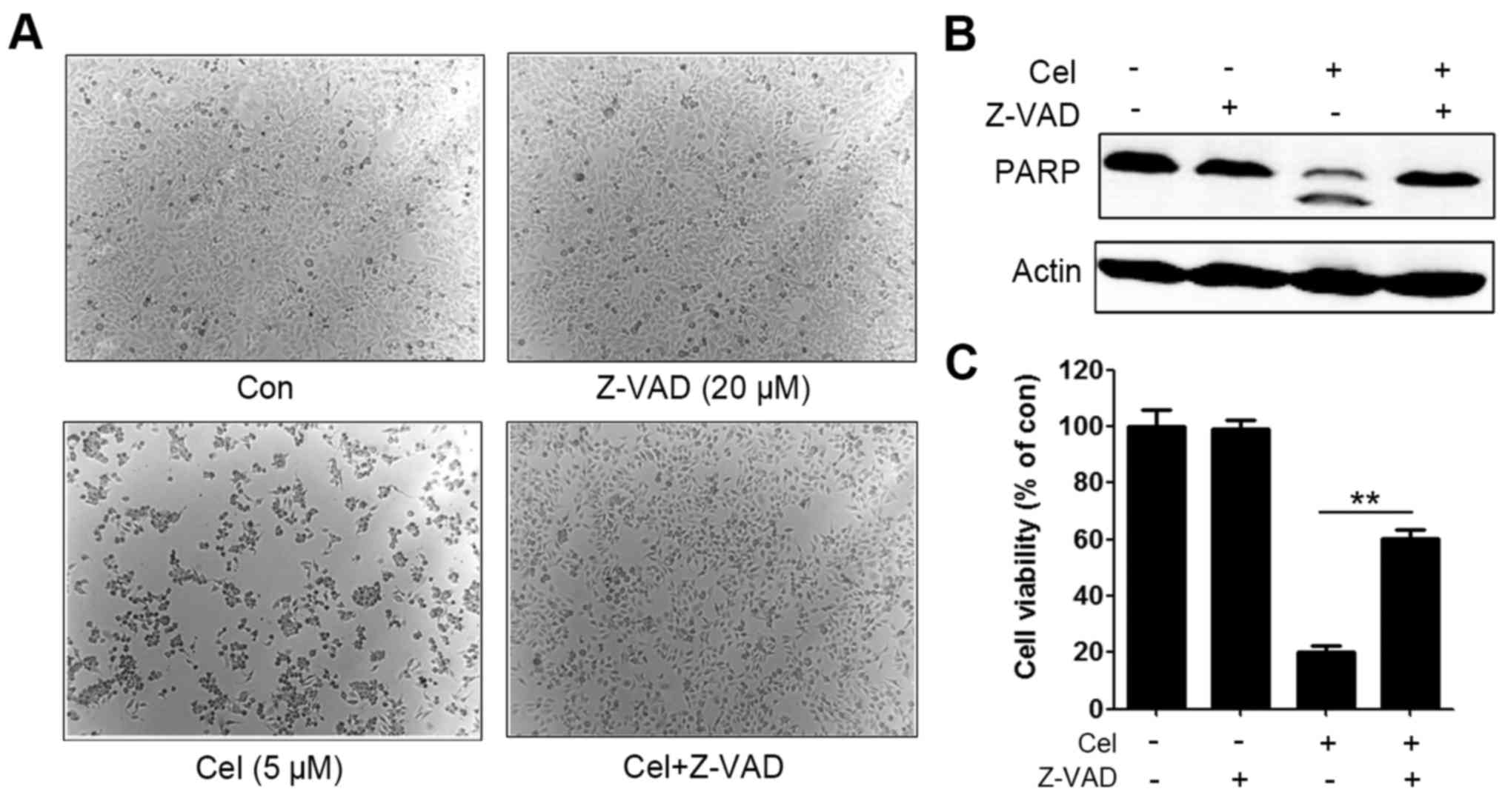 | Figure 3.Caspase activation is involved in
celastrol-induced apoptosis. (A) Representative images of HepG2
cells pretreated with Z-VAD-FMK (20 µM) for 1 h, followed by
treatment with celastrol at 5 µM for 18 h; original magnification,
×40. (B) HepG2 cells were pretreated with Z-VAD-FMK (20 µM) for 1
h, followed by treatment with celastrol at 5 µM for 18 h and lysed,
and the lysates were subjected to western blotting using the
indicated antibodies. (C) HepG2 cells were pretreated with
Z-VAD-FMK (20 µM) for 1 h, followed by treatment with celastrol at
5 µM for 18 h, and cell viability was analyzed by MTT assay,
**P<0.01, celastrol-treated group vs. celastrol and Z-VAD
combination group. Cel, celastrol; Z-VAD,
N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone; PARP, poly(ADP
ribose) polymerase. |
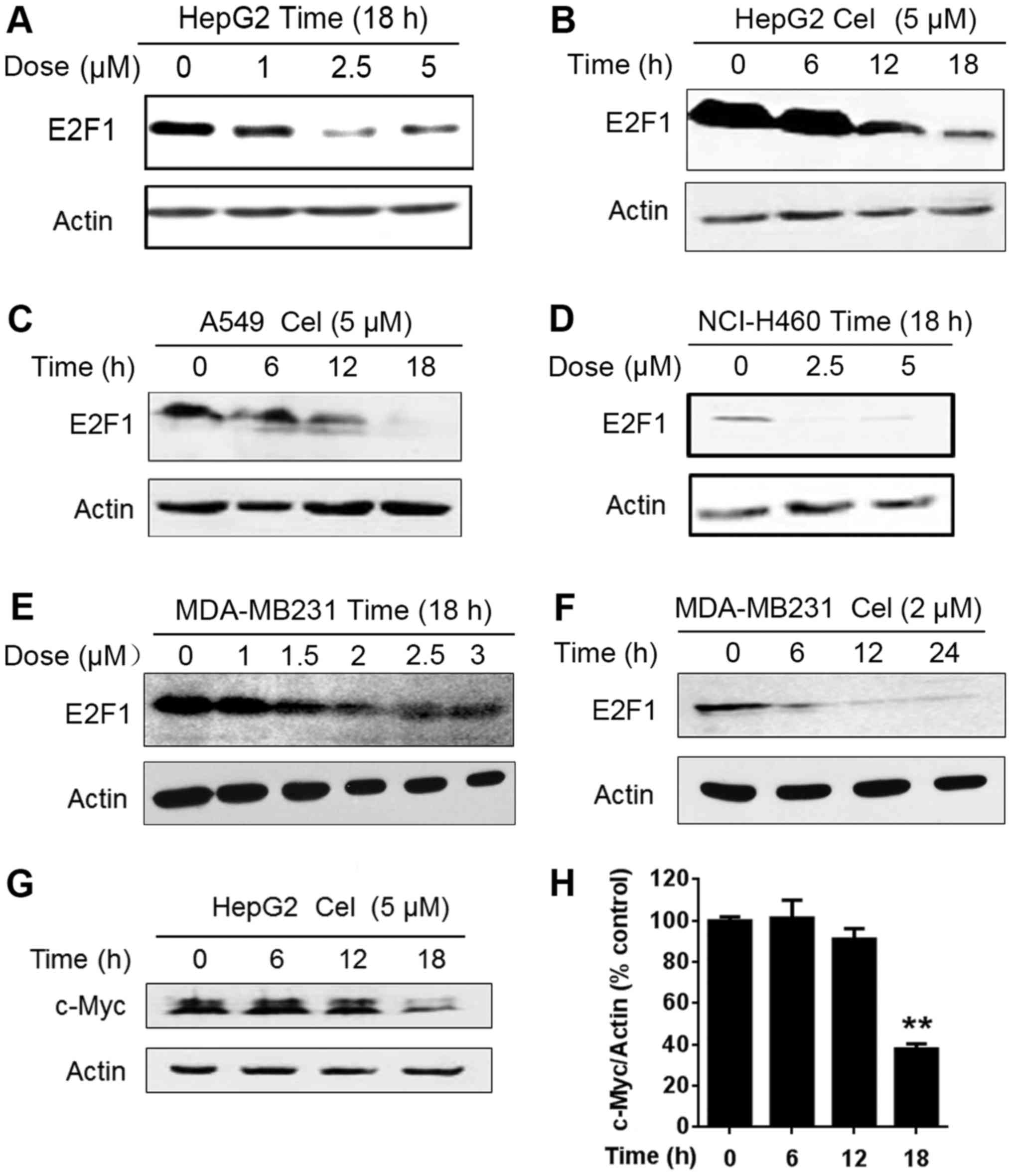
Celastrol downregulates E2F1
expression
E2F1 is reported to be overexpressed in several
types of human tumors (14) and its
inactivation may be a valuable novel potential therapeutic strategy
for cancer treatment (5). This
concept prompted us to detect the effect of celastrol on E2F1
expression. Western blot analysis was performed after the cells
