Introduction
Lung carcinoma is the leading cause of
cancer-related death, for which the 5-year survival rate for lung
cancer patients is less than 20%. Lung cancer can be divided into
small-cell lung cancer (SCLC) and non-small cell lung cancer
(NSCLC). NSCLC comprises ~85% of all lung cancer cases (1). Epidermal growth factor receptor (EGFR)
mutation has been found in lung cancer for years and its kinase
inhibitors are used for clinical treatment. However, it is
disappointing that the efficacy is limited (2). Therefore, research has focused on new
biomarkers for lung cancer diagnosis and therapies are
promising.
δ-Catenin coded by the gene CTNND2 is recognized to
be primarily expressed in the brains of normal people (3,4).
However, in recent years, various research has revealed that
δ-catenin also can be a biomarker for cancers, since it has been
found to be overexpressed in various types of cancers, including
prostate, breast, lung and ovarian cancer (3,5–7).
δ-Catenin can be expressed as different variants in different type
of cancers (3). In some cell lines
of ovarian, breast and esophageal cancer, a full length δ-catenin
transcript is overexpressed. However, in other cancer types, lung
cancer included, δ-catenin species with N- or C-terminal
truncations are expressed. The distributions of different δ-catenin
species are also different. N-terminus of δ-catenin is prominently
associated with the cytoplasmic distribution, whereas the
carboxyl-terminus of δ-catenin can be subject to significant
translocation to the nuclei (3).
Overexpression of full-length or truncated δ-catenin
is usually associated with malignancy and poor prognosis. In
prostate cancer, overexpression of δ-catenin is due to −9 G>A
mutation in 5′-UTR promoters (6).
Moreover, truncated mutations of δ-catenin found in prostate cancer
have been reported to promote cancer cell survival via metabolic
reprogramming, hypoxia pathways and Wnt signaling response
(8). It has been also reported that
δ-catenin promotes the epithelial cell marker, E-cadherin
processing to regulate malignancy in prostate cancer (9). In ovarian cancer, δ-catenin is
overexpressed and associated with advanced stage. δ-catenin
regulates the ovarian cancer cell proliferation, invasion and cell
cycle (7). In non-small cell lung
cancer, δ-catenin has been reported to be expressed much higher in
malignant tissues than in benign tissues. Moreover, δ-catenin is
expressed in the cytoplasm, correlated with high Dvl3, CD31 and
VEGF expression, suggesting that δ-catenin may be related to
angiogenesis and lymphangiogenesis. High expression of δ-catenin in
lung cancer is associated with poor prognosis (10,11).
However, Dai et al reported that δ-catenin may affect lung
cancer prognosis through δ-catenin/Kaiso pathway (5), but the detailed mechanisms of how
δ-catenin enhances the malignancy of lung cancer remain largely
unknown.
In the present study, we investigated the roles of
δ-catenin in the development of lung adenocarcinoma via Lewis lung
cell tumorigenesis and metastasis models. We found that δ-catenin
enhances Lewis lung cell subcutaneous tumorigenesis and metastasis
in vivo. Our data reveal that δ-catenin enhances Lewis lung
cell proliferation and cell cycle progression. δ-Catenin may
contribute to G1-S phase transition in cooperation with canonical
Wnt signaling in Lewis lung cells. Furthermore, δ-catenin promotes
the oncosphere formation of Lewis lung cells. The expression of
cancer stem cell marker, CD133 and Aldh1, may be modulated
by δ-catenin. In the present study, novel mechanisms of how
δ-catenin promotes lung adenocarcinoma malignancy are revealed. Our
data suggest that δ-catenin may be a biomarker and a new therapy
target for lung adenocarcinoma.
Materials and methods
Plasmids, antibodies and reagents
The mouse Ctnnd2 sgRNAs were subcloned into
pLVR-sgRNA-CMV-Cas9-GFP and pU6gR-MCS2-CMV-Cas9-SV40-mCherry
plasmids, separately (GeneCopoeia, Guangzhou, China). The sequences
of sgRNA1 were: forward, 5′-ATCCGCCGGGCGCCAGGGCGGCCC-3′ and
reverse, 5′-AAACGGGCCGCCCTGGCGCCCGGC-3′; sgRNA2 forward,
5′-ATCCGCGGCGGGTGCATGTTCGCC-3′ and reverse,
5′-AAACGGCGAACATGCACCCGCCGC-3′. The human CTNND2 were subcloned
into pEZ-M02 (EX-Z7373-M02-5; GeneCopoeia). Antibodies were used in
the present study: monoclonal anti-δ-catenin (ab54578; Abcam,
Cambridg, UK); anti-CD133 (11-1331-80; eBioscience, San Diego, CA,
USA); anti-β-catenin (844602; BioLegend, San Diego, USA); control
IgG (I5381; Sigma-Aldrich, St. Louis, MO, USA) and anti-β-actin
(A1978; Sigma). Tris-HCl, NaCl and other chemicals were from
Sigma.
CRISP/Cas9 system
The two sgRNAs were subcloned into
pLVR-sgRNA-CMV-Cas9-GFP and pU6gR-MCS2-CMV-Cas9-SV40-mCherry
plasmids, separately (GeneCopoeia). Transfection of plasmids into
Lewis lung cells were performed with polyethylenimine
(Polysciences, Inc., Warminster, PA, USA). G418 (20 µg/ml) (Sigma)
was used to select cells in which pLVR-sgRNA-CMV-Cas9-GFP and
pU6gR-MCS2-CMV-Cas9-SV40-mCherry plasmids were expressed. The Cas9
protein could break the genome of Ctnnd2 at two different
sites guided by the two different sgRNAs, resulting in an ~300 bp
DNA deleted fragment. Monoclone cells were picked out to extract
genome DNAs. Monoclone cells with the targeted genes knocked out
were identified by PCR. The primer sequences were: forward,
5′-CGGGAGGAGCCTCGCTCT-3′ and reverse, 5′-CGGGAGGAGCCTCGCTCT-3′.
Cells and transfection
Lewis lung and A549 cells were purchased from the
American Type Culture Collection (ATCC; Manassas, VA, USA). The
ATCC number of Lewis lung cells was CRL-1642. The ATCC number of
A549 cells was CCL-185. The cells were purchased April 5, 2014.
Lewis lung cells were cultured in RPMI-1640 (Invitrogen, Carlsbad,
CA, USA) with 10% fetal bovine serum (FBS) (HyClone, Logan, UT,
USA); A549 cells in Dulbeccos modified Eagles medium (DMEM)
(Invitrogen) with 10% FBS (HyClone). Transfection of DNA plasmids
into Lewis lung and A549 cells were performed with polyethylenimine
(Polysciences, Inc.), and the stable cell lines were selected with
20 µg/ml G418 (Sigma).
Mice
B6/C57 mice used in the present study were bred and
maintained in a specific pathogen-free animal facility at Fujian
Medical University. Mice were euthanized with carbon dioxide
asphyxiation. All animal experiments were approved by the Animal
Ethical Committee of Fujian Medical University.
