Introduction
It is known that T-cell acute lymphoblastic leukemia
(T-ALL) is an invasive hematological malignancy derived from normal
immature T cells, causing accumulation of leukemia cells in the
bone marrow and suppression of normal hematopoiesis. T-ALL accounts
for ~15% of pediatric and 25% of adult ALL cases (1). Intensive combination chemotherapy has
improved the prognosis of patients with acute leukemia, however the
outcome of patients who fail to respond to conventional
chemotherapy is usually poor and the mortality rate is ~20% in
children and up to 50% in adults with T-ALL (2–4). The
emergence of resistance to chemotherapy is one of the main causes
of treatment failure in acute leukemia (5,6).
Therefore, the development of effective, new therapeutic methods,
such as molecular-targeted therapy and enhancement of sensitivity
to chemotherapy, in order to overcome drug resistance in leukemia
cells may potentially improve the effects of chemotherapy (4,5).
Apoptosis, the natural process of programmed cell
death, is essential for maintaining homeostasis (7,8).
Apoptosis defects results in resistance to chemotherapeutic
medicines (9–11). As cytotoxic agents exert their
anticancer effects by inducing apoptosis, new approaches for
antitumor treatment have focused on targeting mediators of the
apoptosis pathways (12–14).
As a flavin-dependent sulfhydryl oxidase, augmenter
of liver regeneration (ALR) is encoded by the growth factor
erv1-like (GFER) gene (15). ALR is
expressed in a variety of organs and tissues, such as
hepatocellular carcinoma (16),
tubular epithelial cells (17),
human-derived glioma cells (18)
and muscle tissue (19), and plays
a central role in the synthesis of hepatocyte DNA, immune
adjustment, cell cycle regulation and anti-apoptosis (20–24).
Our previous studies revealed that exogenous ALR protected acute
lymphocytic leukemia cells from vincristine (VCR)-induced cell
death, via decreasing activated caspase 8, increasing the ratio of
Bcl-2/Bax, reducing apoptotic cells and attenuating G2/M arrest
(25). It has been reported that
downregulation of ALR expression through antisense oligonucleotides
and small interfering RNA (siRNA) led to cell proliferation
inhibition and increased the sensitivity of cancer cells to
radiation (18,26,27).
To date, there are few studies on whether the suppression of ALR
expression by siRNA could sensitize T-ALL cells to chemotherapeutic
agents.
In the present study, we investigated the effect of
siRNA-induced ALR silencing on apoptosis and cell cycle
distribution and evaluated whether the suppression of ALR could
sensitize the human T-ALL cell line Jurkat to the anti-leukemic
agent VCR.
Materials and methods
Cell culture
Jurkat T leukemia cells (Nanjing KeyGen Biotech,
Nanjing, China) were maintained in suspension in RPMI-1640 medium
(HyClone, Logan, UT, USA) supplemented with 10% fetal bovine serum
(FBS; HyClone), 100 U/ml penicillin and streptomycin. The cells
were grown at 37°C in a humidified atmosphere with 5%
CO2.
RNA interference
The target sequence for ALR was
5′-GTGTGCTGAAGACCTAAGA-3′. Sense siRNA was
5′-GAGTGTGCTGAAGACCTAAGA-3′ and antisense siRNA was
5′-TCTTAGGTCTTCAGCACACTC-3′. Then, the siRNA was cloned into a CMV
promoter-driven lentiviral expression vector GV118 (GeneChem,
Shanghai, China). The GV118-mock vector was set as a negative
control. For infection 5.5×104 Jurkat cells were
collected and resuspended in 1 ml complete RPMI-1640 medium. The
cells were infected with GV118 or GV118-mock at a multiplicity of
infection (MOI) of 1. Twenty-four hours after the infection, the
medium was replaced with 1 ml of fresh culture medium. Then the
cells were grown for another 48 h and fluorescence microscopy
analysis of the GFP expression was performed. The expression of ALR
mRNA was further confirmed by fluorescence quantitative polymerase
chain reaction (FQ-PCR) and the ALR protein was determined by
western blotting and confocal laser scanning microscope.
Real-time reverse transcription
PCR
Total RNA was extracted from Jurkat cells using the
RNeasy Micro kit (Nanjing KeyGen Biotech, Nanjing, China) according
to the manufacturer's instructions. Total RNA (1 µg) from each
sample was used to generate first-strand cDNA synthesis using
PrimeScript RT reagent kit (Takara Biotechnology, Dalian, China).
Reverse transcription reaction started at 37°C for 15 min, followed
by 5 sec at 85°C. Real-time PCR was conducted using the SYBR Premix
Ex Taq™ II kit (Takara) according to the manufacturer's
instructions. cDNA product (1 µl) was used for the real-time PCR in
a final volume of 25 µl containing 12.5 µl of 2X SYBR Premix Ex
Taq™ II and 0.75 µl of the forward and reverse primers (Table I). The thermal cycling conditions
were as follows: initial denaturation step at 95°C for 1 min,
followed by 40 cycles of denaturation for 10 sec at 95°C and
annealing for 30 sec at 60°C. The CFX96 Manager software (Bio-Rad,
Hercules, CA, USA) was used to calculate the relative
concentrations of the PCR products. All the samples were tested in
triplicate and the data were analyzed using the 2−ΔΔCt
method.
 | Table I.Primers of human ALR and β-actin. |
Table I.
Primers of human ALR and β-actin.
| Target gene | Primer sequence |
|---|
| ALR | F: (5′-3′)
AAGGTGAGGCTGGGAATTT |
|
| R: (5′-3′)
GTCTTCATGTCGCGCTTCT |
| β-actin | F: (5′-3′)
TCAGGTCATCACTATCGGCAAT |
|
| R: (5′-3′)
AAAGAAAGGGTGTAAAACGCA |
Western blotting
Briefly, Jurkat cells were collected and then washed
twice with PBS. RIPA (500 µl) was added to the cell pellet to
extract the total protein of the cells. Protein samples were
resolved on 12% SDS-polyacrylamide gels and electroblotted onto
polyvinylidene difluoride (PVDF) membranes. The membranes were
blocked in a Tris-buffered saline/Tween-20 (TBST) solution
containing 2% nonfat dry milk at room temperature for 1 h and
subsequently incubated with the respective primary antibodies
(Epitomics, Burlingame, CA, USA) diluted in PBS for 2 h at room
temperature. Following washing with TBST for three times, the
membranes were incubated with HRP-conjugated secondary antibodies.
