Introduction
The majority (80–85%) of lung cancers are non-small
cell lung cancer (NSCLC), which is the leading cause of
cancer-related death (1). Epidermal
growth factor receptor (EGFR) with primary gain-of-function
mutations is a major molecular driver of NSCLC. EGFR-tyrosine
kinase inhibitors (EGFR-TKIs), such as gefitinib and erlotinib,
have been effective standard therapies for NSCLC with EGFR
mutations. However, almost all patients with NSCLC develop drug
resistance to EGFR-TKIs (1,2).
Multiple mechanisms underlying the resistance to
EGFR-TKIs in NSCLC have been identified. These mechanisms include
the T790M mutation (a threonine to methionine mutation in amino
acid 790), c-MET amplification, activation of alternative signaling
pathways and epithelial to mesenchymal transition (EMT) (1). EMT refers to epithelial cell
transformation, loss of epithelial characteristics and
morphological transformation to a stromal cell phenotype.
Biochemically, EMT is characterized by the reduction of E-cadherin
and the induction of vimentin (2).
EMT is also an important mechanism in the acquired resistance (AR)
to EGFR-TKIs. EMT is detected in ~5% of patients with secondary
resistance to EGFR-TKIs (3).
Much effort has been directed toward overcoming the
AR to EGFR-TKIs by targeting specific mechanisms of resistance.
AZD9291 has been used to treat patients with AR to EGFR-TKIs with
the T790M mutation. However, 50% of patients with AR to EGFR-TKIs
without the T790M mutation need subsequent chemotherapy and
predictive clinical factors to determine sensitivity to
chemotherapy for these patients are still lacking (1,4).
Previous studies have revealed that chemotherapy
efficacy may depend on the underlying mechanisms of AR to
EGFR-TKIs. Rosell et al (5)
observed that in patients with secondary T790M, tumors were more
sensitive to chemotherapeutic drugs, thus prolonging
progression-free survival (PFS) and overall survival (OS). In
contrast, NSCLC cells with EMT and patients with EMT-positive
tumors were less sensitive to chemotherapy or radiotherapy
(6). Furthermore, Kuo et al
(4), revealed that in patients with
AR to EGFR-TKIs, PFS and OS were improved by taxanes, although the
underlying mechanisms of EGFR-TKI resistance are unknown (4).
PC-9 is a NSCLC cell line sensitive to gefitinib.
PC-9 cells have an EGFR-activating mutation, which is a 15-bp
deletion in the EGFR exon 19 (7).
In the present study, we used two PC-9 sublines, PC-9/ZD and
PC-9/GR, which achieve AR through a secondary T790M mutation and
development of EMT, respectively. We studied the correlation
between sensitivity of NSCLC cells to chemotherapeutic drugs and
different mechanisms of EGFR-TKI resistance. We revealed that the
T790M mutation increased NSCLC sensitivity to chemotherapy, whereas
EMT resulted in the opposite effect. Furthermore, we observed that
EMT led to chemotherapeutic resistance by enhancing cancer stem
cell (CSC) properties.
Materials and methods
Cell culture and NSCLC sublines with
AR to EGFR-TKIs
The human lung adenocarcinoma cell line PC-9
(carrying the delE746-A750 mutation in the EGFR gene) was purchased
from The Shanghai Cell Collections of the Chinese Academy of
Sciences (Shanghai, China). The gefitinib-resistant subline PC-9/ZD
was donated by Dr Koizumi at the National Cancer Center Hospital
(Tokyo, Japan). The gefitinib-resistant PC-9/GR subline was
generated as previously described (7). The cancer cells were cultured in
RPMI-1640 supplemented with 10% fetal bovine serum (FBS) (both from
Sigma-Aldrich, St. Louis, MO, USA) at 37°C and 5%
CO2.
Analysis of the EGFR gene mutations
and c-MET amplification
DNA was prepared from cancer cells using the High
Pure PCR Template Preparation kit (Roche Molecular Biochemicals,
Indianapolis, IN, USA) according to the manufacturer's
instructions. We used the amplification refractory mutation system
(ARMS) kit (Zhensheng Biomed, Xiamen, China) to detect point
mutations in the EGFR gene in exons 18, 19, 20 and 21.
Amplification of c-MET was determined by fluorescence in
situ hybridization (FISH) as previously described (7).
Cell Counting Kit-8 (CCK-8) cell
viability assay
Cells in logarithmic growth phase were inoculated
into 96-well plates at a density of 1.5×103 cells/well.
After attachment, the cells were treated with different
concentrations of various drugs. The drugs tested included
gefitinib (10−2-102 µmol/l), paclitaxel
(10−4-104 nmol/l), docetaxel
(10−4-104 nmol/l), pemetrexed
(10−4-104 nmol/l), cisplatin
(10−2-102 µmol/l) and gemcitabine
(10−4-104 nmol/l) (Sigma-Aldrich). After 72
h, cell viability in each well was determined using the CCK-8 kit
(Dojindo Laboratories, Kumamoto, Japan) according to the
manufacturer's instructions.
In vitro cell migration and invasion
assays
Scratch wound assay
Cells at a logarithmic growth phase were inoculated
in 12-well plates at 5.0×104 cells/well. After reaching
80% confluency, the cells were starved overnight in serum-free
medium. Pipette tips (10 µl) were used to generate three scratch
lines in parallel in the cell monolayer. Images were captured at 0,
12, 24 and 48 h post wound under a phase-contrast microscope
(Olympus, Tokyo, Japan). Each experiment was repeated at least
three times.
Invasion assays
QCM™ 24-well Collagen-based Cell Invasion assay
Transwell plates (EMD Millipore, Darmstadt, Germany) were used for
invasion studies. Cells (5×105) were suspended in 200 µl
of serum-free medium and added to the upper chamber. RPMI-1640
medium (500 µl) containing 10% newborn bovine serum was added to
the lower chamber as a chemokine attractant. The cells that
migrated into the lower chamber were stained with crystal violet
for 30 min. Five fields per chamber were imaged using an Olympus
fluorescence inverted microscope (Olympus).
Western blot analysis
Equal amounts of total protein were resolved by
SDS-PAGE and blotted onto polyvinylidene fluoride (PVDF) membranes.
The membranes were probed with primary antibodies against vimentin
(1:1,500; no. SAB1305445), E-cadherin (1:1,000; no. SAB5500022),
EGFR (1:1,000; no. SAB4300352), phosphorylated (p)EGFR (1:1,000;
no. SAB4503760), AKT (1:1,000; no. SAB4500802), pAKT (1:1,000; no.
SAB4504015), extracellular regulated kinase (ERK; 1:1,000; no.
M5670), pERK (1:1,000; no. SAB4301578) and β-actin (1:400; no.
SAB5500001) (all antibodies were rabbit anti-human monoclonal and
were purchased from Sigma-Aldrich). After incubation with primary
antibodies for 24 h, the blots were incubated with HRP-conjugated
secondary antibody (1:2,000; no. A9169, goat anti-rabbit polyclonal
antibody; Sigma-Aldrich). Protein band intensity was analyzed using
Quantity One image analysis software (Bio-Rad Laboratories,
Hercules, CA, USA).
STable expression of E-cadherin and
EGFR T790M-specific siRNA by lentiviral transfection
STable overexpression of cadherin 1 (CDH1) and
T790M-specific siRNA was performed as previously described
(7). Four T790M specific siRNA
sequences (2608, 2597, 2603 and 2600) described by Chen et
al (8) were used to identify
the best sequence to interfere with the expression of T790M, but
minimally affect the expression of wild-type (WT) EGFR for further
studies. The negative shRNA sequence was LV3-NC:
5′TTCTCCGAACGTGTCACGTTTC3′.