Tumorigenesis and metastasis
Cells (5×105) of WT and Ctnnd2
knock out LLCs were subcutaneously injected into the abdomen of
each B6/C57 mouse to make the subcutaneous tumorigenesis mouse
model. Tumor sizes were measured by tumor length and width using
clippers and then tumor volume was calculated using the formula V =
(L × W × W)/2, where V is tumor volume, W is tumor width, and L is
tumor length. Cells (1×106) of LLCs were injected into
the tail veins of B6/C57 mice to make the metastasis model. LLCs
tend usually to transfer to the lungs and bones. Survival curves of
mice were calculated by GraphPad Prism 5 software (GraphPad
Software, Inc., La Jolla, CA, USA).
Hematoxylin and eosin stain
The tissues are embedded in paraffin to be cut into
3 µm tissue sections. Tissue sections were dewaxed with xylene.
Then, sections were rehydrated with 100, 95 and 75% alcohol
gradients. Hematoxylin was stained for 20 min, and then sections
were differentiated with 1% hydrochloric acid for 30 sec. After 15
min of PBS blue staining, eosin was stained for 3 min. After
rinsing, sections were dehydrated with a gradient of 95–100%
alcohol. The sections were cleared with xylene two times. Then,
sections were mounted with a neutral resin.
Flow cytometry
Flow cytometric analysis was performed using BD FACS
C6 Flow Cytometer. To monitor the cell cycle, after cells were
fixed with 70% ethanol overnight, 5 µg/ml propidium iodide was used
for staining. Then, 20,000 cells were collected for analysis. To
monitor cell surface expression of CD133, anti-CD133-FITC antibody
and control IgG were used for staining, and 5,000 cells were
collected for analysis. The percentage of each phase in cell cycle
and the expression of CD133 were analyzed by FlowJo 7.6.1
software.
Quantitative real-time PCR
Total cell RNA was isolated with TRIzol
(Invitrogen), and cDNA was synthesized with ReverTra Ace (Promega,
Madison, USA). Real-time PCR was performed with an ABI QuantStudio
5 system. The expression level of genes was measured using the
comparative Ct method. Expression values were normalized to GAPDH
expression. The primer sequences were as follows. Ctnnd2
(mouse): forward, 5′-CCTCCGAATAGACAATGACC-3′ and reverse,
5′-GAGAAGCAGCCTTGACCAC-3′; CTNND2 (human): forward,
5′-GCTCCGAATAGACAATGACC-3′ and reverse, 5′-GAGATGCAGCCTTGACCAC-3′;
c-myc forward, 5′-ATGCCCCTCAACGTGAACTTC-3′ and reverse,
5′-GTCGCAGATGAAATAGGGCTG-3′; p-21: forward,
5′-GTGATTGCGATGCGCTCATG-3′ and reverse, 5′-TCTCTTGCAGAAGACCAATC-3′;
Gapdh forward, 5′-CATGGCCTTCCGTGTTCCTA-3′ and reverse,
5′-CCTGCTTCACCACCTTCTTGAT-3′.
Immunoblotting and immunoprecipitation
assay
Cells were lysed with TNE buffer (10 mM Tris-HCl,
150 mM NaCl, 1 mM EDTA, 0.5% NP40, pH 7.5). For immunoblotting
assay, cell lysates were mixed with 4X loading buffer (40 mM
Tris-HCl, 200 mM DTT, 4% SDS, 40% glycerol, 0.032% bromophenol
blue, pH 8.0). The samples were run with 4% stacking gel and 10%
separating gels. Then, proteins on the gels were transferred to
nitrocellulose filter membranes for incubation with antibodies. The
membrane exposure was carried out with Thermo Pierce ECL and
FluorChem E (Protein Sample). For immunoprecipitation assay, cell
lysates were incubated with β-catenin antibody or control IgG
overnight. Then, protein A agarose (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) beads were added to bind antibodies and targeted
proteins for 2 h. Protein A agarose bead elution was collected for
immunoblotting.
Cell proliferation assay
Equal amount of cells were placed in the 6-well
plates, and then subjected to different treatments. Every day the
cell numbers were counted.
Matrigel 3D cultures
Cells in 2D cultures were trypsinized and
resuspended in media, and 20,000 cells were plated on each 100 µl
Matrigel (BD BioSciences, New Jersey, NY, USA). Media containing
10% FBS was added for cell growth (12).
Cell migration and wound-healing
assays
Cell migration and wound-healing assays were as
previously described (13).
Statistical analysis
The Student's t-test, one-way ANOVA, Wilcoxon rank
sum and log-rank test were used. P<0.05 were considered
statistically significant.
Results
δ-Catenin enhances Lewis lung cell
tumorigenesis and metastasis in vivo
Lewis lung carcinoma originate from a spontaneous
lung tumor of a B6/C57 mouse (14,15).
In the present study, to study the function of δ-catenin in lung
cancer, we established Ctnnd2 knockout Lewis lung cells
(LLCs) via the CRISPR/Cas9 system (16,17).
SgRNAs were designed in exon1 of Ctnnd2 genome and ~300 bps
DNA fragment may be deleted. According to our results, all four
monoclones of Lewis lung cells contained Ctnnd2 genome DNA
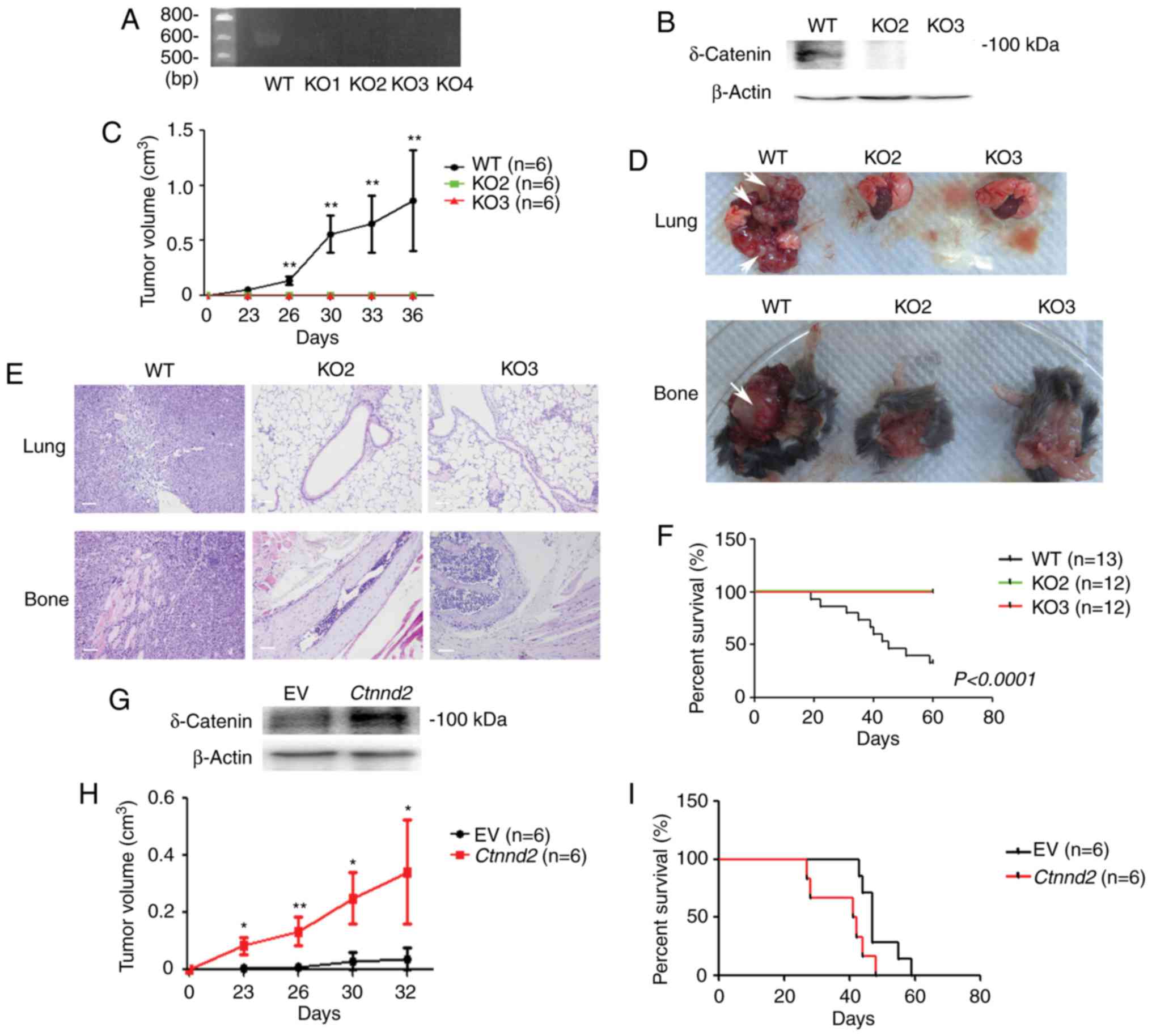
fragment deletion (Fig. 1A).