were exposed to different concentrations of celastrol for 18 h. As
shown in Fig. 4A, the expression of
E2F1 protein was reduced in the HepG2 cells exposed to celastrol at
1 µM and was apparently decreased in the cells treated with
celastrol at 2.5 and 5 µM for 18 h. We further showed that
celastrol caused E2F1 downregulation in a time-dependent manner
(Fig. 4B). Apart from HCC cells,
celastrol triggered a decrease in E2F1 in A549 and NCI-H460 lung
cancer cells (Fig. 4C and D) and
MDA-MB231 breast cancer cells (Fig. 4E
and F). It has been reported that the proliferation factor
c-Myc, as an important downstream target of E2F1 (15,16),
is amplified and an indicator of malignant potential and poor
prognosis in HCC, suggestive of a vital role of c-Myc in HCC
pathogenesis (17). Thus, we
further detected the alteration of c-Myc. As shown in Fig. 4G and H, celastrol decreased c-Myc
expression in a time-dependent manner. These results showed that
celastrol induced time-dependent and dose-dependent downregulation
of E2F1 protein, and the decrease was not cell type-specific.
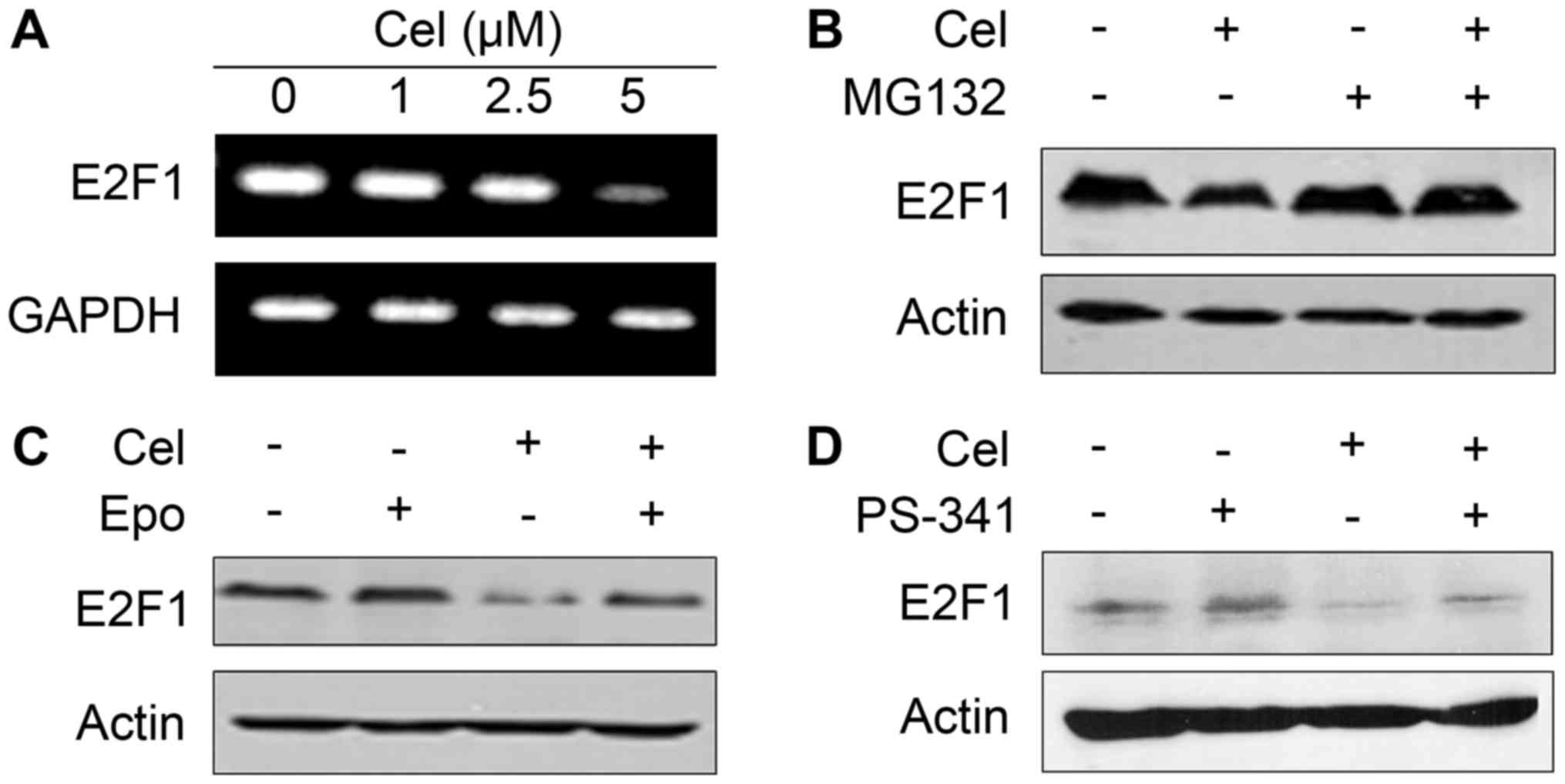
Celastrol decreases E2F1 at both the
mRNA and protein levels