Real-time PCR was employed to determine the
expression level of EGFR mRNA using the following oligonucleotides:
EGFR forward and EGFR reverse for the WT allele (9) and T790M forward,
5′TCCTCGATGAAGCCTACGTGAT3′ and reverse, 5′CAAGCGACGGTCCTCCAAGTAG3′
(10).
CSC analysis
The expression of CD133 and CD44, putative
biomarkers for the NSCLC CSCs was determined by flow cytometry.
Cells (106) collected in a 1.5 ml EP tube were incubated
with 100 µl diluted fluorescent-conjugated antibodies against CD133
and CD44 (both from BD Biosciences, Franklin Lakes, NJ, USA), or in
a control group with 100 µl phosphate buffered solutions with
albumin (PBA), on ice for 1 h in the dark. The cells were then
washed three times with (PBS). Following centrifugation (600 × g,
4°C, 10 min), the precipitated cells were re-suspended in 300 µl
PBS, followed by flow cytometric analysis (BD, Biosciences,
Franklin Lakes, NJ, USA).
Side population assay
Cells (106) were incubated with (test
group) or without (control group) 50 µmol/l verapamil. Hoechst
33342 (Sigma-Aldrich) was added to a final concentration of 5 µg/ml
followed by addition of pyridine iodide at a final concentration of
2 µg/ml to screen for dead cells. The cells were then subjected to
flow cytometric analysis.
In vitro tumor spheroid formation
assay
Cells at 5×103/well were inoculated in a
24-well culture plate and cultured in serum-free DMEM/F12 medium
(Invitrogen, Carlsbad, CA, USA) with (experimental group) or
without (control group) 10 ng/ml basic fibroblast growth factor
(bFGF) and 20 ng/ml epidermal growth factor (EGF) (both from Gibco,
Grand Island, NY, USA). The medium was changed every three days.
The culture was terminated after two weeks and tumor spheroids
exceeding 75 µm were counted.
In vivo tumor formation assay
Cells were sorted into four groups: i)
CD133+, ii) CD133−, iii) CD44+ and
iv) CD44−. For each group of four female nude (NOD/SCID)
mice, 103, 104, 105 and
106 cells were injected subcutaneously in the front
flanks of the mice. Tumors exceeding 50 mm3 were counted
to determine the minimum number of inoculated cells required for
tumor formation in vivo. The mice (~100) were purchased from
Vital River Laboratories (Beijing, China). All animal handling and
procedures were approved by the Animal Care and Use Committee of
the General Hospital of Guangzhou Military Command of PLA.
Statistical analysis
Data are expressed as the mean ± standard deviation.
SPSS 20.0 software (IBM Corp., Armonk, NY, USA) was used for
statistical analysis. The t-test was used to compare data between
two groups. Single factor analysis of variance (ANOVA) was used to
compare data among groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
Characterization of NSCLC sublines
with AR to gefitinib
The sensitivity of PC-9/ZD and PC-9/GR cells to
gefitinib was significantly lower compared to PC-9 cells
(P<0.05), while the sensitivity of PC-9/ZD and PC-9/GR to
gefitinib was similar (P>0.05). The inhibitory concentrations at
50% (IC50) of growth in PC-9/ZD, PC-9/GR and PC-9 cells
were 7.85±0.21, 10.62±0.19 and 0.05±0.005 µmol/l, respectively.
PC-9/ZD cells carried the T790M mutation in addition to the
delE746-A750 in exon 19. T790M was not detected in the PC-9/GR
cells. No c-MET amplification was detected in any of the cell lines
as determined by FISH assays (data not shown).
Sensitivity of resistant sublines to
various chemotherapeutic drugs
Cytotoxicity assays revealed that PC-9/ZD cells had
higher sensitivity to paclitaxel and docetaxel compared to PC-9
cells (P<0.05; Table I). PC-9/ZD
and PC-9 cells were similar in sensitivity to cisplatin,
gemcitabine and pemetrexed (P>0.05; Table I). PC-9/GR cells were less sensitive
to all chemotherapeutic drugs tested, including cisplatin,
gemcitabine, pemetrexed, paclitaxel and docetaxel, compared to PC-9
and PC-9/ZD cells (P<0.05; Table
I).
 | Table I.IC50 values of various
chemotherapeutic agents in NSCLCs with AR to EGFR-TKIs. |
Table I.
IC50 values of various
chemotherapeutic agents in NSCLCs with AR to EGFR-TKIs.
|
IC50 | PC-9 | PC-9/GR | PC-9/ZD |
|---|
| Cisplatin
(µmol/l) |
3.877±1.346 |
20.283±4.316a |
2.956±1.685b |
| Gemcitabine
(µmol/l) |
0.145±0.724 |
2.647±0.117a |
0.130±0.028b |
| Pemetrexed
(µmol/l) |
30.517±4.69 |
399.530±0.903a |
36.100±4.615b |
| Paclitaxel
(nmol/l) |
1.532±0.777 |
16.710±4.023a |
0.124±0.231a,b |
| Docetaxel
(nmol/l) |
5.226±0.648 |
64.560±8.454a |
0.800±0.109a,b |
T790M increases sensitivity of PC-9
cells to chemotherapy
A total of four siRNAs targeting T790M (2597, 2600,
2603 and 2608) and one control (scrambled sequence) were examined.
