Introduction
Activating cyclin-dependent kinases (CDKs) promotes
cell cycle progression, and abnormal cell cycle regulation lies at
the heart of tumorigenesis (1),
leading to uncontrolled cell cycle progression and cell
proliferation. Activated CDKs lead to genomic instability (GIN),
dysregulated proliferation, and chromosomal instability (CIN),
resulting in proliferative advantages and susceptibility to genetic
alterations (1). During cell cycle
progression, the restriction point from G1-S phase is regulated by
CDK4/6 and plays a crucial role in tumorigenesis; thus, inhibitors
targeting this transition show promise as clinical therapy
(2). Recently, therapeutic regimens
targeting a dysregulated cell cycle caused by misregulated CDKs
have shown promise in the treatment of diverse cancers. While the
first generation of CDK4/6 inhibitors was quickly abandoned because
pan-CDK inhibition was toxic to normal cells and led to
unacceptable side effects (3), new
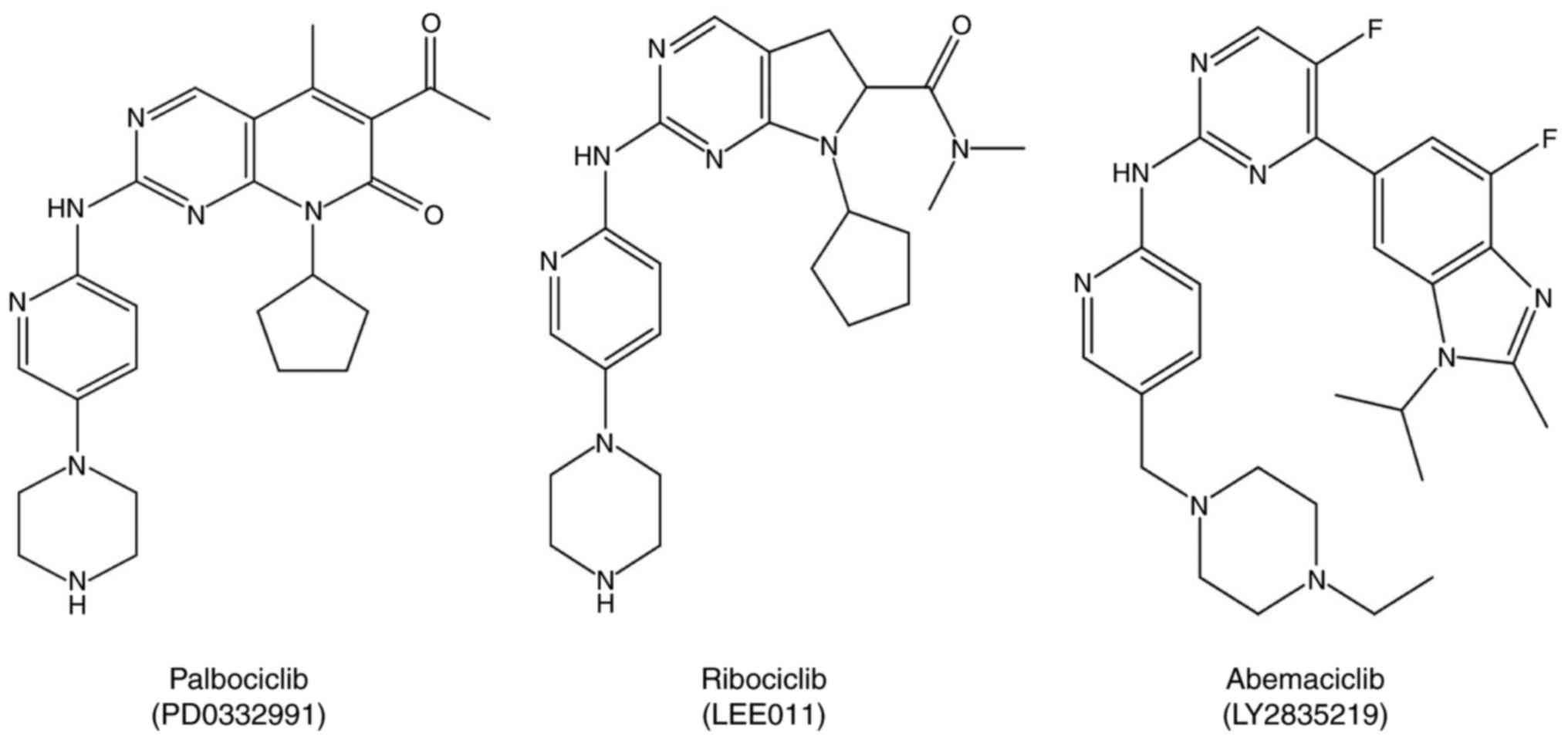
CDK4/6 inhibitors, such as palbociclib, abemaciclib, and ribociclib
(Fig. 1), overcame the
disadvantages of the first generation of CDK4/6 inhibitors, showing
promising anticancer effects and manageable toxicity. Of these
three drugs, palbociclib and ribociclib have recently been approved
by the Food and Drug Administration (FDA) as a new treatment
strategy in combination with letrozole for hormone receptor
(HR)-positive, advanced-stage breast cancer, showing promising
results for these patients (4).
Additionally, palbociclib has been successfully applied in multiple
myeloma therapy. However, further research is required to define
the patients who would receive the greatest benefit, test possible
combination treatments in other cancers, and interrogate acquired
resistance. Finally, to make efforts to establish new treatment
pattern, cell cycle inhibitor+molecular targeted drug. In this
review, we discuss normal cell cycle regulation associated with
CDK4/6 inhibitors as well as the use of palbociclib to treat
different cancers and its advantages in clinical application, and
propose a new cancer treatment pattern.
CDKs and cell cycle regulation
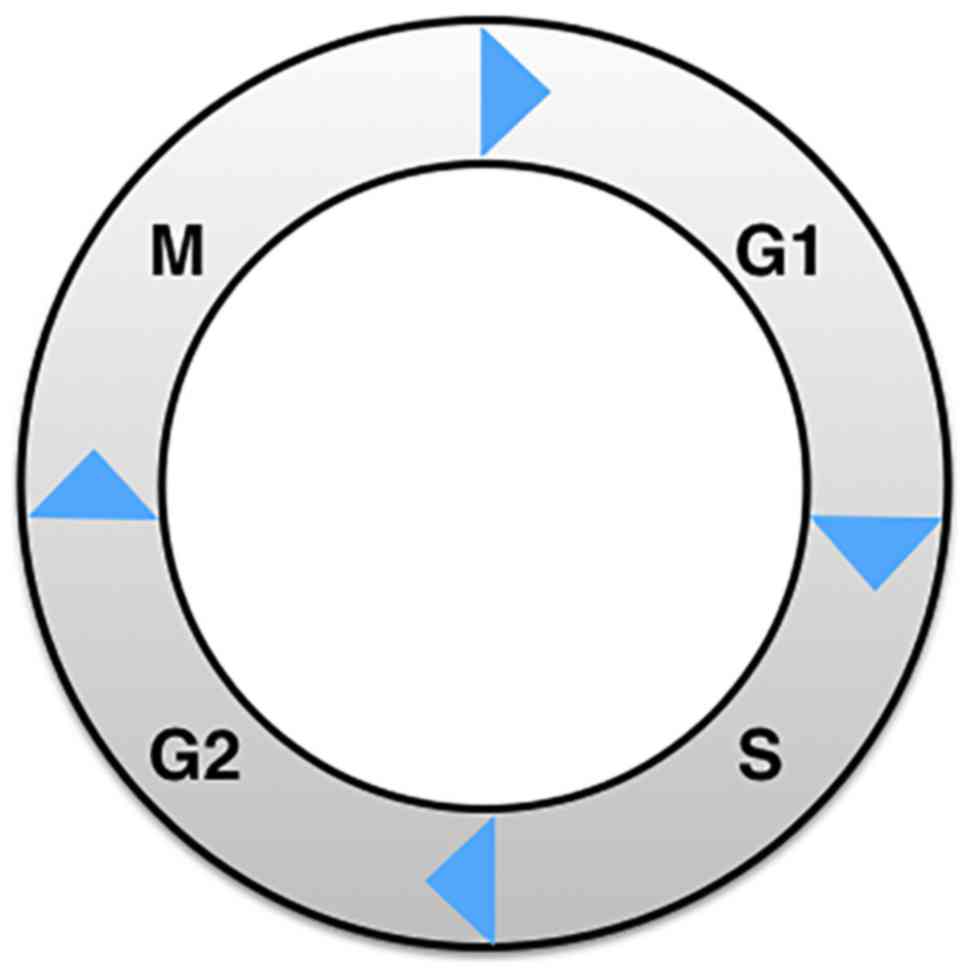
Cell cycle regulation is complex and comprises two
major phases: interphase and mitosis (M phase; Fig. 2). The former can be subdivided into
three phases: G1 phase for preparing for DNA synthesis, S phase for
DNA synthesis, and finally G2 phase for preparing for cell
division. The M phase includes prophase, metaphase, anaphase, and
telophase. After M phase, one cell will have divided into two
daughter cells. During interphase, the G1-S transition is a
critical restriction point, resulting in one of three fates for the
cell: continue cycling, exit active proliferation, or enter a
quiescent (G0) state. Many growth factors and inhibitors interact
to coordinate cell cycle progression, and the CDK4/6-Rb pathway
(5,6) plays a central role in regulating the
G1 to S phase transition.
