Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the
fourth leading cause of cancer-related mortality (1) and it is estimated to become the second
by 2030 (2). Less than 20% of PDAC
patients are eligible for surgical resection (3) and, since chemotherapy and radiotherapy
only marginally improve survival (4), the 5-year survival rate for patients
is approximately 5% (1).
Since its approval by the FDA in 1996, the standard
treatment for advanced PDAC in the past two decades has been
chemotherapy with gemcitabine, a nucleoside analogue of
deoxycytidine that has been also extensively used for the treatment
of other solid tumors. Gemcitabine, however, offers only a small
improvement of survival to patients with advanced PDAC compared to
5-fluorouracil (5-FU) (5). The
relative lack of response to gemcitabine treatments is attributed
to mechanisms of either primary or acquired resistance, many of
which have been investigated extensively (6). For instance, resistance to gemcitabine
can be acquired through mechanisms related to its transport,
cellular uptake and metabolism within tumor cells. Furthermore, the
activation of pro-survival signaling pathways and the expression of
specific microRNAs can also influence the response to this drug
(6). Recently, we have highlighted
the impact of alternative splicing on both short-term and long-term
resistance to gemcitabine. Upon brief exposure to the drug,
upregulation of the oncogenic splicing factor SRSF1 induces
splicing of the MNK2b protein kinase variant and phosphorylation of
the translation factor eIF4E, which promote PDAC cell survival
under genotoxic stress (7).
Conversely, selection of gemcitabine-resistant PDAC clones after
chronic exposure to the drug, correlated with increased expression
of the polypyrimidine-tract binding protein (PTBP1) and alternative
splicing of the pyruvate kinase gene (PKM) resulting in the
promotion of the PKM2 isoform (8).
The expression of PKM2 was required for the maintenance of
gemcitabine-resistance in PDAC cell lines and correlated with worse
recurrence-free survival in operated patients treated with adjuvant
gemcitabine (8).
Recently, the standard treatment for patients with
advanced PDAC has improved due to the positive results of trials
with the combination of
fluorouracil-leucovorin-irinotecan-oxaliplatin (FOLFIRINOX)
(9) and the addition of
nanoparticle albumin-bound paclitaxel (nab-paclitaxel) to
gemcitabine (10,11). These combined regimens are now
considered the standard care for patients with advanced PDAC.
However, toxicity limits FOLFIRINOX use to patients with a good
performance status, while the combination of gemcitabine and
nab-paclitaxel is usually more tolerable. Single-agent gemcitabine
therapy is still an acceptable treatment in patients with advanced
disease and reduced performance status, as well as in the adjuvant
setting after surgical resection (12).
Nab-paclitaxel (Abraxane®; Celgene Inc.,
Odenton, MD, USA) is a specific formulation of paclitaxel that was
developed to improve its solubility and to overcome resistance due
to the desmoplastic stroma surrounding PDAC cells (13). Paclitaxel is a taxane and acts by
reversibly binding to tubulin, causing defects in mitotic functions
that lead to blockage of the cell cycle and eventually to
apoptosis, with mechanisms that differ from those of gemcitabine.
While the clinical use of nab-paclitaxel and gemcitabine has been
investigated extensively (14), the
available data on the activity of nab-paclitaxel as a single-agent
therapy in PDAC both in clinical trials and preclinical models are
poor. Therefore, the aim of the present study was to assess the
activity of nab-paclitaxel alone or in combination with gemcitabine
in PDAC cell lines displaying different degrees of sensitivity to
gemcitabine treatment.
Materials and methods
Cell cultures and drugs
All cell lines were obtained from the Centre for
Molecular Oncology, Barts Cancer Institute (London, UK) in 2004 and
authenticated in 2012. The HPAF-II, Pt45P1, PANC-1 and PANC-1 DR
cell lines were cultured in RPMI-1640 (Lonza, Basel, Switzerland)
and MiaPaCa-2 cell line was cultured in Dulbecco's modified Eagle's
medium (DMEM; Lonza). All media were supplemented with 10% fetal
bovine serum (FBS; Gibco, Gaithersburg, MD, USA), gentamycin,
penicillin, streptomycin and non-essential amino acids and the
cells were maintained at 37°C with 5% CO2.
Nab-paclitaxel (Abraxane®; kindly
provided by Celgene Inc.) was dissolved in physiological solution.
Gemcitabine (Eli Lilly and Company, Clinton, IN, USA) was dissolved
in water. The cells were plated at 50% confluency. Twenty-four
hours after plating, the cells were treated with nab-paclitaxel
and/or gemcitabine at the indicated concentrations for 24, 48 and
72 h before being collected for further analyses.
Cell viability assays
The cells were plated at 50% confluency in 96 wells
and, after 24 h, treated with nab-paclitaxel at the concentrations
indicated in Fig. 1. After 72 h of
treatment, the cell viability was evaluated by MTS assay (Promega,
Madison, WI, USA) following the manufacturer's instructions and by
assessing the optical density (OD) at 490 nm. The results are
represented as the mean ± standard deviation (SD) of three
experiments.
For cell death, the cells were plated at 70%
confluency and, after 24 h, treated with gemcitabine and/or
nab-paclitaxel at the indicated doses. After an additional 48 h,
the cells were washed in phosphate-buffered saline (PBS),
trypsinized and incubated with 0.4% Trypan Blue stain
(Sigma-Aldrich, St. Louis, MO, USA). Blue positive cells were then
counted using the Countess II Automated Cell Counter (Invitrogen
Life Technologies, Carlsbad, CA, USA) and the percentage of cell
death was determined. The results are represented as the mean ± SD
of three experiments.
