Introduction
The discovery of human induced pluripotent stem
cells (hiPSCs) has revolutionized the field of pluripotent stem
cell research (1). Expression of
OCT4 and SOX2, genes involved in early development, in concert with
MYC and KLF4 oncogenes, can induce the transformation of adult
somatic cells into hiPSCs that adopt morphological and functional
characteristics of pluripotency indistinguishable from human
embryonic stem cells (hESCs) (2,3). The
importance of obtaining hiPSCs that possess the same phenotype as
hESCs, resides in the fact that such cells can be differentiated
into tissues derived from the three germ layers when appropriate
differentiation protocols are applied (4–6).
Although hiPSCs represent an important source for therapeutic
applications such as regenerative and personalized medicine
(7–9), one of the major hindrances in
translating the hiPSC technology into the clinic is the development
of teratoma-like tumors that originate from the in vivo
growth of hiPSCs (10). For this
reason, before hiPSCs can be successfully used in innovative
therapeutic designs, it is mandatory to understand their karyotypic
stability. Notably, forced expression of OCT4, SOX2, MYC and KLF4
reprogramming factors has been revealed to predispose hiPSCs to
chromosomal aberrations (11).
Impairment of the cell cycle machinery has an important role in the
propensity of hESCs and hiPSCs to acquire chromosomal defects. It
has been demonstrated that aberrant cell cycle regulation of hESCs
is linked to numerical centrosomal abnormalities during mitosis,
which may account, for their enhanced chromosomal instability
(CIN), and thus increase their tumorigenicity (12). Supporting these findings, the tumor
suppressor p53 plays a key role as a ‘guardian of reprogramming’,
safeguarding genomic integrity during reprogramming at the cost of
a reduced efficiency of the process (13). Notably, the mitotic kinase Aurora-A,
that induces centrosome amplification and CIN in cancer (14), facilitated pluripotency through
phosphorylation-mediated inhibition of p53-directed ectodermal and
mesodermal gene expression (15).
Phosphorylation of p53 not only impaired p53-induced hESC
differentiation but also p53-mediated suppression of hiPSC
reprogramming. Although these studies demonstrated a critical role
for the Aurora-A/p53 axis in the regulation of self-renewal,
chromosomal stability and somatic cell reprogramming, it remains to
be explored whether concurrent aberrant Aurora-A activity and loss
of p53 function increases the tumorigenicity of hiPSCs.
In the present study we analyzed the development of
CIN during hiPSCs reprogramming and demonstrated that chromosomal
and mitotic aberrations were linked to centrosome amplification,
Aurora-A overexpression, abrogation of p53-mediated G1/S cell cycle
checkpoint and loss of Rb function. Notably, hiPSCs with
CIN-developed high-grade teratomas harboring centrosome
abnormalities in immunocompromised mice and ex vivo teratoma
cells exhibited high self-renewal capacity in vitro that was
linked to Aurora-A kinase activity and development of lung
metastasis. Collectively, these findings demonstrated a previously
undisclosed linkage between Aurora-A overexpression, CIN and
development of aggressive teratomas derived from hiPSCs.
Materials and methods
Generation of human iPS cells
(hiPSCs)
hiPSCs from skin-derived keratinocytes (N1-hiPSCs)
and blood-derived cells (DS1-hiPSCs) were established by
transduction of 4 reprogramming lentiviral vectors as previously
described (16). hiPSCs were
maintained in Pluriton Reprogramming Medium (Stemgent, Cambridge,
MA, USA) supplemented with 25% (v/v) mTeSR™-1 maintenance media
(Stemcell Technologies, Vancouver, BC, Canada) on BD
Matrigel-coated cell culture plates (BD Biosciences, San Jose, CA,
USA).
Spectral karyotyping (SKY)
analysis
Hybridization, wash, and detection of the human
SKYPaint® probe (Applied Spectral Imaging, Vista, CA,
USA) were performed as recommended by the manufacturer. Image
acquisition and spectral analysis of metaphase cells were achieved
by using the SD200 SpectraCube™ Spectral Imaging system (Applied
Spectral Imaging) mounted on a Zeiss Axioplan2 microscope (Carl
Zeiss MicroImaging, Inc., Thornwood, NY, USA). Images were analyzed
using HiSKY analysis software (Applied Spectral Imaging).
Immunoblotting, immunofluorescence and
FACS assays
Immunoblotting, immunofluorescence and FACS assays
were performed as previously described (14). Antibodies that were employed to
perform these studies were the following: p53 (1:500; mouse
monoclonal; cat. no. PIMA512557; Invitrogen/Thermo Fisher
Scientific, Waltham, MA, USA), p21 (1:500; mouse monoclonal; cat.
no. AHZ0422; Thermo Fisher Scientific), centrin 20H5 (1:1,000;
mouse monoclonal; Mayo Clinic, Rochester, MN, USA), pericentrin
(1:500; rabbit polyclonal; cat. no. ab4448l) and p-retinoblastoma
(1:400; rabbit polyclonal; cat. no. ab47763; both from Abcam,
Cambridge, MA, USA), γ-tubulin (1:4,000; mouse monoclonal; cat. no.