However, only KO2 and KO3 cell lines were chosen for the following
research, since compared with KO1 and KO4 cell lines, mRNA levels
of KO2 and KO3 cell lines were reduced (data not shown).
Furthermore, proteins of δ-catenin were abolished in KO2 and KO3
cell lines (Fig. 1B). Next, WT and
Ctnnd2 knockout LLCs were subcutaneously injected into
B6/C57 mice to examine their tumor formation ability. WT LLCs
formed tumors with average volumes to ~1 cm3 within 40
days, but Ctnnd2 knockout LLCs could not form tumors,
suggesting significant roles of δ-catenin in tumorigenesis
(Fig. 1C). Furthermore, we injected
WT LLCs into tail veins of B6/C57 mice, leading to lung and bone
metastasis. However, Ctnnd2 knockout LLCs did not have
metastatic ability (Fig. 1D). Lung
and bone metastasis were confirmed by tissue anatomy and histology
analyses (Fig. 1D and E). Moreover,
lung and bone metastasis of WT LLCs caused mice to die in <20
days but mice injected with Ctnnd2 knockout LLCs survived
for a long time (Fig. 1F). Results
above indicated that loss of δ-catenin inhibited the tumorigenesis
and metastasis of Lewis lung cells.
Consistently, overexpression of Ctnnd2
enhances subcutaneous tumorigenesis and metastasis of LLCs. We
established a stable LLC cell line with Ctnnd2 overexpressed
driven by CMV promoter (Fig. 1G).
We used the human CTNND2 gene cDNA to transfect mouse lung tumor
cells, since the mouse Ctnnd2 cDNA and human CTNND2 cDNA are
highly conserved, particularly the key regions. Human δ-catenin and
mouse δ-catenin have alike functions. Ctnnd2 overexpressed
LLCs could form larger tumors subcutaneously than empty vector
expressed LLCs at the same time point (Fig. 1H). Tail vein injection of either
empty vector expressed LLCs or Ctnnd2 overexpressed LLCs led
to lung or bone metastasis. However, the metastasis of
Ctnnd2 overexpressed LLCs was much faster, resulting in
shorter survival time of mice (Fig.
1I). Taken together, our data suggested that Ctnnd2
coded δ-catenin promoted tumorigenesis and metastasis of Lewis lung
cells in vivo.
δ-Catenin enhances Lewis lung cell
proliferation
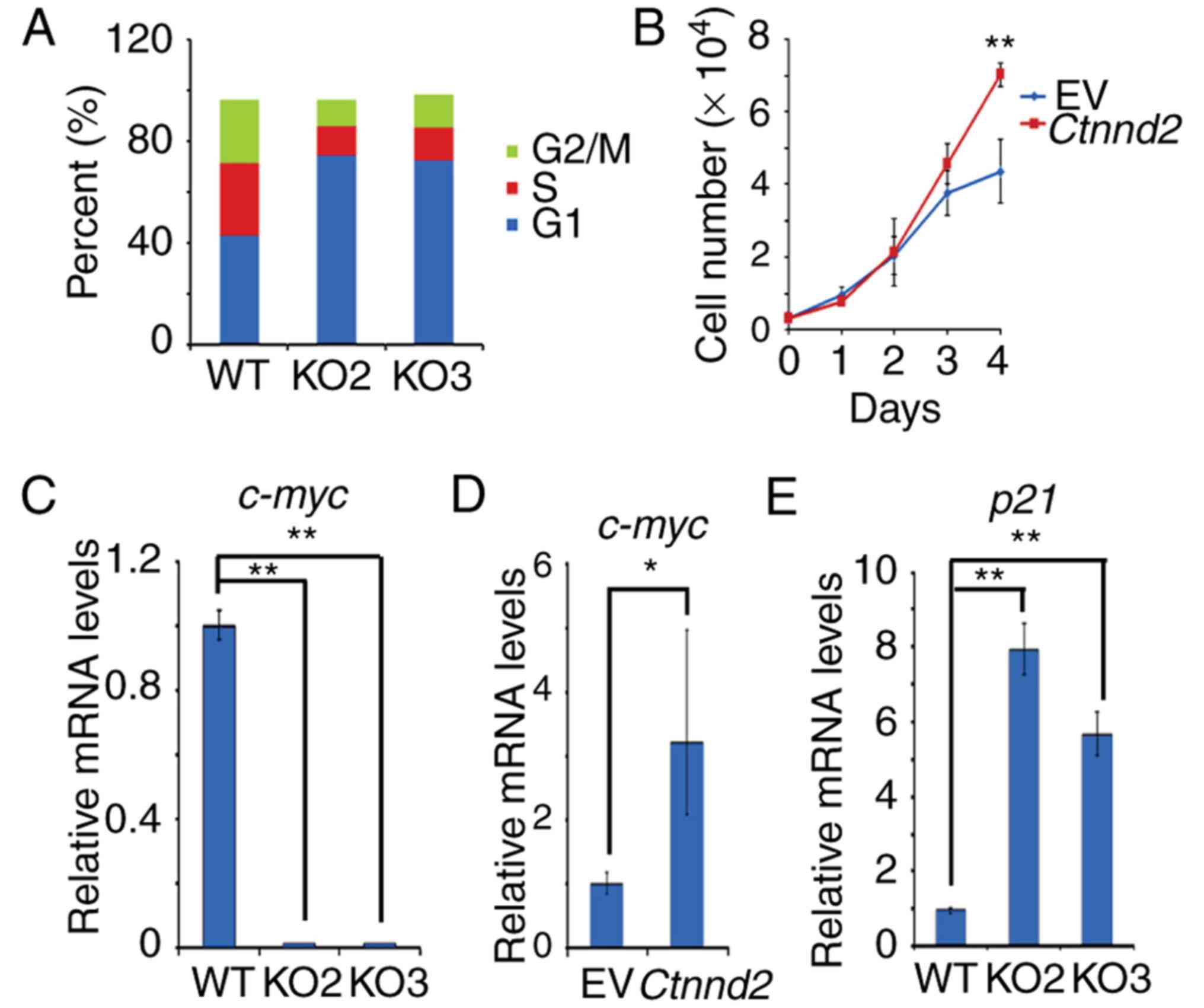
According to the results above, δ-catenin promotes
tumor growth of Lewis lung cells in vivo. Therefore, we
sought to exam whether δ-catenin enhanced the proliferation or cell
cycle progression of Lewis lung cells. Our results showed that
Ctnnd2 knockout could make the cell cycle of Lewis lung
cells arrest in G1 phase but Ctnnd2 overexpression could
enhance the growth of Lewis lung cells (Fig. 2A and B). Furthermore, c-myc, which
promotes cell proliferation and G1-S phase transition (18–20),
was downregulated when Ctnnd2 was knocked out while it was
upregulated with Ctnnd2 overexpressed (Fig. 2C and D). However, the CDK inhibitor,
p21 (21–23), was upregulated when Ctnnd2
was knocked out (Fig. 2E). Taken
together, δ-catenin promoted cell cycle progression and
proliferation via c-myc and p21 in Lewis lung cells.