So far, 21 cyclin-dependent kinases (CDKs) have been
identified in mammalian cells by the HUGO Gene Nomenclature
Committee (HGNC) and the Mouse Genomic Nomenclature Committee
(7). These CDKs can be categorized
into cell cycle related subfamilies and transcriptional subfamilies
on a functional basis (8). The
former group includes CDK1 (9),
CDK4 (10), CDK6 and CDK5 (11). The latter group includes CDK7
(12), CDK8 (13), CDK9 (14), CDK11 (15–18)
and CDK20 (19). This
transcriptional group can phosphorylate the c-terminal domain (CTD)
of RNAPII. CDKs can also regulate cell cycle progression at
different levels (12). For
example, CDK7 is a member of the CDK Activating Kinase (CAK,
CDK7/CycH/MAT1) complex and is involved in phosphorylating many
CDKs (12).
Among the 21 CDKs, some are critical to drive cells
through G1 phase into S phase, such as CDK2, CDK4 and CDK6. CDK2,
consisting of 298 amino acids, is an important CDK that is involved
in the transition from G1 to S phase and S phase progression
(20). Abnormal CDK2 activation
leads to cell cycle disorders and may induce tumorigenesis. It has
two associated cyclins that can regulate the cell cycle: cyclins A
and E (21). The cyclin A-CDK2
complex is active during S phase and the cyclin E-CDK2 complex is
involved in the G1/S transition by phosphorylating the Rb protein
(22,23). Thus, CDK2 inhibition can disrupt the
proliferation of cancer cells like prostate cancer (24), breast cancer (25) and non-small cell lung cancer
(26).
Two important cell cycle regulating CDKs are CDK4
and CDK6, which have pivotal roles in the transition from G1 to S
phase. CDK4 and CDK6 are similar in the ircrystal structures and
functions (27). External growth
factors regulate CDK4 and CDK6, facilitating their combination with
cyclin D (D1, D2, D3) to form CDK4/6-cyclin D complexes. These
complexes regulate progression through the checkpoint from G1 to S
phase in mammalian cells (10).
Recent studies have shown that CDK4 and CDK6 can interact outside
of the context of cell cycle regulation, leading to tumorigenesis.
For example, CDK4 and CDK6 affect vascular endothelial growth
factor (VEGF)-B and VEGF-A, respectively. They both have also been
identified as regulatorsof inflammatory cytokines through
activation of the nuclear factor (NF)-κB pathway (28). CDK4 and CDK6 are also associated
with DNA repair (29), senescence
(30) and metabolism (31). In these non-cell cycle functions,
the kinase functions of CDK4 and CDK6 play important roles; thus,
selective molecules such as palbociclib (PD 0332991), ribociclib
(LEE011), and abemaciclib (LY2835219) can disrupt CDK-associated
pathways (Fig. 1).
Other functions of CDK family members are shown in
Table I. These CDKs all contribute
to the progression of tumorigenesis, and some are critical cell
cycle modulators. Thus, they may be promising targets for novel
cancer treatments.
 | Table I.CDK family members. |
Table I.
CDK family members.
| Name of CDKs | Synonym | Major partner | Functions |
|---|
| CDK1 | Cell division
control protein 2 (Cdc2) | A-type and B-type
cyclins | It is essential for
mitosis in all cells and can promote centrosome maturation and
separation, chromosome condensation, and mitotic entry |
| CDK2 |
| E-type cyclins | Initiates DNA
replication and repair, but is dispensable for the mitotic cell
cycle. It is also crucial for the first meiotic division of male
and female germ cells |
| CDK3 |
| E-type and A-type
cyclins, Cdk5 and Abl1 enzyme substrate subfamily | Inactivation of
pRb |
| CDK4 | PSK-J3 | Cyclin D | CDK4/6 is involved
in the transition from G1 to S phase and associated with
cytoskeletal rearrangement and cell migration |
| CDK5 | PSSALRE | Cdk5R1 (P35) Cdk5R2
(P39) No corresponding cyclin protein | Associated with
transcription, neuronal function, migration and synaptic
transmission |
| CDK6 | PLSTIRE | Cyclin D | CDK4/6 is involved
in the transition from G1 to S phase and associated with
cytoskeletal rearrangement and cell migration |
| CDK7 |
|
| A component of the
Cdk-activating kinase (CAK, which phosphorylates and presumably
activates all cell cycle Cdks |
| CDK8 |
| Cyclin C | A component of the
RNA polymerase holoenzyme involved in transcription that can
phosphorylate cyclin H to inhibit CAK activity |
| CDK9 |
| Cyclin T cyclin
K | Forms positive
transcription elongation factor b (P-TEFb) |
| CDK10 |
| Not found | Its antisense and
dominant-negative mutants arrest cells in G2-M; it also inhibits
transactivation of the Ets2 transcription factor |
| CDK11 | PITSLRE | Cyclin L | An important factor
involved in pre-mRNA splicing in combination with the splicing
protein 9G8 |
The Rb-E2F pathway and tumorigenesis
The retinoblastoma tumor suppressor gene (Rb) was
first discovered in human retinoblastoma (1). It is an ancestral protein that
regulates cell cycle progression by promoting G1 to S phase entry.
Multiple studies have shown that Rb functions in diverse cellular
pathways, including apoptosis and the cell cycle. Inactivation of
the Rb protein is an event that can occur in most cancers. The
mechanisms underlying this inactivation include mutation of Rb
itself, Rb loss by methylation or chromosomal deletion, or via
upstream signaling molecule interactions such as INK4 and K-Ras
(32).
Previous studies have identified three Rb family
members: Rb, p107, and p130. Not all organisms have all three
members: most lower and unicellular organisms have only one
Rb-related protein that functions in cell cycle progression
(33). However, in mammalian cells,
Rb as well as the Rb-like proteins p130 and p107 can all be
detected and regulate the cell cycle with BMYB (MYBL2) and Forkhead
box M1(FOXM1) (34). In the past
few years, many experts have asserted that Rb is stronger than p107
or p130 with respect to a tumor suppressive function in the cell
cycle. Rb gene knockout in mice results in embryonic lethality,
which is not seen in p107 or p130 deficient mice (35,36).
The precise mechanisms underlying differences in Rb, p107 and p130
functions, however, remain unclear.
The other member of the Rb-E2F pathway is E2Fs, a
family of transcription factors that regulate cell cycle
progression, cellular proliferation, and DNA synthesis (32). E2F is a large family, members of
which have many conserved DNA binding domains that bind target
promoters and could promote or restrain target gene expression
(37,38). The E2F family includes ten members,
most of which function as cell cycle regulators. The first group
includes the strong transcriptional activators E2F1-E2F3a, which
are regulated by Rb. The second group comprises the passive
repressors E2F3b-E2F5, which interact with the other Rb family
members, p107 and p130, to modulate transcriptional activity of
target gene promoters. The third group, E2F6, 7a, 7b and 8, have
been identified as active repressors of transcription (39,40).
The first group of E2Fs is responsible for promoting proliferation
by inhibiting the expression of growth inhibitory proteins like
p16, p21 and p27 as well as promoting genes associated with
proliferation-related expression in the G1-S transition (41–43).
Unlike E2F1-3, E2F4 and E2F5 can be detected uniformly in quiescent
(G0) cells, and they function as repressors of E2F-responsive genes
(44–46). E2F7 and E2F8 are both important
during embryonic development and regulating the E2F1-p53 apoptotic
axis as repressors (47). They are
highly expressed in mid- to late S phase, and repress target gene
transcription in G1/S (48).
The Rb-E2F pathway is almost always disrupted in
human cancers. It is believed that this is the initial hit of most
cancers, including lung cancer. The Rb-E2F pathway can regulate
tumor progression, influencing angiogenesis and metastasis.
Usually, Rb is inactivated through mutation or deletion and E2F
transcription factors are activated, thus leading to increased
proliferation in almost all tumors.
In early G1 phase, endogenous or exogenous mitogenic
stimuli function induce expression of cyclin D (D1, D2 and D3), and
activated cyclin D form complexes with CDK4 and CDK6, termed the
cyclin D-CDK4/6 complex. The active complex will then phosphorylate
the C-terminal region of Rb or other members of the Rb family,
depending on cell type (49,50).