MA1-850; Thermo Fisher Scientific), Aurora-A (1:500; mouse
monoclonal; cat. no. ab13824; Abcam), p-Aurora-A (1:400; rabbit
monoclonal; cat. no. 3079S; Cell Signaling Technology, Inc.,
Danvers, MA, USA), p-retinoblastoma (1:500; mouse monoclonal; cat.
no. R6878-1ML) and β-actin (1:5,000; mouse monoclonal, cat. no.
A2228-100UL; both from Sigma-Aldrich, St. Louis, MO, USA). Results
were derived from three independent experiments with comparable
outcomes.
Generation of tumorspheres
One thousand N1-hiPSCs 1GX (first generation
xenografts) were cultured under non-adherent conditions using a
defined stem cell medium (Stem Cell Technologies) for 10 days. The
formation of tumorspheres was monitored and recorded using a Zeiss
light microscope. After 48 h separate groups of tumorspheres were
treated with the selective Aurora-A inhibitor, alisertib (0.5 µM)
for 8 days. Three independent experiments were performed with
comparable outcomes.
Animal studies
Procedures established by the Institutional Animal
Care and Use Committee (IACUC) of Mayo Clinic, based on the US NIH
guidelines for the care and use of laboratory animals were adhered
to for all experiments. Four-week-old SCID/beige mice were
anesthetized by exposure to 3% isoflurane and 1×106
N1-hiPSCs were injected into their kidney capsule (three mice per
each group). After 12 weeks, the mice were sacrificed and the tumor
xenografts were processed for histology. To re-establish cell
cultures from tumor explants, tumors tissues were excised from
sacrificed animals, minced using sterile scissors, transferred to
complete culture medium and fibroblast-free tumor cells were
established by serial passages in culture. Animals were examined