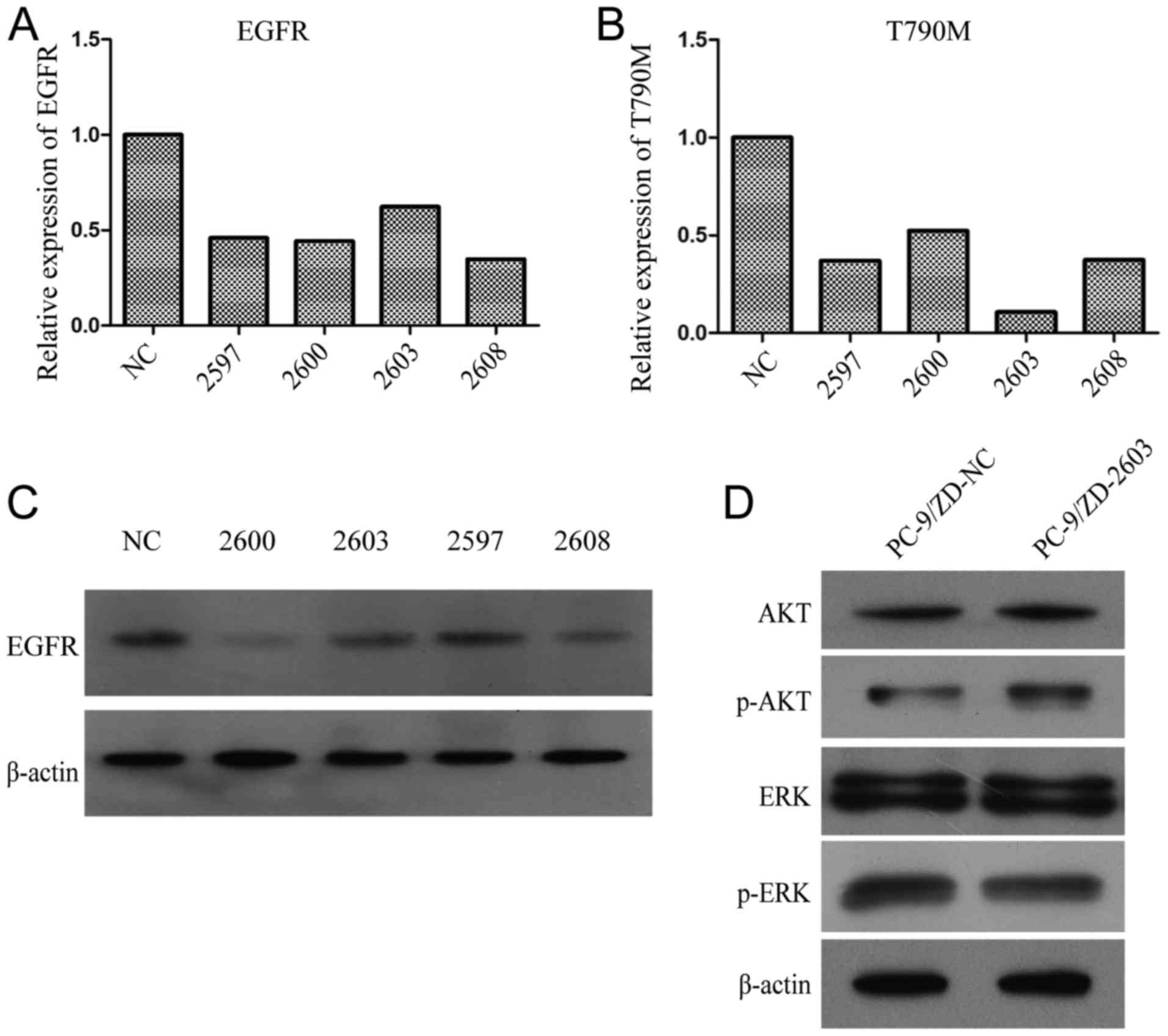
We observed that siRNA-2603 minimally inhibited WT EGFR (Fig. 1A), but it had the greatest
inhibitory effect on T790M EGFR (Fig.
1B). Western blot analysis demonstrated that siRNA-2603 had the
least inhibitory effect on EGFR (Fig.
1C). Consequently 2603 and the empty vector control were
transfected into PC-9/ZD cells to assess the effect on downstream
EGFR signaling pathways. The transfection of 2603 did not affect
the expression of AKT, p-AKT, ERK and p-ERK (P>0.05; Fig. 1D), indicating that 2603 had no
effect on the AKT and ERK pathways. The sensitivity of 2603-treated
PC-9/ZD cells to gefitinib was significantly higher compared to the
control. The IC50 value of gefitinib was 0.016 µmol/l
vs. 1.91 µmol/l (P<0.05, data not shown).
Treatment with 2603 markedly decreased the
IC50 values in PC-9/ZD cells from 15.762±1.673 to
0.124±0.231 µmol/l (P<0.05) for paclitaxel and from 21.110±2.165
to 0.800±0.109 nmol/l for docetaxel (P<0.05), indicating that
knockdown of T790M EGFR significantly reduced sensitivity to
taxanes (data not shown).
PC-9/GR cells display EMT
properties
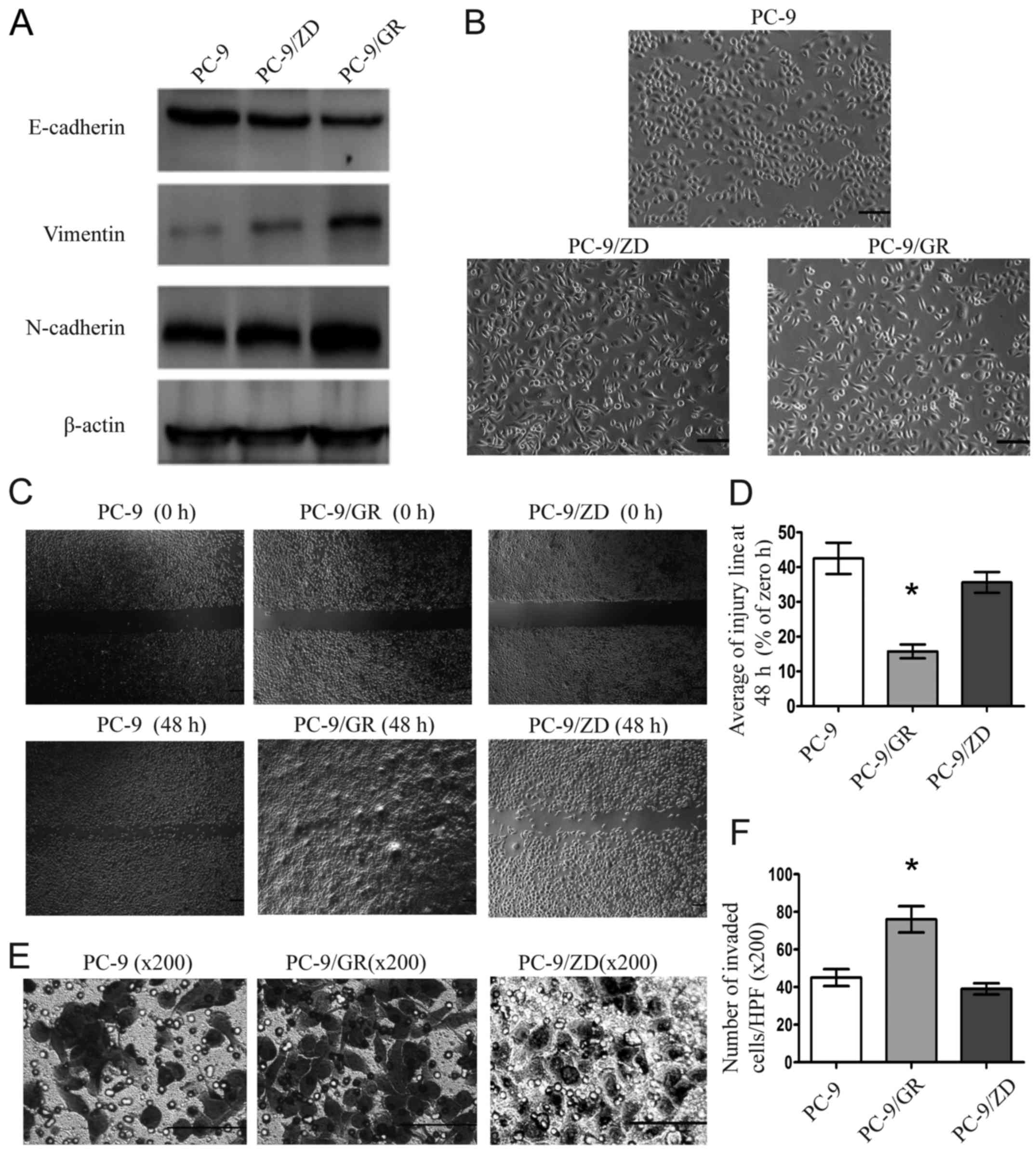
Western blot analysis revealed that E-cadherin was
decreased and vimentin and N-cadherin were increased in PC-9/GR
cells, but not in PC-9/ZD cells, indicating that PC-9/GR cells but
not PC-9/ZD cells acquired the EMT phenotype (P<0.05, Fig. 2A). In addition, PC-9/GR cells
exhibited mesenchymal morphology, with cells being long, polarized
and sparsely connected (Fig. 2B).