Hyper-phosphorylated Rb proteins release E2Fs, allowing them to
participate in synthesizing S phase initiating proteins. While in
the late G1 phase, the previous activation in the early G1 will
also participate in the generation of molecules like cyclin E that
will bind and activate CDK2 in the G1/S transition (51,52).
The CDK2-cyclin E complex and activated CDK2 can potently
phosphorylate and inactivate Rb, simultaneously regulating the
protein switch controlled by E2Fs to participate in the transition
from G1 to S and DNA replication.
Clearly understanding the mechanism of cell cycle
regulation can inform tumor treatment strategies. In the past few
years, many targeted therapies for cancer, such as Herceptin and
Tarceva, have emerged and changed clinical treatments; however,
patient responses are imperfect. Growth signaling pathways are
web-like, and blocking all possible cross-talking pathways and
feedback loops using one or two drugs is difficult or impossible.
The cell cycle, however, is not like a growth pathway: it is a
one-way street (53). Thus,
inhibitors of cell cycle proteins may be promising in a therapeutic
setting.
CDK2 inhibitors are promising and they can be
divided into two types according to their binding sites:
ATP-competitive and non-ATP-competitive inhibitors. ATP-competitive
inhibitors of CDK2 have evolved into second-generation inhibitors,
although first generation inhibitors have low inhibitory effects
and potent side effects. Second-generation inhibitors of CDK2 have
strong anti-proliferative effects, and some are being evaluated in
ongoing preclinical and clinical trials, including AT7519 (54), NU2058 (55) and P276-00 (56). The other type of CDK2 inhibitor,
non-ATP competitive inhibitors Spa310 (58) and CIP (59), have high specificity and show
promise in clinical applications in the treatment of cancer. The
mechanisms of these inhibitors center around CDK2-cyclin complexes
and binding sites.
The CDK4/6 inhibitors palbociclib (PD0332991),
abemaciclib (LY2835219), and ribociclib (LEE011) have also been
studied in many types of cancers including breast cancer, multiple
myeloma, and lung cancer. Of these CDK inhibitors, palbociclib in
particular has been used in phase III clinical trial development
(60).
Palbociclib and its applications in
different cancers
Palbociclib (PD0332991, Ibrance®, Pfizer
Inc.) is an oral, reversible small molecule inhibitor of
cyclin-dependent kinases 4 and 6 (CDK4/6). While CDK4/6 can bind
cyclin D1, resulting in Rb hyperphosphorylation, palbociclib can
separate CDK4/6-cyclin D1 complexes, blocking Rb phosphorylation
and preventing E2F1 release, thus leading to G1 phase arrest and
tumor growth suppression (61). The
elimination half-life of palbociclib is over 29 h. The primary
metabolic region is the liver, being metabolized by SULT2A1 and
CYP3A, and the primary excretion routes are feces (74.1%) and
kidneys (17.5%).
Applications of palbociclib in different
cancers
Since the 1990s, most cell cycle inhibitors produced
by Roche, Bristol-Myers Squibb, Kyowa Hakko Kirin, and others have
been used in clinical trials. However, these pioneering pan-CDK
inhibitors were halted soon thereafter due to a narrow therapeutic
window, lack of efficacy in solid tumors, toxicity issues, and
challenges with dosing schedules. After years of development, the
current CDK4/6 inhibitors became more specific and/or more potent.
Specific inhibitors of CDK4/6 appear to be less toxic, have mild
bone marrow suppression, are able to be orally administered, and
facilitate dosing schedules. These merits have led to their
applications in different cancers (62). In 2004, a molecular biologist at
Weill Cornell Medical College, Selina Chen-Kiang, demonstrated that
palbociclib functions in blood cancer. In almost all cancers,
including breast cancer, mutation of Rb1 or components regulating
the CDK-RB-E2F pathway is often observed. Thus, the use of
cyclin-dependent kinase (CDK) inhibitors to re-establish cell cycle
control has been an attractive therapy in the treatment of breast
cancer.
Breast cancer
Breast cancer is the most commonly-diagnosed tumor
in women. More than 75% of patients diagnosed with metastatic
breast cancer are estrogen receptor and progesterone receptor
positive (HMR-positive). Compared with other types of metastatic
breast cancers, HMR-positive metastatic breast cancer has a more
favorable median overall survival (24–36 months) from time of
diagnosis (63). When treating this
type of breast cancer, the goal is to ensure quality of life,
prolonging survival and postponing death. Previously, chemotherapy
was thought to be the first-line therapy for HMR-positive breast
cancer. In February 2015, however, the Food and Drug Administration
(FDA) approved palbociclib as an initial endocrine-based therapy in
combination with letrozole in postmenopausal women with
HMR-positive, Her2-negative advanced breast cancer (64). Palbociclib is also effective in
HR-positive metastatic breast cancer patients with fulvestrant
(4).
Until now, the use of the selective CDK4/6 inhibitor
palbociclib combined with endocrine therapy to treat estrogen
receptor-positive breast cancer patients was the most successful
application of CDK inhibition. The data from the PALOMA-I trials
showed that letrozole plus palbociclib confers longer median
progression-free survival (PFS) than letrozole alone. This study
included two phases: phase I and phase II. Slamon et al
conducted the phase I portion of PALOMA-I. They assessed the safety
and tolerability of palbociclib in combination with letrozole; the
most common side effects are leucopenia, neutropenia, and fatigue.
The two combined drugs have no pharmacokinetic interaction. Twelve
patients were enrolled in this portion, nine of whom had stable
disease for ≥6 months, and three of whom had a partial response.
Finn et al conducted the phase II portion including two
cohorts. The members enrolled in cohort 1 comprise an unselected
population to identify the safety and efficacy of the combination
of palbociclib and letrozole. In cohort 2, patients with cyclin D1
amplification and/or loss of p16 were enrolled with the goal of
identifying the endpoint of investigator-assessed PFS. The median
PFS was 20.2 months for the combination as compared to 10.2 months
for letrozole alone [hazard ratio (HR)=0.488; 95% confidence
interval (CI), 0.319–0.748; 1-sided P=0.0004]. Both PALOMA-I phase
studies showed that palbociclib combined with letrozole confers
more benefits to breast cancer patients than letrozole alone, and
that biomarker-selected patients do not show improved OS.
In recent years, the results of PALOMA-2 showed that
in 666 postmenopausal patients with untreated, HR-positive,
HER2-negative, metastasis breast cancer treated with palbociclib
plus letrozole had longer PFS (24.8 months) and ORR (42.1%) than
letrozole with placebo (14.5 months and 34.7% respectively). The
primary side effects observed in this randomized, double-blind
phase 3 trial are myelotoxic effects; other side effects are
identical to those observed in PALOMA-1 (65). Biochemical analysis of this trial
showed that in the ER-positive, Rb-negative and p16-negative
subgroup, PFS is also longer in patients treated with palbociclib
and letrozole than with palbociclib and placebo. Thus, the
combination of palbciclib and letrozole shows significant results
in clinical trials.
PALOMA-3 (66),
another important trial, was performed to assess palbociclib in
combination with fulvestrant in asecond-line setting. Five hundred
and twenty-one patients were enrolled in this clinical trial; among
them, two-thirds received 125 mg palbociclib for 3 weeks on and 1
week off, and one-third received a placebo. All patients in this
study were also administered 500 mg fulvestrant on days 1, 14, and
28, then every 28 days thereafter. After 195 PFS events, the
results of this double-blind trial were analyzed. Median PFS of the
palbociclib-fulvestrant and placebo-fulvestrant groups were 9.2 and
3.8 months, respectively (HR=0.42; 95% CI, 0.32–0.56; P<0.001).
The five most commonly-experienced side effects in this trial were
neutropenia, leukopenia, anemia, thrombocytopenia and fatigue.
Aside from these adverse effects, febrile neutropenia was observed
in both arms at an incidence of 0.6%. PE was only seen in the
palbociclib-fulvestrant groups at an incidence of 0.9% (67).
Some investigators have also tested CDK4/6 inhibitor
monotherapy. A phase I study on palbociclib monotherapy indicated
that this drug has promising clinical efficacy and a well-tolerated
toxicity profile in patients with Rb-positive advanced solid tumors
and non-Hodgkin lymphoma (68). In
this study, 33 patients were enrolled and treated with palbociclib
(once daily for 14 days on and 7 days off). Among them, nine
patients presented with stable disease and one exhibited a partial
response. Another CDK4/6 inhibitor, LEE011, was also investigated
in a phase I study in patients with advanced solid tumors. LEE011
was well tolerated and almost 40% of tested patients presented with
stable disease; two exhibited a partial response. Similarly, a
single-agent study of abemaciclib showed delayed disease
progression and activity in metastatic ER+ breast
cancer. While neutropenia is the principal dose-limiting toxicity
of palbociclib and LEE011, it is also a common side effect of
cytotoxic agents. As opposed to the neutropenia caused by
cytotoxicity, palbociclib and LEE011-induced neutropenia is rapidly
reversible (52). While the
principle side effect of abemaciclib is gastrointestinal-associated
toxicity, neutropenia is also seen, to a lesser extent. The
mechanisms of these differences in toxicity are not clear, and
require further research.