everyday and body weight and primary tumor size were assessed at
least 1–2 times/week. Consistent distress and potential pain (>1
day) were alleviated by euthanasia. If some of the animals were
losing >10% of their body weight, if blood was consistently
observed in the urine or around the genitals of the mice, the mice
were appropriately euthanized. When typical signs of distress
including labored breathing and inactivity were consistently
observed for >1 day, the animals were appropriately euthanized.
When the primary tumor was >2 cm, the animals were sacrificed.
Animals were euthanized using Pentobarbital (i.p., 100 mg/kg)
followed by cervical dislocation. The IACUC approved this
study.
Experimental lung metastases
Four-week-old SCID/beige mice were anesthetized by
exposure to 3% isoflurane and 1×106 N1-hiPSCs 1GX were
injected into their tail vein (three mice per each group) as
previously described (14). After 4
weeks, the mice were sacrificed and lungs were isolated to detect
metastatic lesions by employing a human-mitochondria specific
antibody.
Results and Discussion
To determine the extent to which hiPSC reprogramming
of somatic cells may induce the development of CIN, we generated
hiPSCs from human keratinocytes and blood cells as previously
described (16) and termed them
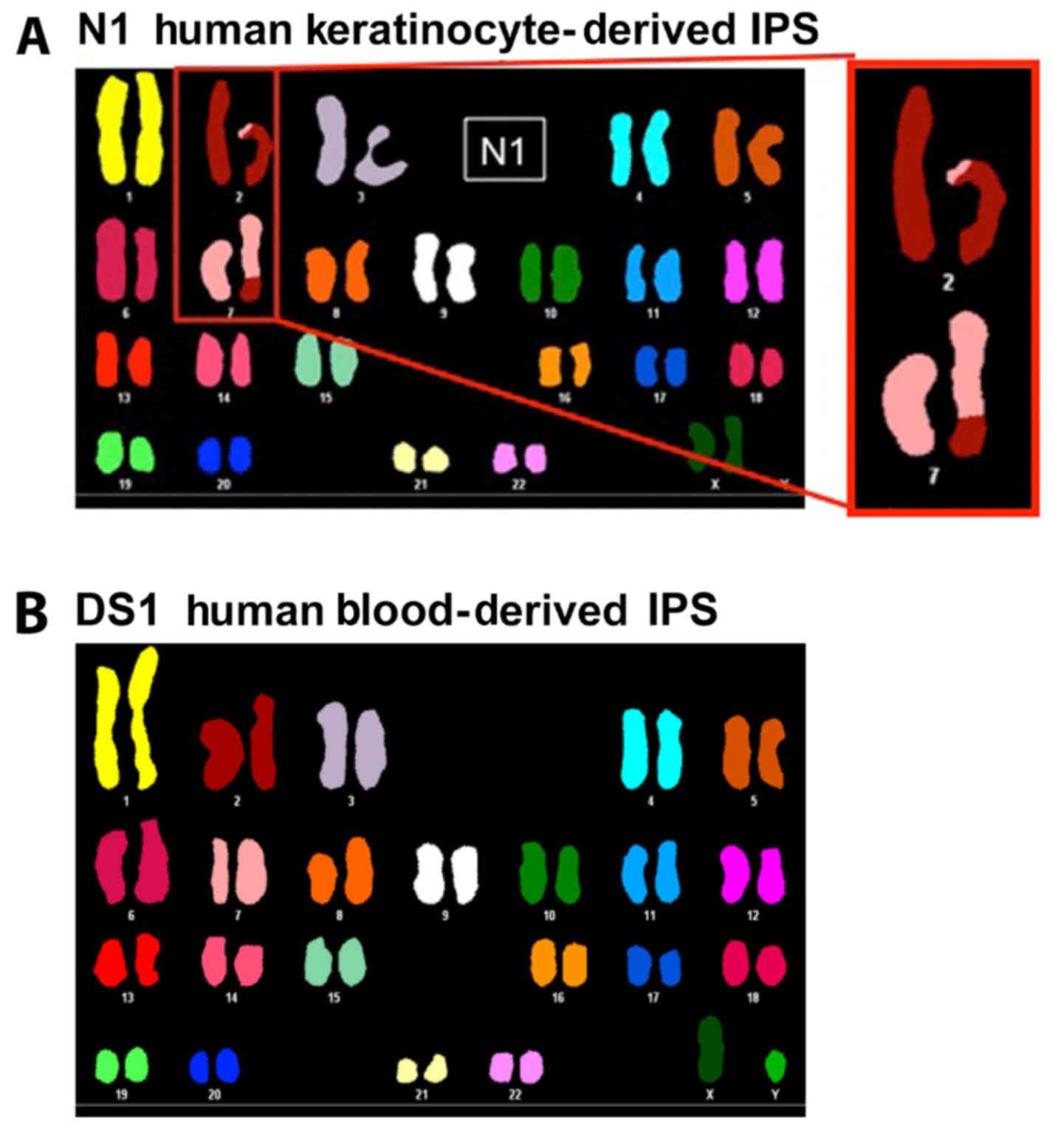
N1-hiPSCs and DS1-hiPSCs, respectively. Chromosome analysis using
spectral karyotyping (SKY) technology revealed that N1-hiPSCs were
characterized by a translocation between chromosomes 2 and 7
(Fig. 1A), while DS1-hiPSCs
exhibited a normal karyotype (Fig.
1B). This unique chromosome 2 and 7 translocation identified in
N1-hiPSCs was uncommon since the predominant genetic changes found