The expression of GAPHD was used as an internal loading control.
Immunoblot quantification was carried out by densitometry using
Image Lab statistical software (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Cell proliferation assay
To investigate the effects of ALR silencing on
Jurkat leukemia cell growth and sensitivity to VCR, cells were
seeded at 1×105/well in 96-well microtiter plates. After
culturing for 1, 2, 3 and 4 days, the quantity of viable cells was
detected. Afterwards, 20 µl of MTS solution was added into each
well and the cells were incubated for 3 h. Complete solubilization
of the dye was achieved by vortexing the plate, and then absorbance
was read at 490 nm using SpectraMax M2 plate reader. Wells
containing medium but no cells, served as blank controls.
Meanwhile, after 48 h of infection, cells were incubated with
various concentrations of VCR (0.0625–10 µg/ml) for another 24 h at
37°C. The viable cell count in each well was assessed by the OD
value. The percentage of surviving cells was calculated and the
IC50 value was determined by nonlinear regression
analysis using SPSS 17.0 software (SPSS, Inc., Chicago, IL,
USA).
Cell cycle analysis
After 72 h of infection, cell samples were harvested
and washed once in cold PBS, then fixed and stained with PI
solution. The cell cycle was detected using the cellular DNA flow
cytometric analysis kit (Nanjing KeyGen Biotech) according to the
manufacturer's protocol. The percentages of cells within the G1/G0,
S and G2/M phases were determined by flow cytometry. The results
were analyzed using CellQuest software (BD Biosciences, San Jose,
CA, USA). For the chemotherapeutic drug tests, after 48 h of
infection, the cells were cultured with VCR 1 µg/ml for an
additional 24 h and then analyzed as aforementioned.
Analysis of apoptosis
Apoptosis was assessed using the Annexin-V-PE/7-AAD
Apoptosis Detection kit (Nanjing KeyGen Biotech). After 48 h of
infection, the cells were incubated with 1 µg/ml VCR or alone, for
an additional 24 h. Then cell samples were collected and washed
twice with cold PBS. Subsequently, the cells were stained with 1 µl
Annexin V-PE and 5 µl 7-AAD. Flow cytometric analysis was performed
on a FACScan flow cytometer (BD Biosciences). Results were analyzed
using CellQuest PRO software (BD Biosciences).
Statistical analysis
All data are reported as the means ± SEM.
Comparisons between pairs of groups were made by Student's t-test
or one way ANOVA for multiple group comparisons. A P-value <0.05
was considered to indicate a statistically significant difference.
Statistical analysis was performed using SPSS 17.0 software (SPSS,
Inc.).
Results
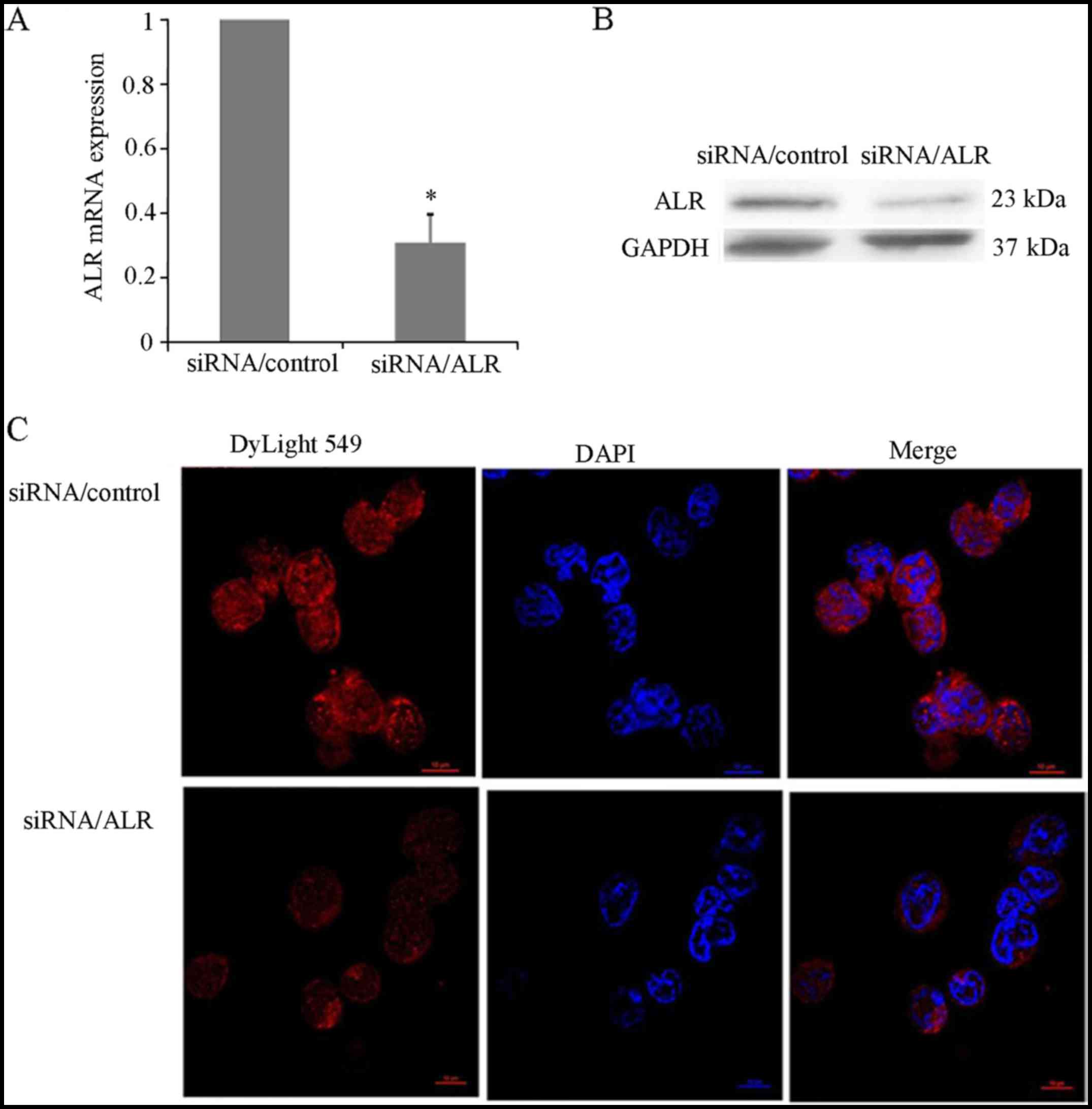
ALR siRNA effectively decreases ALR
expression in Jurkat cells
The expression level of ALR in the T-ALL Jurkat cell
line was detected by real-time PCR, western blot analysis and
confocal laser scanning microscopy. ALR-specific siRNA induced a
marked decrease of ALR mRNA expression in Jurkat cells (Fig. 1A). The protein level of ALR was
greatly downregulated in the ALR siRNA-infected group compared with
the level in the mock control group (Fig. 1B). After 72 h of infection, the
relative expression of ALR mRNA was 28.17±2.63%, while the relative
expression of ALR protein was 21.45±1.98%, respectively. Compared
with the control group, a significant reduction of the ALR protein
in the ALR siRNA-infected group was determined by confocal
microscopic immunodetection (Fig.