ER+ breast cancer patients should be
treated with endocrine therapy such as aromatase inhibitors (AIs),
fulvestrant, ortamoxifen as a first line treatment. If a patient
treated with an endocrine agent then progresses, other types of
endocrine agents may confer benefits (53). However, nearly all patients will
experience an initial effect and eventually develop drug
resistance. Although the mechanism of acquired resistance to
endocrine therapy has been described in many reports, recent
studies have found mutations in the ER gene (ESR1) (69,70),
which occur most frequently in post-aromatase inhibitor
(AI)-treated breast cancer patients, termed acquired resistance,
but rarely occurs in primary breast cancer, which may be an
important point. It is hypothesized that the combination of
palbociclib and letrozole may reverse acquired resistance in breast
cancer patients.
Multiple myeloma
Multiple myeloma (MM), the second most common
hematopoietic cancer, represents another successful application of
CDK4/6 inhibition. MM is a malignancy characterized by the
uncontrolled proliferation of clonal plasma cells with an incidence
of about 20,000 per year in the United States (71). The standard treatment of alkylating
agents combined with steroids has been used for over 30 years
(72). Other treatments include
stem cell transplantation with immunomodulatory therapies and
proteasome inhibitors. Although the appearance of new therapies has
reversed the poor outcomes associated with relapsed/refractory
multiple myeloma (RRMM) (73),
overall survival is far from optimal.
In MM, as in other cancers, dysregulation of the
cell cycle contributes to disease progression, and CDK4/6 disorders
are often seen in MM. Thus, palbociclib has been successfully
applied in recent years. A multicenter, open-label, phase I/II
study of palbociclib with bortezomib and dexamethasone in RRMM has
been completed (74). This study
included two phases: phase 1 and phase 2. The phase 1 study
enrolled Rb protein-positive patients with relapsed and/or
refractory MM after ≥1 previous treatments and a life expectancy
>3 months to determine the maximum tolerated dose (MTD) and
recommended dose of palbociclib to be used in phase 2. Two
schedules were evaluated. For schedule A, palbociclib was given
orally once daily for days 1–21 of each 28-day cycle, with 7 days
off. For schedule B, palbociclib was given orally once daily for 12
days of a 21-day cycle, with 9 days off. For both schedules A and
B, bortezomib was administered intravenously on days 8, 11, 15, and
18 in each tested cycle. Aproximately 30 min before the
administration of bortezomib, 20 mg dexamethasone was given orally.
Of the 21 patients who were enrolled in phase 1, nine were grouped
in Schedule A (three patients were administered 100 mg palbociclib
and the remaining patients were administered 75 mg at the onset),
and 12 patients grouped in Schedule B (seven patients were
administered 100 mg palbociclib and the remaining patients were
administered 125 mg at the onset). Observed side effects are shown
in Table II.
| Table II.Treatment-related adverse events in
phase 1a and phase
2b. |
Table II.
Treatment-related adverse events in
phase 1a and phase
2b.
| A, |
|---|
|
|---|
|
| Schedule A
(n=9) | Schedule B
(n=12) | Total (N=21) |
|---|
|
|
|
|
|
|---|
| Phase 1 event, n
(%) | Grade 1/2 | Grade 3 | Grade 4 | Grade 1/2 | Grade 3 | Grade
4c | Grade 1/2 | Grade 3 | Grade 4 |
|---|
|
Thrombocytopenia | 0 | 1 (11) | 7 (78) | 0 | 3 (25) | 5 (42) | 0 | 4 (19) | 12 (57) |
| Neutropenia | 2 (22) | 4 (44) | 1 (11) | 1 (8) | 2 (17) | 3 (5) | 3 (14) | 6 (29) | 4 (19) |
| Lymphopenia | 0 | 0 | 0 | 4 (33) | 0 | 1 (8) | 4 (19) | 0 | 1 (15) |
| Neutrophils
decreased | 0 | 0 | 0 | 4 (33) | 1 (8) | 0 | 4 (19) | 1 (5) | 0 |
| Hemoglobin
decreased | 0 | 0 | 0 | 1 (8) | 2 (17) | 0 | 1 (5) | 2 (10) | 0 |
| Leukopenia | 0 | 0 | 0 | 2 (17) | 1 (8) | 0 | 2 (10) | 1 (5) | 0 |
| White blood cells
decreased | 0 | 0 | 0 | 2 (17) | 1 (8) | 0 | 2 (10) | 1 (5) | 0 |
| Anemia | 1 (11) | 1 (11) | 0 | 0 | 2 (17) | 0 | 1 (5) | 3 (14) | 0 |
| Rash | 0 | 0 | 0 | 2 (17) | 0 | 0 | 2 (10) | 0 | 0 |
|
| B, |
|
|
|
|
|
| Schedule B
(n=30) |
|
|
|
|
|
|
| Phase 2 event, n
(%) | Schedule A (not
assessed) | Grade
1/2 | Grade
3d | Grade 4 |
|
|
Thrombocytopenia | – | – | – | 3 (10) | 4 (13) | 8 (27) |
| Anemia | – | – | – | 4 (13) | 8 (27) | 0 |
| Fatigue | – | – | – | 10 (33) | 2 (7) | 0 |
| Nausea | – | – | – | 9 (30) | 0 | 0 |
| Diarrhea | – | – | – | 6 (20) | 0 | 0 |
| Neutropenia | – | – | – | 1 (3) | 3 (10) | 2 (7) |
| Dyspnea | – | – | – | 4 (13) | 0 | 0 |
| Headache | – | – | – | 3 (10) | 0 | 0 |
| Peripheral
edema | – | – | – | 3 (10) | 0 | 0 |
|
Nasopharyngitis | – | – | – | 3 (10) | 0 | 0 |
| Peripheral
neuropathy | – | – | – | 4 (13) | 0 | 0 |
| Leukopenia | – | – | – | 0 | 2 (7) | 2 (7) |
| Dizziness | – | – | – | 4 (13) | 1 (3) | 0 |
| Pyrexia | – | – | – | 6 (20) | 0 | 0 |
For Schedule A, the objective response rate (ORR)
was 16.7%. One patient who received 100 mg palbociclib had
progressive disease. Of the patients who received 75 mg
palbociclib, one had a very good partial response (20%; duration,
6.1 months), one had stable disease (20%; duration, 1 month), and
the last three had progressive disease (60%). For Schedule B, the
ORR was 8%. In the 100 mg palbociclib group, four patients had
stable disease [57%; median (range)duration, 3.2 (2.3–5.6) months]
and three had progressive disease (43%). In the 125 mg palbociclib
group, one patient had a very good partial response (20%; duration,
2.1 months), two had stable disease (40%; duration, 6.0 and 10.9
months), one had progressive disease (20%), and one had an
indeterminate response (20%). According to phase 1 analyses
(Schedule A and B), the dose schedule applied in phase 2 was 100 mg
palbociclibin combination with 1.0 mg/m2 bortezomib and
20 mg dexamethasone.
Phase 2 of the study included two stages. Forty-two
patients were enrolled in phase 2, with 25 enrolled in stage 1 and
17 in stage 2. The primary endpoint was antitumor activity based on
overall response rate (ORR). Safety, duration of response in
objective response patients, progression-free survival (PFS), time
to tumor progression, and overall survival (OS) were secondary
endpoints. Upon completion of phase 2, 32 patients remained and 30
received ≥1 dose of study treatment; only one patient completed the
entire study treatment. Reasons for discontinuation in the 31
patients who did not complete treatment included disease
progression (n=16; 53%), AE (n=4; 13%), global health status
deterioration (n=2; 7%); withdrawal of consent (n=2; 7%), and other
[n=5; 17%: moderate response (n=2; 7%); lack of clinical benefit
(n=1); and lack of efficacy (n=1)]. Five (20%) of the 25 evaluable
patients reached the objective response. The objective response
median (range) time was 2.8 (0.7–3.5) months within four cycles.
Eleven of 25 patients had stable disease lasting a median of 3.9
months. Another seven patients had responses that preceded to stage
2. Twenty-six percent of the patients maintained PFS at 12 months,
17 patients had objective progression, 13 were censored, and six
ceased treatment before progression.
Phase 1 of this study identified that the treatment
plan of 100 mg palbociclib + 1.0 mg/m2 bortezomib + 20
mg dexamethasone is safe for patients. The most common side effect
seen in this study was thrombocytopenia, and the total burden of
side effects of this treatment regimen was light. In phase 2 of the
study, the ORR was much higher in patients who had not received
bortezomib treatment than in those who had received prior treatment
with bortezomib (75).
