Introduction
Endometrial carcinoma (EC) is one of the most common
gynecological malignant tumors in western countries. EC is the
fourth most common cancer in women, accounting for 7% of all
cancers in women in the US (1). The
American Cancer Society estimated that 61,380 new cases of EC and
10,920 deaths from EC will occur in 2017 (1). Surgery and adjuvant chemo-radiotherapy
are the first-line therapies used for most patients with EC
(2). However, for patients with
advanced or relapsed disease, and for patients who want to retain
reproductive function, hormonal regimens, including progestins,
luteinizing hormone releasing hormone agonists, anti-estrogens and
aromatase inhibitors, are mainly applied. Additionally, progestins
have been used as the first option in various protocols (2). Gunderson et al analyzed the
contemporary literature on women with complex atypical hyperplasia
and grade 1 EC undergoing medical management with progestins from
2004 to 2011 (3). Forty-five
studies and 391 patients were included, and 34% of the endometrial
hyperplasia patients and 52% of the grade 1 EC patients failed to
respond to progestins (3). A
retrospective analysis was conducted by Hahn et al to
analyze the response to therapy among 35 patients with early-stage
grade 1 endometrioid endometrial adenocarcinoma who were treated
with progestins from January 1996 to December 2006; 34.3% of the
patients exhibited no response to the progestins (4). Thus, new therapeutic targets and drugs
are needed.
Sterol regulatory element-binding proteins (SREBPs)
are critical regulators of lipid homeostasis that function by
transcriptionally activating genes that are involved in fatty acid
and cholesterol homeostasis (5). In
mammalian cells, three isoforms of SREBP (SREBP-1a, SREBP-1c and
SREBP-2), which are coded by two genes (SREBF1 and SREBF2) have
been identified (5). Studies have
suggested that SREBP-1 is the main regulator of fatty acid
metabolism, while SREBP-2 predominantly regulates cholesterol
metabolism (5). Additionally,
several studies have reported that SREBPs function as oncogenes in
various malignant tumors and that SREBPs can promote tumor
progression by regulating lipogenesis (6–9). Li
et al demonstrated that the expression level of SREBP-1 was
significantly elevated in EC compared with that in healthy
endometrium and that the expression levels were positively
correlated with cancer progression (10). Eberhard et al demonstrated
that the silencing of SREBP-1 or inhibition of fatty acid synthase
sensitized resistant tumor cells to death ligands (11). Therefore, these studies revealed
that blocking SREBP-regulated metabolic pathways via
pharmacological intervention may be a novel therapeutic approach
for treating EC.
Fatostatin is a chemical inhibitor of the SREBP
pathway and inhibits the maturation and nuclear translocation of
SREBPs (12,13). Kamisuki et al demonstrated
that fatostatin increased fatty acid mobilization and oxidation and
reduced lipogenesis in obese ob/ob mice while exhibiting low
cytotoxicity (12). In addition, Li
et al revealed that fatostatin demonstrated high antitumor
activity against prostate cancer by blocking SREBP-regulated
metabolic pathways and androgen receptor signaling in vitro
and in vivo (14,15). Furthermore, Siqingaowa et al
demonstrated that fatostatin decreased pancreatic cancer cell
viability and proliferation (16).
In our present study, we demonstrated that fatostatin suppressed EC
growth and tumorigenesis by blocking SREBP-regulated metabolic
pathways in EC. Our findings revealed that inhibition of SREBPs may
be a potential therapeutic strategy for EC treatment.
Materials and methods
Cell lines and culture conditions
The human EC cell lines Ishikawa and HEC-1A were
kindly provided by Professor Beihua Kong (Qilu Hospital, Shandong
University, Jinan, China). Ishikawa cells were cultured in
RPMI-1640 medium (with 10% FBS). HEC-1A cells were cultured in
Dulbecco's modified Eagle's medium (DMEM) (with 10% FBS). The cells
were cultured in a 37°C incubator with 5% CO2.