in hiPSCs involve structural and numerical changes in chromosomes
1, 12, 17 and 20 (17,18). Due to the fact that chromosomal
abnormalities are linked to mitotic defects during cell division,
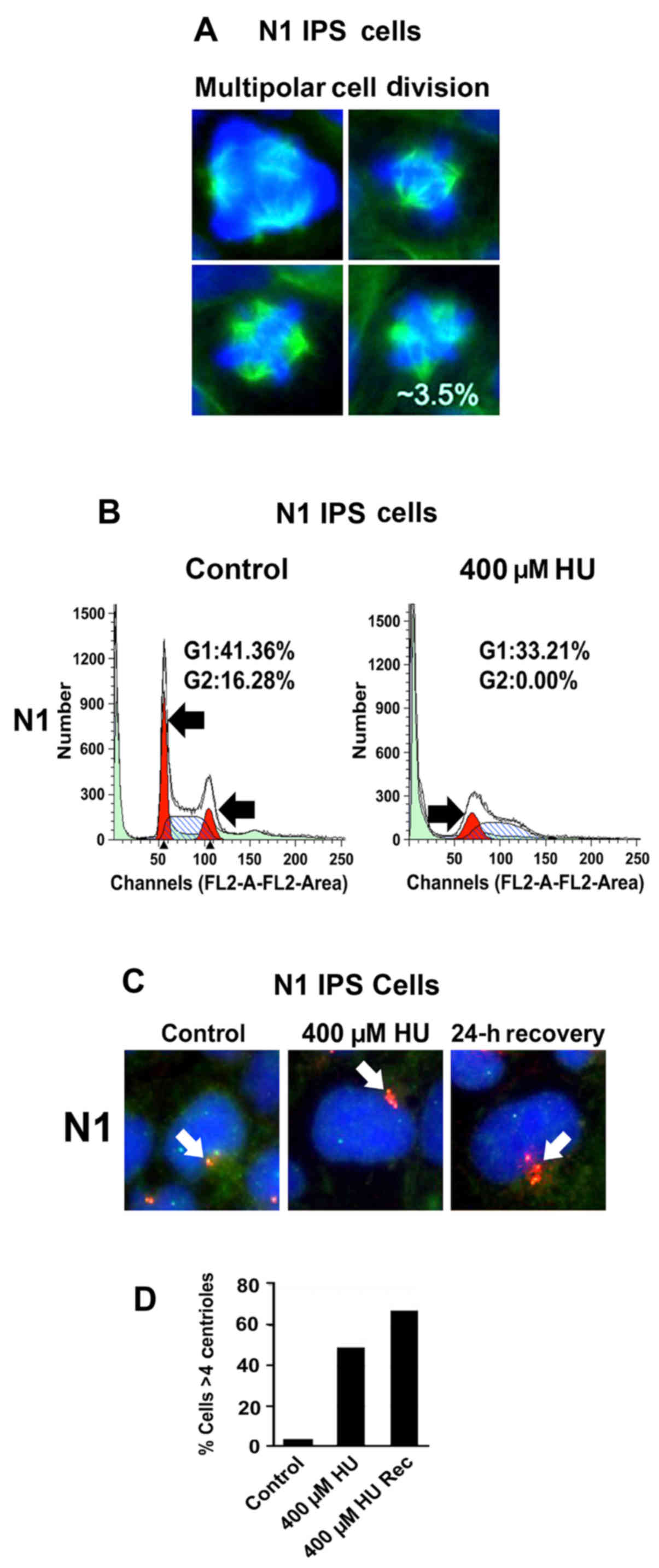
we analyzed the percentage of normal and aberrant mitoses in
N1-hiPSCs. The counting of mitotic images revealed that 3.5% of
total mitoses were characterized by multipolar mitotic spindles
(Fig. 2A) that promote unequal
chromosome segregation and CIN (19). Since hiPSC reprogramming is
characterized by induction of genotoxic stress (20), we established whether development of
multipolar mitoses was linked to genotoxic stress-induced
centrosome amplification. N1-hiPSCs were treated with hydroxyurea
(HU), a genotoxic agent that induces G1/S cell cycle arrest and
centrosome amplification in cancer cells lacking the p53-mediated
G1/S cell cycle checkpoint (21).
Following treatment with HU for 48 h, N1-hiPSCs were arrested in
the G1/S phase of the cell cycle (Fig.
2B). To determine whether G1/S cell cycle arrest was uncoupled
from centrosome duplication, we analyzed the centrosome phenotype
in N1-hiPSCs before and after HU treatment. The percentage of cells
exhibiting centrosome amplification (>4 centrioles) was
increased in hiPSCs treated with HU (Fig. 2C and D), indicating that N1-hiPSCs
exhibited a defective G1/S cell cycle checkpoint. Notably, after
recovery from HU, N1-hiPSCs exhibited an exacerbation of centrosome
amplification (Fig. 2C and D). One
possible explanation is that after recovery from HU, N1-hiPSCs
re-entered the cell cycle with amplified centrosomes leading to an
increase of centrosome over-duplication. In view of the fact that
development of centrosome amplification after genotoxic stress is