Furthermore, PC-9/GR cells undergoing EMT exhibited higher
migration and invasion abilities than PC-9 cells without EMT
(P<0.05), whereas no significant difference of the migration and
invasion abilities was observed between PC-9/ZD and PC-9 cells
(P>0.05; Fig. 2C-F).
EMT decreases sensitivity to
chemotherapy in AR-cell lines
Application of exogenous TGF-β1 (10 ng/ml) to
parental PC-9 cells successfully induced EMT after 72 h and
decreased the sensitivity to gefitinib. The sensitivity of PC-9
cells treated with exogenous TGF-β1 (10 ng/ml) to different
chemotherapeutic agents was assessed 72 h after treatment. TGF-β1
significantly inhibited the chemotherapeutic effect of gemcitabine,
paclitaxel, docetaxel and cisplatin, as evidenced by decreased
IC50 values (P<0.05; Table II).
| Table II.IC50 values of various
chemotherapeutic drugs in PC-9 cells before and after treatment
with TGF-β1. |
Table II.
IC50 values of various
chemotherapeutic drugs in PC-9 cells before and after treatment
with TGF-β1.
|
IC50 | PC-9 | PC-9/TGF-β1 |
|---|
| Cisplatin
(µmol/l) |
3.877±1.346 |
15.617±0.811a |
| Gemcitabine
(µmol/l) |
0.145±0.724 |
1.667±0.101a |
| Pemetrexed
(µmol/l) |
30.517±4.69 |
246.197±4.883a |
| Paclitaxel
(nmol/l) |
1.532±0.777 |
10.377±0.891a |
| Docetaxel
(nmol/l) |
5.226±0.648 |
36.110±3.360a |
PC-9/GR cells that overexpressed E-cadherin
following CDH1 transfection had significantly increased sensitivity
to gemcitabine, paclitaxel, pemetrexed, docetaxel and cisplatin, as
evidenced by the increased IC50 values (P<0.05,
Table III).
| Table III.IC50 values of
chemotherapeutic agents in PC-9/GR cells after the overexpression
of E-cadherin by CDH1 transfection. |
Table III.
IC50 values of
chemotherapeutic agents in PC-9/GR cells after the overexpression
of E-cadherin by CDH1 transfection.
|
IC50 | PC-9/GR-CDH1 |
PC-9/GR-control |
|---|
| Cisplatin
(µmol/l) |
3.987±0.818a |
17.28±0.445 |
| Gemcitabine
(µmol/l) |
0.267±0.098a |
3.546±0.120 |
| Pemetrexed
(µmol/l) |
35.497±2.075a |
348.856±1.875 |
| Paclitaxel
(nmol/l) |
2.484±0.908a |
19.429±0.937 |
| Docetaxol
(nmol/l) |
7.838±2.885a |
73.676±2.840 |
EMT is associated with increased CSCs
in NSCLC sublines
Previous studies have used CD44 and CD133 as markers
for CSCs in NSCLC cells (11–13).
Thus, we determined the expression of CD44 and CD133 in PC-9 cell
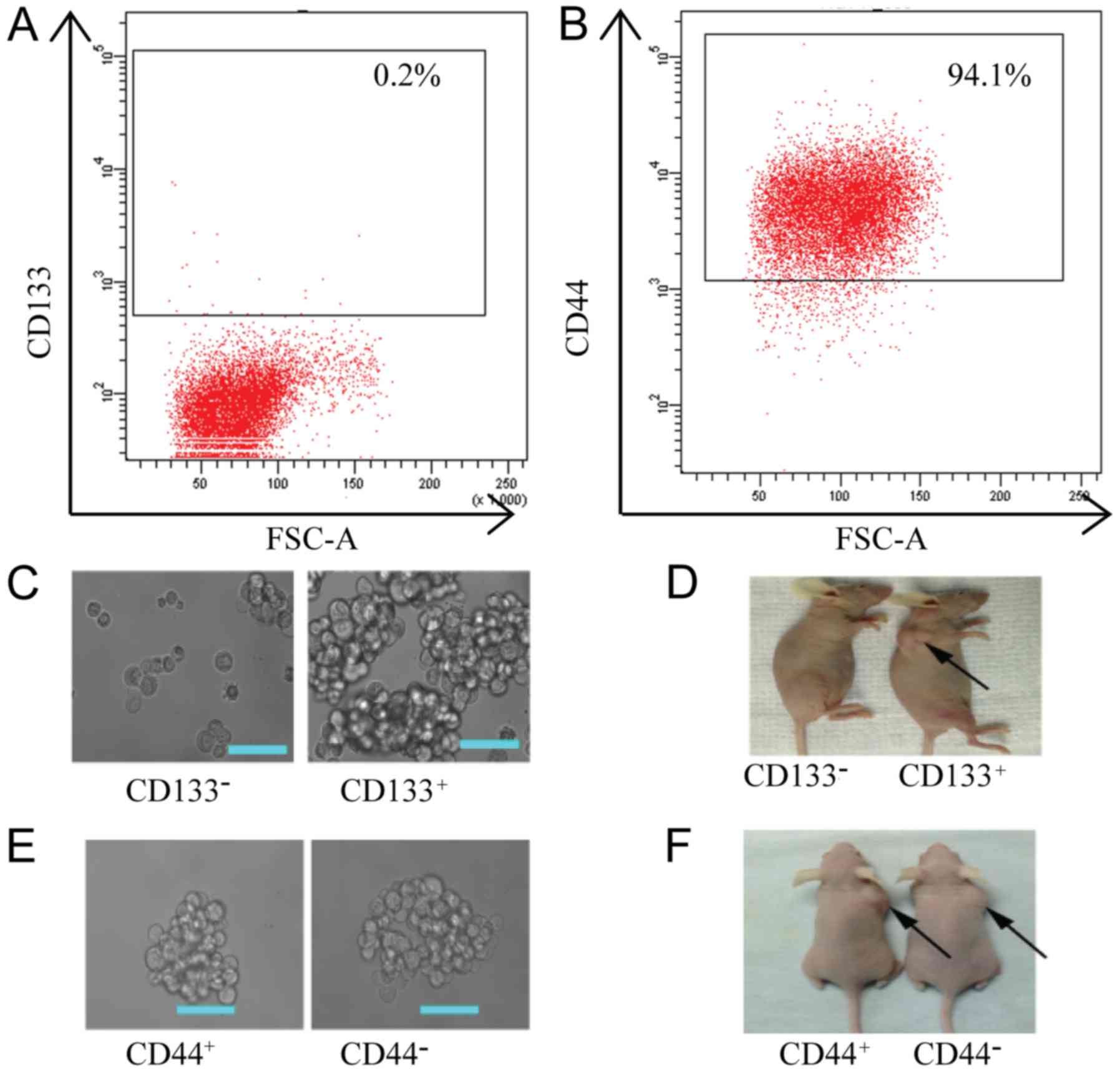
lines by flow cytometry. The percentage of CD133+ cells
in the PC-9 cell line was 0.2% (Fig.
3A) and the percentage of CD44+ cells was 94.1%
(Fig. 3B). To further confirm
whether CD44 and CD133 can be used as CSC surface markers in PC-9
derived cells, we first separated PC-9/GR cells into
CD133+, CD133−, CD44+ and
CD44− cell groups using MACS and assessed their tumor
formation activity both in vitro and in vivo. The
number of spheroids formed in the in vitro assays was
significantly higher in CD133+ cells (452±10.60)
compared to CD133− cells (4.67±0.88) (P<0.05,
Fig. 3C). In vivo,
tumorigenicity in CD133+ cells was also significantly
higher compared to CD133− cells (Fig. 3D). The CD133+ cells
required only 103 cells to form tumors, while the
CD133− cells did not form tumors even when injected with
106 cells (Fig. 3D).