BrdU-PI staining and cell cycle
analysis
For the cell cycle analysis, the cells were treated
with 10 µM BrdU (Sigma-Aldrich) in the final 30 min of treatments.
Subsequently, the cells were trypsinized, washed in chilled PBS and
resuspended in PBS/ethanol 70%. The samples were incubated at −20°C
until use. The cells were then centrifuged at 2,000 rpm for 5 min,
washed with PBS and incubated with 2 N HCl/0.5% Triton X-100 at
room temperature (RT) for 30 min. The cells were centrifuged at
2,000 rpm for 5 min and then resuspended with 0.1 M
NaB4O. After incubation for 2 min at RT, the cells were
washed with PBS/1% BSA and incubated for 1 h at RT in a solution of
0.5% Tween-20/1% BSA in PBS containing 10 µl of anti-BrdU 1 mM
(Becton-Dickinson and Company, Franklin Lakes, NJ, USA).
Subsequently, the cells were washed with PBS/1% BSA and incubated
in a solution of PBS/0.5% Tween-20/1% BSA containing 5 µl of Alexa
Flour 488 anti-mouse IgG-FITC (polyclonal; cat. no. A-11001; Thermo
Fisher Scientific, Waltham, MA, USA) for 30 min at RT. The cells
were washed with PBS/1% BSA and incubated with PBS containing 1
mg/ml RNAse A (Roche, Basel, Switzerland) and 20 µg/ml propidium
iodide (PI; Sigma-Aldrich) for 30 min at 37°C. Subsequently the
cells stained with BrDU-PI were analyzed by FACS.
Cell extracts and western blot
analysis
MiaPaCa-2 cells were resuspended in lysis buffer (50
mM HEPES pH 7,4, 10% glycerol, 15 mM MgCl2, 150 mM NaCl;
15 mM EGTA; 20 mM β-glycerophosphate; 1 mM dithiothreitol, 0.5 mM
NaVO4, 1 mM NaF and protease inhibitor cocktail)
supplemented with 1% Triton X-100, sonicated for 5 sec and
centrifuged for 10 min at 13,000 rpm at 4°C. Supernatants were
collected, diluted in sodium dodecyl sulphate (SDS) sample buffer
and boiled for 5 min. The proteins were separated on 8 or 12%
SDS-PAGE gel and transferred onto PVDF blotting membranes (Amersham
Hybond; GE Healthcare, Little Chalfont, UK). The membranes were
saturated in 5% non-fat dry milk in PBS plus 0.1% Tween-20 for 1 h
at RT and incubated overnight at 4°C with the following primary