In order to investigate the mechanism underlying the
downregulation of E2F1 protein, we firstly analyzed the expression
of E2F1 at the mRNA level by RT-PCR assay. We found that E2F1 was
reduced apparently in response to celastrol treatment at 5 µM for
18 h (Fig. 5A). Celastrol at 2.5 µM
significantly suppressed E2F1 expression as detected by western
blot assay (Fig. 4A), indicating
that celastrol may also reduce E2F1 at the protein level. The
ubiquitin-proteasome pathway is responsible for the degradation of
most intracellular proteins in eukaryotic cells (18). We then tried to use classic
proteasome inhibitors in vitro to examine the effect of
these inhibitors on the degradation of E2F1 induced by celastrol.
As shown in Fig. 5B-D, while
proteasome inhibitor MG132, epoxomycin and PS-341 (19) did not influence E2F1 stability,
pretreatment of HepG2 cells with these compounds markedly inhibited
celastrol-triggered E2F1 degradation. These data indicated that
celastrol downregulated E2F1 at both the mRNA and protein levels,
and the decrease in E2F1 protein through a ubiquitin-proteasome
pathway played a more important role in celastrol-induced E2F1
downregulation.
E2F1 downregulation is involved in the
celastrol-induced inhibitory effect on HCC cells
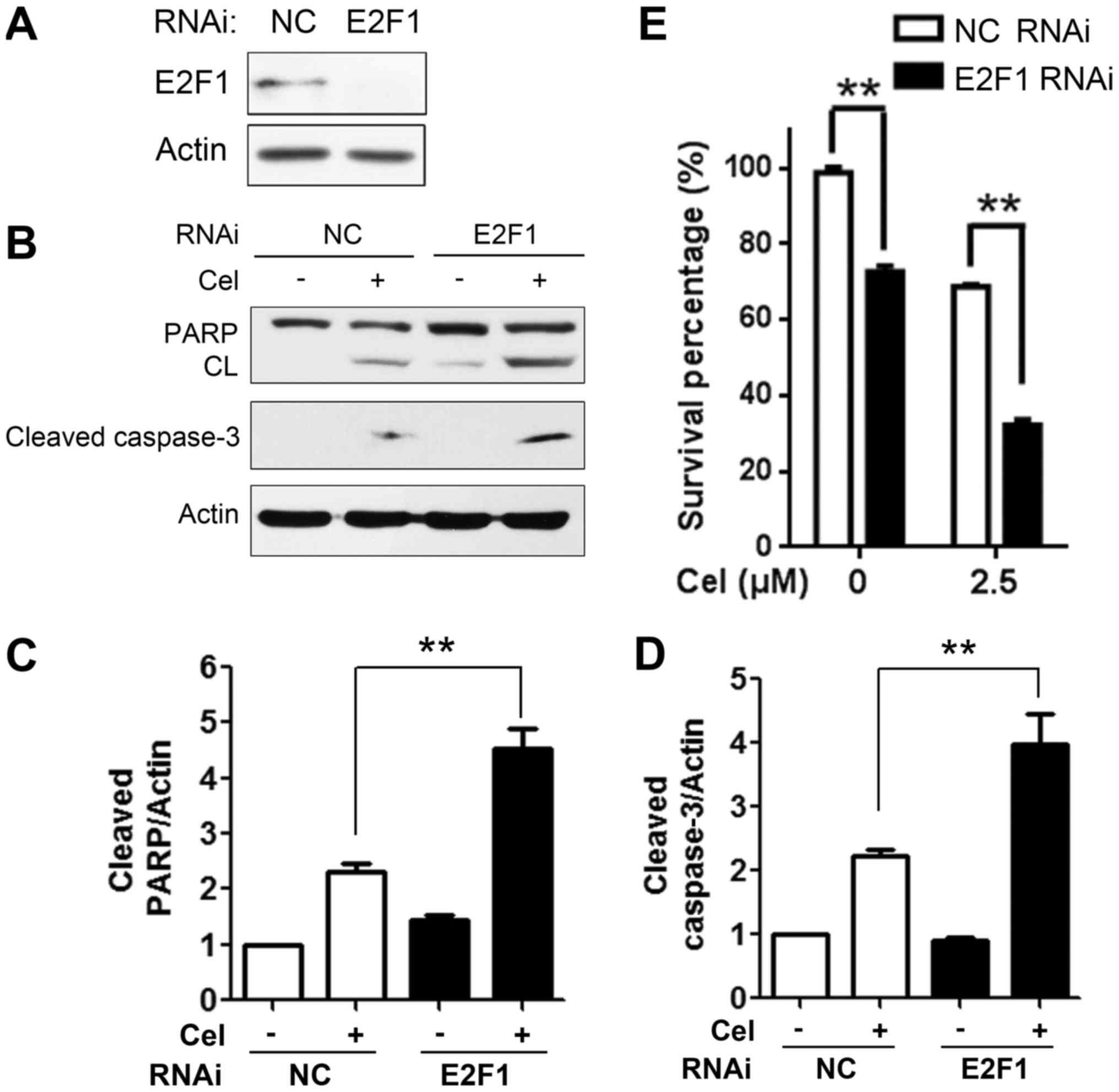
E2F1-specific siRNA (Fig. 6A) was employed to evaluate its role
in the celastrol-mediated inhibitory effects on HepG2 cells. Cells
were transfected with siRNA targeting E2F1 for 48 h, followed by
celastrol treatment for an additional 18 h, and apoptotic proteins
were measured using western blot analysis. As presented in Fig. 6B-D, compared with the NC
siRNA-treated cells, depletion of E2F1 resulted in potentiated
cleavage of PARP and caspase-3 in the HepG2 cells upon celastrol
treatment, suggestive of the enhancement of the apoptotic effect.
In addition, cell proliferation was assessed by MTT assay. The
result showed that knockdown of E2F1 alone partially inhibited the
cell viability of HepG2 cells. Moreover, in responding to celastrol
treatment, E2F1 depletion significantly enhanced celastrol-induced
suppression of cell viability (Fig.
6E; n=3, P<0.01). These results indicated that E2F1 played
an important role in the celastrol-induced antiproliferative and
pro-apoptotic effects on HCC cells.
Discussion
Hepatocellular carcinoma (HCC) is the most common
primary liver malignancy ranking fifth in incidence and third in
mortality worldwide (1,20). Current therapies result in minimal
survival advantage and are linked with drug resistance. Thus, a
great challenge lies in identifying novel agents for HCC treatment.
Natural compounds have been important sources of new drugs.
Numerous successful anticancer drugs currently in use are derived
from nature, and some of their analogues are under clinical trials
(21). Celastrol, the main active
ingredient of Tripterygium wilfordii, reveals a wide array
of antitumor activity against various types of cancers, containing
HCC (22). In HCC C3A cells,
celastrol was found to suppress growth and induce apoptosis through
the modulation of STAT3/JAK2 signaling cascade in vitro and
in vivo (6). In HCC Bel-7402
cells, celastrol triggered apoptosis via activation of the
mitochondria-mediated pathway (23). Celastrol showed synergism combined
with lapatinib in HCC HepG2 cells (24) and with ABT-737 in HepG2 and Bel-7402
cells (7). However, the molecular
mechanism of action of celastrol in HCC therapy has not been well
understood. The present study revealed for the first time that
celastrol exerted its antiproliferative and pro-apoptotic effects
partially mediated through modulation of E2F1 expression in
HCC.