1C).
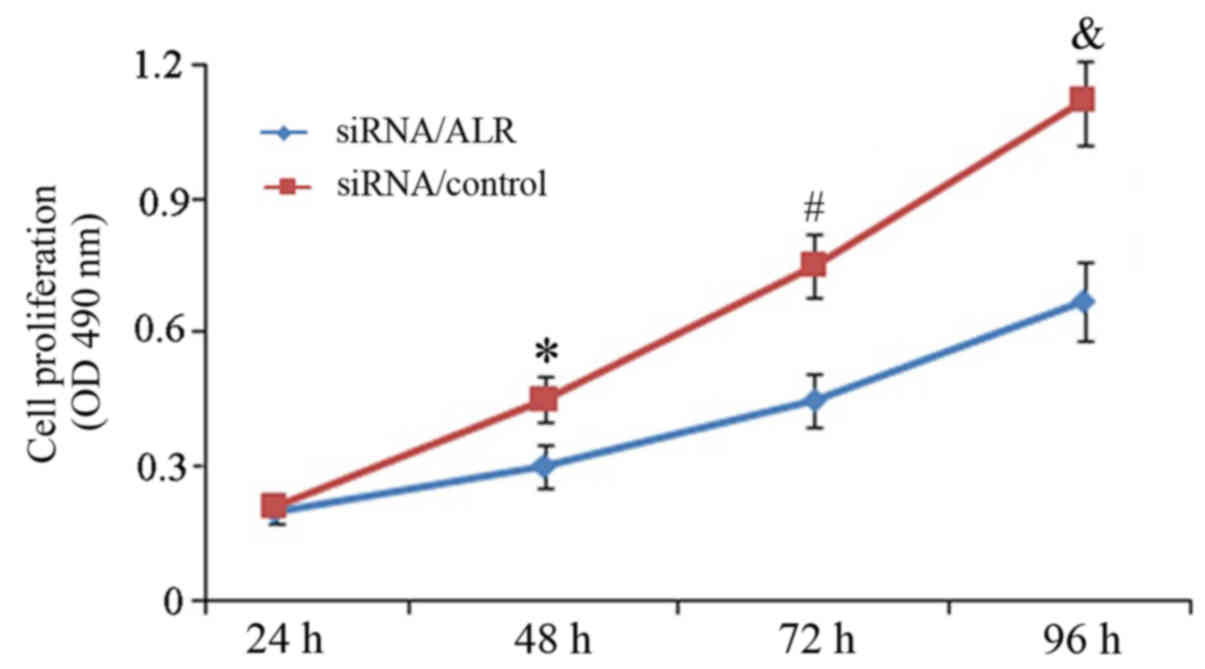
Downregulation of ALR expression
inhibits proliferation of Jurkat cells
As extraneous ALR is associated with the survival of
Jurkat T leukemia cells, the effect of ALR siRNA on cell
proliferation was detected. Twenty-four, 48, 72 and 96 h after
infection with siRNA, cell growth was determined using MTS assay.
The cell proliferation curve demonstrated that compared to the
control group, ALR siRNA significantly reduced cell growth in a
time-dependent manner (Fig. 2). At
24 h post-infection, the cell growth was 96.84±5.76% and rapidly
dropped to 49.38±7.21% at 96 h in the ALR siRNA group. The results