antibodies: Rabbit anti-PARP1 (1:500; Cell Signaling Technology,
Inc., Danvers, MA, USA), mouse anti-actin (1:1,000; Santa Cruz
Biotechnology, Dallas, TX, USA), rabbit anti-cyclin E2 (1:1,000;
Cell Signaling Technology), rabbit anti-cyclin A2 (1:1,000), rabbit
anti-cyclin B1 (1:1,000), mouse anti-cyclin D1 (1,000; cat. no.
A-12) (all from Santa Cruz Biotechnology).
Results
Nab-paclitaxel exerts cytotoxic
effects in PDAC cells displaying different primary sensitivity to
gemcitabine
In order to assess the efficacy of nab-paclitaxel on
cell proliferation and viability, we analyzed the dose-response to
nab-paclitaxel of the PDAC cells displaying different sensitivity
to gemcitabine, with the MiaPaCa-2 and Panc-1 cells demonstrating
the highest resistance to gemcitabine, thus offering in
vitro models of primary resistance to this drug (7,8,15).
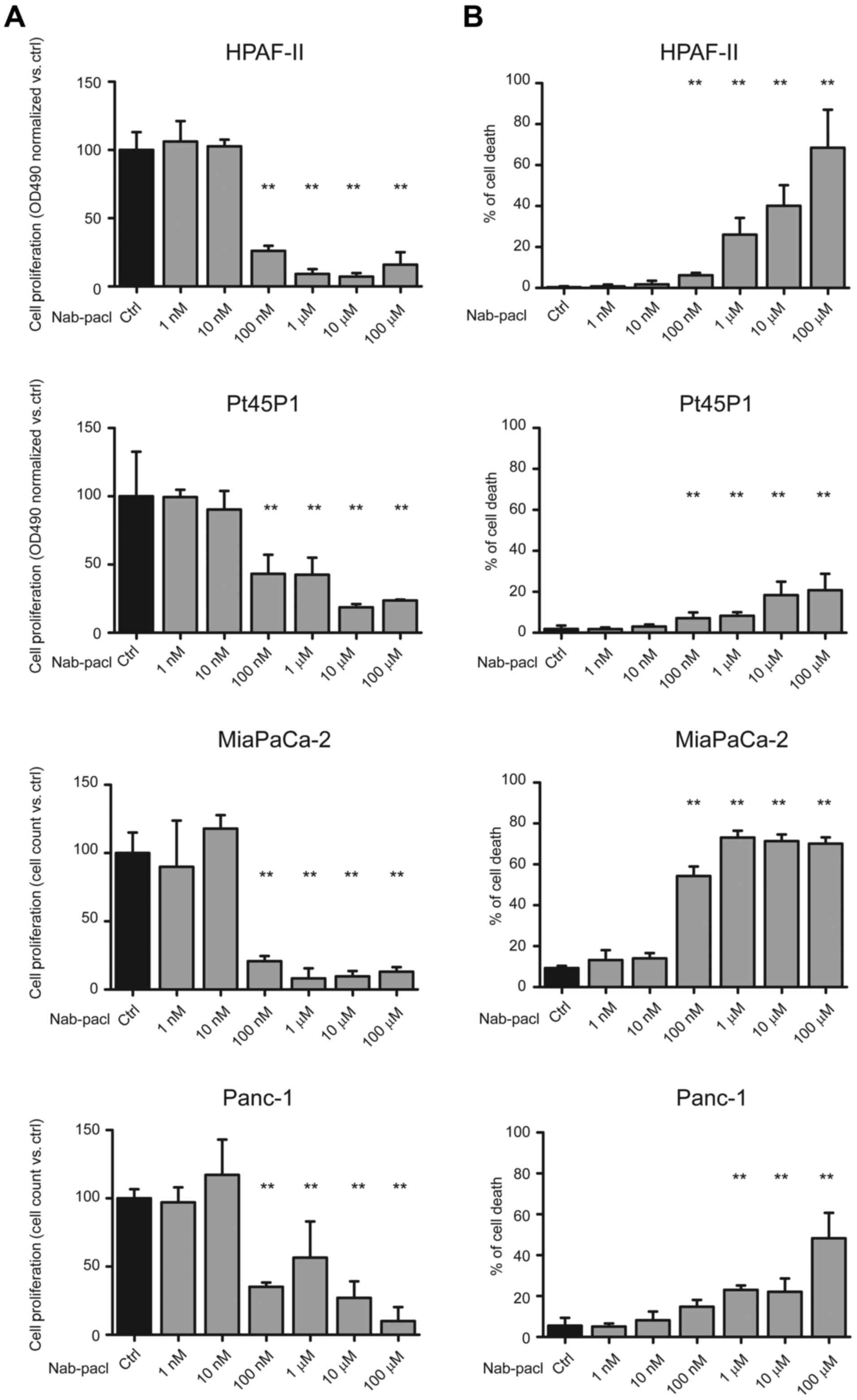
Nab-paclitaxel induced a significant reduction of
cell proliferation (60–65%) starting from the dose of 100 nM
compared to controls in all PDAC cells (Fig. 1A). Furthermore, at this dose,
nab-paclitaxel induced a significant increase of cell death in all
cell lines with the exception of Panc-1 cells (Fig. 1B). Notably, the increase of cell
death at 100 nM was modest in HPAF-II (6%) and Pt45P1 (7%) cells,
whereas it was very high in MiaPaCa-2 cells (54%) (Fig. 1B), which displayed higher resistance
to gemcitabine (15). Conversely,
nab-paclitaxel significantly reduced Panc-1 cell proliferation at
this dose without inducing cell death, whereas cell viability was
affected only at micromolar doses of the drug (Fig. 1A and B). At these higher doses
(1–100 µM), nab-paclitaxel led to substantial induction of cell
death in the HPAF-II, MiaPaCa-2 and Panc-1 cell lines, while cell
death remained at 20% in Pt45P1 even at the highest dose (Fig. 1B).
These results revealed that, regardless of their
sensitivity to gemcitabine, the PDAC cells demonstrated similar
sensitivity to nab-paclitaxel in terms of inhibition of cell
proliferation, however, different response in terms of cell