δ-Catenin promotes G1-S phase
transition of Lewis lung cells in cooperation with canonical Wnt
signaling
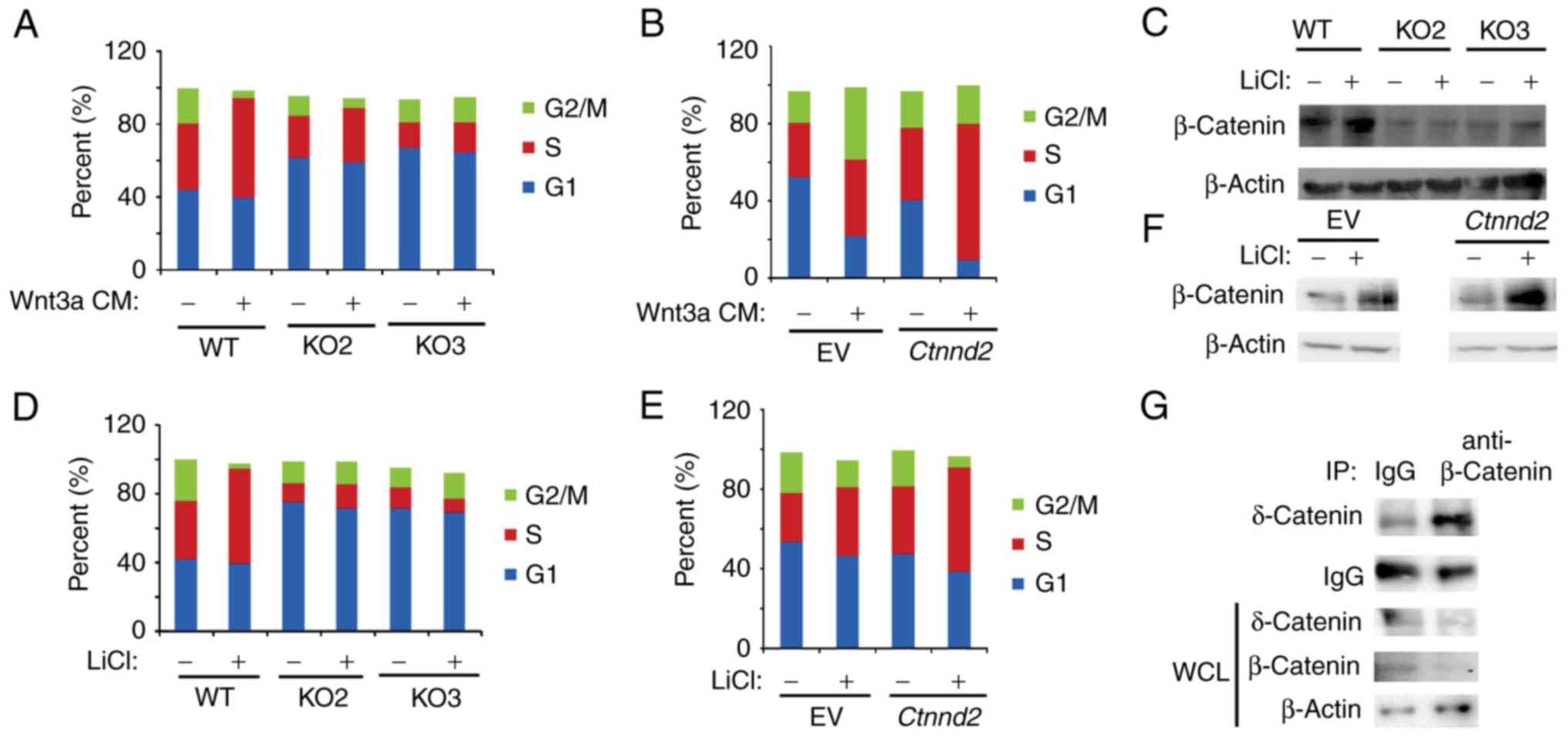
δ-Catenin was reported to promote prostate cancer
malignancy by enhancing Wnt signaling pathway (8). Wnt signaling enhances cell
proliferation by promoting cell cycle progression (24). Thus, we sought to analyze whether
δ-catenin regulated the cell cycle of Lewis lung cells in
cooperation with Wnt3a signaling. We found that Ctnnd2
knockout attenuated G1-S phase transition induced by Wnt3a CM
(Fig. 3A). Consistently,
overexpression of Ctnnd2 facilitated Wnt3a signaling to
promote cell cycle progression (Fig.
3B). In our results, sub-G1 and sub-G2 phase were deducted,
thus, the sum of G1, S and G2 was <100%. The activation of Wnt3a
signaling results in the nuclear accumulation of β-catenin, which
regulates the expression of downstream target genes (25). GSK3β is known to be a classical Wnt
signaling inhibitor by phosphorylating β-catenin to degradation
(26). GSK3β inhibitor, lithium
chloride (LiCl), induces the accumulation of β-catenin (27). Our data demonstrated that
Ctnnd2 knockout could abolish the accumulation of β-catenin
induced by LiCl in Lewis lung cells (Fig. 3C). Such as Wnt3a CM, LiCl enhanced
G1-S phase progression in Lewis lung cells, which was diminished
when Ctnnd2 was knocked out (Fig. 3D). However, overexpression of
Ctnnd2 resulted in β-catenin proteins and S phase
accumulation of Lewis lung cells induced by LiCl (Fig. 3E and F). δ-Catenin may enhance the
Lewis lung cell response to Wnt3a signaling through the interaction
between β-catenin and δ-catenin (Fig.
3G), which needs further investigation.