Compounds
Fatostatin A (chemical name:
4-[4-(4-methylphenyl)-2-thiazolyl]-2-propyl-pyridine, hydrobromide;
synonym: 125B11; formula: C18H18N2S) was purchased from MedChem
Express (Monmouth Junction, NJ, USA). The stock solution (10,000
µM) was prepared with dimethyl sulfoxide (DMSO) and stored at
−20°C. The working concentration of fatostatin was diluted in the
respective medium, and the final concentration of DMSO was <0.1%
(v/v). The control groups were treated with an equal volume of
DMSO.
Cell viability assays
We analyzed cell viability with MTT assays and
determined the 50% inhibitory concentration (IC50). EC
cells were seeded in 96-well plates at a density of
8×103 (Ishikawa) or 6×103 (HEC-1A)
cells/well. Following overnight incubation, the cells were treated
with either the vehicle control or different concentrations of
fatostatin (5, 10, 15, 20, 30, 40 and 50 µM for Ishikawa cells and
2.5, 5.0, 7.5, 10.0, 15.0 and 20.0 µM for HEC-1A cells) for 24, 48
and 72 h. Finally, cell viability was assessed with the infinite
M200 PRO (Bio-Rad Laboratories, Hercules, CA, USA) after adding 20
µl of MTT into the culture medium, incubating the cells for 4 h at
37°C, and dissolving the formazan product in 100 µl of DMSO. Data
were collected from at least three independent experiments with
triplicate wells. Based on the readings, we calculated the
IC50 values using GraphPad Prism 5 for further study.
For the growth curve assays, cells were seeded in 96-well plates at
a density of 4×103 (Ishikawa) or 3×103
(HEC-1A) cells/well and treated with either the vehicle control or
different concentrations of fatostatin (5, 10 and 20 µM for
Ishikawa cells and 1.25, 2.50 and 5.00 µM for HEC-1A cells) for 5
days. The OD value of each well was read daily with the infinite
M200 PRO. Data were collected from at least three independent
experiments with triplicate wells.
Colony formation assays
For the clonogenic assays, cells in the logarithmic
growth phase (200) were seeded in 6-well plates in culture medium
containing either the vehicle control or different concentrations
of fatostatin (2.5, 5.0 and 10.0 µM for Ishikawa cells and 0.625,
1.250 and 2.500 µM for HEC-1A cells) for two weeks. Each group was
allotted three plates. The colonies that formed on each plate were
stained with crystal violet, and the number of colonies was
determined quantitatively with Gel-Pro Analyzer (Media Cybernetics,
Inc., Rockville, MD, USA).
Invasion and migration assays
The in vitro EC cell invasion and migration
assays were performed with 24-well Boyden chambers (8-µm pore size;
Corning Costar, Cambridge, MA, USA). The undersides of the upper
chambers were precoated with Matrigel (BD Biosciences, San Jose,
CA, USA) for the invasion assays. Following treatment with either
the vehicle control or different concentrations of fatostatin (10,
20 and 40 µM for Ishikawa cells and 2.5, 5.0 and 10.0 µM for HEC-1A
cells) for 48 h, Ishikawa cells (1.5×105/chamber) and
HEC-1A cells (1.5×105/chamber) were seeded into the
upper chambers. After a period of incubation (48 h for the invasion
assay and 24 h for the migration assay), the invading or migrated
cells were stained with crystal violet, images were captured with
an Olympus IX51 inverted microscope and quantified using ImageJ
software.