death.
Nab-paclitaxel exerts a cytotoxic
effect in PDAC cells with secondary gemcitabine resistance
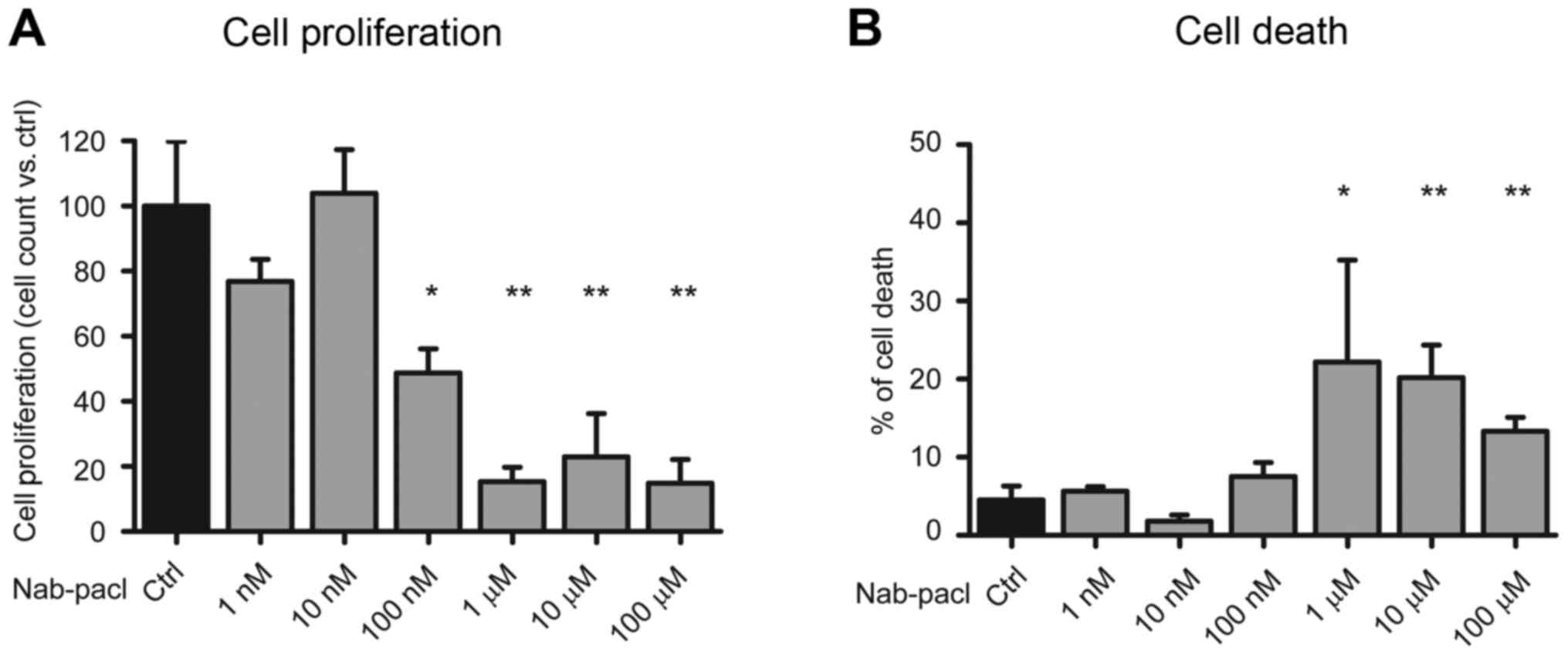
As aforementioned we selected PDAC cells which
acquired resistance to gemcitabine after chronic exposure to the
drug (8). Notably, these cells were
also more resistant to cisplatin (8), another drug exerting genotoxic stress.
To examine whether these drug-resistant (DR) cells were still
sensitive to nab-paclitaxel, a dose-response study was performed.
We found that DR-Panc-1 cells maintained the same sensitivity to
nab-paclitaxel as the parental cell line, with significant
inhibition of cell proliferation starting at the dose of 100 nM,
while cell death increased significantly at the dose of 1 µM
(Fig. 2A and B). These results
confirmed that nab-paclitaxel sensitivity did not correlate with
gemcitabine sensitivity and suggested that nab-paclitaxel may
overcome acquired resistance to gemcitabine in PDAC cells.
Combined treatment with Nab-paclitaxel
and gemcitabine exerts additive effects on the inhibition of cell
proliferation
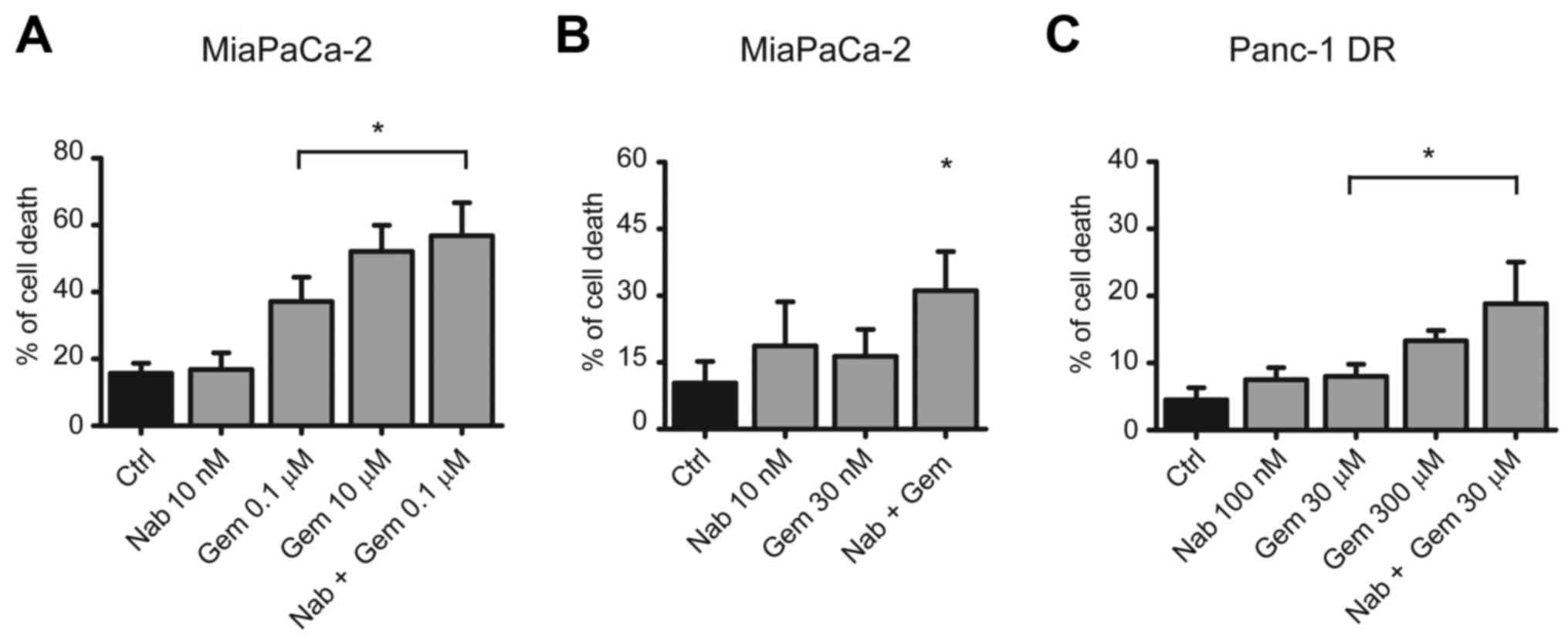
In order to understand whether nab-paclitaxel in
combination with gemcitabine enhances the cytostatic and cytotoxic
effects of the chemotherapeutic treatment, we tested their combined
action on cell proliferation and death in MiaPaCa-2 cells, a cell
line demonstrating relatively high resistance to gemcitabine
(15). The combination of
gemcitabine (100 nM) and nab-paclitaxel (10 nM) exerted a
significant increase in cell death compared to gemcitabine alone
(56 vs. 37%) (Fig. 3A). Notably,
the effect of the combined treatment was similar to that exerted by
gemcitabine alone at a dose 100 times higher (i.e. 10 µM; Fig. 3A). The combination of gemcitabine
(30 nM) and nab-paclitaxel (10 nM) elicited a significant additive
effect even when used at a suboptimal dosage (Fig. 3B). Furthermore, similar effects were
also obtained with the highly resistant DR-Panc-1 cells, albeit at
considerably higher doses (Fig.