These results indicated that CD133+ cells had a strong
self-renewal and differentiation ability. Thus, CD133+
can be used as a CSC marker in PC-9-derived cells. However, there
was no significant difference between CD44+ and
CD44− cells in tumor formation capability both in
vitro and in vivo (P>0.05, Fig. 3E and F).
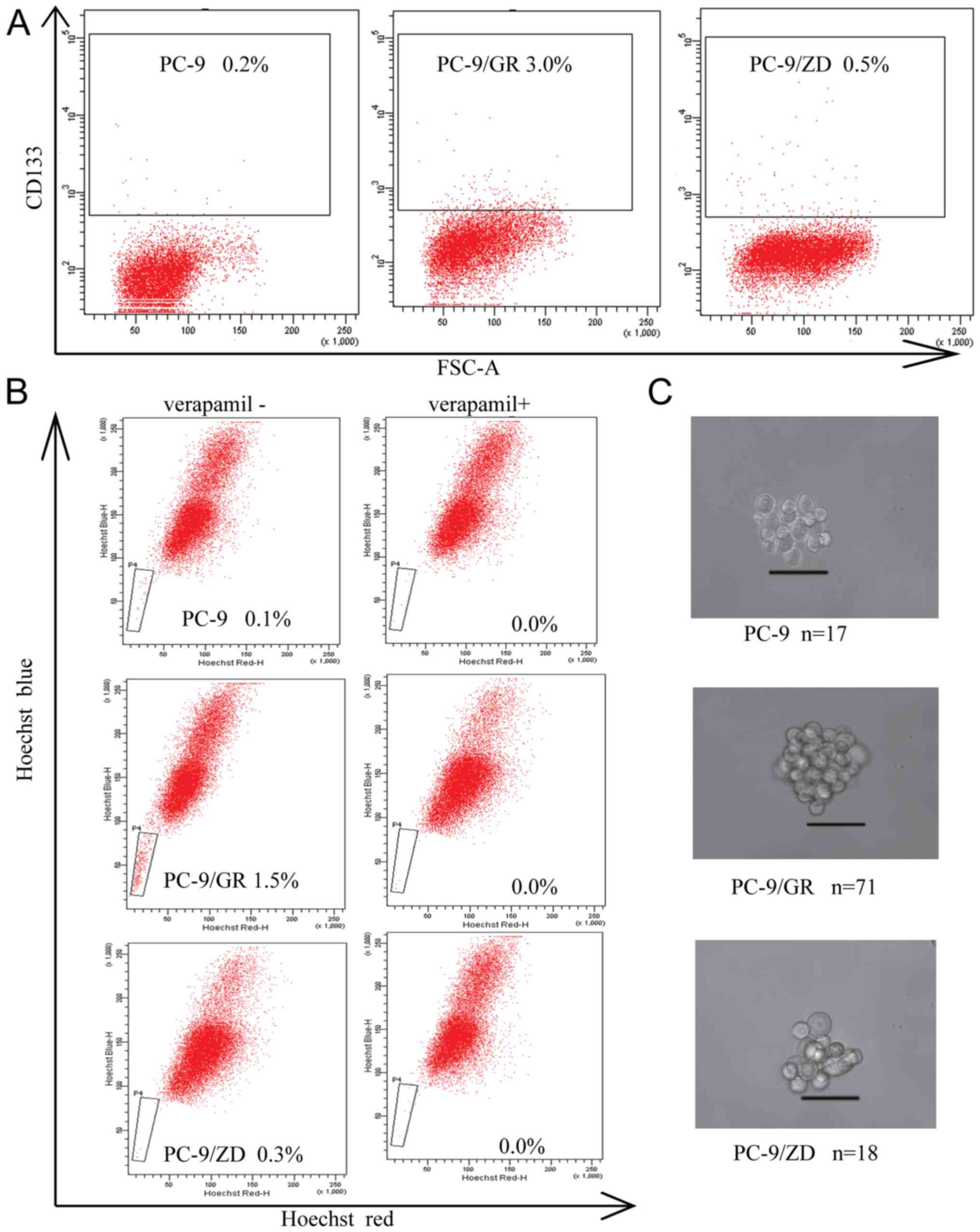
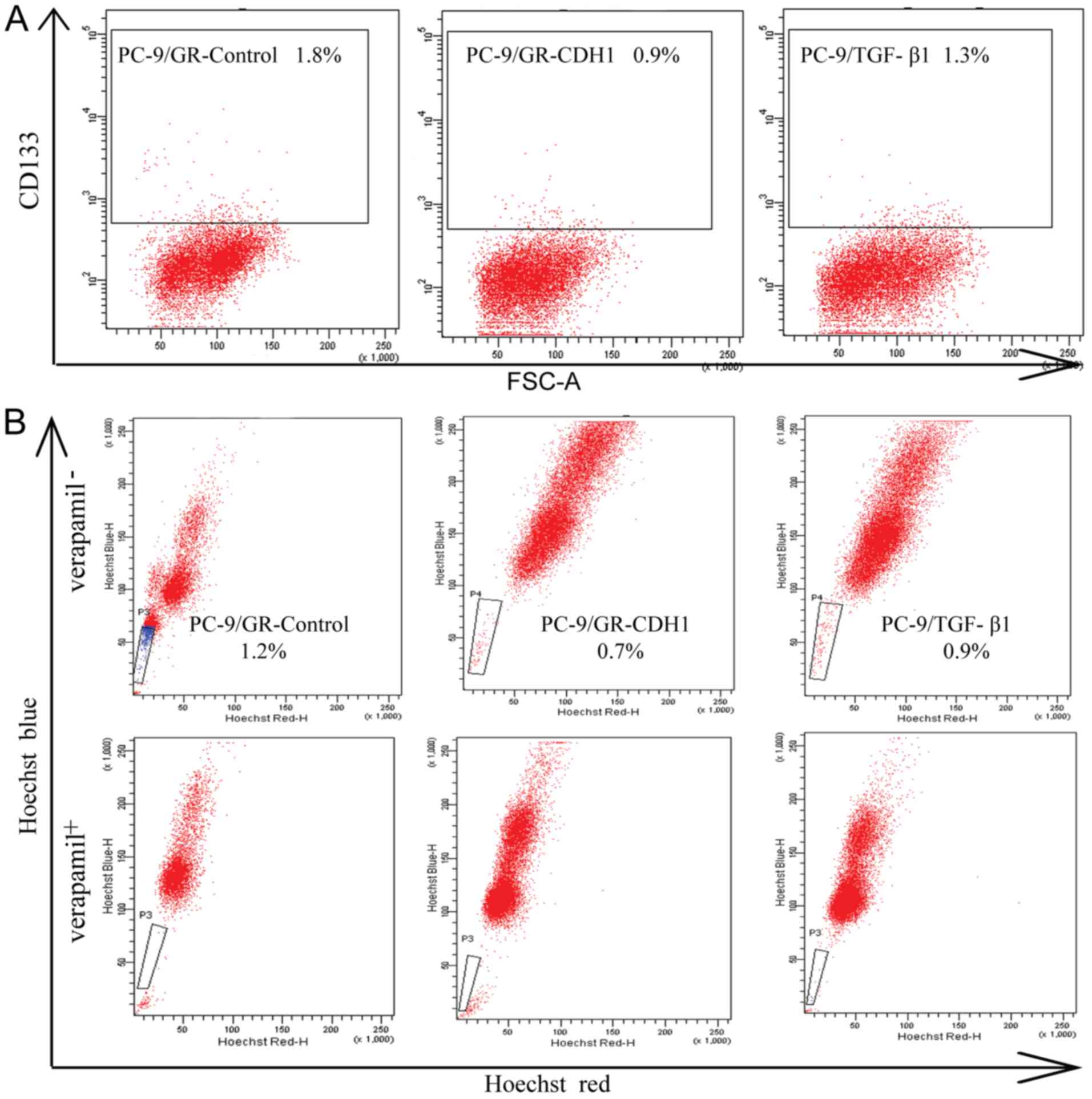
PC-9/GR cells expressed a much higher percentage of
CD133+ compared to PC-9 and PC-9/ZD cells (P<0.05,
Fig. 4A). The percentage of
CD133+ cells was 0.20±0.02% in PC-9, 0.50±0.04% in
PC-9/ZD and 3.00±0.22% in PC-9/GR cells. Side population assays
revealed that the number of cells in the PC-9/GR cell line was
increased (1.50±0.24%) compared to the PC-9 and PC-9/ZD cell lines
(P<0.05, Fig. 4B). Furthermore,
the side-population cells were essentially eliminated by verapamil.