Cell cycle and apoptosis analysis
Following treatment with either the vehicle control
or different concentrations of fatostatin (10, 20 and 40 µM for
Ishikawa cells and 2.5, 5.0 and 10.0 µM for HEC-1A cells) for 48 h,
Ishikawa and HEC-1A cells were fixed, stained with a PI/RNase
staining buffer (Pharmingen; BD Biosciences, San Diego, CA, USA)
and analyzed with a FACS flow cytometer (BD Biosciences, Franklin
Lakes, NJ, USA) on the basis of 2N and 4N DNA content. The data
were analyzed by ModFit LT 3.2 software (Verity Software House,
Topsham, ME, USA). Following treatment with either the vehicle
control or fatostatin for 48 h, cell apoptosis analysis was
performed with a FACS flow cytometer using the FITC Annexin V
Apoptosis Detection Kit I (Pharmingen; BD Biosciences) according to
the manufacturer's instructions. The data were analyzed by
CellQuest software (BD Biosciences, Franklin Lakes, NJ, USA).
Protein extraction and western blot
analysis
After treatment with either the vehicle control or
different concentrations of fatostatin (10, 20 and 40 µM for
Ishikawa cells and 2.5, 5.0 and 10.0 µM for HEC-1A cells) for 24 h,
the cells were collected and lysed in a mixed RIPA buffer,
containing PMSF and NaF (100:1:1; Beyotime Institute of
Biotechnology, Haimen, China). Protein concentrations were
determined with the BCA protein assay kit (Tiangen Biotech Co.,
Ltd., Beijing, China). The proteins were then separated on a 10% or
12% polyacrylamide gel and transferred to a polyvinylidene
difluoride (PVDF) membrane (Immobilon-P; Millipore, Bedford, MA,
USA). Following incubation with the appropriate primary antibodies
for 10–16 h at 4°C and the secondary antibodies for 2 h at room
temperature, the protein bands were detected using horseradish
peroxidase luminescence solution (Millipore, Bedford, MA, USA) and
ImageQuant LAS4000 (General Electric Company, Boston, MA, USA) and
quantified with ImageJ software. The GAPDH band served as the
loading control. The primary antibodies used in the experiments
were as follows: SREBP-1 (dilution 1:200; rabbit polyclonal; cat.
no. sc-8984; Santa Cruz Biotechnology, Inc., Dallas, TX, USA); FASN
(dilution 1:1,000; rabbit polyclonal; cat. no. ab96866; Abcam,
Cambridge, UK); SREBP-2 (dilution 1:200; mouse monoclonal; cat. no.
sc-13552); HMGCR (dilution 1:500; mouse monoclonal; cat. no.
sc-271595); caspase-9 (dilution 1:500; mouse monoclonal; cat. no.
sc-56076; all from Santa Cruz Biotechnology, Inc.), caspase-3
(dilution 1:500; rabbit monoclonal; cat. no. ab32042; Abcam),
PARP-1 (dilution 1:500; mouse monoclonal; cat. no. sc-8007; Santa
Cruz Biotechnology, Inc.); cleaved-PARP (dilution 1:1,000; rabbit
monoclonal; cat. no. 5625S); and GAPDH (dilution 1:1,000; rabbit
monoclonal; cat. no. 2118S; both from Cell Signaling Technology,
Inc., Danvers, MA, USA). Horseradish peroxidase-conjugated
secondary antibodies: m-IgGκ BP-HRP (dilution 1:2,000; cat. no.
sc-516102; Santa Cruz Biotechnology, Inc.) and goat anti-rabbit IgG
H&L (HRP) (dilution 1:2,000; cat. no. ab205718; Abcam) were
used.
Quantitative real-time RT-PCR
(qRT-PCR) analysis
Total RNA from the cells treated for 24 h with
either the vehicle control or different concentrations of
fatostatin (10, 20 and 40 µM for Ishikawa cells and 2.5, 5.0 and
10.0 µM for HEC-1A cells) was extracted using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Carlsbad, CA, USA) following
the manufacturers instructions. After assessing the concentrations
using the NanoPhotometer Pearl (Implen GmbH, Munich, Germany), 3 µg
of total RNA was reverse-transcribed into cDNA using the
SuperScript™ II Reverse Transcriptase kit (Invitrogen; Thermo
Fisher Scientific). qRT-PCR reactions were performed on an Applied
Biosystems 7900HT Fast Real-Time PCR system with SYBR Premix Ex Taq
(Tli RNaseH Plus) (cat. no. RR420A; Takara Bio, Inc., Otsu, Japan)
in a 10-µl reaction system, and GAPDH was used as the control. The
primers used in the present study included ATP citrate lyase (ACL),
stearoyl-CoA desaturase-1 (SCD-1), fatty acid synthase (FASN),
3-hydroxy-3-methyl-glutaryl-CoA synthase 1 (HMGCS1),
3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGCR), mevalonate
5-pyrophosphate decarboxylase (MVD), mevalonate kinase (MVK),
low-density lipoprotein receptor (LDLR), insulin-induced gene 1
(INSIG1), SREBP cleavage activating protein (SCAP) and GAPDH. The
primer sequences are listed in Table
I.