3C). These data indicated that combined treatment with
nab-paclitaxel and gemcitabine enhanced the cytotoxic effects of
both drugs, allowing to lower their doses, thus possibly limiting
adverse effects.
Combination of suboptimal doses of
nab-paclitaxel and gemcitabine induces a significant increase in
apoptosis
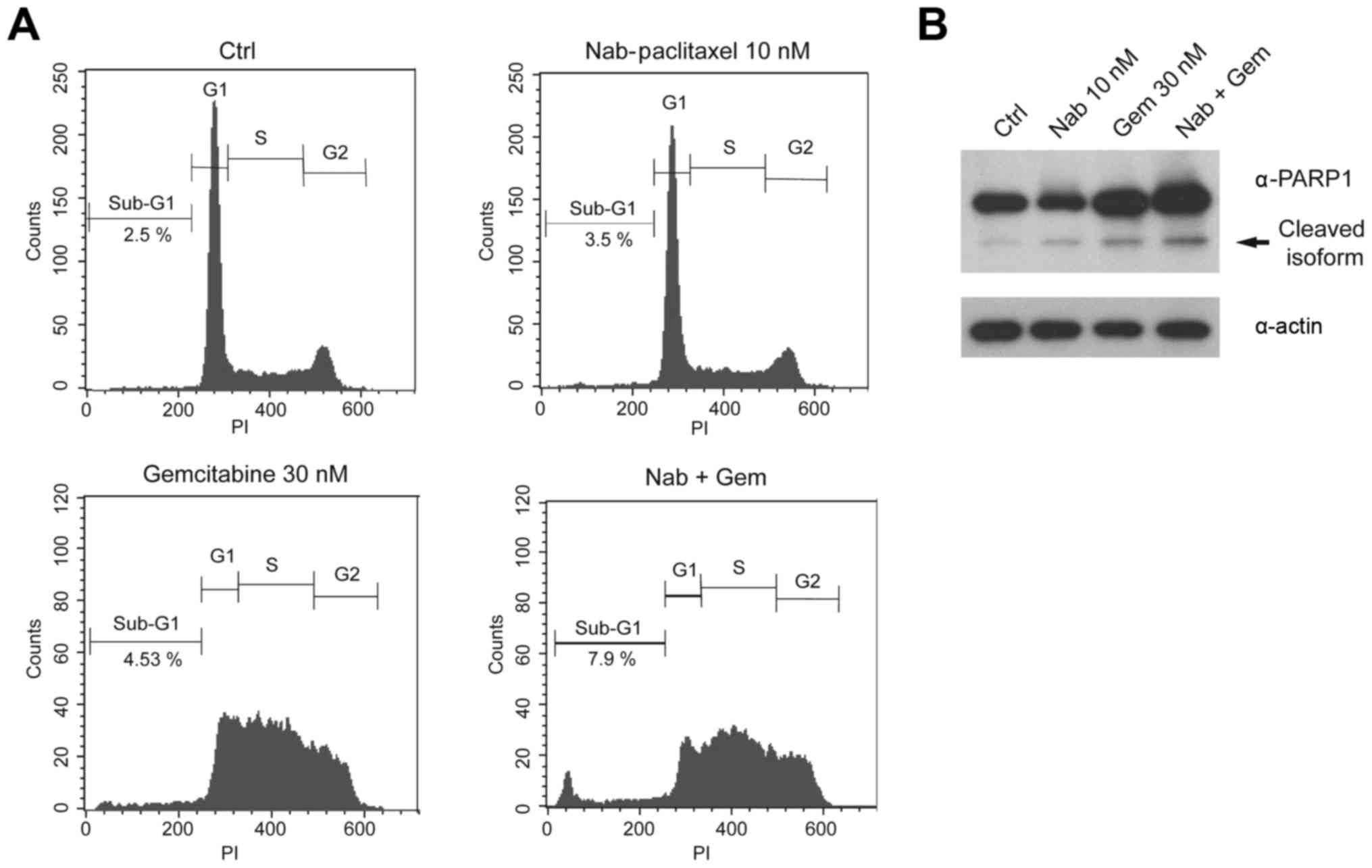
To investigate the nature of the additive effect of
gemcitabine and nab-paclitaxel on PDAC cell death, we analyzed cell
cycle progression and cell death in more detail in MiaPaCa-2 cells.