functionally linked to abrogation of p53-mediated G1/S cell cycle
checkpoint (21), we analyzed the
integrity of p53 tumor-suppressor signaling before and after HU
treatment in N1-hiPSCs and DS1-hiPSCs (used as a control).
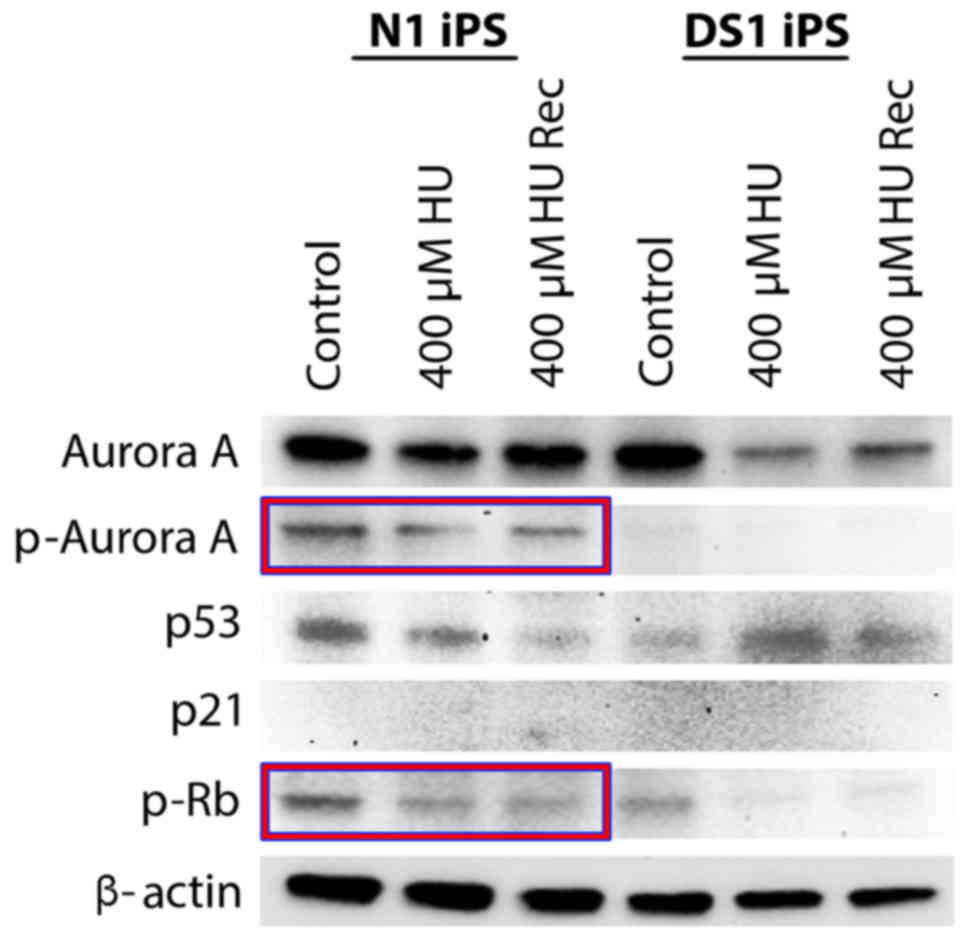
N1-hiPSCs treated with HU exhibited low expression of p53 and did
not exhibit a significant decrease of Rb phosphorylation (Fig. 3) that is critical to induce Rb
activation, G1/S cell cycle arrest and inhibition of centrosome
amplification (19,21). In contrast, DS1-hiPSCs treated with
HU exhibited increased p53 expression and a significant decrease of
Rb phosphorylation, indicating activation of the G1/S cell cycle
checkpoint (Fig. 3). Due to neither
N1-hiPSCs nor DS1-hiPSCs exhibiting increased expression of the p53
downstream target p21, we investigated the expression of the
mitotic kinase Aurora-A that induces p53 degradation and centrosome
amplification in cancer cells (14,22).
Notably, N1-hiPSCs exhibited high levels of total and
phosphorylated (active kinase) Aurora-A before and after HU
treatment, while Aurora-A levels were reduced in DS1-hiPSCs after
HU treatment (Fig. 3), suggesting
that aberrant expression/activation of Aurora-A is linked to
abrogation of p53-mediated G1/S cell cycle checkpoint resulting in
centrosome amplification, mitotic defects and CIN in hiPSCs. To
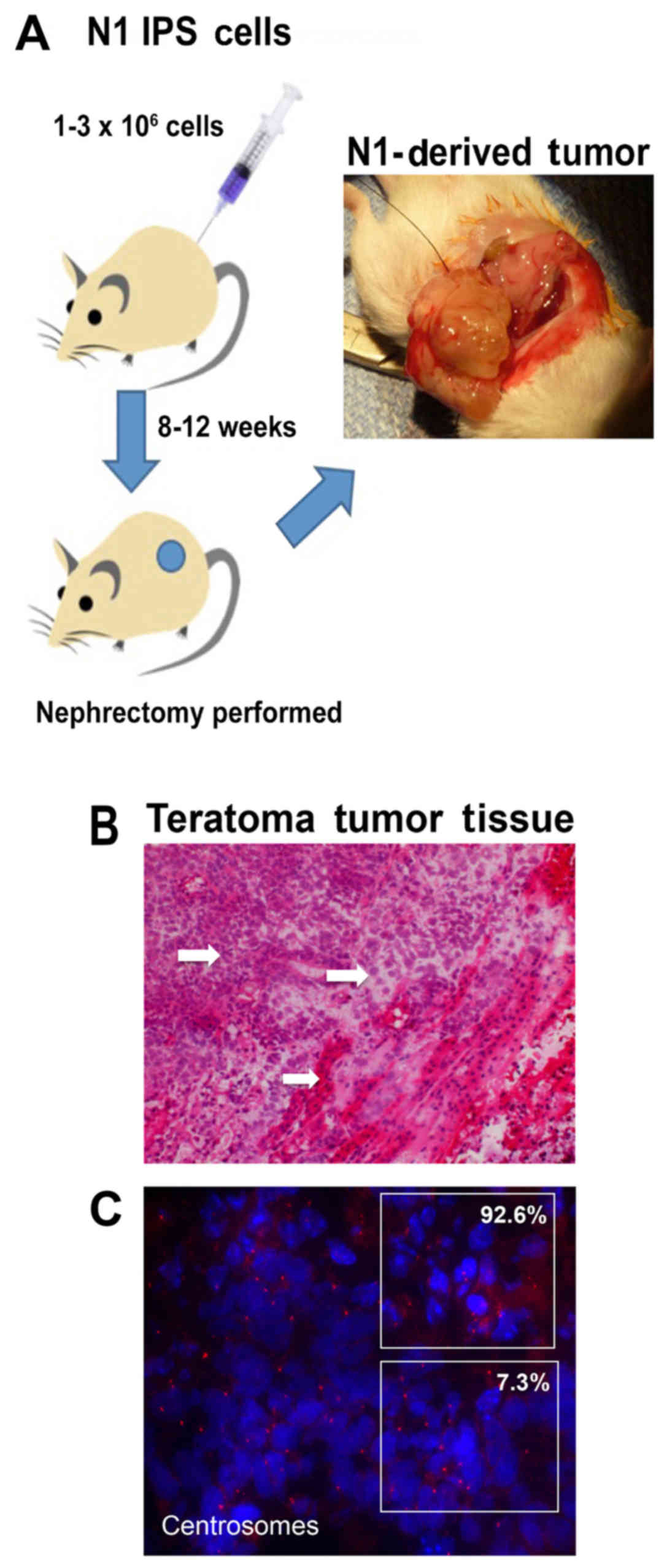
establish the extent to which N1-hiPSCs overexpressing endogenous
Aurora-A give rise to aggressive teratomas, N1-hiPSCs were injected
into the kidney capsule of immunocompromised mice (Fig. 4A). After 12 weeks of injections, the
animals were sacrificed and the tumors were isolated for
histopathological analysis (Fig.