revealed that suppression of the ALR expression by siRNA
significantly inhibited cell proliferation in Jurkat cells.
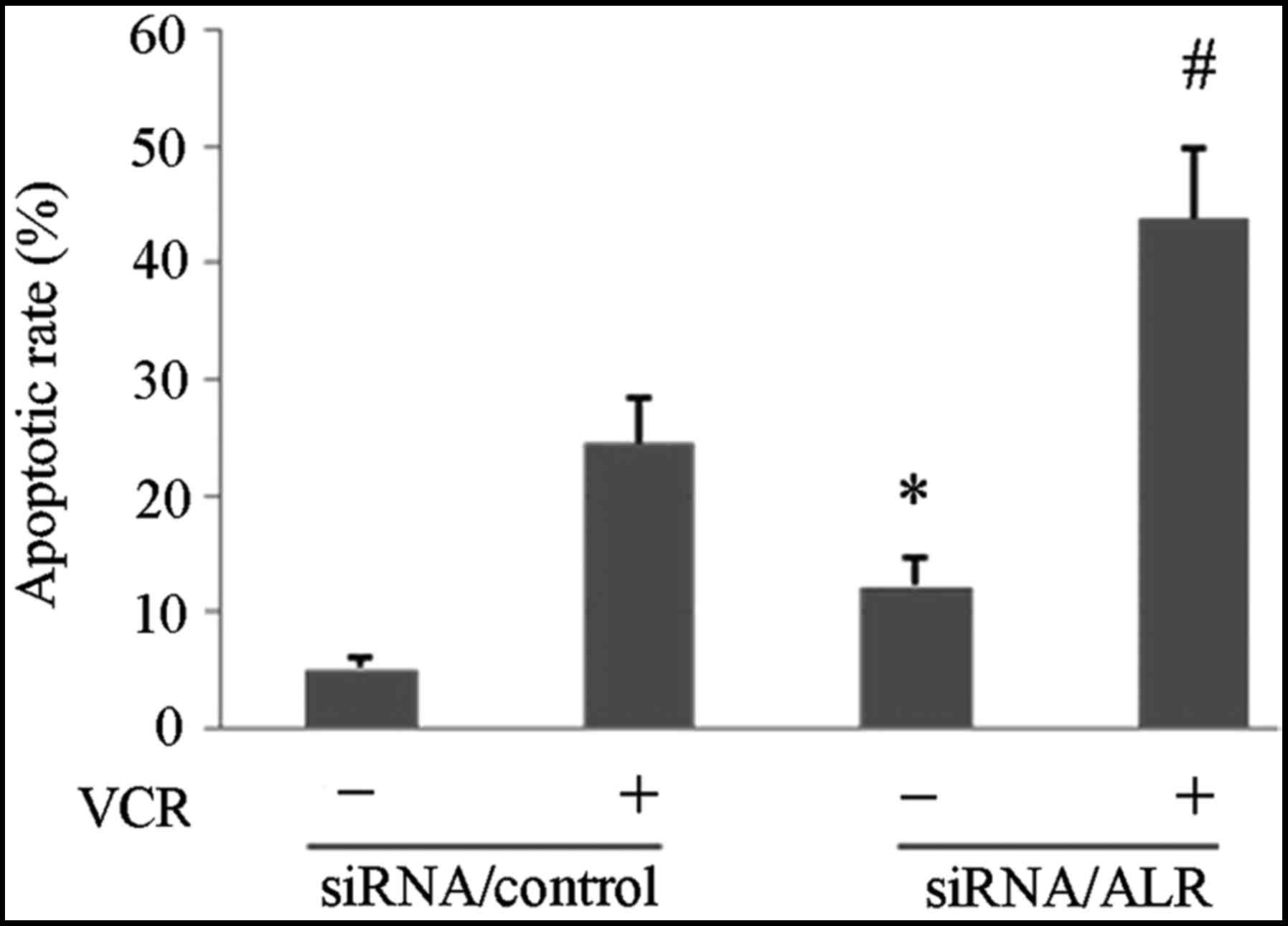
Downregulation of ALR expression
induces apoptosis in Jurkat cells
We next investigated whether the ALR siRNA-induced
proliferation suppression was related to apoptosis. The FCM
analysis revealed that the apoptosis rate was 19.85±1.74% in the
ALR siRNA-infected group, compared with 7.48±0.83% in the control
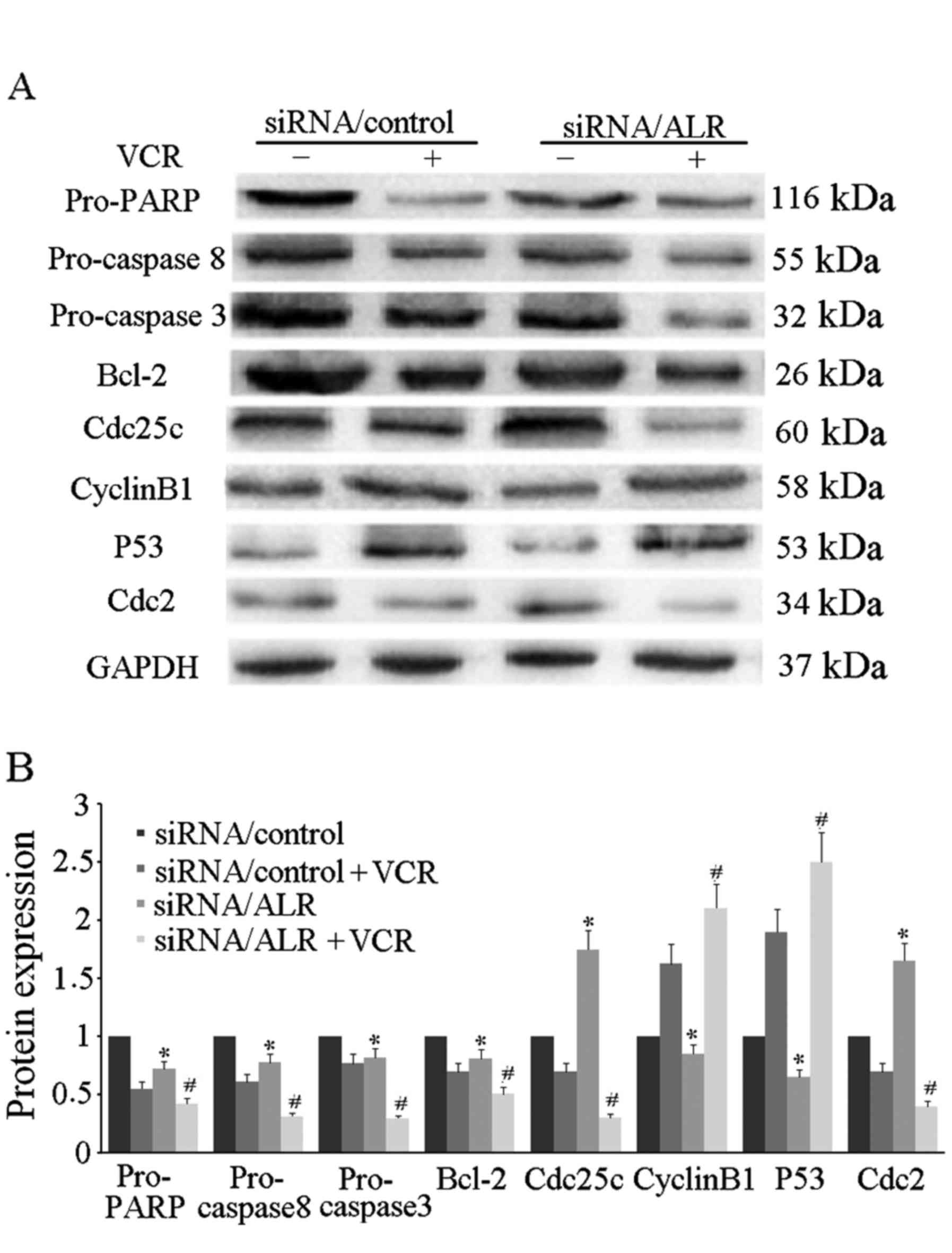
group (Fig. 3). Western blot
analysis revealed that after ALR siRNA infection, pro-PARP,
pro-caspase 8, pro-caspase 3 and Bcl-2 expression were
downregulated in the Jurkat cells (Fig.