Flow cytometry analysis with propidium iodide (PI) of cells treated
with suboptimal doses of gemcitabine (30 nM) and nab-paclitaxel (10
nM) for 48 h indicated that gemcitabine strongly affected the cell
cycle progression, leading to cell accumulation in S phase, whereas
nab-paclitaxel elicited very mild effects. Notably, however, the
addition of nab-paclitaxel to gemcitabine led to the appearance of
a defined peak in the sub-G1 population of MiaPaCa-2 cells
(Fig. 4A), indicating cell death by
apoptosis. To confirm the effect of the combined treatment on cell
apoptosis, we monitored cleavage of poly(ADP-ribose) polymerase
(PARP1) by western blot analysis. Consistent with the appearance of
the sub-G1 peak, PARP1 cleavage was noticeably increased in
MiaPaCa-2 cells treated with the combination of the two drugs
(Fig. 4B).
Addition of nab-paclitaxel to
gemcitabine induces a stronger cell cycle blockage in S phase
The PI profile indicated that co-treatment with
nab-paclitaxel enhanced the accumulation of cells in the S phase of
the cycle compared to gemcitabine alone (Fig. 4A). To further investigate this
possibility, we analyzed the incorporation of BrdU as a precise
marker of DNA duplication in S phase. A short pulse of BrdU was
administered to MiaPaCa-2 cells 30 min before harvesting, following
24 h of incubation with the drugs. We observed that both
gemcitabine and nab-paclitaxel, used as single agents, caused an
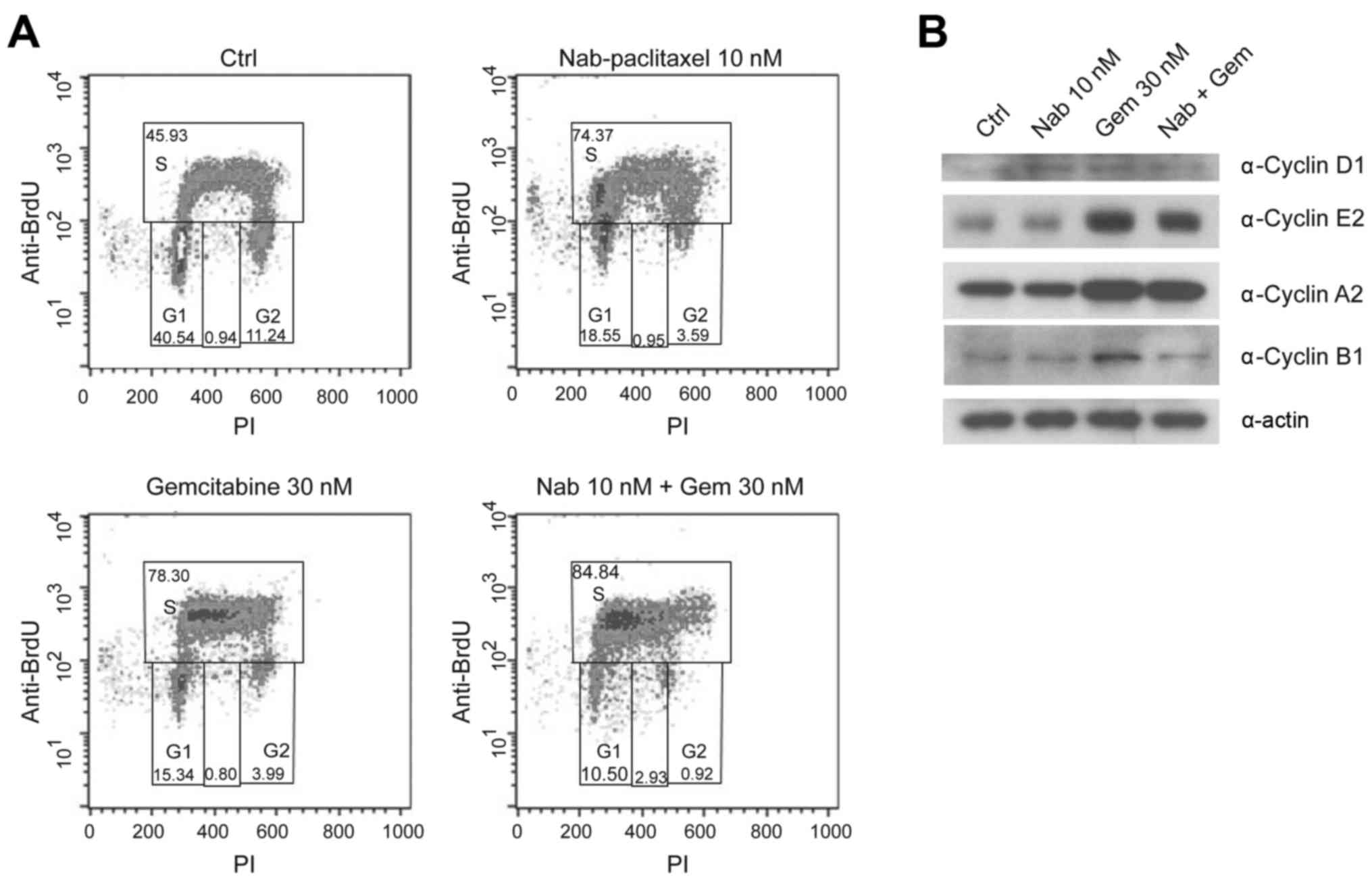
accumulation of cells in the S phase (from 45,93 to 78,30 and
74,37%) (Fig. 5A). Notably, the
combination of both drugs resulted in an additive effect on the
accumulation of cells in S phase, which reached 84.84%. As a
consequence of this blockage in cell cycle progression,
co-treatment with gemcitabine and nab-paclitaxel resulted in a
sharp reduction of cells transiting in the G2 phase (Fig. 5A).