The response topalbociclibin clinical settings in
recent years has been modest. This inhibitor can not target other
critical myeloma oncogenic kinases. Recently, Perumal et al
discovered that a new dual inhibitor of CDK4 and ARK5, ON123300,
can rapidly induce cell cycle arrest and apoptosis in vitro
and can effectively shrink xenografted tumors in vivo
(76). AMPK-related protein kinase
5 (ARK5), which is expressed in nearly all myeloma cell lines, is a
member of the AMPK family that is involved in tumor growth and
invasion (77). Thus, these results
provide promising evidence for ARK5 inhibition and lay a solid
foundation for developing the next generation of CDK
inhibitors.
Lung cancer
The application of CDK4/6 inhibitors in lung cancer
has also increased in recent years. Although the emergence of
targeted therapies in lung adenocarcinoma (LUAD) have led to
positive initial effects for patients with EGFR mutation or
EML4-ALK rearrangements, recent data have shown that patients with
LUAD have longer PFS when they are treated with targeted therapy as
compared to traditional chemotherapy (78). Unfortunately, despite the success of
targeted therapy in LUAD patients, almost all cases eventually
recur after a median of approximately 10 months from the onset of
treatment. This phenomenon may be the greatest challenge for the
application of targeted therapy in lung cancer.
Our group has found that the combination of EGFR
tyrosine kinase inhibitors (EGFR-TKIs) and palbociclib can reduce
proliferation and induce cell apoptosis and G0/G1 cell cycle arrest
in EGFR-TKI sensitive and resistant cell lines to a greater extent
than gefitinib alone. In vivo experiments, we found that
mice treated with palbociclib and gefitinib had more rapid tumor
regression and a delayed relapse pattern as compared to mice
treated with gefitinib alone. Tumors from the experimental mice
showed significantly reduced proliferation, increased apoptosis,
and reduced angiogenesis (79).
In our department, many LUAD patients are treated
with EGFR-TKIs and eventually develop drug resistance. Three of
these patients are receiving palbociclib in an attempt to treat
their resistant disease, with fully informed consent. Two of these
patients developed stable disease and one had a partial response to
palbociclib. We previously reported a 63-year-old woman with bone
and lung metastases who had received gefitinib (250 mg per
os (PO) daily) and zoledronic acid (0.4 mg infusion monthly).
After treatment, her metastasis was controlled. However, a brain
metastasis was found, indicating that her disease had progressed
and that she had developed drug resistance to EGFR-TKI. The patient
was then administered palbociclib (100 mg PO) and gefitinib
continued. Three weeks later, an MRI examination revealed the brain
metastasis had disappeared, and the pain caused by bone metastasis
was reduced (79).
In KRAS-mutant non-small cell lung cancer (NSCLC)
patients, accounting for almost 20% of NSCLC patients, KRAS
mutation can constitutively activate mitogen-activated protein
kinase (MAPK) signaling, leading to increased proliferation. This
mutation can also decrease efficacy and resistance to
chemotherapies and radiotherapy (80). Blocking KRAS activity with small
molecules remains difficult because K-RAS can activate multiple
pathways such as MEK/ERK, PI3K/AKT and NF-κB (81); thus, targeted therapy in KRAS-mutant
NSCLC has not been widely employed. Recently, Lu et al found
that the combination of MEK (trametinib, GSK112012) and CDK4/6
inhibition (palbociclib) can strongly reduce cell proliferation in
KRAS-mutant NSCLCs that were previous resistant to MEK inhibitor
in vitro and in vivo (82). Lu et al elucidated that
resistance to MEK inhibition is associated with p16 mutation
status. The application of palbociclib in KRAS-mutant NSCLC cells
can trigger radio-sensitizing effects, apoptosis, and cell cycle
arrest with trametinib.
Aside from research on palbociclib use in lung
cancer, other CDK4/6 inhibitors such as abeamciclib (LY2835219) and
ribociclib (LEE011) are also in different research stages in
squamous cell lung cancer (LUSQ). No effective small molecule
inhibitors have been discovered for the treatment of LUSQ. However,
a study profiling 178 patients showed that CDK4/6-Rb-E2F pathway
alterations are commonly observed in LUSQ (83). Thus, CDK4/6 inhibitor maybe an
attractive target point in the treatment of LUSQ. In vitro,
both palbociclib and LEE011 can potently inhibit Rb phosphorylation
at S780, a canonical substrate of CDK4, in H157 and other cell
lines. Compared with LEE011, palbociclib may be more effective.
Both CDK4/6 inhibitors can inhibit CDK4/6 and CDK9, and palbociclib
can also affect several other kinases, such as case in kinase 2 and
PIK3R4, that are associated with autophagy, and several lipid
kinases such as PIK3CD and PIP4K2A/B/C. Palbociclib, but not
LEE011, was observed to modulate autophagy and AKT pathway
inhibition. The application of CDK4/6 inhibitors requires
significant research before clinical use, however. Additionally to
these three CDK4/6 inhibitors, other cell cycle inhibitors like
enterolactone, a rich source of the plant lignan
secoisolariciresinol diglucoside, could inhibit the growth of NSCLC
cell lines by arresting the cell cycle in G1 phase (84). Many mechanisms underlying the
effects of such drugs are not clear and the targeted persons have
not been identified, so there is a long way to go.
Some research has shown that the CDK4-Rb-E2F pathway
can regulate pancreatic β-cell size and function, skeletal muscle
metabolism, and white adipose cell function. Currently, palbociclib
is in clinical trials for the treatment of primary brain tumors.
According to previous research, primary brain tumors like
glioblastoma multiforme (GBM) also have dysregulated CDK4/6
activity. Thus, palbociclib may be a promising clinical drug that
can be used in brain tumor treatment. One problem to overcome for
clinical palbociclib use may be the blood-brain barrier (BBB),
which can limit the delivery of palbociclib to invasive regions of
GBM. A team of researchers have found that palbociclib is a
substrate for both P-glycoprotein and breast cancer resistance
protein, which are both efflux transporters involved in limiting
brain distribution of palbociclib. In murine experiments, they have
used the transporter-deficient and wild-type models to compare
palbociclib delivery to the brain: palbociclib delivery in the
transporter-deficient model was 115-fold higher than in the
wild-type model. These experimental results have positive
implications for the clinical use of palbociclib.
Though the future of CDK4/6 inhibitors is bright,
many significant problems remain. Which patients would most benefit
from such treatment need to be identified, acquired resistance of
CDK4/6 inhibitors must be interrogated, and a clear mechanism of
action must be determined. Once these problems have been resolved,
its combination with traditional therapy may prove a safe and
effective method to apply in cancer.
Future directions
Presently, the use of targeted therapy is thought to
be promising for many kinds of cancers. However, the big hurdle
that must be resolved immediately is acquired resistance. We found
that acquired resistance is likely caused by mutation in or
amplification of the targeted kinase itself or activation of a
compensatory kinase (85–88). Both of these alterations can lead to
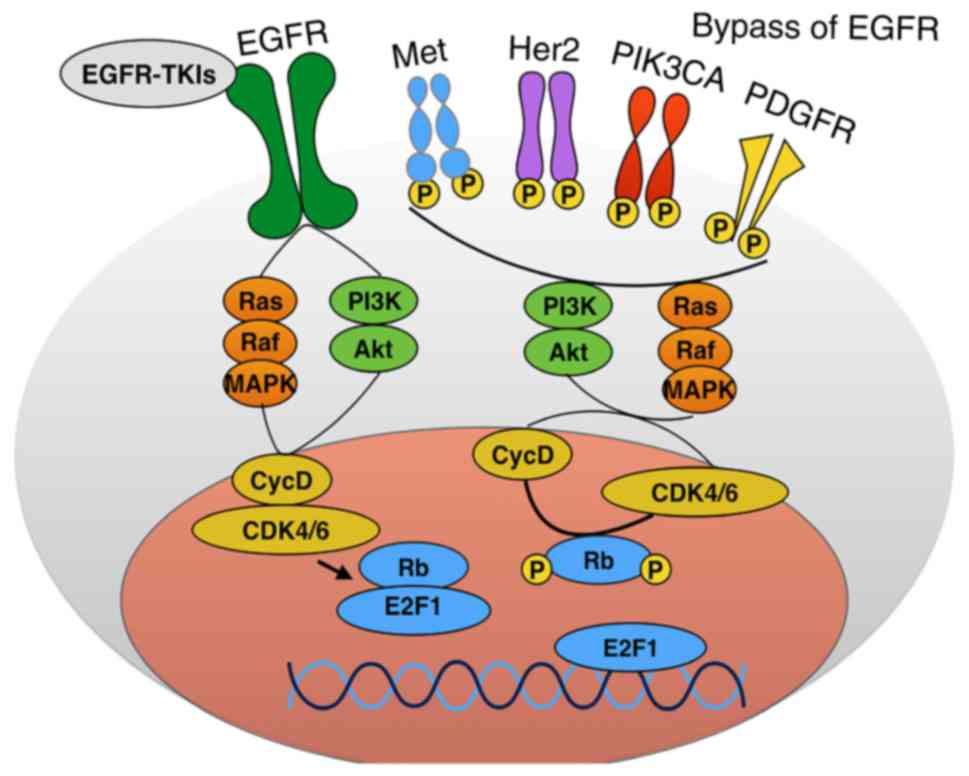
substantial tumor cell proliferation, as shown in Fig. 3. Regarding acquired resistance, we
hypothesize that blocking one or two growth pathways is
insufficient to inhibit tumor cell proliferation. However, all
cells must progress through the cell cycle in order to proliferate;
thus, cell cycle inhibitors may be the promising agents that can be
used to treat cancer. As shown in Fig.