It is well known that inducing apoptosis in tumor
cells is an important mechanism of action for many antitumor drugs.
In the present study, antiproliferative evaluation showed that
celastrol inhibited the growth of HCC HepG2 and Bel-7402 cells in a
time- and dose-dependent manner. Flow cytometry revealed that
apoptosis occurred in HepG2 cells after treatment of celastrol. The
extrinsic and intrinsic apoptotic pathways are two common apoptotic
pathways that lead to activation of initiator caspases (typically
caspase-8 in the extrinsic pathway and caspase-9 in the intrinsic
pathway) and ultimately effector caspases (caspase-3), which in
turn cleaves downstream substrates, such as PARP (25,26).
Accordingly, activation of apoptotic proteins indicative of cell
apoptosis, including apparent cleavage of caspase-9 and −3, and
PARP in a dose- and time-dependent way, was clearly observed by
western blot analysis in HepG2 cells, suggesting that the intrinsic
apoptotic pathway may be activated in celastrol-treated cells. In
addition, we observed a rescue in celastrol-induced cell death in
cells pretreated with a caspase inhibitor. These results indicate
that caspase activation mediates celastrol-induced apoptosis.
The E2F1 transcription factor has been identified as
a tumor-suppressor gene enhancing apoptosis by DNA damage in tumors
lacking p53 (27). However,
findings from transgenic models indicate that increased expression
of E2F-1 occurs in c-myc/TGFα double transgenic mice during
hepatocarcinogenesis and E2F-1 overexpression in the liver causes
dysplasia and tumors (28).
Moreover, abnormalities in E2F1 gene expression and/or E2F1 gene
amplification have been described in various cancer cell lines and
tumor types, including HCC (4,29,30).
Notably, overexpression of E2F1 is frequently associated with
high-grade tumors and poor patient survival prognosis (31). These data indicate that E2F1 has
oncogenic functions in several types of cancer and may represent a
rational therapeutic target. In the present study, we identified a
natural compound celastrol that was capable of inducing a
significant decrease in E2F1 in HCC HepG2 cells, as well as in lung
cancer cells (A549 and NCI-H460) and breast cancer MDA-MB231 cells,
implying that E2F1 suppression by celastrol is not a cell
type-specific event, and celastrol could be a promising lead
compound with potential anticancer activity in other
E2F1-overexpressing human cancers, such as lung (32–34)
and breast cancer (35). The
proliferation factor c-Myc, an important downstream target of E2F1,
was also decreased following celastrol treatment in a
time-dependent manner, which may contribute to the observed
antiproliferative effect of celastrol. Understanding of the
mechanism by which E2F1 undergoes proteolytic breakdown is
important to develop effective strategies for inactivation of E2F1
for HCC therapy. We demonstrated that celastrol not only
downregulated E2F1 mRNA, but also reduced E2F1 protein via a
ubiquitin-proteasome pathway. In addition, we first reported the
role of E2F1 in the celastrol-mediated inhibitory effect as the
siRNA assay revealed that depletion of E2F1 potentiated
celastrol-mediated antiproliferative and pro-apoptotic activity in
HepG2 cells. Pagliarini et al (5) reported that downregulation of E2F1 is
required for ER-stress mediated apoptosis. ER stress was activated
upon celastrol treatment in HCC cells (36), thus, the decrease in E2F1 may have
been mediated by ER stress in celastrol-triggered apoptosis. This
question warrants further investigation.
In conclusion, the present study provides initial
evidence of an effect of E2F1 inhibition on the onset of
celastrol-induced inhibitory activity in HCC cells, and celastrol
may serve as a lead compound for the development of an E2F1
inhibitor.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (nos. 81402511 and
81201577), and the Student Research Training Program of Anhui
University of Technology (nos. 201510360171 and 2015024Z).