4B). N1-hiPSC-derived teratomas exhibited high tumor grade
based on cells that exhibited non-uniform shapes and sizes and high
nuclear pleomorphism. In addition, we identified in some tumor
sections extensive regions of necrotic foci that are characteristic
of proliferative malignant tumor cells (23). Notably, the majority of
N1-hiPSC-derived teratoma cells exhibited duplicated and amplified
centrosomes (Fig. 4C),
demonstrating the linkage between an aggressive teratoma phenotype
and dysregulation of the centrosome cycle responsible for CIN. Due
to the fact that the tumor tissue analysis previously
aforementioned revealed that N1-hiPSC-derived tumors were more
malignant than benign teratomas, we aimed to characterize their
self-renewal and metastatic properties. N1-hiPSC-derived teratomas
were excised and re-cultured cells were termed N1-hiPSCs 1GX (first
generation xenografts). One thousand N1-HiPSCs 1GX were grown under
non-adherent conditions for 10 days and successfully formed
tumorspheres that represented an in vitro surrogate of
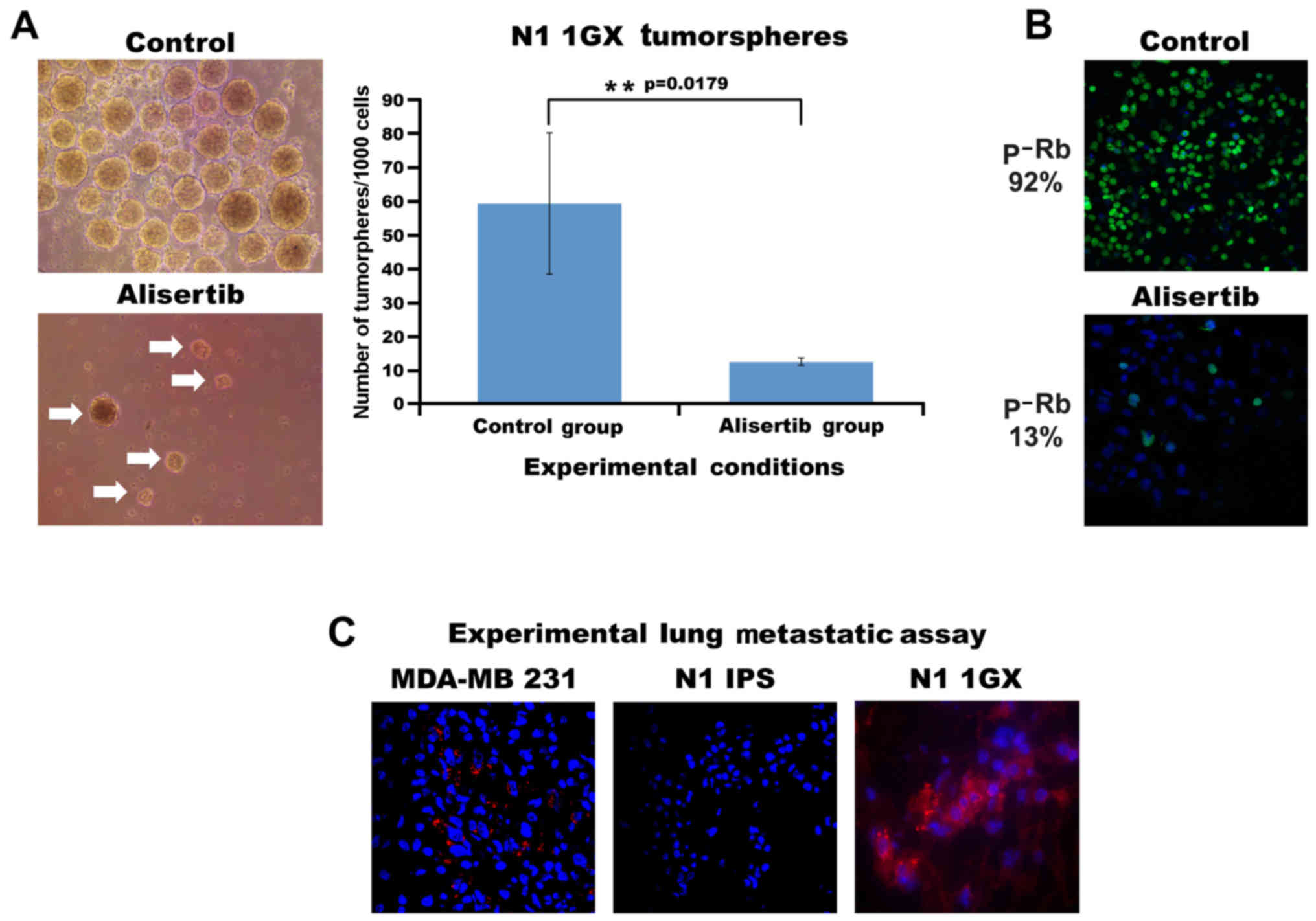
self-renewal activity (Fig. 5A). To
define the causal role of Aurora-A kinase activity in inducing
N1-hiPSCs 1GX self-renewal capacity, N1-hiPSCs 1GX tumorspheres
were treated with the selective Aurora-A inhibitor alisertib.