δ-Catenin enhances the oncosphere
formation of Lewis lung cells
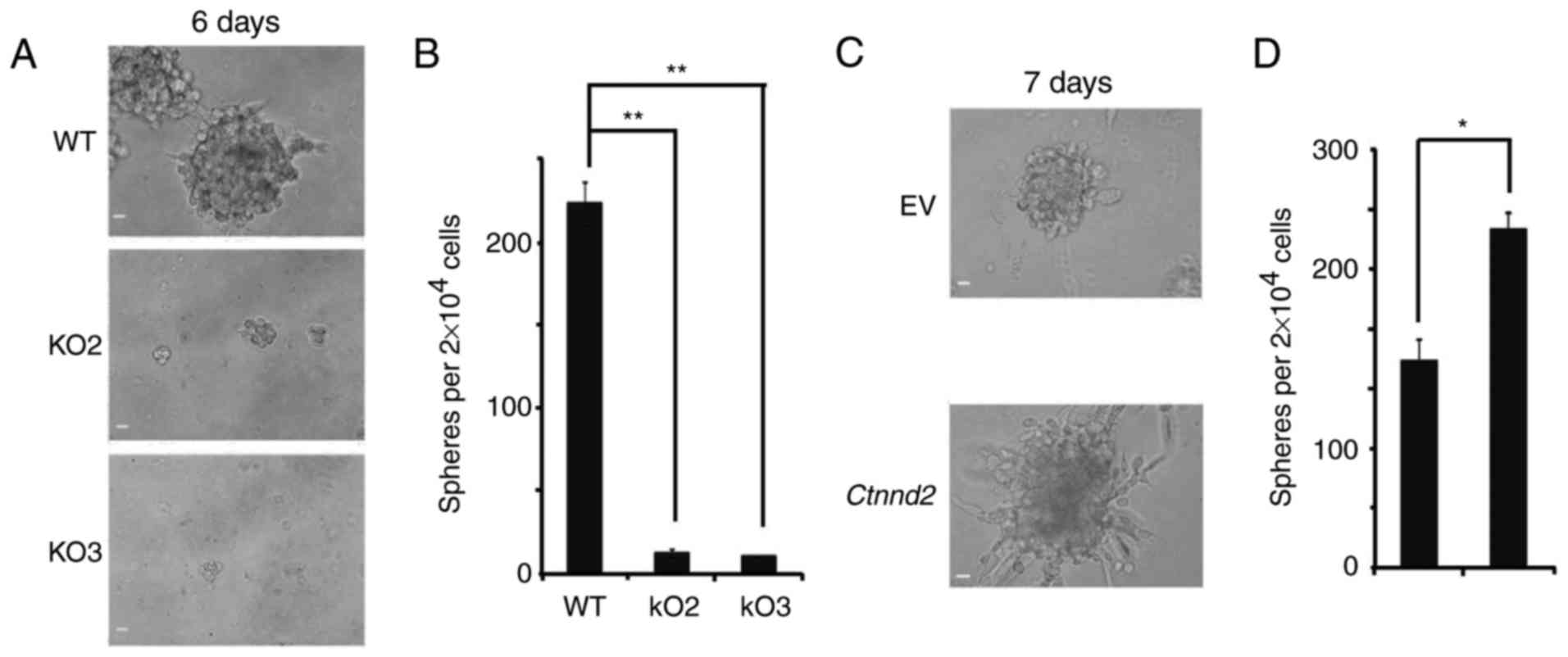
Tumor cells are highly heterogeneous in both
phenotypes and functions. Implanting tumors usually derive from
subpopulations in the highly tumorigenic state. These subsets of
tumor cells, called cancer stem cells, can proliferate
asymmetrically, sustain tumorigenesis and inherent heterogeneity
(28,29). As Lewis lung cells totally lost
their tumorigenic and metastatic ability after Ctnnd2 was
knocked out, we supposed that δ-catenin protein contributed to the
maintenance of cancer stem cells. 3D cultures of tumor cells can
mimic the in vivo growth environment for tumors. Oncospheres
in 3D cultures can show the activity of cancer stem cell subsets
(30,31). WT Lewis lung cells could form
oncospheres under non-adhesive 3D culture conditions, but when
Ctnnd2 was knocked out, Lewis lung cells formed hardly any
oncospheres in Matrigel (Fig. 4A and
B). With Ctnnd2 overexpressed, Lewis lung cells could
form more and larger oncospheres (Fig.
4C and D). Moreover, we observed that overexpression of
Ctnnd2 made Lewis lung cells form highly protrusive
structures with a compact spherical core, indicating Ctnnd2
overexpressed cancer cells had higher invasive ability (32) (Fig.
4C). Results above revealed that, δ-catenin enhanced Lewis lung
cell colonization and invasion. Overexpression of δ-catenin
indicated higher tumorigenic and metastatic ability in Lewis lung
cells.
δ-Catenin enhances the expression of
cancer stem cell markers in Lewis lung cells
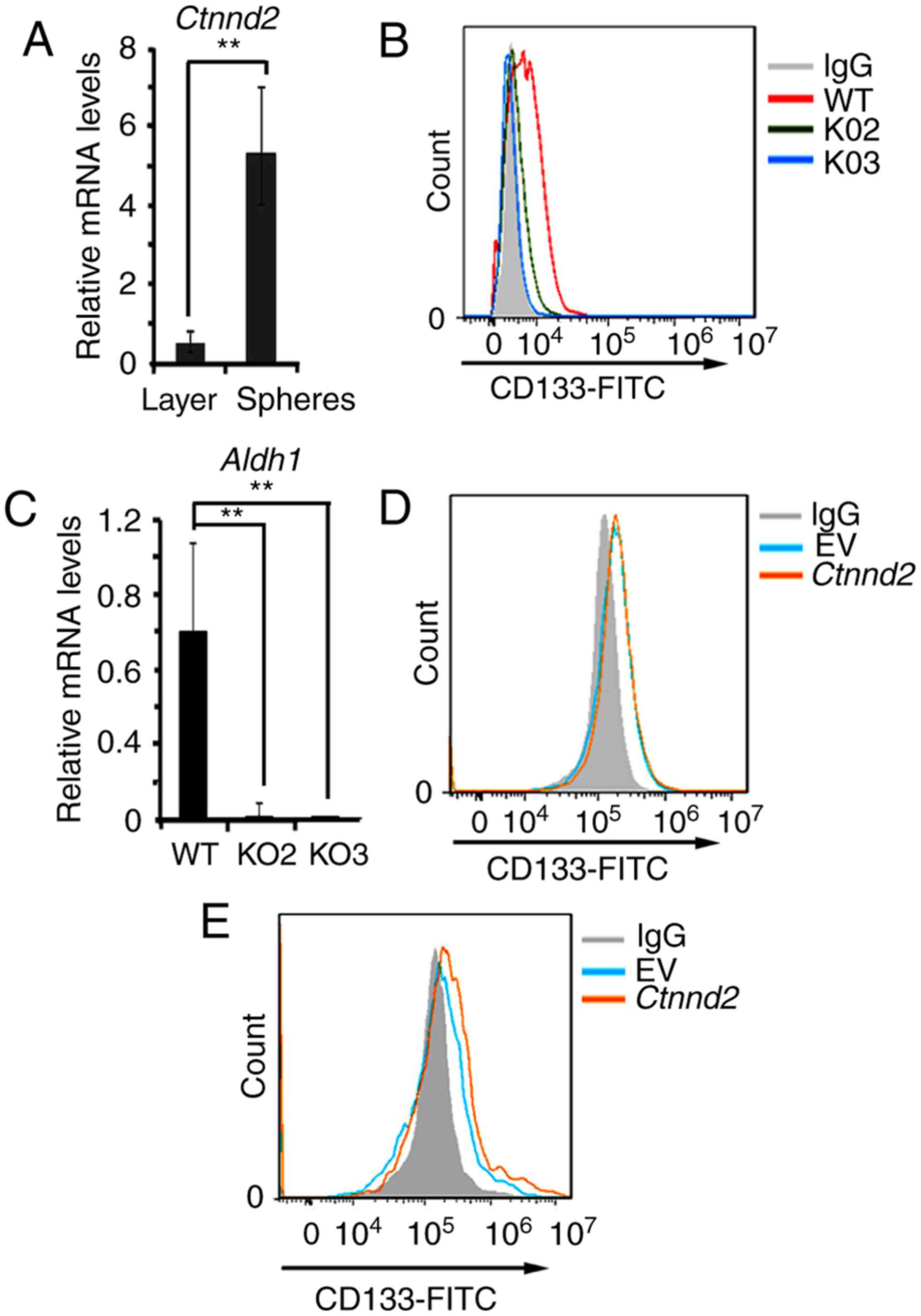
Notably, Ctnnd2 was upregulated in the
oncospheres compared to monolayer cultures of the same samples
(Fig. 5A). Oncospheres in 3D
cultures can show the activity of cancer stem cell subsets