4). These results revealed that the increased apoptosis induced
by ALR siRNA was partially responsible for the inhibition of cell
proliferation in the Jurkat cells.
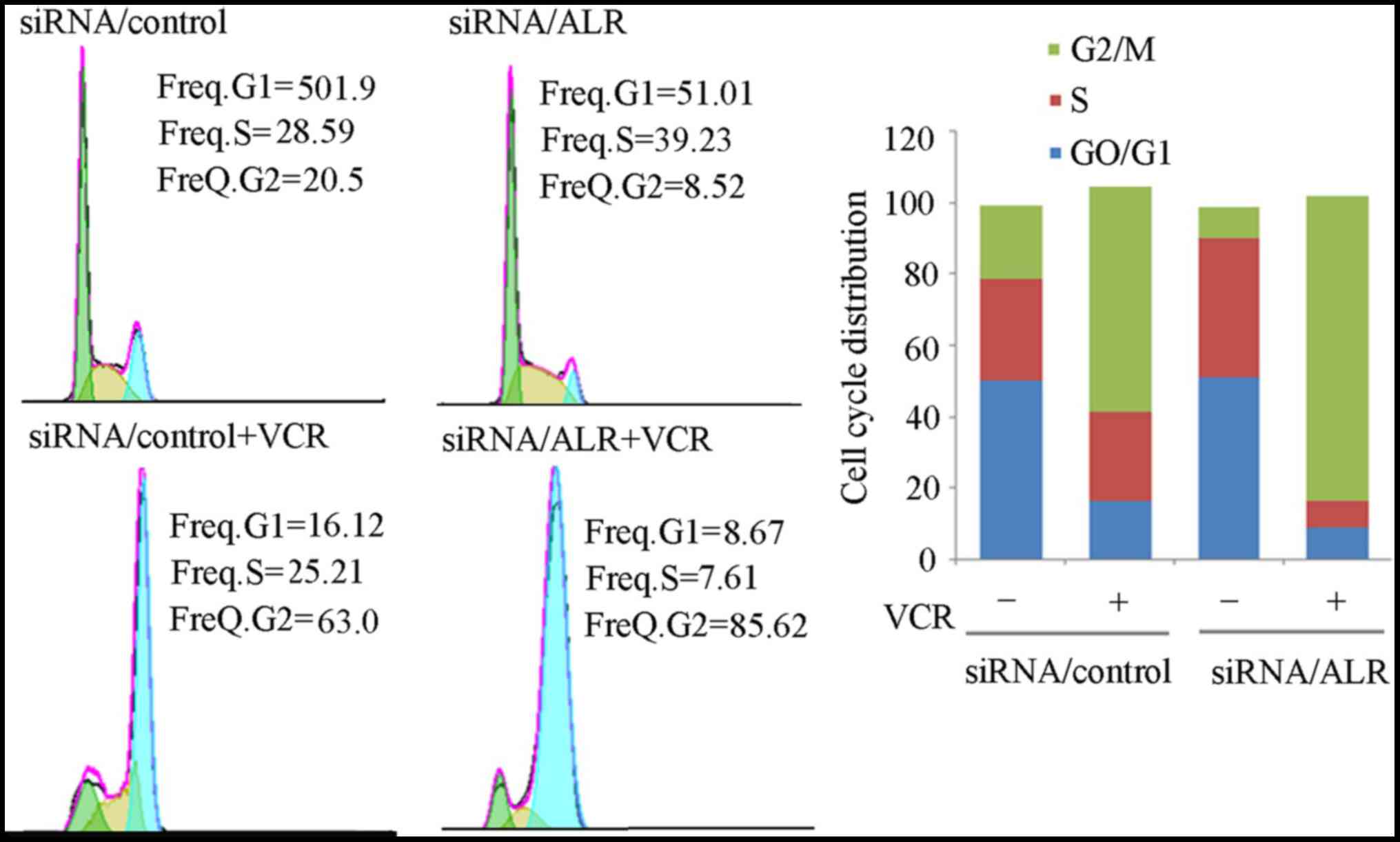
Downregulation of ALR expression
reduces the G2/M ratio in Jurkat cells
It is known that cell cycle progression influences
cell proliferation, thus we detected the effects of ALR siRNA
infection on cell cycle distribution. Data revealed that
suppression of ALR expression induced a decreased percentage of
G2/M-phase cells (Fig. 5). The
percentage of G0/G1-phase cells was 51%, of S-phase cells was
39.23% and of G2/M-phase cells was 8.53% in typical ALR
siRNA-infected cells, compared with 50.19%, 28.59% and 20.5% in the
control cells, respectively. Then we detected the G2/M-phase
regulatory factors, including cyclin B1, P53, cdc2 and cdc25c
proteins, for their molecular mechanisms. Western blot analysis
revealed that cyclin B1 and P53 expression was downregulated, while
cdc25c and cdc2 expression was upregulated (Fig. 4).