Treatment with alisertib significantly reduced the number and size
of tumorspheres, demonstrating that Aurora-A kinase activity was
required for the self-renewal capacity of teratoma cells (Fig. 5A). Subsequently, to determine the
extent to which alisertib-mediated inhibition of self-renewal
capacity was linked to impairment of Rb phosphorylation. Treatment
of N1-hiPSCs 1GX tumorspheres with alisertib for 48 h reduced
nuclear Rb phosphorylation (Fig.
5B), indicating that Aurora-A kinase promoted N1-hiPSCs 1GX
tumorsphere self-renewal capacity through phosphorylation and
inactivation of the Rb tumor suppressor. Finally, to assess the
malignant phenotype of N1-hiPSC-derived teratomas, N1-hiPSCs and
N1-HiPSCs 1GX were injected into the tail vein of immunocompromised
mice to develop experimental lung metastasis. Highly metastatic
MDA-MB 231 breast cancer cells were used as a positive control
(14). Only MDA-MB 231 cells and
N1-HiPSCs 1GX developed experimental lung metastasis after 4 weeks
of tail vein injections (Fig. 5C),
demonstrating that hiPSCs with CIN give rise to aggressive
teratomas in vivo. Collectively, these findings demonstrated
a high risk for malignancy of human keratinocyte-derived hiPSCs
that exhibited Aurora-A overexpression, centrosome amplification,
loss of Rb function and CIN. Notably, rigorous quality-control
tests, including comprehensive genomic integrity validation,
analysis of p53/Rb tumor-suppressor pathways and Aurora-A kinase
activity should be conducted before the clinical application of
hiPSCs. Based on these findings, we propose that Aurora-A-targeted
therapy could represent a promising prophylactic therapeutic
strategy to decrease the likelihood of CIN and development of
aggressive teratomas derived from hiPSCs.
Acknowledgements
The present study was supported by the USAMRMC
BC022276, Intramural RECDA and The Nan Sayner Awards to A.B.D., NCI
CA72836 to J.L.S., the Mayo Clinic Breast Cancer Specialized
Program of Research Excellence (SPORE) NIH CA116201 to J.I., the
Prospect Creek Foundation to E.G. and the Mayo Clinic Comprehensive
Cancer Center. We also wish to acknowledge the Pathology Research
Core facility of the Mayo Clinic School of Medicine and Science,
for performing IHC assays and assisting us with the interpretation
of the results.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Takahashi K and Yamanaka S: Induction of
pluripotent stem cells from mouse embryonic and adult fibroblast
cultures by defined factors. Cell. 126:663–676. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Takahashi K, Tanabe K, Ohnuki M, Narita M,
Ichisaka T, Tomoda K and Yamanaka S: Induction of pluripotent stem
cells from adult human fibroblasts by defined factors. Cell.
131:861–872. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yu J, Vodyanik MA, Smuga-Otto K,
Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA,
Ruotti V, Stewart R, et al: Induced pluripotent stem cell lines
derived from human somatic cells. Science. 318:1917–1920. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pettinato G, Wen X and Zhang N: Formation
of well-defined embryoid bodies from dissociated human induced
pluripotent stem cells using microfabricated cell-repellent
microwell arrays. Sci Rep. 4:74022014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pettinato G, Vanden Berg-Foels WS, Zhang N
and Wen X: ROCK inhibitor is not required for embryoid body
formation from singularized human embryonic stem cells. PLoS One.