The tumor-forming ability of PC-9/GR cells was significantly higher
compared to PC-9 and PC-9/ZD cells (P<0.05, Fig. 4C).
These results indicated that PC-9/GR cells with EMT
displayed more prominent stem cell characteristics. To further
demonstrate the effect of EMT on CSCs, we repeated the assays by
overexpressing CDH1 in the PC-9/GR cells to reverse EMT, as well as
by treating PC-9 cells with TGF-β1 to induce EMT. The percentage of
CD133+ and side population cells significantly decreased
when PC-9/GR cells were transfected with CDH1 (P<0.05, Fig. 5A and B). Furthermore, the number of
both CD133+ and side population cells increased in PC-9
cells in response to TGF-β1 (Fig. 5A
and B).
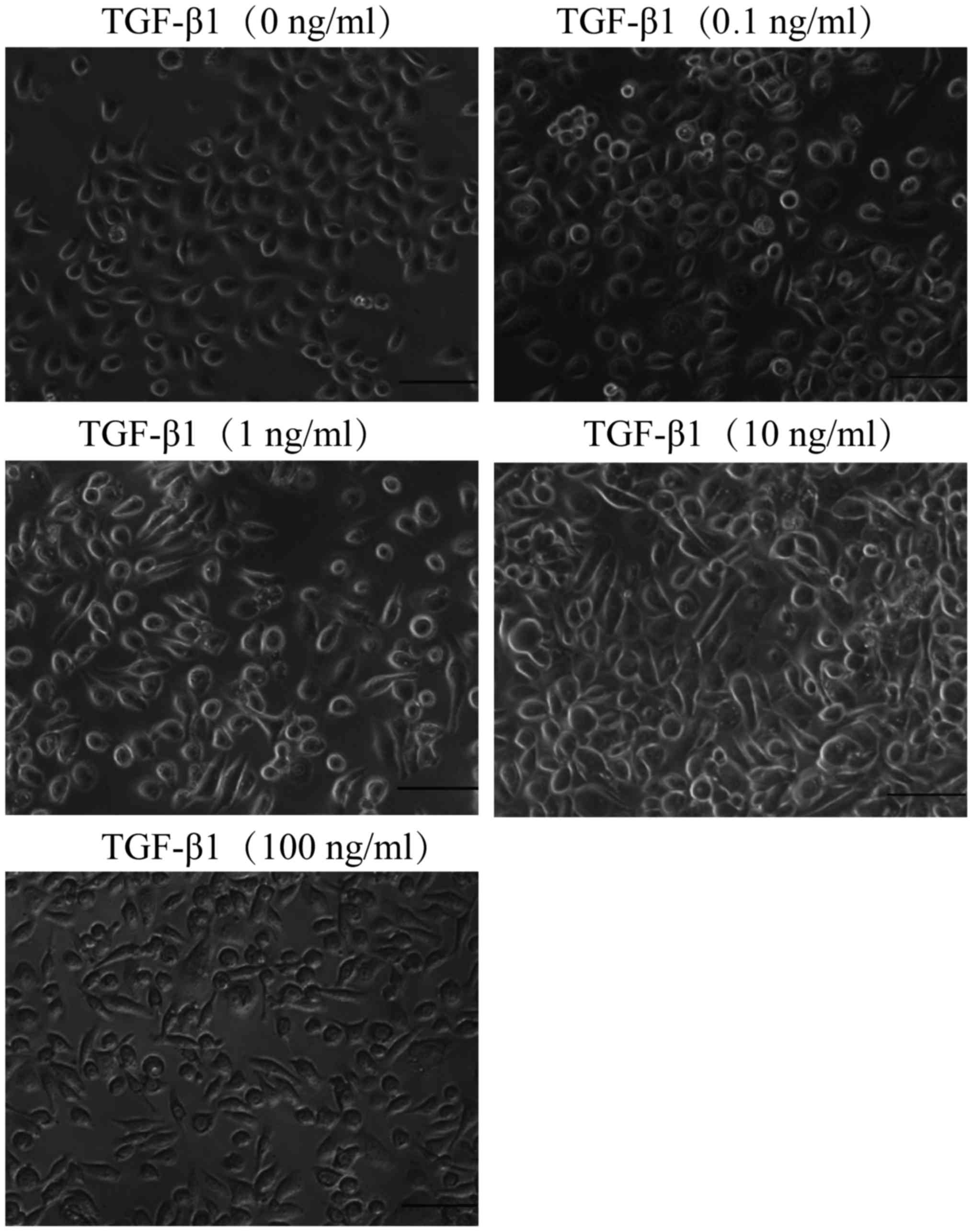
TGF-β1 induces EMT of NSCLC cells
To demonstrate the EMT of the NSCLC cells, PC-9
cells were treated with a potent EMT inducer TGF-β1 at different
concentrations ranging from 0.1 to 100 ng/ml. As displayed in
Fig. 6, vehicle-treated PC-9 cells
maintained a flattened shape which is a prominent feature of the
epithelial phenotype. In contrast, TGF-β1-treated cells exhibited a
spindle-shaped morphology and 10 ng/ml of TGF-β1 induced the most
pronounced mesenchymal phenotype while having no effect on cell
viability. These results indicated that TGF-β1 induces EMT in a
dose-dependent manner.
Discussion
In the present study, we investigated the
association between sensitivity of gefitinib-resistant NSCLC cell
lines to chemotherapeutic drugs and the underlying molecular
mechanisms of resistance to gefitinib, using two distinct PC-9
derived gefitinib-resistant sublines, PC-9/ZD and PC-9/GR. We
confirmed that increased sensitivity of the PC-9/ZD cell line to
taxanes was associated with the T790M mutation and decreased
sensitivity of the PC-9/GR cell line to various chemotherapeutics
was associated with EMT. Furthermore, we revealed that the
promotion of CSC properties may be one of the mechanisms for the
increased resistance to chemotherapeutics in the EMT phenotype in
NSCLC cells.