3, when EGFR or other growth pathways, such as Met, Her2 or
PDGFR, are mutated and activated, all downstream pathways will
activate cell cycle progression. Thus, we propose that cancer
treatment should focus on combining cell cycle inhibitors with
small molecule inhibitors.
The FDA has approved the CDK4/6 inhibitor PD 0332991
for combination use in treating postmenopausal
ER-positive/HER2-negative advanced breast cancer with letrozole as
first-line therapy. Other combination therapies including a cell
cycle inhibitor and targeted therapy have been described. In
colorectal cancer (CRC) with KRAS mutation, the combination of
CDK4/6 and MEK inhibitors are effective in CRC cells and will be
studied in planned phase II clinical trials (89). In cutaneous melanoma, PD 0332991 can
be combined with MEK inhibitor (89). CDK4/6 inhibitors can be combined
with phosphatidylinositol 3-kinase (PI3K) pathway inhibitors for
HR+ breast cancer, and with RAF and MEK inhibitors for
melanoma with MAPK-activating mutations (2).
In addition to CDK4/6 inhibitors, other CDK and cell
cycle inhibitors (Table III) are
being actively pursued, such the first covalent inhibitor of CDK7,
THZ1. This inhibitor is a promising and effective drug that can
suppress cancer progression, and Kwiatkowski et al have
asserted that it is an effective anti-proliferative agent in blood
cancers and Jurkat cells (90).
Thus, in almost every kind of tumor, dysregulation of the cell
cycle is often seen and leads to tumor progression. We therefore
propose that combination therapy with a cell cycle inhibitor and
targeted drug may prove effective in reversing acquired resistance
and potentiate the effect of targeted therapy.
| Table III.Cell cycle inhibitors. |
Table III.
Cell cycle inhibitors.
| Name | Mechanism of cell
cycle inhibitor |
|---|
| BIX-01294 | Could upregulate
P21 and induces cell cycle arrest in the phase G0/G1 in acute T
lymphoblastic leukemia cells |
| OAMDP | Induces S or G2/M
phase arrest by modulating cycle regulatory proteins in hepatoma
HepG2 cells |
| MEK162 | Downregulates and
dephosphorylates the cell cycle checkpoint proteins CDK1/CDK2/WEE1
in glioblastoma |
| Tinospora
cordifolia | Arrests the cell
cycle in G0/G1-phase in human oral squamous cell carcinoma
cells |
| Yangyinjiedu
(YYJD) | Induces G2/M phase
arrest in lung cancer cells |
|
Prim-O-glucosylcimifugin | Arrests acute
lymphoblastic leukemia cells in G2/M phase by downregulating
phosphorylated CDK1 levels |
| Glucosamine | Arrests renal cell
carcinoma cells in G0/G1 phase by downregulating cyclin D1, CDK4
and CDK6 and upregulating p21 and p53 |
Though the future of CDK4/6 inhibition is bright,
many questions remain including how to identify optimal patients,
understanding mechanisms of acquired resistance to CDK4/6
inhibitors, and overcoming acquired resistance. Nonetheless, we
assert that its combination with traditional therapy may be a safe
and effective means to treat cancer.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (to Jun Chen, no. 81773207, to
Hongyu Liu, no. 81372306) and the Tianjin key project of the
Natural Science Foundation (to Jun Chen, 16JCZDJC34200), Special
support program for High Tech Leader & Team of Tianjin (to Jun
Chen), Tianjin Natural Science Foundation (to Hongyu Liu,
13JCYBJC22600, 16PTSYJC00160), and the Ph.D. Programs Foundation
from the Ministry of Education of China (to Jun Chen,
20131202110004).
References
|
1
|
Minton K: Cancer immunotherapy: Cell cycle
inhibitors boost tumour immunogenicity. Nat Rev Drug Dis.
16:6792017. View Article : Google Scholar
|
|
2
|
Hamilton E and Infante JR: Targeting
CDK4/6 in patients with cancer. Cancer Treat Rev. 45:129–138. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhou J, Zhang S, Chen X, Zheng X, Yao Y,
Lu G and Zhou J: Palbociclib, a selective CDK4/6 inhibitor,
enhances the effect of selumetinib in RAS-driven non-small cell
lung cancer. Cancer Lett. 408:130–137. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bilgin B, Sendur MA, Şener Dede D, Akıncı
MB and Yalçın B: A current and comprehensive review of
cyclin-dependent kinase ınhibitors for the treatment of metastatic
breast cancer. Curr Med Res Opin. 33:1559–1569. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen L and Pan J: Dual cyclin-dependent
kinase 4/6 inhibition by PD-0332991 induces apoptosis and
senescence in oesophageal squamous cell carcinoma cells. Br J
Pharmacol. 174:2427–2443. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Patel P, Asbach B, Shteyn E, Gomez C,
Coltoff A, Bhuyan S, Tyner AL, Wagner R and Blain SW: Brk/Protein
tyrosine kinase 6 phosphorylates p27KIP1, regulating the activity
of cyclin D-cyclin-dependent kinase 4. Mol Cell Biol. 35:1506–1522.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Malumbres M, Harlow E, Hunt T, Hunter T,
Lahti JM, Manning G, Morgan DO, Tsai LH and Wolgemuth DJ:
Cyclin-dependent kinases: A family portrait. Nat Cell Biol.
11:1275–1276. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Malumbres M: Cyclin-dependent kinases.
Genome Biology. 15:1222014. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ubersax JA, Woodbury EL, Quang PN, Paraz
M, Blethrow JD, Shah K, Shokat KM and Morgan DO: Targets of the
cyclin-dependent kinase Cdk1. Nature. 425:859–864. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Baker SJ and Reddy EP: CDK4: A key player
in the cell cycle, development, and cancer. Genes Cancer.
3:658–669. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shah K and Lahiri DK: Cdk5 activity in the
brain-multiple paths of regulation. J Cell Sci. 127:2391–2400.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pinhero R and Yankulov K: Expression and
purification of recombinant CDKs: CDK7, CDK8, and CDK9. Methods Mol
Biol. 1336:13–28. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Galbraith MD, Donner AJ and Espinosa JM:
CDK8: A positive regulator of transcription. Transcription. 1:4–12.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Krystof V, Baumli S and Fürst R:
Perspective of cyclin-dependent kinase 9 (CDK9) as a drug target.
Curr Pharm Des. 18:2883–2890. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hu D, Mayeda A, Trembley JH, Lahti JM and
Kidd VJ: CDK11 complexes promote pre-mRNA splicing. J Biol Chem.
278:8623–8629. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chi Y, Huang S, Peng H, Liu M, Zhao J,
Shao Z and Wu J: Critical role of CDK11(p58) in human breast cancer
growth and angiogenesis. BMC Cancer. 15:7012015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bajić VP, Su B, Lee HG, Kudo W, Siedlak
SL, Zivković L, Spremo-Potparević B, Djelic N, Milicevic Z, Singh
AK, et al: Mislocalization of CDK11/PITSLRE, a regulator of the
G2/M phase of the cell cycle, in Alzheimer disease. Cell Mol Biol
Lett. 16:359–372. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhou Y, Han C, Li D, Yu Z, Li F, Li F, An
Q, Bai H, Zhang X, Duan Z and Kan Q: Cyclin-dependent kinase
11(p110) (CDK11(p110)) is crucial for human breast cancer cell
proliferation and growth. Sci Rep. 5:104332015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Malumbres M and Barbacid M: Mammalian
cyclin-dependent kinases. Trends Biochem Sci. 30:630–641. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shapiro GI: Cyclin-dependent kinase
pathways as targets for cancer treatment. J Clin Oncol.