Downregulation of ALR expression
increases sensitivity to VCR in Jurkat cells
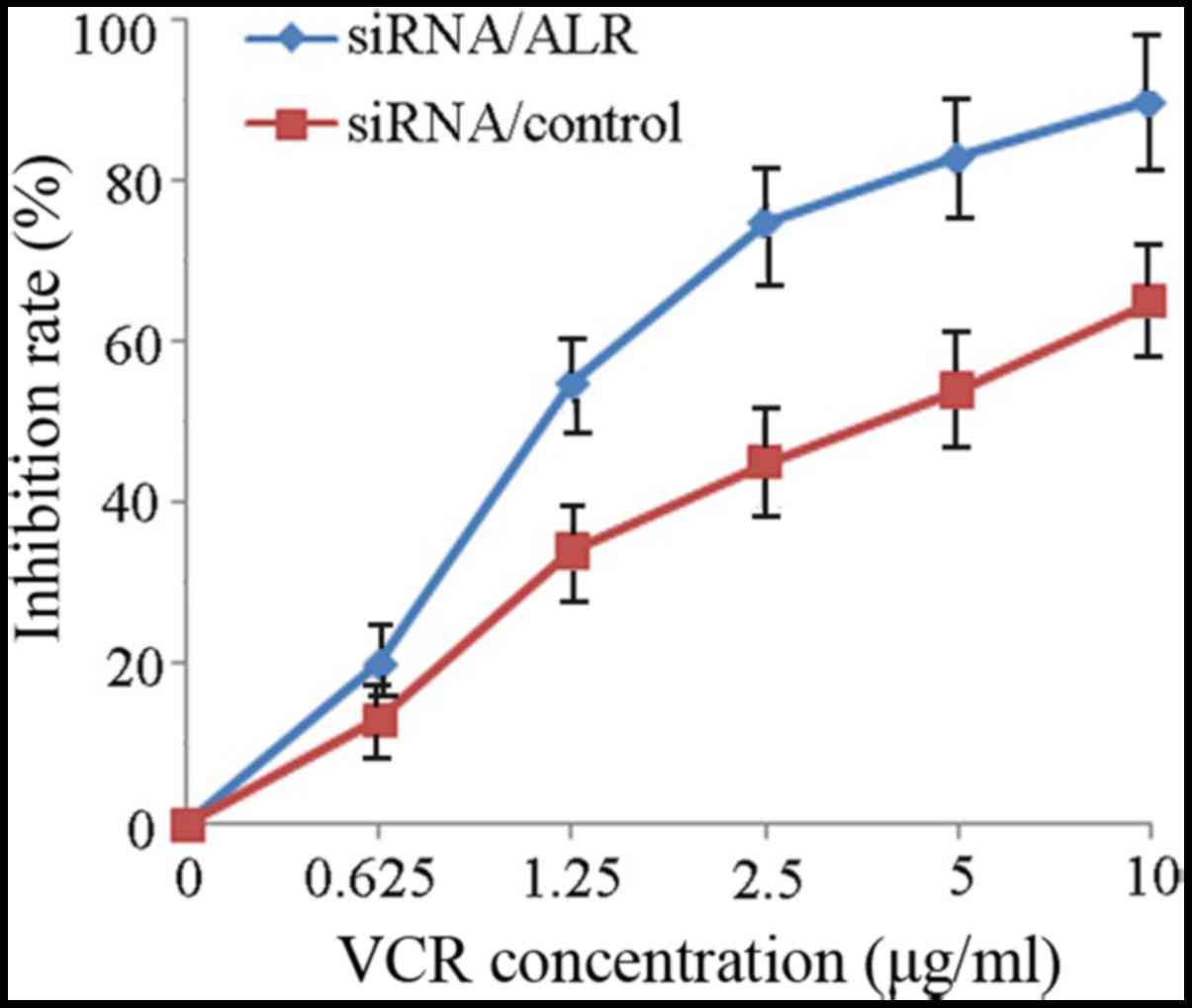
To investigate the effect of ALR downregulation on
the response of Jurkat cells to a chemotherapeutic agent, we
suppressed the ALR expression by siRNA in Jurkat cells and then
detected cell proliferation and apoptosis and cell cycle change
induced by VCR. Data revealed that the IC50 value for
VCR in the control siRNA group was 4.87±0.52 µg/ml. Jurkat cells
infected with siRNA/ALR were more sensitive and the IC50
value decreased to 1.32±0.17 µg/ml (Fig. 6). Subsequently, we examined the
apoptotic rate induced by VCR after the siRNA/ALR infection. FCM
analysis revealed that after the siRNA infection, VCR induced a
higher apoptotic rate in the siRNA/ALR-infected group (43.75±5.96%)
than that in the control siRNA infected group (24.59±3.76%)
(Fig. 3). The western blot analysis
revealed that the process of VCR-induced apoptosis in the
siRNA/ALR-infected cells was accompanied by the decreased
expression of the pro-PARP, pro-caspase 8, pro-caspase 3 and Bcl-2
proteins (Fig. 4). Furthermore,
cell cycle stage analysis revealed that in comparison with the
control siRNA group, VCR prolonged G2/M arrest in the siRNA/ALR
group (Fig. 5). Then we detected
the expression of several G2/M-phase cell cycle regulatory factors
to explore the molecular mechanisms. Western blot analysis revealed
that the expression of cyclin B1 and P53 was increased while that
of cdc25c and cdc2 was decreased (Fig.
4).
Discussion
ALR was first isolated from neonatal mouse
hepatocytes in 1994 (28). ALR
exhibits numerous biological activities, such as anti-apoptosis,
immunoregulation, antioxidant and preserving mitochondrial function
(20–24,29).
Our previous study revealed that exogeneous ALR could protect
Jurkat T leukemia cells from VCR-induced cell death, by decreasing
apoptotic cells and attenuating G2/M arrest (25). The present study revealed that ALR
may play an important role in Jurkat cell resistance to
chemotherapeutic drugs. Cao et al found that ALR could
enhance radiation sensitivity in hepatoma cells (26). Thus we further investigated whether
or not the suppression of ALR expression could enhance the
sensitivity of Jurkat cells to an anti-leukemic agent. In the
current study, we investigated the effect of ALR silencing by siRNA
on the sensitivity of Jurkat cells to VCR and explored the related
mechanism.
RT-PCR, western blot analysis and confocal laser
scanning microscopy revealed that ALR siRNA infection significantly
reduced ALR gene mRNA and protein levels at 72 h post-infection in
Jurkat cells. These data revealed that ALR-specific siRNA
translationally suppressed the ALR mRNA. The MTS assay for cell
proliferation revealed that the suppression of ALR expression
markedly restrained the proliferation of the Jurkat cells, proving
that ALR can exert a positive effect on leukemic cell growth. Most
prominently, the data from the cell proliferation assay revealed
that with ALR siRNA infection, the IC50 value of VCR
obviously decreased, which led to a significant synergistic
anti-leukemia effect. This supports the hypothesis that
downregulation of ALR can increase the sensitivity of leukemia
cells to VCR and possibly improve chemotherapy efficiency in
vivo.
To further investigate the influence of ALR on
survival of Jurkat leukemia cells in vitro, we detected the
effect of downregulation of the ALR expression on cell apoptosis.
These experiment findings indicated that siRNA-mediated suppression
of ALR expression exerted an obvious effect on cell apoptosis. ALR
siRNA pretreatment in combination with the chemotherapeutic agent
VCR resulted in obvious apoptosis in Jurkat cells and in
enhancement of chemotherapy sensitivity. On the contrary, cells
infected with non-targeting siRNA in the control group had no
alteration of the ALR gene expression level. Subsequently, cell
growth and toxicity of VCR were not changed. These findings
illustrated the specific impact of ALR siRNA in Jurkat cells.