T790M accounts for ~50% of AR cases in patients with
NSCLC. After prolonged exposure to EGFR-TKIs, the T790M mutant
allele becomes enriched, resulting in AR to EGFR-TKIs (1). Rosell et al (5) reported that among AR NSCLC patients,
T790M-positive patients had better outcomes (longer PFS and OS)
than T790M-negative patients when treated with chemotherapy,
indicating that tumors with AR due to the T790M mutation are more
sensitive to chemotherapy. Kuo et al (4) observed that patients with AR to
EGFR-TKIs achieved better response rates and longer PFS and OS in
response to taxane chemotherapy. Although the underlying mechanisms
remained unknown in their study, the study indicated that the
improved outcomes following taxane chemotherapy were due to higher
sensitivity of T790M positive tumors.
In the present study, we first revealed the
relationship between the T790M mutation and the sensitivity to
chemotherapy in vitro. We identified that PC-9/ZD cells with
the T790M mutation were resistant to gefitinib and had a higher
sensitivity to taxanes (paclitaxel and docetaxel). Our findings
were different from the ones of the study by Cheng et al
(12), which revealed no
significant change in sensitivity to paclitaxel before and after AR
to gefitinib. The discrepancy is probably due to differences in the
ratio of the T790M allele to the WT allele. Our resistant PC-9/ZD
cells were pure and were derived from a single T790M mutant cell,
while only 30% of the cells used by Cheng et al were T790M
positive. To further confirm the correlation between the T790M
mutation and sensitivity to chemotherapy, we used siRNA to
specifically knock down T790M (8)
and found that sensitivity of the PC-9/ZD cells to paclitaxel and
docetaxel decreased, indicating that the T790M mutation is a useful
marker for taxane chemotherapy sensitivity.
Numerous previous studies have revealed that EMT
contributes to AR to EGFR-TKIs in NSCLC (2,14–16).
It has also been reported that NSCLC patients with EMT-positive
tumors are resistant to chemotherapy and such patients have poor
prognoses (2,6,13). In
the present study, we revealed that PC-9/GR cells with EMT were
significantly less sensitive to chemotherapeutic agents
(gemcitabine, paclitaxel, pemetrexed, docetaxel and cisplatin)
compared to parental PC-9 cells. Furthermore, we revealed that
TGF-β1-induced EMT in PC-9 cells conferred resistance to
chemotherapy. This is consistent with the study by Shintani et
al (6) who found that the
sensitivity of cells to the chemotherapeutic drugs cisplatin and
paclitaxel decreased after TGF-β1-induced EMT in NSCLC cells,
indicating that EMT plays an important role in tumor cell
chemotherapeutic resistance (6).
Furthermore, we demonstrated that when EMT in PC-9/GR cells was
reversed by the overexpression of CDH1 the cells became less
resistant to chemotherapeutic agents. These results further
confirmed that EMT in NSCLC cells is directly associated with
chemotherapeutic resistance. Previous studies have also
demonstrated that EMT in other types of tumor cells, such as
pancreatic cancer (17), colon
carcinoma (18) and ovarian cancer
(19), lead to chemotherapeutic
resistance.
Previous studies have revealed that EMT promotes the
formation of cells with the CSC phenotype (20–22).
The expression profiles of stem cell-related genes, including Oct4,
Wnt, Notch, Hedgehog and Nanog (23,24),
are altered in the process of EMT. This alteration is associated
with the proliferation of CSCs, leading to increased resistance to
chemotherapy (25). The present
study revealed that PC-9/GR cells with EMT had an increase in the
number of CD133+ and side population cells. These cells
had tumor stem cell characteristics, indicating that the increased
resistance to chemotherapeutic drugs in EMT-positive cells may be
related to the increase in CSCs. Shien et al (20) also revealed that in cells with AR to
EGFR-TKIs, CSC characteristics of the sub-groups led to resistance
to subsequent chemotherapy. We further confirmed the correlation
between EMT and CSCs. We revealed that the number of CSCs was
increased in the PC-9 cells in response to TGF-β1-induced EMT. This
finding indicated that the production of CSCs may be one of the
contributing factors to the resistance of EMT-positive cells to
chemotherapy. However, it is worth noting that only 3.0% of the
PC-9/GR cells were CSCs in the present study. Thus, further
investigation is necessary to determine if CSCs are a primary
factor in chemotherapy resistance.
NSCLC patients with EGFR-activating mutations
initially respond very well to EGFR-TKIs, such as gefitinib and
erlotinib, when administered as first-line treatments (2). However, all patients eventually
develop resistance to EGFR-TKIs (1,2). Thus,
it is critical to develop treatments that can overcome EGFR-TKI
resistance by targeting the underlying mechanisms responsible for
the resistance (26–29). The majority of NSCLC patients
develop resistance that is mediated by the T790M mutation. Thus,
much effort has been made to develop a third generation of
EGFR-TKIs that target both the activating mutations, such as
deletion in exon 19, and T790M. Third generation inhibitors include
AZD9291, HM61713 and CO-1686. These inhibitors have shown early
promise in clinical trials, with objective response rates ranging
from 58–66% (2). However, there is
no doubt that NSCLC patients will quickly develop resistance, since
resistance to CO-1686 has already been demonstrated by an EMT
mechanism (2). To target the
mechanisms associated with EMT, histone deacetylase (HDAC)
inhibitors and MAPK/ERK kinase (MEK) inhibitors are being developed
to delay the onset or reverse EMT (2). Such efforts have only achieved limited
success, therefore subsequent chemotherapy remains an important
choice of treatment after AR to EGFR-TKIs. In the present study, we
confirmed that NSCLC cells with AR mediated by T790M had higher
sensitivity to taxanes and those mediated by EMT were resistant to
various chemotherapeutic drugs including taxanes and non-taxanes.
These results provide a rationale for the selection of appropriate
chemotherapeutic regimens to maximally benefit patients with
intrinsic or acquired EGFR-TKI resistance.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (nos. 81472172 and
81172225), and The Science and Technology Planning Project of
Guangdong Province (no. 2014A020212254).
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Stewart EL, Tan SZ, Liu G and Tsao MS:
Known and putative mechanisms of resistance to EGFR targeted
therapies in NSCLC patients with EGFR mutations - a review. Transl
Lung Cancer Res. 4:67–81. 2015.PubMed/NCBI
|
|
2
|
Jakobsen KR, Demuth C, Sorensen BS and
Nielsen AL: The role of epithelial to mesenchymal transition in
resistance to epidermal growth factor receptor tyrosine kinase
inhibitors in non-small cell lung cancer. Transl Lung Cancer Res.
5:172–182. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Suda K, Tomizawa K, Fujii M, Murakami H,
Osada H, Maehara Y, Yatabe Y, Sekido Y and Mitsudomi T: Epithelial
to mesenchymal transition in an epidermal growth factor
receptor-mutant lung cancer cell line with acquired resistance to
erlotinib. J Thorac Oncol. 6:1152–1161. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kuo CH, Lin SM, Lee KY, Chung FT, Hsieh
MH, Fang YF, Yu CT and Kuo HP: Subsequent chemotherapy improves
survival outcome in advanced non-small-cell lung cancer with
acquired tyrosine kinase inhibitor resistance. Clin Lung Cancer.