24:1770–1783. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sakurikar N and Eastman A: Critical
reanalysis of the methods that discriminate the activity of CDK2
from CDK1. Cell Cycle. 15:1184–1188. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sherr CJ: G1 phase progression: Cycling on
cue. Cell. 79:551–555. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li Y, Zhang J, Gao W, Zhang L, Pan Y,
Zhang S and Wang Y: Insights on structural characteristics and
ligand binding mechanisms of CDK2. Int J Mol Sci. 16:9314–9340.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Flores O, Wang Z, Knudsen KE and Burnstein
KL: Nuclear targeting of cyclin-dependent kinase 2 reveals
essential roles of cyclin-dependent kinase 2 localization and
cyclin E in vitamin D-mediated growth inhibition. Endocrinology.
151:896–908. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ali S, Heathcote DA, Kroll SH, Jogalekar
AS, Scheiper B, Pate H, Brackow J, Siwicka A, Fuchter MJ,
Periyasamy M, et al: The development of a selective
cyclin-dependent kinase inhibitor that shows antitumor activity.
Cancer Res. 69:6208–6215. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kawana H, Tamaru J, Tanaka T, Hirai A,
Saito Y, Kitagawa M, Mikata A, Harigaya K and Kuriyama T: Role of
p27Kip1 and cyclin-dependent kinase 2 in the proliferation of
non-small cell lung cancer. Am J Pathol. 153:505–513. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Morgan DO: Cyclin-dependent kinases:
Engines, clocks, and microprocessors. Annu Rev Cell Dev Biol.
13:261–291. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Clark AS, Karasic TB, DeMichele A, Vaughn
DJ, O'Hara M, Perini R, Zhang P, Lal P, Feldman M, Gallagher M and
O'Dwyer PJ: Palbociclib (PD0332991) - a selective and potent
cyclin-dependent kinase inhibitor: A review of pharmacodynamics and
clinical development. JAMA Oncol. 2:253–260. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dean JL, McClendon AK and Knudsen ES:
Modification of the DNA damage response by therapeutic CDK4/6
inhibition. J Biol Chem. 287:29075–29087. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rader J, Russell MR, Hart LS, Nakazawa MS,
Belcastro LT, Martinez D, Li Y, Carpenter EL, Attiyeh EF, Diskin
SJ, et al: Dual CDK4/CDK6 inhibition induces cell-cycle arrest and
senescence in neuroblastoma. Clin Cancer Res. 19:6173–6182. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee Y, Dominy JE, Choi YJ, Jurczak M,
Tolliday N, Camporez JP, Chim H, Lim JH, Ruan HB, Yang X, et al:
Cyclin D1-Cdk4 controls glucose metabolism independently of cell
cycle progression. Nature. 510:547–551. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Weijts BGMW, Westendorp B, Hien BT,
Martínez-López LM, Zijp M, Thurlings I, Thomas RE, Schulte-Merker
S, Bakker WJ and de Bruin A: Atypical E2Fs inhibit tumor
angiogenesis. Oncogene. Sep 18–2017.(Epub ahead of print).
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wirt SE and Sage J: p107 in the public
eye: An Rb under study and more. Cell Div. 5:92010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sadasivam S and DeCaprio JA: The DREAM
complex: Master coordinator of cell cycle-dependent gene
expression. Nat Rev Cancer. 13:585–595. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee MH, Williams BO, Mulligan G, Mukai S,
Bronson RT, Dyson N, Harlow E and Jacks T: Targeted disruption of
p107: Functional overlap between p107 and Rb. Genes Dev.
10:1621–1632. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cobrinik D, Lee MH, Hannon G, Mulligan G,
Bronson RT, Dyson N, Harlow E, Beach D, Weinberg RA and Jacks T:
Shared role of the pRB-related p130 and p107 proteins in limb
development. Genes Dev. 10:1633–1644. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shen Y, Nar R, Fan AX, Aryan M, Hossain
MA, Gurumurthy A, Wassel PC, Tang M, Lu J, Strouboulis J and
Bungert J: Functional interrelationship between TFII-I and E2F
transcription factors at specific cell cycle gene loci. J Cell
Biochem. 119:712–722. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kent LN, Bae S, Tsai SY, Tang X,
Srivastava A, Koivisto C, Martin CK, Ridolfi E, Miller GC, Zorko
SM, et al: Dosage-dependent copy number gains in E2f1 and E2f3
drive hepatocellular carcinoma. J Clin Invest. 127:830–842. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Conklin JF and Sage J: Keeping an eye on
retinoblastoma control of human embryonic stem cells. J Cell
Biochem. 108:1023–1030. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dyson N: The regulation of E2F by
pRB-family proteins. Genes Dev. 12:2245–2262. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lukas J, Petersen BO, Holm K, Bartek J and
Helin K: Deregulated expression of E2F family members induces
S-phase entry and overcomes p16INK4A-mediated growth suppression.
Mol Cell Biol. 16:1047–1057. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Asano M, Nevins JR and Wharton RP: Ectopic
E2F expression induces S phase and apoptosis in Drosophila imaginal
discs. Genes Dev. 10:1422–1432. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
DeGregori J, Leone G, Ohtani K, Miron A
and Nevins JR: E2F-1 accumulation bypasses a G1 arrest resulting
from the inhibition of G1 cyclin-dependent kinase activity. Genes
Dev. 9:2873–2887. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Allen KE, La Luna de S, Kerkhoven RM,
Bernards R and La Thangue NB: Distinct mechanisms of nuclear
accumulation regulate the functional consequence of E2F
transcription factors. J Cell Sci. 110:2819–2831. 1997.PubMed/NCBI
|
|
45
|
Müller H, Moroni MC, Vigo E, Petersen BO,
Bartek J and Helin K: Induction of S-phase entry by E2F
transcription factors depends on their nuclear localization. Mol
Cell Biol. 17:5508–5520. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wu Z, Zheng S and Yu Q: The E2F family and
the role of E2F1 in apoptosis. Int J Biochem Cell Biol.
41:2389–2397. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Li J, Ran C, Li E, Gordon F, Comstock G,
Siddiqui H, Cleghorn W, Chen HZ, Kornacker K, Liu CG, et al:
Synergistic function of E2F7 and E2F8 is essential for cell
survival and embryonic development. Dev Cell. 14:62–75. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Westendorp B, Mokry M, Groot Koerkamp MJ,
Holstege FC, Cuppen E and de Bruin A: E2F7 represses a network of
oscillating cell cycle genes to control S-phase progression.
Nucleic Acids Res. 40:3511–3523. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lundberg AS and Weinberg RA: Functional
inactivation of the retinoblastoma protein requires sequential
modification by at least two distinct cyclin-cdk complexes. Mol
Cell Biol. 18:753–761. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ezhevsky SA, Ho A, Becker-Hapak M, Davis
PK and Dowdy SF: Differential regulation of retinoblastoma tumor
suppressor protein by G(1) cyclin-dependent kinase complexes in
vivo. Mol Cell Biol. 21:4773–4784. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Harbour JW, Luo RX, Dei Santi A, Postigo
AA and Dean DC: Cdk phosphorylation triggers sequential
intramolecular interactions that progressively block Rb functions
as cells move through G1. Cell. 98:859–869. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
van den Heuvel S and Harlow E: Distinct
roles for cyclin-dependent kinases in cell cycle control. Science.
262:2050–2054. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Garber K: The cancer drug that almost
wasn't. Science. 345:865–867. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Dolman ME, Poon E, Ebus ME, den Hartog IJ,
van Noesel CJ, Jamin Y, Hallsworth A, Robinson SP, Petrie K,
Sparidans RW, et al: Cyclin-dependent kinase inhibitor AT7519 as a
potential drug for MYCN-dependent neuroblastoma. Clin Cancer Res.
21:5100–5109. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Rigas AC, Robson CN and Curtin NJ:
Therapeutic potential of CDK inhibitor NU2058 in
androgen-independent prostate cancer. Oncogene. 26:7611–7619. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Joshi KS, Rathos MJ, Mahajan P, Wagh V,
Shenoy S, Bhatia D, Chile S, Sivakumar M, Maier A, Fiebig HH and
Sharma S: P276-00, a novel cyclin-dependent inhibitor induces G1-G2
arrest, shows antitumor activity on cisplatin-resistant cells and
significant in vivo efficacy in tumor models. Mol Cancer Ther.
6:926–934. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Joshi KS, Rathos MJ, Joshi RD, Sivakumar
M, Mascarenhas M, Kamble S, Lal B and Sharma S: In vitro antitumor
properties of a novel cyclin-dependent kinase inhibitor, P276-00.
Mol Cancer Ther. 6:918–925. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Giordano A, Rossi A, Romano G and Bagella
L: Tumor suppressor pRb2/p130 gene and its derived product Spa310
spacer domain as perspective candidates for cancer therapy. J Cell
Physiol. 213:403–406. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
De Azevedo WF, Leclerc S, Meijer L,
Havlicek L, Strnad M and Kim SH: Inhibition of cyclin-dependent
kinases by purine analogues: Crystal structure of human cdk2
complexed with roscovitine. Eur J Biochem. 243:518–526. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lee B, Sandhu S and McArthur G: Cell cycle
control as a promising target in melanoma. Curr Opin Oncol.