In addition, we checked the changes in cell cycle
progression by monitoring the expression levels of phase-specific
cyclins. As dislplayed in Fig. 5B,
cyclin D1 levels were not affected by treatments, whereas cyclin A2
and E2 levels increased after gemcitabine administration either
alone or in combination with nab-Paclitaxel, confirming that the
cells are mainly blocked in the S phase. Treatment with
nab-paclitaxel alone did not cause accumulation of S phase cyclins
(Fig. 5B), even though the cells
were blocked at this stage of the cycle. Since we noticed that
nab-paclitaxel caused accumulation of cells in the left-most region
of S phase (Fig. 5A), indicating
very little duplication of DNA, it is probable that this drug
blocks cells before the accumulation of cyclins E2 and A2.
Additionally, we observed that the combined treatment with both
drugs reduced the expression of cyclin B1 compared to gemcitabine
alone. Since this cyclin is involved in the S-G2 cell cycle
transition, its levels reflect the reduction of cells in G2 phase,
which was observed in flow cytometric analyses (Figs. 4A and 5A).
Discussion
The aim of the present study was to examine the
activity of nab-paclitaxel alone or in combination with gemcitabine
in PDAC cell lines displaying different degree of primary
resistance to gemcitabine and in a previously described model of
secondary resistance to the drug (7,8).
The results of the present study revealed that
nab-paclitaxel is effective in PDAC cells irrespective of their
sensitivity to gemcitabine and to the status of primary or
secondary (acquired) resistance (Figs.
1 and 2). Notably, both drugs
demonstrated an addictive effect at suboptimal doses in cell lines
with primary or secondary resistance to gemcitabine (Fig. 3).
To investigate the underlying mechanisms of the
observed efficacy of nab-paclitaxel, we explored the changes
occurring in the cell cycle (Figs.
4 and 5). Our results indicated
that nab-paclitaxel blocked cell proliferation in a different
manner compared to gemcitabine. Although both drugs caused an
arrest in S phase, the cells treated with gemcitabine exhibited a
different extent of DNA duplication, whereas the peak of cells
treated with nab-paclitaxel is present in the left region of the
graph, indicating that cells arrest as soon as they start
duplicating their DNA. This difference is also illustrated by the
accumulation of S phase cyclins, which is evident in gemcitabine-
but not in nab-paclitaxel-treated cells. While the blockage in S
phase is expected after gemcitabine exposure, due to depletion of
the nucleotide pool required for DNA duplication, cells treated
with nab-paclitaxel were expected to arrest in mitosis or late G2
phase due to defects in spindle elongation. However, recent data
have revealed that cells treated with paclitaxel often proceeded
through mitosis into the next interphase, where the majority of
cell deaths occurred (16). In
particular, nab-paclitaxel seemed to interfere with the very early
stages of the S phase in PDAC cells (Fig. 5A). This may explain why it was
previously found that the interference with the DNA replication
origin activity enhanced the response of cells to paclitaxel
(16).
The different mechanism of S phase blockage by the
two drugs may explain the additive affect observed in the combined
treatment. Markedly, such additive effect was observed both at the
cell cycle and the cell death level, indicating a causal
relationship between the two events. Although the molecular
mechanisms involved in such effect need further investigation, our
results indicated that nab-paclitaxel strongly enhances the
cytotoxicity of gemcitabine and may help to overcome both primary
and acquired resistance to this drug.
The in vitro results of the present study
revealed that nab-paclitaxel, alone or in combination with
gemcitabine, is an active drug in preclinical models of
gemcitabine-resistant PDAC. Hence, our observations indicated that,
in certain clinical scenarios, nab-paclitaxel could be active in
patients with PDAC that are not responding to gemcitabine, even in
monotherapy. However, clinical data on the use of nab-paclitaxel as
a single agent in patients with PDAC that were previously treated
with gemcitabine, are limited. In a small phase II trial, 19
patients received nab-paclitaxel after progression under
gemcitabine-based therapy (17).
One of them (5.3%) had a confirmed partial response and 6 (32%)
exhibited stable disease as the best response. Another
single-center retrospective study evaluated the use of
nab-paclitaxel in 20 patients with advanced PDAC who previously
exhibited progression under gemcitabine, 40% of whom also received
FOLFIRINOX. Notably, about two thirds of patients had stable
disease as best response, although the median OS was only of 5.2
months (18). Further studies are
needed to elucidate whether this approach may be beneficial,
possibly in patients with less advanced disease.
The present study is one of the few that aimed to
evaluate the efficacy of nab-paclitaxel in preclinical settings,
using cell lines that are models of both primary and secondary
resistance to gemcitabine. As the investigation is limited to in
vitro models, the results should be interpreted with caution
and the mechanisms of the activity observed in the present study
need further experiments to be elucidated. In particular, our data
are useful to generate hypotheses that need to be confirmed in
other models, such as animal ones, that may better recapitulate the
human pathology. Conversely, the additive effect of nab-paclitaxel
and gemcitabine observed in these experiments cannot be due to
factors that have been extensively investigated in animal models
and that are related with tumor stroma and penetration of drugs.