However, our findings are similar with other research which focused
on the biological effects of ALR in a variety of cell lines
(17,18,30).
Collectively, these results revealed that ALR plays a significant
role in cell survival, proliferation and chemotherapy sensitivity
of malignant cells. All of the observations above, ascertained that
the presence of the multifunctional ALR protein is critical to the
survival and chemotherapeutic agent resistance in Jurkat cells.
Therefore, ALR gene silencing could induce cell apoptosis and
sensitize leukemia cells to cytotoxic drugs.
The findings of the signaling pathway analysis
revealed that targeted suppression of ALR expression could promote
apoptotic signaling and lead to an increase of chemotherapeutic
efficiency. Consequently, Jurkat ALR-knockdown cells exhibited more
sensitivity to the chemotherapeutic drug. Enhanced cell death was
mediated by a higher percentage of apoptotic cells, which was
accompanied by decreased expressions of the pro-PARP, pro-caspase
8, pro-caspase 3 and Bcl-2.
Previous findings have demonstrated that ALR acts as
a regulatory factor for cell cycle regulation (31–33).
In the present study, we found that downregulation of ALR prevented
Jurkat cell transition from the S to the G2/M phase which resulted
in a decrease of the G2/M ratio, while it prolonged VCR-induced
G2/M arrest. The targeted suppresion of the ALR expression
increased the protein levels of cdc25c and cdc2, inhibited the
dissociation of cyclin B1 and decreased G2/M ratio. Finally, it led
to prevention of cell mitosis and induced Jurkat cell apoptosis.
Treatment with siRNA/ALR and VCR significantly downregulated the
expression of cdc25c and cdc2, promoted dissociation of cyclin B1,
resulted in prolonged G2/M arrest and enhanced apoptosis. The
evidence revealed that ALR knockdown may exert a bidirectional
regulatory effect on Jurkat cell cycle progression. However, more
research is needed to identify the roles of ALR in the regulation
of leukemia cell cycle and apoptosis.
In conclusion, our findings ascertained that ALR
plays important roles in both cell growth and drug susceptibility
of Jurkat cells in vitro. Targeted inhibition of the ALR
expression by siRNA triggered cell apoptosis and enhanced
sensitivity of leukemic cells to VCR in a synergistic manner. Our
study focused on the capability of ALR siRNA to sensitize the
leukemia cells to chemotherapeutic agent and decrease the
occurrence of anti-leukemia drug resistance. Therefore, siRNA
silencing of ALR gene expression may offer a new strategy for the
treatment of drug-resistant T-ALL.
Acknowledgements
This study was supported by the Medical Research
Foundation of Chongqing Health Bureau (no. 2011-1-051), and partly
by the Natural Science Foundation Project of CQ CSTC (cstc2017
jcyjAX0239).
References
|
1
|
Pieters R and Carroll WL: Biology and
treatment of acute lymphoblastic leukemia. Hematol Oncol Clin North
Am. 24:1–18. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marks DI, Paietta EM, Moorman AV, Richards
SM, Buck G, DeWald G, Ferrando A, Fielding AK, Goldstone AH,
Ketterling RP, et al: T-cell acute lymphoblastic leukemia in
adults: Clinical features, immunophenotype, cytogenetics, and
outcome from the large randomized prospective trial (UKALL XII/ECOG
2993). Blood. 114:5136–5145. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gianfelici V, Chiaretti S, Demeyer S, Di
Giacomo F, Messina M, La Starza R, Peragine N, Paoloni F, Geerdens
E, Pierini V, et al: RNA sequencing unravels the genetics of
refractory/relapsed T-cell acute lymphoblastic leukemia. Prognostic
and therapeutic implications. Haematologica. 101:941–950. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ko RH, Ji L, Barnette P, Bostrom B,
Hutchinson R, Raetz E, Seibel NL, Twist CJ, Eckroth E, Sposto R, et
al: Outcome of patients treated for relapsed or refractory acute
lymphoblastic leukemia: A Therapeutic Advances in Childhood
Leukemia Consortium study. J Clin Oncol. 28:648–654. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Paszel-Jaworska A, Rubiś B,
Bednarczyk-Cwynar B, Zaprutko L and Rybczyńska M: Proapoptotic
activity and ABCC1-related multidrug resistance reduction ability
of semisynthetic oleanolic acid derivatives DIOXOL and HIMOXOL in
human acute promyelocytic leukemia cells. Chem Biol Interact.
242:1–12. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Soverini S, De Benedittis C, Papayannidis
C, Paolini S, Venturi C, Iacobucci I, Luppi M, Bresciani P,
Salvucci M, Russo D, et al: Drug resistance and BCR-ABL kinase
domain mutations in Philadelphia chromosome-positive acute
lymphoblastic leukemia from the imatinib to the second-generation
tyrosine kinase inhibitor era: The main changes are in the type of
mutations, but not in the frequency of mutation involvement.
Cancer. 120:1002–1009. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kiraz Y, Adan A, Yandim M Kartal and Baran
Y: Major apoptotic mechanisms and genes involved in apoptosis.
Tumour Biol. 37:8471–8486. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pistritto G, Trisciuoglio D, Ceci C,
Garufi A and D'Orazi G: Apoptosis as anticancer mechanism: Function
and dysfunction of its modulators and targeted therapeutic
strategies. Aging (Albany NY). 8:603–619. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fodale V, Pierobon M, Liotta L and
Petricoin E: Mechanism of cell adaptation: When and how do cancer
cells develop chemoresistance? Cancer J. 17:89–95. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang S, Li G, Ma X, Wang Y, Liu G, Feng
L, Zhao Y, Zhang G, Wu Y, Ye X, et al: Norcantharidin enhances
ABT-737-induced apoptosis in hepatocellular carcinoma cells by
transcriptional repression of Mcl-1. Cell Signal. 24:1803–1809.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li G, Chang H, Zhai YP and Xu W: Targeted
silencing of inhibitors of apoptosis proteins with siRNAs: A
potential anti-cancer strategy for hepatocellular carcinoma. Asian
Pac J Cancer Prev. 14:4943–4952. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Karami H, Baradaran B, Esfehani A,
Sakhinia M and Sakhinia E: Down-regulation of Mcl-1 by small
interference RNA induces apoptosis and sensitizes HL-60 leukemia
cells to etoposide. Asian Pac J Cancer Prev. 15:629–635. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
High LM, Szymanska B, Wilczynska-Kalak U,
Barber N, O'Brien R, Khaw SL, Vikstrom IB, Roberts AW and Lock RB:
The Bcl-2 homology domain 3 mimetic ABT-737 targets the apoptotic
machinery in acute lymphoblastic leukemia resulting in synergistic
in vitro and in vivo interactions with established drugs. Mol
Pharmacol. 77:483–494. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Akagi H, Higuchi H, Sumimoto H, Igarashi
T, Kabashima A, Mizuguchi H, Izumiya M, Sakai G, Adachi M,
Funakoshi S, et al: Suppression of myeloid cell leukemia-1 (Mcl-1)
enhances chemotherapy-associated apoptosis in gastric cancer cells.