11:51–56. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rosell R, Molina-Vila MA, Taron M,
Bertran-Alamillo J, Mayo C, Vergnenegre A, De Marinis F, Massuti B,
De Castro J, Gervais R, et al: EGFR compound mutants and survival
on erlotinib in non-small cell lung cancer (NSCLC) patients (p) in
the EURTAC study. J Clin Oncol. 30 15 Suppl:S75222012.
|
|
6
|
Shintani Y, Okimura A, Sato K, Nakagiri T,
Kadota Y, Inoue M, Sawabata N, Minami M, Ikeda N, Kawahara K, et
al: Epithelial to mesenchymal transition is a determinant of
sensitivity to chemoradiotherapy in non-small cell lung cancer. Ann
Thorac Surg. 92:1794–1804. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhou J, Wang J, Zeng Y, Zhang X, Hu Q,
Zheng J, Chen B, Xie B and Zhang WM: Implication of
epithelial-mesenchymal transition in IGF1R-induced resistance to
EGFR-TKIs in advanced non-small cell lung cancer. Oncotarget.
6:44332–44345. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen G, Kronenberger P, Teugels E, Umelo
IA and De Grève J: Effect of siRNAs targeting the EGFR T790M
mutation in a non-small cell lung cancer cell line resistant to
EGFR tyrosine kinase inhibitors and combination with various
agents. Biochem Biophys Res Commun. 431:623–629. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen G, Kronenberger P, Teugels E, Umelo
IA and De Grève J: Targeting the epidermal growth factor receptor
in non-small cell lung cancer cells: The effect of combining RNA
interference with tyrosine kinase inhibitors or cetuximab. BMC Med.
10:282012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen G, Kronenberger P, Umelo IA, Teugels
E and De Grève J: Quantification of epidermal growth factor
receptor T790M mutant transcripts in lung cancer cells by real-time
reverse transcriptase-quantitative polymerase chain reaction. Anal
Biochem. 398:266–268. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ogino A, Kitao H, Hirano S, Uchida A,
Ishiai M, Kozuki T, Takigawa N, Takata M, Kiura K and Tanimoto M:
Emergence of epidermal growth factor receptor T790M mutation during
chronic exposure to gefitinib in a non small cell lung cancer cell
line. Cancer Res. 67:7807–7814. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cheng H, An SJ, Dong S, Zhang YF, Zhang
XC, Chen ZH, Jian Su and Wu YL: Molecular mechanism of the
schedule-dependent synergistic interaction in EGFR-mutant non-small
cell lung cancer cell lines treated with paclitaxel and gefitinib.
J Hematol Oncol. 4:52011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rho JK, Choi YJ, Lee JK, Ryoo BY, Na II,
Yang SH, Kim CH and Lee JC: Epithelial to mesenchymal transition
derived from repeated exposure to gefitinib determines the
sensitivity to EGFR inhibitors in A549, a non-small cell lung
cancer cell line. Lung Cancer. 63:219–226. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mittal V: Epithelial mesenchymal
transition in aggressive lung cancers. Adv Exp Med Biol. 890:37–56.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yin H, Wang Y, Chen W, Zhong S, Liu Z and
Zhao J: Drug-resistant CXCR4-positive cells have the molecular
characteristics of EMT in NSCLC. Gene. 594:23–29. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rastogi I, Rajanna S, Webb A, Chhabra G,
Foster B, Webb B and Puri N: Mechanism of c-Met and EGFR tyrosine
kinase inhibitor resistance through epithelial mesenchymal
transition in non-small cell lung cancer. Biochem Biophys Res
Commun. 477:937–944. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang Z, Li Y, Kong D, Banerjee S, Ahmad A,
Azmi AS, Ali S, Abbruzzese JL, Gallick GE and Sarkar FH:
Acquisition of epithelial-mesenchymal transition phenotype of
gemcitabine-resistant pancreatic cancer cells is linked with
activation of the notch signaling pathway. Cancer Res.
69:2400–2407. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang AD, Fan F, Camp ER, van Buren G, Liu
W, Somcio R, Gray MJ, Cheng H, Hoff PM and Ellis LM: Chronic
oxaliplatin resistance induces epithelial-to-mesenchymal transition
in colorectal cancer cell lines. Clin Cancer Res. 12:4147–4153.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kajiyama H, Shibata K, Terauchi M,
Yamashita M, Ino K, Nawa A and Kikkawa F: Chemoresistance to
paclitaxel induces epithelial-mesenchymal transition and enhances
metastatic potential for epithelial ovarian carcinoma cells. Int J
Oncol. 31:277–283. 2007.PubMed/NCBI
|
|
20
|
Shien K, Toyooka S, Yamamoto H, Soh J,
Jida M, Thu KL, Hashida S, Maki Y, Ichihara E, Asano H, et al:
Acquired resistance to EGFR inhibitors is associated with a
manifestation of stem cell-like properties in cancer cells. Cancer
Res. 73:3051–3061. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Polyak K and Weinberg RA: Transitions
between epithelial and mesenchymal states: Acquisition of malignant
and stem cell traits. Nat Rev Cancer. 9:265–273. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Floor S, van Staveren WC, Larsimont D,
Dumont JE and Maenhaut C: Cancer cells in epithelial-to-mesenchymal
transition and tumor-propagating-cancer stem cells: Distinct,
overlapping or same populations. Oncogene. 30:4609–4621. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chiou SH, Wang ML, Chou YT, Chen CJ, Hong
CF, Hsieh WJ, Chang HT, Chen YS, Lin TW, Hsu HS and Wu CW:
Coexpression of Oct4 and Nanog enhances malignancy in lung
adenocarcinoma by inducing cancer stem cell-like properties and
epithelial-mesenchymal transdifferentiation. Cancer Res.
70:10433–10444. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pirozzi G, Tirino V, Camerlingo R, Franco
R, La Rocca A, Liguori E, Martucci N, Paino F, Normanno N and Rocco
G: Epithelial to mesenchymal transition by TGFβ-1 induction
increases stemness characteristics in primary non small cell lung
cancer cell line. PLoS One. 6:e215482011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hu J, Qiu M, Jiang F, Zhang S, Yang X,
Wang J, Xu L and Yin R: MiR-145 regulates cancer stem-like
properties and epithelial-to-mesenchymal transition in lung
adenocarcinoma-initiating cells. Tumour Biol. 35:8953–8961. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kuwano M, Sonoda K, Murakami Y, Watari K
and Ono M: Overcoming drug resistance to receptor tyrosine kinase
inhibitors: Learning from lung cancer. Pharmacol Ther. 161:97–110.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pirazzoli V, Ayeni D, Meador CB,
Sanganahalli BG, Hyder F, de Stanchina E, Goldberg SB, Pao W and
Politi K: Afatinib plus cetuximab delays resistance compared to
single-agent erlotinib or afatinib in mouse models of TKI-Naïve
EGFR L858R-induced lung adenocarcinoma. Clin Cancer Res.
22:426–435. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang Y, Singh R, Wang L, Nilsson M,
Goonatilake R, Tong P, Li L, Giri U, Villalobos P, Mino B, et al:
Polo-like kinase 1 inhibition diminishes acquired resistance to
epidermal growth factor receptor inhibition in non-small cell lung
cancer with T790M mutations. Oncotarget. 7:47998–48010.
2016.PubMed/NCBI
|
|
29
|
Leung EL, Fan XX, Wong MP, Jiang ZH, Liu
ZQ, Yao XJ, Lu LL, Zhou YL, Yau LF, Tin VP and Liu L: Targeting
tyrosine kinase inhibitor-resistant Non-small cell lung cancer by
inducing epidermal growth factor receptor degradation via
methionine 790 oxidation. Antioxid Redox Signal. 24:263–279. 2016.
View Article : Google Scholar : PubMed/NCBI
|