Another limitation of the present study concerns the lack of a
defined mechanism for the observed effects. Following the revision
process, we tested some common signal transduction pathways that
could be involved in the response to these chemotherapeutic agents,
such as the PI3K-mTOR and ERK pathways (data not shown). However,
we did not find a direct correlation with the cytotoxic effect.
Thus, further studies are needed to analyze the mechanisms
underlying the observed effects. Considering the above-mentioned
limitations, our results revealed that treatment with
nab-paclitaxel may overcome resistance to gemcitabine and may
represent a potentially valuable therapeutic approach for advanced
PDAC.
Acknowledgements
The present study was supported by grants from the
Associazione Italiana per la Ricerca sul Cancro (AIRC; IG18790 for
C.S. and IG 17177 for G.C.).
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
Statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rahib L, Smith BD, Aizenberg R, Rosenzweig
AB, Fleshman JM and Matrisian LM: Projecting cancer incidence and
deaths to 2030: The unexpected burden of thyroid, liver, and
pancreas cancers in the United States. Cancer Res. 74:2913–2921.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fogel EL, Shahda S, Sandrasegaran K,
DeWitt J, Easler JJ, Agarwal DM, Eagleson M, Zyromski NJ, House MG,
Ellsworth S, et al: A multidisciplinary approach to pancreas cancer
in 2016: A review. Am J Gastroenterol. 112:537–554. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Neuzillet C, Tijeras-Raballand A, Bourget
P, Cros J, Couvelard A, Sauvanet A, Vullierme MP, Tournigand C and
Hammel P: State of the art and future directions of pancreatic
ductal adenocarcinoma therapy. Pharmacol Ther. 155:80–104. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Burris HA III, Moore MJ, Andersen J, Green
MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo
AM, Tarassoff P, et al: Improvements in survival and clinical
benefit with gemcitabine as first-line therapy for patients with
advanced pancreas cancer: A randomized trial. J Clin Oncol.
15:2403–2413. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Binenbaum Y, Na'ara S and Gil Z:
Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug
Resist Updat. 23:55–68. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Adesso L, Calabretta S, Barbagallo F,
Capurso G, Pilozzi E, Geremia R, Delle Fave G and Sette C:
Gemcitabine triggers a pro-survival response in pancreatic cancer
cells through activation of the MNK2/eIF4E pathway. Oncogene.
32:2848–2857. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Calabretta S, Bielli P, Passacantilli I,
Pilozzi E, Fendrich V, Capurso G, Fave GD and Sette C: Modulation
of PKM alternative splicing by PTBP1 promotes gemcitabine
resistance in pancreatic cancer cells. Oncogene. 35:2031–2039.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Conroy T, Desseigne F, Ychou M, Bouché O,
Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de
la Fouchardière C, et al: FOLFIRINOX versus gemcitabine for
metastatic pancreatic cancer. N Engl J Med. 364:1817–1825. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Von Hoff DD, Ramanathan RK, Borad MJ,
Laheru DA, Smith LS, Wood TE, Korn RL, Desai N, Trieu V, Iglesias
JL, et al: Gemcitabine plus nab-paclitaxel is an active regimen in
patients with advanced pancreatic cancer: A phase I/II trial. J
Clin Oncol. 29:4548–4554. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Von Hoff DD, Ervin T, Arena FP, Chiorean
EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et
al: Increased survival in pancreatic cancer with nab-paclitaxel
plus gemcitabine. N Engl J Med. 369:1691–1703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vaccaro V, Sperduti I, Vari S, Bria E,
Melisi D, Garufi C, Nuzzo C, Scarpa A, Tortora G, Cognetti F, et
al: Metastatic pancreatic cancer: Is there a light at the end of
the tunnel? World J Gastroenterol. 21:4788–4801. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lemstrova R, Melichar B and
Mohelnikova-Duchonova B: Therapeutic potential of taxanes in the
treatment of metastatic pancreatic cancer. Cancer Chemother
Pharmacol. 78:1101–1111. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Neesse A, Algül H, Tuveson DA and Gress
TM: Stromal biology and therapy in pancreatic cancer: A changing
paradigm. Gut. 64:1476–1484. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Arumugam T, Ramachandran V, Fournier KF,
Wang H, Marquis L, Abbruzzese JL, Gallick GE, Logsdon CD, McConkey
DJ and Choi W: Epithelial to mesenchymal transition contributes to
drug resistance in pancreatic cancer. Cancer Res. 69:5820–5828.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Koh SB, Mascalchi P, Rodriguez E, Lin Y,
Jodrell DI, Richards FM and Lyons SK: A quantitative FastFUCCI
assay defines cell cycle dynamics at a single-cell level. J Cell
Sci. 130:512–520. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hosein PJ, de Lima Lopes G Jr, Pastorini
VH, Gomez C, Macintyre J, Zayas G, Reis I, Montero AJ, Merchan JR
and Rocha Lima CM: A phase II trial of nab-Paclitaxel as
second-line therapy in patients with advanced pancreatic cancer. Am
J Clin Oncol. 36:151–156. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Peddi PF, Cho M, Wang J, Gao F and
Wang-Gillam A: Nab-paclitaxel monotherapy in refractory pancreatic
adenocarcinoma. J Gastrointest Oncol. 4:370–373. 2013.PubMed/NCBI
|