 | Table I.The primer sequences used for
qRT-PCR. |
Table I.
The primer sequences used for
qRT-PCR.
| Gene | Sequence
(5′-3′) |
|---|
| ACL | F |
TGTAACAGACCAGGAACCC |
|
| R |
CTGTACCCCAGTGGCTGTTT |
| SCD-1 | F |
CACTTGGGAGCCCTGTATGG |
|
| R |
AGCCGAGCTTTGTAAGAGCG |
| FASN | F |
CGGTACGCGACGGCTGCCTG |
|
| R |
GCTGCTCCACGAACTCAAACACCG |
| HMGCS1 | F |
GAGGGCTTCGTGGGACACATA |
|
| R |
GCCACTGGGATGGATCTTT |
| HMGCR | F |
GTCATTCCAGCCAAGGTTGT |
|
| R |
GGGACCACTTGCTTCCATTA |
| MVD | F |
ACCACGGGGACACCACGGT |
|
| R |
CCACACAGCAGCCACAAACTC |
| MVK | F |
CCTTGTGGCTGGCGTCAGAAA |
|
| R |
CGAGGGCATTCAGATGGTGCT |
| LDLR | F |
CAACGGCTCAGACGAGCAAG |
|
| R |
AGTCACAGACGAACTGCCGAGA |
| INSIG1 | F |
GGACGACAGTTAGCTATGGGTGTT |
|
| R |
GAGTCATTTGTACAGTCAGCCCGA |
| SCAP | F |
TATCTCGGGCCTTCTACAACCA |
|
| R |
ACACAACTCCTCCAAGCTCCTG |
| GAPDH | F |
TGCACCACCAACTGCTTAGC |
|
| R |
GGCATGGACTGTGGTCATGAG |
Quantification of fatty acids and
total cholesterol
Following treatment with either the vehicle control
or fatostatin (20 µM for Ishikawa cells and 5 µM for HEC-1A cells)
for 48 h, the amounts of fatty acids and total cholesterol were
assessed using a free fatty acid quantification detection kit and a
total cholesterol quantification detection kit (Solarbio Science
& Technology Co., Ltd., Beijing, China) following the
manufacturer's instructions. The data were analyzed using the
calculation formula provided in the instruction manual.
Statistical analysis
All experiments were repeated three times. The data
were expressed as the mean ± standard deviation (SD). Relative
quantification of RNA expression was assessed using the
2−ΔΔCt method. All statistical analyses were performed
using GraphPad Prism version 5.01. Statistically significant
differences between cell viabilities were analyzed by two-way
ANOVA, and comparisons of the other quantitative data were analyzed
by Student's t-test. Statistical significance was defined as
P<0.05.