(30,31). We supposed that the expression of
Ctnnd2 may be associated with the expression of cancer stem
cell markers. Later, we measured the expression of lung cancer stem
cell maker CD133 by flow cytometry when Ctnnd2 was knocked
out or overexpressed (33). CD133
in Ctnnd2 knockout cells was downregulated (Fig. 5B). Aldh1, another cancer stem
cell marker, was also downregulated in Ctnnd2 knockout cells
(Fig. 5C). In monolayer cultures,
Ctnnd2 overexpression could not increase the expression of
CD133 (Fig. 5D). However, the
expression of CD133 in Ctnnd2 overexpressed cells was
increased in 3D cultures (Fig. 5E).
Collectively, Ctnnd2 coded δ-catenin was associated with
maintenance of cancer stem cell markers in Lewis lung cells.
δ-catenin promotes the malignancy of
human lung adenocarcinoma
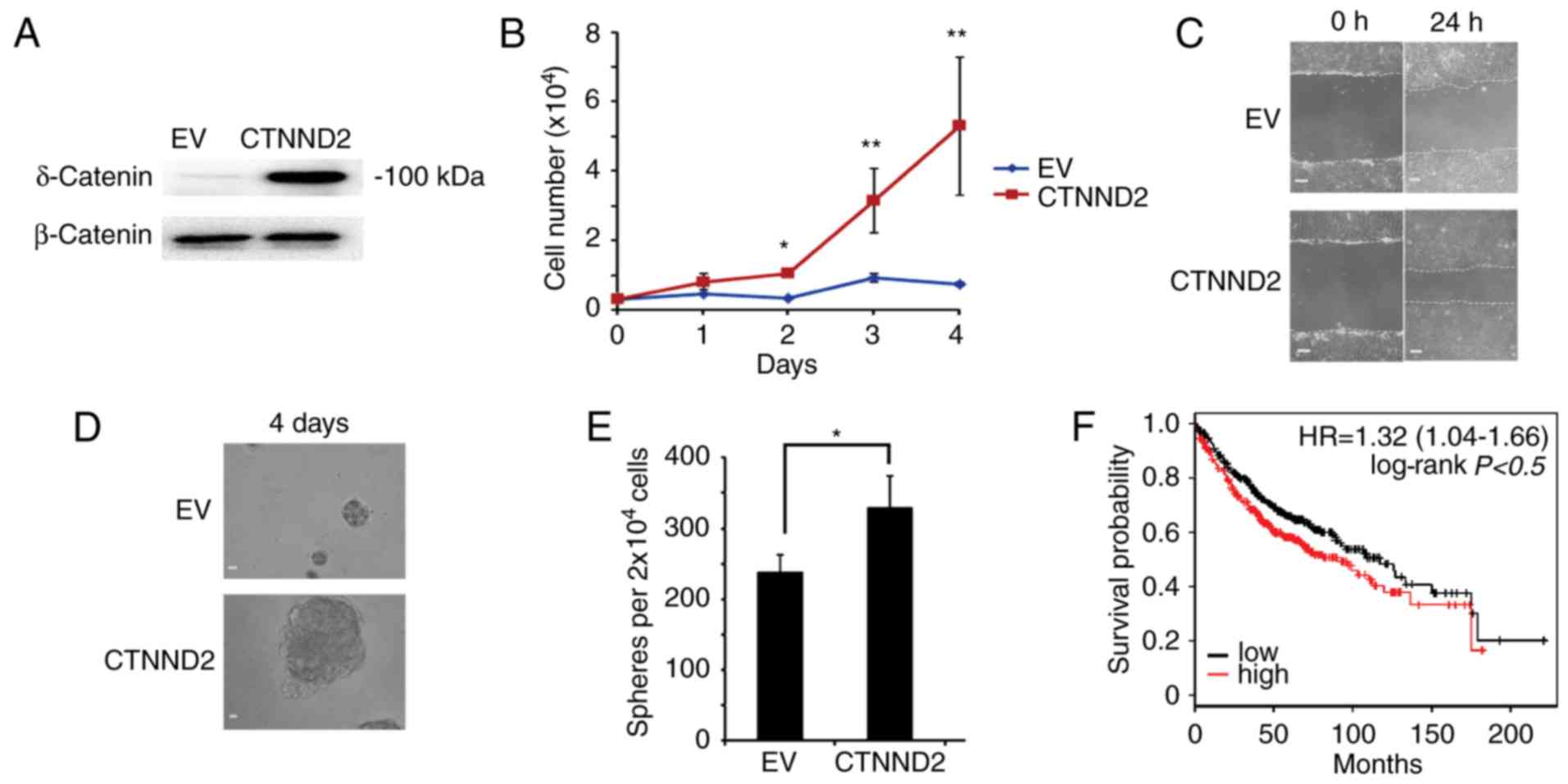
To test whether δ-catenin played roles in human lung
adenocarcinoma as it did in mice, we overexpressed CTNND2 in human
lung adenocarcinoma cell line A549 (34) (Fig.
6A). With CTNND2 overexpressed, A549 cells grew faster,
indicating that δ-catenin enhanced the proliferation of A549 cells
(Fig. 6B). Furthermore, we measured
the migration of A549 cells affected by δ-catenin via scratch wound
assay. We found that the wound shrunk rapidly when CTNND2 was
overexpressed, suggesting that CTNND2 benefited cell migration
(Fig. 6C). Moreover, ectopic
expression of CTNND2 caused more and larger oncosphere formation by
A549 cells (Fig. 6D and E).
Collectively, CTNND2 contributed to the proliferation, migration
and oncosphere formation of A549 cells, indicating that CTNND2
played similar roles in human lung adenocarcinoma cells. In
general, our results demonstrated that δ-catenin promoted the
malignancy of human lung adenocarcinoma.