References
|
1
|
Attwa MH and El-Etreby SA: Guide for
diagnosis and treatment of hepatocellular carcinoma. World J
Hepatol. 7:1632–1651. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al SHARP Investigators Study Group, : Sorafenib in advanced
hepatocellular carcinoma. N Engl J Med. 359:378–390. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Thorgeirsson SS and Grisham JW: Molecular
pathogenesis of human hepatocellular carcinoma. Nat Genet.
31:339–346. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nakajima T, Yasui K, Zen K, Inagaki Y,
Fujii H, Minami M, Tanaka S, Taniwaki M, Itoh Y, Arii S, et al:
Activation of B-Myb by E2F1 in hepatocellular carcinoma. Hepatol
Res. 38:886–895. 2008.PubMed/NCBI
|
|
5
|
Pagliarini V, Giglio P, Bernardoni P, De
Zio D, Fimia GM, Piacentini M and Corazzari M: Downregulation of
E2F1 during ER stress is required to induce apoptosis. J Cell Sci.
128:1166–1179. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rajendran P, Li F, Shanmugam MK, Kannaiyan
R, Goh JN, Wong KF, Wang W, Khin E, Tergaonkar V, Kumar AP, et al:
Celastrol suppresses growth and induces apoptosis of human
hepatocellular carcinoma through the modulation of STAT3/JAK2
signaling cascade in vitro and in vivo. Cancer Prev Res. 5:631–643.
2012. View Article : Google Scholar
|
|
7
|
Feng L, Zhang D, Fan C, Ma C, Yang W, Meng
Y, Wu W, Guan S, Jiang B, Yang M, et al: ER stress-mediated
apoptosis induced by celastrol in cancer cells and important role
of glycogen synthase kinase-3β in the signal network. Cell Death
Dis. 4:e7152013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fribley AM, Miller JR, Brownell AL,
Garshott DM, Zeng Q, Reist TE, Narula N, Cai P, Xi Y, Callaghan MU,
et al: Celastrol induces unfolded protein response-dependent cell
death in head and neck cancer. Exp Cell Res. 330:412–422. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ma L, Wen ZS, Liu Z, Hu Z, Ma J, Chen XQ,
Liu YQ, Pu JX, Xiao WL, Sun HD, et al: Overexpression and small
molecule-triggered downregulation of CIP2A in lung cancer. PLoS
One. 6:e201592011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu Z, Ma L, Wen ZS, Cheng YX and Zhou GB:
Ethoxysanguinarine induces inhibitory effects and downregulates
CIP2A in lung cancer cells. ACS Med Chem Lett. 5:113–118. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu JL, Zeng GZ, Liu XL, Liu YQ, Hu ZG,
Liu Y, Tan NH and Zhou GB: Small compound bigelovin exerts
inhibitory effects and triggers proteolysis of E2F1 in multiple
myeloma cells. Cancer Sci. 104:1697–1704. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu Z, Ma L, Wen ZS, Hu Z, Wu FQ, Li W,
Liu J and Zhou GB: Cancerous inhibitor of PP2A is targeted by
natural compound celastrol for degradation in non-small-cell lung
cancer. Carcinogenesis. 35:905–914. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Diefenbach J and Bürkle A: Introduction to
poly(ADP-ribose) metabolism. Cell Mol Life Sci. 62:721–730. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhao Y, Tan J, Zhuang L, Jiang X, Liu ET
and Yu Q: Inhibitors of histone deacetylases target the Rb-E2F1
pathway for apoptosis induction through activation of pro-apoptotic
protein Bim. Proc Natl Acad Sci USA. 102:pp. 16090–16095. 2005;
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Matsumura I, Tanaka H and Kanakura Y: E2F1
and c-Myc in cell growth and death. Cell Cycle. 2:333–338. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lam SK, Li YY, Zheng CY, Leung LL and Ho
JC: E2F1 downregulation by arsenic trioxide in lung adenocarcinoma.
Int J Oncol. 45:2033–2043. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kawate S, Fukusato T, Ohwada S, Watanuki A
and Morishita Y: Amplification of c-myc in hepatocellular
carcinoma: Correlation with clinicopathologic features,
proliferative activity and p53 overexpression. Oncology.
57:157–163. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ciechanover A: Proteolysis: From the
lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol.