9:e1007422014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pettinato G, Wen X and Zhang N:
Engineering strategies for the formation of embryoid bodies from
human pluripotent stem cells. Stem Cells Dev. 24:1595–1609. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pettinato G, Ramanathan R, Fisher RA,
Mangino MJ, Zhang N and Wen X: Scalable differentiation of human
iPSCs in a multicellular spheroid-based 3D culture into
hepatocyte-like cells through direct Wnt/β-catenin pathway
inhibition. Sci Rep. 6:328882016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ramanathan R, Pettinato G, Beeston JT, Lee
DD, Wen X, Mangino MJ and Fisher RA: Transplantation of human stem
cell-derived hepatocytes in an animal model of acute liver failure.
Surgery. 158:349–359. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Takahashi K and Yamanaka S: A decade of
transcription factor-mediated reprogramming to pluripotency. Nat
Rev Mol Cell Biol. 17:183–193. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Riegler J, Ebert A, Qin X, Shen Q, Wang M,
Ameen M, Kodo K, Ong SG, Lee WH, Lee G, et al: Comparison of
magnetic resonance imaging and serum biomarkers for detection of
human pluripotent stem cell-derived teratomas. Stem Cell Reports.
6:176–187. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Buganim Y, Markoulaki S, van Wietmarschen
N, Hoke H, Wu T, Ganz K, Akhtar-Zaidi B, He Y, Abraham BJ, Porubsky
D, et al: The developmental potential of iPSCs is greatly
influenced by reprogramming factor selection. Cell Stem Cell.
15:295–309. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Holubcová Z, Matula P, Sedláčková M,
Vinarský V, Doležalová D, Bárta T, Dvořák P and Hampl A: Human
embryonic stem cells suffer from centrosomal amplification. Stem
Cells. 29:46–56. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marión RM, Strati K, Li H, Murga M, Blanco
R, Ortega S, Fernandez-Capetillo O, Serrano M and Blasco MA: A
p53-mediated DNA damage response limits reprogramming to ensure iPS
cell genomic integrity. Nature. 460:1149–1153. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
D'Assoro AB, Liu T, Quatraro C, Amato A,
Opyrchal M, Leontovich A, Ikeda Y, Ohmine S, Lingle W, Suman V, et
al: The mitotic kinase Aurora: A promotes distant metastases by
inducing epithelial-to-mesenchymal transition in ERα+
breast cancer cells. Oncogene. 33:599–610. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee DF, Su J, Ang YS, Carvajal-Vergara X,
Mulero-Navarro S, Pereira CF, Gingold J, Wang HL, Zhao R, Sevilla
A, et al: Regulation of embryonic and induced pluripotency by
aurora kinase-p53 signaling. Cell Stem Cell. 11:179–194. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ohmine S, Squillace KA, Hartjes KA, Deeds
MC, Armstrong AS, Thatava T, Sakuma T, Terzic A, Kudva Y and Ikeda
Y: Reprogrammed keratinocytes from elderly type 2 diabetes patients
suppress senescence genes to acquire induced pluripotency. Aging
(Albany NY). 4:60–73. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Draper JS, Moore HD, Ruban LN, Gokhale PJ
and Andrews PW: Culture and characterization of human embryonic
stem cells. Stem Cells Dev. 13:325–336. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Maitra A, Arking DE, Shivapurkar N, Ikeda
M, Stastny V, Kassauei K, Sui G, Cutler DJ, Liu Y, Brimble SN, et
al: Genomic alterations in cultured human embryonic stem cells. Nat
Genet. 37:1099–1103. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
D'Assoro AB, Busby R, Acu ID, Quatraro C,
Reinholz MM, Farrugia DJ, Schroeder MA, Allen C, Stivala F, Galanis
E and Salisbury JL: Impaired p53 function leads to centrosome
amplification, acquired ERalpha phenotypic heterogeneity and
distant metastases in breast cancer MCF-7 xenografts. Oncogene.
27:3901–3911. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ruiz S, Lopez-Contreras AJ, Gabut M,
Marion RM, Gutierrez-Martinez P, Bua S, Ramirez O, Olalde I,
Rodrigo-Perez S, Li H, et al: Limiting replication stress during
somatic cell reprogramming reduces genomic instability in induced
pluripotent stem cells. Nat Commun. 6:80362015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
D'Assoro AB, Busby R, Suino K, Delva E,
Almodovar-Mercado GJ, Johnson H, Folk C, Farrugia DJ, Vasile V,
Stivala F, et al: Genotoxic stress leads to centrosome
amplification in breast cancer cell lines that have an inactive
G1/S cell cycle checkpoint. Oncogene. 23:4068–4075. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Katayama H, Sasai K, Kawai H, Yuan ZM,
Bondaruk J, Suzuki F, Fujii S, Arlinghaus RB, Czerniak BA and Sen
S: Phosphorylation by aurora kinase A induces Mdm2-mediated
destabilization and inhibition of p53. Nat Genet. 36:55–62. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou J, Schmid T, Schnitzer S and Brüne B:
Tumor hypoxia and cancer progression. Cancer Lett. 237:10–21. 2006.
View Article : Google Scholar : PubMed/NCBI
|