Discussion
δ-Catenin has been reported to be involved in the
malignancy of various types of cancers. It may be a promising
biomarker for the clinical diagnosis and therapy of cancer
(3,5–7).
According to our results, δ-catenin contributes to the malignancy
of lung adenocarcinoma, owing to its important effects on
tumorigenesis and metastasis. Previous research has proved the
higher expression of δ-catenin in lung cancer (5). The preliminary functional mechanisms
of δ-catenin were also proposed. However, our results throw light
upon new working mechanisms of δ-catenin. δ-catenin promotes cell
proliferation and cell cycle progression via canonical Wnt
signaling pathway in Lewis lung cells. Moreover, the deletion of
δ-catenin results in almost complete loss of cancer stem cells in
Lewis lung cells, indicating a significant role of δ-catenin in
tumorigenesis and metastasis (28,33).
From a public clinical microarray database of lung adenocarcinoma
from 1,157 patients (35), we
estimated that the prognosis of the patients with higher CTNND2
expression was worse than that of these with lower CTNND2
expression (Fig. 6F). In summary,
our current research provides more evidence to prove that δ-catenin
is a promising biomarker and therapy target for lung cancer.
Knockdown of genes by siRNA or shRNA has been used
for years to silence genes. The shortcoming of knockdown is that it
can only partly silence the expression of one gene but the residual
mRNAs and proteins can still have partial functions (36,37).
The CRISP/Cas9 system, developed in recent years, is a new method
for the gene knockout in mammalian cells (38,39).
Compared with knockdown by siRNA or shRNA, knockout via the
CRISP/Cas9 system can absolutely eliminate residual expression of
proteins. Therefore, the gene knockout via CRISP/Cas9 system is a
more reliable method for research on the loss of the gene function.
In our research, the CRISP/Cas9 system was used for Ctnnd2
knockout. The following experiments were based on the Ctnnd2
knockout cell lines. Thus, the roles of δ-catenin in lung
adenocarcinoma cells can be revealed more directly and reliably.
Compared with WT Lewis lung cells, empty vector overexpressed Lewis
lung cells in our experiments had slightly weaker tumorigenesis,
stemness and proliferation ability (Figs. 1H, 3B
and E, and 4C and D), which may
be due to the load of vectors or the G418 selection process. As the
gene Ctnnd2 was subcloned to the same vector and
Ctnnd2 overexpressed Lewis lung cells underwent the same
selection process, we came to the opinion that results from
Ctnnd2 overexpressed Lewis lung cells should be compared
with empty vector overexpressed Lewis lung cells in our
experiments.
Dai et al reported that the expression of
δ-catenin is increased in lung cancer tissues (5), but our research provide direct
evidence that δ-catenin contributes to tumorigenesis and metastasis
of lung adenocarcinoma in mouse models. Moreover, in their data
from 70 patients, higher expression levels of δ-catenin alone
cannot indicate worse prognosis (5). However, in our data from 1,157
patients in Kaplan-Meier plotter database, the higher expression
levels of δ-catenin indicate worse patient prognosis. The
differences may be due to the size of samples. Furthermore, the
effects of δ-catenin on cell proliferation and invasion, which can
support δ-catenin to promote metastasis at the cellular level, are
seriously missing in previous reports. In our research, δ-catenin
was proved to have effects on cell proliferation and cancer stem
cell maintenance.
Different variants with modifications of δ-catenin
are expressed in different cancers (3). It has been reported, in lung cancer
cell line, NIC-H1299, that fragments with low molecular weight of
δ-catenin are expressed (3).
According to our data, δ-catenin expressed in Lewis lung cells and
A549 cells migrate faster than the 100 kDa protein marker (Figs. 1B and G, and 6A). Furthermore, the CMV promoter-driven
exogenously expressed δ-catenin has the same molecular weight with
the endogenous expressed δ-catenin. Collectively, probably
post-translational modifications regulate the expression of
δ-catenin fragments in lung cancer cells.
The mechanisms how δ-catenin promotes the malignant
progression of cancer may be complicated. Mutations of δ-catenin
have been proven to contribute to its oncogenic functions. In
prostate cancer, −9 G>A mutation in 5′-UTR promotes δ-catenin
expression (6). Moreover, truncated
mutations of CTNND2 enhance metabolic reprogramming, hypoxia
pathways and Wnt signaling response to promote cancer cell survival
in prostate cancer (8). In our
research, mutations of CTNND2 in lung cancer were not tested.
Oncogenic roles of CTNND2 mutations in lung cancer development may
be a spot to be focused on in the future. However, based on our
research, CTNND2 coded protein, δ-catenin enhanced canonical Wnt3a
signaling by reinforcing the accumulation of β-catenin, but the
detailed mechanism is not clear. CTNND1 has been reported to
facilitate β-catenin to shuttle from the cytoplasm to the nuclear
(40). The interaction between
β-catenin and δ-catenin is confirmed in our research. Other
mechanisms can be further investigated in the future.
δ-Catenin can be a clinical diagnosis marker and
therapy target for cancer (3).
According to our results and former reports, mutants or high
expression of δ-catenin indicate poorer prognosis (5–8).
Therefore, examination of CTNND2 mutations or immunohistochemistry
of δ-catenin in tumor tissues can be applied to anticipate the
patients prognosis or select treatment plans. Deletion of CTNND2
attenuates tumor growth and metastasis. Thus, inhibitors or
antibodies of δ-catenin can be developed for cancer therapy.
Currently, neither inhibitors nor antibodies have been developed
for the abolishment of δ-catenin, which can be a promising future
research area.
Acknowledgements
We thank Ruiqing Chen and Jingan Lin for excellent
technical assistance. The present study was supported by grants
from the Innovative Project of Young Scientists and Technicians in
Fujian Province (2616J05075), and the Fujian Platform for Medical
Research, First Affiliated Hospital, Fujian Medical University.
References
|
1
|
Chen Z, Fillmore CM, Hammerman PS, Kim CF
and Wong KK: Non-small-cell lung cancers: A heterogeneous set of
diseases. Nat Rev Cancer. 14:535–546. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sharma SV, Bell DW, Settleman J and Haber
DA: Epidermal growth factor receptor mutations in lung cancer. Nat
Rev Cancer. 7:169–181. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lu Q, Lanford GW, Hong H and Chen YH:
δ-Catenin as a potential cancer biomarker. Pathol Int. 64:243–246.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Medina M, Marinescu RC, Overhauser J and
Kosik KS: Hemizygosity of delta-catenin (CTNND2) is associated with
severe mental retardation in cri-du-chat syndrome. Genomics.