6:79–87. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Adams J: The proteasome: A suitable
antineoplastic target. Nat Rev Cancer. 4:349–360. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huynh H: Molecularly targeted therapy in
hepatocellular carcinoma. Biochem Pharmacol. 80:550–560. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Newman DJ and Giddings LA: Natural
products as leads to antitumor drugs. Phytochem Rev. 13:123–137.
2014. View Article : Google Scholar
|
|
22
|
Liu Z, Ma L and Zhou GB: The main
anticancer bullets of the Chinese medicinal herb, thunder god vine.
Molecules. 16:5283–5297. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li PP, He W, Yuan PF, Song SS, Lu JT and
Wei W: Celastrol induces mitochondria-mediated apoptosis in
hepatocellular carcinoma Bel-7402 cells. Am J Chin Med. 43:137–148.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yan YY, Guo Y, Zhang W, Ma CG, Zhang YX,
Wang C and Wang HX: Celastrol enhanced the anticancer effect of
lapatinib in human hepatocellular carcinoma cells in vitro. J BUON.
19:412–418. 2014.PubMed/NCBI
|
|
25
|
Nicholson DW: Caspase structure,
proteolytic substrates, and function during apoptotic cell death.
Cell Death Differ. 6:1028–1042. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Johnstone RW, Ruefli AA and Lowe SW:
Apoptosis: A link between cancer genetics and chemotherapy. Cell.
108:153–164. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Engelmann D, Knoll S, Ewerth D, Steder M,
Stoll A and Pützer BM: Functional interplay between E2F1 and
chemotherapeutic drugs defines immediate E2F1 target genes crucial
for cancer cell death. Cell Mol Life Sci. 67:931–948. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Conner EA, Lemmer ER, Omori M, Wirth PJ,
Factor VM and Thorgeirsson SS: Dual functions of E2F-1 in a
transgenic mouse model of liver carcinogenesis. Oncogene.
19:5054–5062. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rabbani F, Richon VM, Orlow I, Lu ML,
Drobnjak M, Dudas M, Charytonowicz E, Dalbagni G and Cordon-Cardo
C: Prognostic significance of transcription factor E2F-1 in bladder
cancer: Genotypic and phenotypic characterization. J Natl Cancer
Inst. 91:874–881. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zacharatos P, Kotsinas A, Evangelou K,
Karakaidos P, Vassiliou LV, Rezaei N, Kyroudi A, Kittas C,
Patsouris E, Papavassiliou AG, et al: Distinct expression patterns
of the transcription factor E2F-1 in relation to tumour growth
parameters in common human carcinomas. J Pathol. 203:744–753. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Engelmann D and Pützer BM: The dark side
of E2F1: In transit beyond apoptosis. Cancer Res. 72:571–575. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wu LC, Wen ZS, Qiu YT, Chen XQ, Chen HB,
Wei MM, Liu Z, Jiang S and Zhou GB: Largazole arrests cell cycle at
G1 phase and triggers proteasomal degradation of E2F1 in lung
cancer cells. ACS Med Chem Lett. 4:921–926. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gorgoulis VG, Zacharatos P, Mariatos G,
Kotsinas A, Bouda M, Kletsas D, Asimacopoulos PJ, Agnantis N,
Kittas C and Papavassiliou AG: Transcription factor E2F-1 acts as a
growth-promoting factor and is associated with adverse prognosis in
non-small cell lung carcinomas. J Pathol. 198:142–156. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Eymin B, Gazzeri S, Brambilla C and
Brambilla E: Distinct pattern of E2F1 expression in human lung
tumours: E2F1 is upregulated in small cell lung carcinoma.
Oncogene. 20:1678–1687. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Han S, Park K, Bae BN, Kim KH, Kim HJ, Kim
YD and Kim HY: E2F1 expression is related with the poor survival of
lymph node-positive breast cancer patients treated with
fluorouracil, doxorubicin and cyclophosphamide. Breast Cancer Res
Treat. 82:11–16. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhu H, Yang W, He LJ, Ding WJ, Zheng L,
Liao SD, Huang P, Lu W, He QJ and Yang B: Upregulating Noxa by ER
stress, celastrol exerts synergistic anti-cancer activity in
combination with ABT-737 in human hepatocellular carcinoma cells.
PLoS One. 7:e523332012. View Article : Google Scholar : PubMed/NCBI
|