Gastric cancer. 16:100–110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lisowsky T, Lee JE, Polimeno L,
Francavilla A and Hofhaus G: Mammalian augmenter of liver
regeneration protein is a sulfhydryl oxidase. Dig Liver Dis.
33:173–180. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu HY, Xiang DR, Huang HJ, Li J and Sheng
JF: Expression level of augmenter of liver regeneration in patients
with hepatic failure and hepatocellular carcinoma. Hepatobiliary
Pancreat Dis Int. 9:492–498. 2010.PubMed/NCBI
|
|
17
|
Liao XH, Zhang L, Liu Q, Sun H, Peng CM
and Guo H: Augmenter of liver regeneration protects kidneys from
ischaemia/reperfusion injury in rats. Nephrol Dial Transplant.
25:2921–2929. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Polimeno L, Pesetti B, De Santis F, Resta
L, Rossi R, De Palma A, Girardi B, Amoruso A and Francavilla A:
Decreased expression of the augmenter of liver regeneration results
in increased apoptosis and oxidative damage in human-derived glioma
cells. Cell Death Dis. 3:e2892012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Polimeno L, Pesetti B, Giorgio F, Moretti
B, Resta L, Rossi R, Annoscia E, Patella V, Notarnicola A,
Mallamaci R, et al: Expression and localization of augmenter of
liver regeneration in human muscle tissue. Int J Exp Pathol.
90:423–430. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yan R, Zhang L, Xia N, Liu Q, Sun H and
Guo H: Knockdown of augmenter of liver regeneration in HK-2 cells
inhibits inflammation response via the mitogen-activated protein
kinase signaling pathway. Inflamm Res. 64:453–462. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang N, Sun H, Shen Y, Li XF, Pan T, Liu
GL and Liu Q: Augmenter of liver regeneration inhibits apoptosis of
activated human peripheral blood lymphocytes in vitro.
Immunopharmacol Immunotoxicol. 35:257–263. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Han LH, Dong LY, Yu H, Sun GY, Wu Y, Gao
J, Thasler W and An W: Deceleration of liver regeneration by
knockdown of augmenter of liver regeneration gene is associated
with impairment of mitochondrial DNA synthesis in mice. Am J
Physiol Gastrointest Liver Physiol. 309:G112–G122. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kumar S, Wang J, Rani R and Gandhi CR:
Hepatic deficiency of augmenter of liver regeneration exacerbates
alcohol-induced liver injury and promotes fibrosis in mice. PLoS
One. 11:e01478642016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mu M, Zhang Z, Cheng Y, Liu G, Chen X, Wu
X, Zhuang C, Liu B, Kong X and You S: Augmenter of liver
regeneration (ALR) restrains concanavalin A-induced hepatitis in
mice. Int Immunopharmacol. 35:280–286. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shen Y, Liu Q, Sun H, Li X, Wang N and Guo
H: Protective effect of augmenter of liver regeneration on
vincristine-induced cell death in Jurkat T leukemia cells. Int
Immunopharmacol. 17:162–167. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cao Y, Fu YL, Yu M, Yue PB, Ge CH, Xu WX,
Zhan YQ, Li CY, Li W and Wang XH: Human augmenter of liver
regeneration is important for hepatoma cell viability and
resistance to radiation-induced oxidative stress. Free Radic Biol
Med. 47:1057–1066. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li Y, Farooq M, Sheng D, Chandramouli C,
Lan T, Mahajan NK, Kini RM, Hong Y, Lisowsky T and Ge R: Augmenter
of liver regeneration (alr) promotes liver outgrowth during
zebrafish hepatogenesis. PLoS One. 7:e308352012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hagiya M, Francavilla A, Polimeno L, Ihara
I, Sakai H, Seki T, Shimonishi M, Porter KA and Starzl TE: Cloning
and sequence analysis of the rat augmenter of liver regeneration
(ALR) gene: Expression of biologically active recombinant ALR and
demonstration of tissue distribution. Proc Natl Acad Sci USA.
91:pp. 8142–8146. 1994; View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Todd LR, Damin MN, Gomathinayagam R, Horn
SR, Means AR and Sankar U: Growth factor erv1-like modulates Drp1
to preserve mitochondrial dynamics and function in mouse embryonic
stem cells. Mol Biol Cell. 21:1225–1236. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Polimeno L, Pesetti B, Lisowsky T, Iannone
F, Resta L, Giorgio F, Mallamaci R, Buttiglione M, Santovito D,
Vitiello F, et al: Protective effect of augmenter of liver
regeneration on hydrogen peroxide-induced apoptosis in SH-SY5Y
human neuroblastoma cells. Free Radic Res. 43:865–875. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li Y, Wei K, Lu C, Li Y, Li M, Xing G, Wei
H, Wang Q, Chen J, Wu C, et al: Identification of hepatopoietin
dimerization, its interacting regions and alternative splicing of
its transcription. Eur J Biochem. 269:3888–3893. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Giorda R, Hagiya M, Seki T, Shimonishi M,
Sakai H, Michaelson J, Francavilla A, Starzl TE and Trucco M:
Analysis of the structure and expression of the augmenter of liver
regeneration (ALR) gene. Mol Med. 2:97–108. 1996.PubMed/NCBI
|
|
33
|
Lisowsky T: ERV1 is involved in the
cell-division cycle and the maintenance of mitochondrial genomes in
Saccharomyces cerevisiae. Curr Genet. 26:15–20. 1994. View Article : Google Scholar : PubMed/NCBI
|