27:141–150. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Dange Y, Bhinge S and Salunkhe V:
Optimization and validation of RP-HPLC method for simultaneous
estimation of palbociclib and letrozole. Toxicol Mech Methods 1–8.
2017.
|
|
62
|
Guha M: Cyclin-dependent kinase inhibitors
move into Phase III. Nat Rev Drug Discov. 11:892–894. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Cardoso F, Bischoff J, Brain E, Zotano ÁG,
Lück HJ, Tjan-Heijnen VC, Tanner M and Aapro M: A review of the
treatment of endocrine responsive metastatic breast cancer in
postmenopausal women. Cancer Treat Rev. 39:457–465. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Sammons SL, Topping DL and Blackwell KL:
HR+, HER2 advanced breast cancer and CDK4/6 inhibitors:
mode of action, clinical activity, and safety profiles. Current
Cancer Drug Targets. 17:637–649. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Costa R, Costa RB, Talamantes SM,
Helenowski I, Peterson J, Kaplan J, Carneiro BA, Giles FJ and
Gradishar WJ: Meta-analysis of selected toxicity endpoints of
CDK4/6 inhibitors: Palbociclib and ribociclib. Breast. 35:1–7.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Iwata H, Im SA, Masuda N, Im YH, Inoue K,
Rai Y, Nakamura R, Kim JH, Hoffman JT, Zhang K, et al: PALOMA-3:
Phase III trial of fulvestrant with or without palbociclib in
premenopausal and postmenopausal women with hormone
receptor-positive, human epidermal growth factor receptor
2-negative metastatic breast cancer that progressed on prior
endocrine therapy-safety and efficacy in Asian patients. J Global
Oncol. 3:289–303. 2017. View Article : Google Scholar
|
|
67
|
Loibl S, Turner NC, Ro J, Cristofanilli M,
Iwata H, Im SA, Masuda N, Loi S, André F, Harbeck N, et al:
Palbociclib combined with fulvestrant in premenopausal women with
advanced breast cancer and prior progression on endocrine therapy:
PALOMA-3 Results. Oncologist. 22:1028–1038. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Schwartz GK, LoRusso PM, Dickson MA,
Randolph SS, Shaik MN, Wilner KD, Courtney R and O'Dwyer PJ: Phase
I study of PD 0332991, a cyclin-dependent kinase inhibitor,
administered in 3-week cycles (Schedule 2/1). Br J Cancer.
104:1862–1868. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Fribbens C, OLeary B, Kilburn L, Hrebien
S, Garcia-Murillas I, Beaney M, Cristofanilli M, Andre F, Loi S,
Loibl S, et al: Plasma ESR1 mutations and the treatment of estrogen
receptor-positive advanced breast cancer. J Clin Oncol.
34:2961–2968. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Gelsomino L, Gu G, Rechoum Y, Beyer AR,
Pejerrey SM, Tsimelzon A, Wang T, Huffman K, Ludlow A, Andò S and
Fuqua SAW: ESR1 mutations affect anti-proliferative responses to
tamoxifen through enhanced cross-talk with IGF signaling. Breast
Cancer Res Treat. 157:253–265. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Kuehl WM and Bergsagel PL: Molecular
pathogenesis of multiple myeloma and its premalignant precursor. J
Clin Invest. 122:3456–3463. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Ocio EM, Mitsiades CS, Orlowski RZ and
Anderson KC: Future agents and treatment directions in multiple
myeloma. Expert Rev Hematol. 7:127–141. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Castelli R, Gualtierotti R, Orofino N,
Losurdo A, Gandolfi S and Cugno M: Current and emerging treatment
options for patients with relapsed myeloma. Clin Med Insights
Oncol. 7:209–219. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Niesvizky R, Badros AZ, Costa LJ, Ely SA,
Singhal SB, Stadtmauer EA, Haideri NA, Yacoub A, Hess G, Lentzsch
S, et al: Phase 1/2 study of cyclin-dependent kinase (CDK)4/6
inhibitor palbociclib (PD-0332991) with bortezomib and
dexamethasone in relapsed/refractory multiple myeloma. Leuk
Lymphoma. 56:3320–3328. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Richardson PG, Barlogie B, Berenson J,
Singhal S, Jagannath S, Irwin D, Rajkumar SV, Srkalovic G, Alsina
M, Alexanian R, et al: A phase 2 study of bortezomib in relapsed,
refractory myeloma. N Engl J Med. 348:2609–2617. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Perumal D, Kuo PY, Leshchenko VV, Jiang Z,
Divakar SK, Cho HJ, Chari A, Brody J, Reddy MV, Zhang W, et al:
Dual targeting of CDK4 and ARK5 using a novel kinase inhibitor
ON123300 exerts potent anticancer activity against multiple
myeloma. Cancer Res. 76:1225–1236. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Liu L, Ulbrich J, Müller J, Wüstefeld T,
Aeberhard L, Kress TR, Muthalagu N, Rycak L, Rudalska R, Moll R, et
al: Deregulated MYC expression induces dependence upon AMPK-related
kinase 5. Nature. 483:608–612. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Sequist LV, Yang JC, Yamamoto N, O'Byrne
K, Hirsh V, Mok T, Geater SL, Orlov S, Tsai CM, Boyer M, et al:
Phase III study of afatinib or cisplatin plus pemetrexed in
patients with metastatic lung adenocarcinoma with EGFR mutations. J
Clin Oncol. 31:3327–3334. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Liu M, Xu S, Wang Y, Li Y, Li Y, Zhang H,
Liu H and Chen J: PD 0332991, a selective cyclin D kinase 4/6
inhibitor, sensitizes lung cancer cells to treatment with epidermal
growth factor receptor tyrosine kinase inhibitors. Oncotarget.
7:84951–84964. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Shaw AT, Winslow MM, Magendantz M, Ouyang
C, Dowdle J, Subramanian A, Lewis TA, Maglathin RL, Tolliday N and
Jacks T: Selective killing of K-ras mutant cancer cells by small
molecule inducers of oxidative stress. Proc Natl Acad Sci USA.
108:pp. 8773–8778. 2011; View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Montagut C and Settleman J: Targeting the
RAF-MEK-ERK pathway in cancer therapy. Cancer Lett. 283:125–134.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Tao Z, Le Blanc JM, Wang C, Zhan T, Zhuang
H, Wang P, Yuan Z and Lu B: Coadministration of trametinib and
palbociclib radiosensitizes KRAS-mutant non-small cell lung cancers
in vitro and in vivo. Clin Cancer Res. 22:122–133. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Cancer Genome Atlas Research Network, .
Comprehensive genomic characterization of squamous cell lung
cancers. Nature. 489:519–525. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Chikara S, Lindsey K, Dhillon H, Mamidi S,
Kittilson J, Christofidou-Solomidou M and Reindl KM: Enterolactone
induces G1-phase cell cycle arrest in nonsmall cell lung cancer
cells by downregulating cyclins and cyclin-dependent kinases. Nutr
Cancer. 69:652–662. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Chen DH and Zhang XS: Targeted therapy:
Resistance and re-sensitization. Chin J Cancer. 34:496–501. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Lee JE, Park HS, Lee D, Yoo G, Kim T, Jeon
H, Yeo MK, Lee CS, Moon JY, Jung SS, et al: Hippo pathway effector
YAP inhibition restores the sensitivity of EGFR-TKI in lung
adenocarcinoma having primary or acquired EGFR-TKI resistance.
Biochem Biophys Res Commun. 474:154–160. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Cross DA, Ashton SE, Ghiorghiu S, Eberlein
C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ,
et al: AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated
resistance to EGFR inhibitors in lung cancer. Cancer Discov.
4:1046–1061. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Luque-Cabal M, García-Teijido P,
Fernández-Pérez Y, Sánchez-Lorenzo L and Palacio-Vázquez I:
Mechanisms behind the resistance to trastuzumab in her2-amplified
breast cancer and strategies to overcome it. Clin Med Insights
Oncol. 10 Suppl 1:S21–S30. 2016.
|
|
89
|
Teh JL, Purwin TJ, Greenawalt EJ,
Chervoneva I, Goldberg A, Davies MA and Aplin AE: An in vivo
reporter to quantitatively and temporally analyze the effects of
CDK4/6 inhibitor-based therapies in melanoma. Cancer Res.
76:5455–5466. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Kwiatkowski N, Zhang T, Rahl PB, Abraham
BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar
B, et al: Targeting transcription regulation in cancer with a
covalent CDK7 inhibitor. Nature. 511:616–620. 2014. View Article : Google Scholar : PubMed/NCBI
|