63:157–164. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dai SD, Wang Y, Zhang JY, Zhang D, Zhang
PX, Jiang GY, Han Y, Zhang S, Cui QZ and Wang EH: Upregulation of
δ-catenin is associated with poor prognosis and enhances
transcriptional activity through Kaiso in non-small-cell lung
cancer. Cancer Sci. 102:95–103. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang T, Chen YH, Hong H, Zeng Y, Zhang J,
Lu JP, Jeansonne B and Lu Q: Increased nucleotide polymorphic
changes in the 5′-untranslated region of δ-catenin (CTNND2) gene in
prostate cancer. Oncogene. 28:555–564. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fang Y, Li Z, Wang X and Zhang S:
Expression and biological role of δ-catenin in human ovarian
cancer. J Cancer Res Clin Oncol. 138:1769–1776. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nopparat J, Zhang J, Lu JP, Chen YH, Zheng
D, Neufer PD, Fan JM, Hong H, Boykin C and Lu Q: δ-catenin, a
Wnt/β-catenin modulator, reveals inducible mutagenesis promoting
cancer cell survival adaptation and metabolic reprogramming.
Oncogene. 34:1542–1552. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim H, He Y, Yang I, Zeng Y, Kim Y, Seo
YW, Murnane MJ, Jung C, Lee JH, Min JJ, et al: δ-Catenin promotes
E-cadherin processing and activates β-catenin-mediated signaling:
Implications on human prostate cancer progression. Biochim Biophys
Acta. 1822:509–521. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu XL, Liu LD, Zhang SG, Dai SD, Li WY
and Zhang L: Correlation between expression and significance of
δ-catenin, CD31, and VEGF of non-small cell lung cancer. Genet Mol
Res. 14:13496–13503. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li XY, Liu SL, Cha N, Zhao YJ, Wang SC, Li
WN, Wang EH and Wu GP: Transcription expression and clinical
significance of dishevelled-3 mRNA and δ-catenin mRNA in pleural
effusions from patients with lung cancer. Clin Dev Immunol.
2012:9049462012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Viloria-Petit AM and Wrana JL: The
TGFbeta-Par6 polarity pathway: Linking the Par complex to EMT and
breast cancer progression. Cell Cycle. 9:623–624. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zuo W and Chen YG: Specific activation of
mitogen-activated protein kinase by transforming growth factor-beta
receptors in lipid rafts is required for epithelial cell
plasticity. Mol Biol Cell. 20:1020–1029. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bertram JS and Janik P: Establishment of a
cloned line of Lewis lung carcinoma cells adapted to cell culture.
Cancer Lett. 11:63–73. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
O'Reilly MS, Holmgren L, Shing Y, Chen C,
Rosenthal RA, Moses M, Lane WS, Cao Y, Sage EH and Folkman J:
Angiostatin: A novel angiogenesis inhibitor that mediates the
suppression of metastases by a Lewis lung carcinoma. Cell.
79:315–328. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mali P, Yang L, Esvelt KM, Aach J, Guell
M, DiCarlo JE, Norville JE and Church GM: RNA-guided human genome
engineering via Cas9. Science. 339:823–826. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cong L, Ran FA, Cox D, Lin S, Barretto R,
Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al: Multiplex
genome engineering using CRISPR/Cas systems. Science. 339:819–823.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
O'Donnell KA, Wentzel EA, Zeller KI, Dang
CV and Mendell JT: c-Myc-regulated microRNAs modulate E2F1
expression. Nature. 435:839–843. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dang CV: c-Myc target genes involved in
cell growth, apoptosis, and metabolism. Mol Cell Biol. 19:1–11.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Evan GI and Littlewood TD: The role of
c-myc in cell growth. Curr Opin Genet Dev. 3:44–49. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Claassen GF and Hann SR: A role for
transcriptional repression of p21CIP1 by c-Myc in overcoming
transforming growth factor beta-induced cell-cycle arrest. Proc
Natl Acad Sci USA. 97:pp. 9498–9503. 2000; View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chin YE, Kitagawa M, Su WC, You ZH,
Iwamoto Y and Fu XY: Cell growth arrest and induction of
cyclin-dependent kinase inhibitor p21WAF1/CIP1 mediated by STAT1.
Science. 272:719–722. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Macleod KF, Sherry N, Hannon G, Beach D,
Tokino T, Kinzler K, Vogelstein B and Jacks T: p53-dependent and
independent expression of p21 during cell growth, differentiation,
and DNA damage. Genes Dev. 9:935–944. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Reya T and Clevers H: Wnt signalling in
stem cells and cancer. Nature. 434:843–850. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang Y: Wnt signaling in development and
disease. Cell Biosci. 2:142012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Salic A, Lee E, Mayer L and Kirschner MW:
Control of beta-catenin stability: Reconstitution of the
cytoplasmic steps of the wnt pathway in Xenopus egg extracts. Mol
Cell. 5:523–532. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cohen P and Goedert M: GSK3 inhibitors:
Development and therapeutic potential. Nat Rev Drug Discov.
3:479–487. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Visvader JE and Lindeman GJ: Cancer stem
cells: Current status and evolving complexities. Cell Stem Cell.
10:717–728. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Meacham CE and Morrison SJ: Tumour
heterogeneity and cancer cell plasticity. Nature. 501:328–337.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yamada KM and Cukierman E: Modeling tissue
morphogenesis and cancer in 3D. Cell. 130:601–610. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bissell MJ and Labarge MA: Context, tissue
plasticity, and cancer: Are tumor stem cells also regulated by the
microenvironment? Cancer Cell. 7:17–23. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Viloria-Petit AM, David L, Jia JY, Erdemir
T, Bane AL, Pinnaduwage D, Roncari L, Narimatsu M, Bose R, Moffat
J, et al: A role for the TGFbeta-Par6 polarity pathway in breast
cancer progression. Proc Natl Acad Sci USA. 106:pp. 14028–14033.
2009; View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Eramo A, Lotti F, Sette G, Pilozzi E,
Biffoni M, Di Virgilio A, Conticello C, Ruco L, Peschle C and De
Maria R: Identification and expansion of the tumorigenic lung
cancer stem cell population. Cell Death Differ. 15:504–514. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Giard DJ, Aaronson SA, Todaro GJ, Arnstein
P, Kersey JH, Dosik H and Parks WP: In vitro cultivation of human
tumors: Establishment of cell lines derived from a series of solid
tumors. J Natl Cancer Inst. 51:1417–1423. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Győrffy B, Surowiak P, Budczies J and
Lánczky A: Online survival analysis software to assess the
prognostic value of biomarkers using transcriptomic data in
non-small-cell lung cancer. PLoS One. 8:e822412013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Reynolds A, Leake D, Boese Q, Scaringe S,
Marshall WS and Khvorova A: Rational siRNA design for RNA
interference. Nat Biotechnol. 22:326–330. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Silva JM, Li MZ, Chang K, Ge W, Golding
MC, Rickles RJ, Siolas D, Hu G, Paddison PJ, Schlabach MR, et al:
Second-generation shRNA libraries covering the mouse and human
genomes. Nat Genet. 37:1281–1288. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cho SW, Kim S, Kim JM and Kim JS: Targeted
genome engineering in human cells with the Cas9 RNA-guided
endonuclease. Nat Biotechnol. 31:230–232. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hsu PD, Scott DA, Weinstein JA, Ran FA,
Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al: DNA
targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol.
31:827–832. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yamada N, Noguchi S, Mori T, Naoe T, Maruo
K and Akao Y: Tumor-suppressive microRNA-145 targets catenin δ-1 to
regulate Wnt/β-catenin signaling in human colon cancer cells.
Cancer Lett. 335:332–342. 2013. View Article : Google Scholar : PubMed/NCBI
|