Results
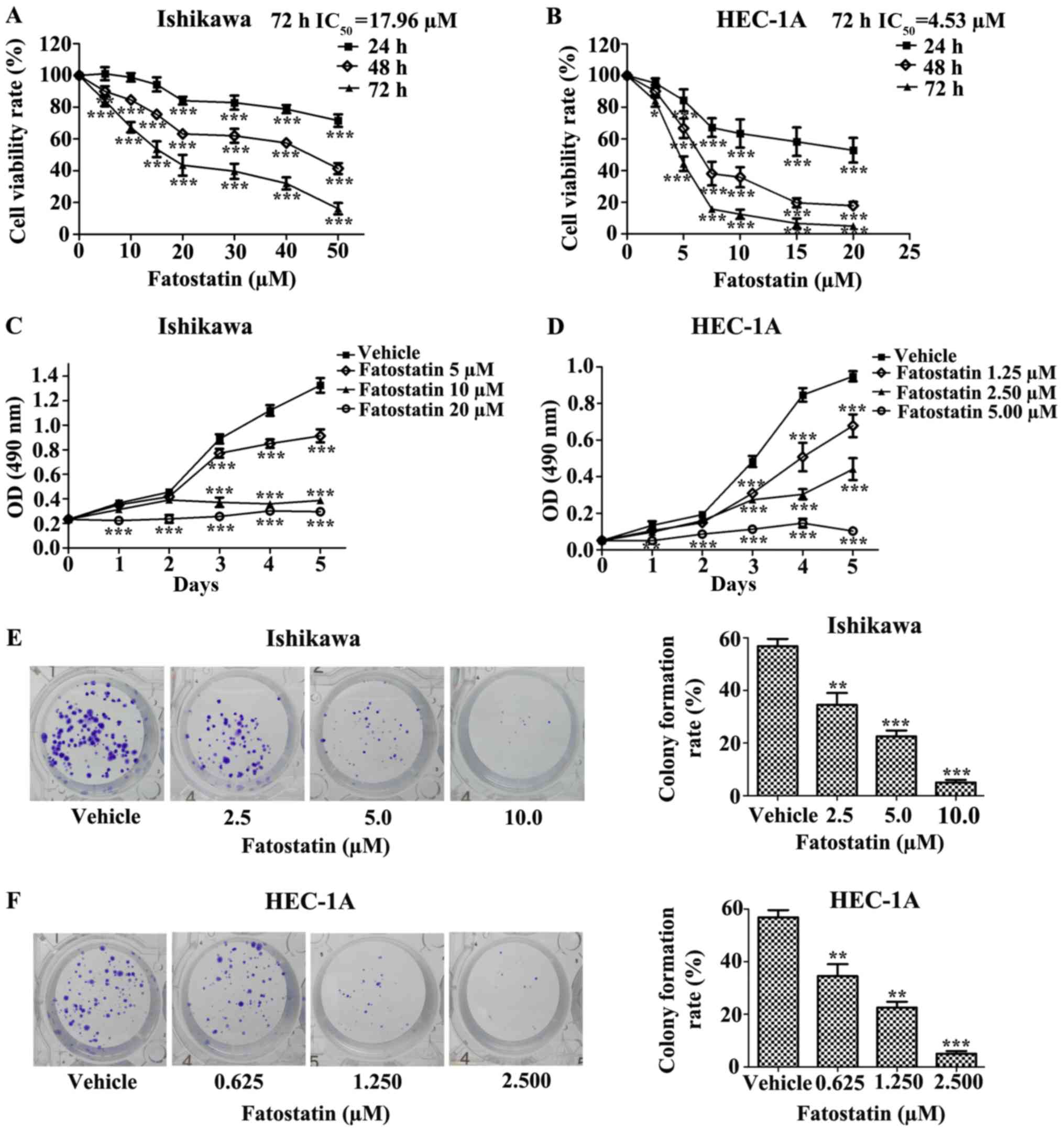
Fatostatin inhibits EC cell viability
and colony formation
To determine the effect of fatostatin on EC cells,
we first investigated the effect of fatostatin on EC cell
viability. We treated Ishikawa and HEC-1A EC cells with different
concentrations of fatostatin for 24, 48 and 72 h, as described in
Materials and methods. We found that fatostatin significantly
inhibited the viability of both cell lines in a dose- and
time-dependent manner. In addition, the IC50 values
(72-h treatment) for fatostatin in the Ishikawa and HEC-1A cells
were 17.96 and 4.53 µmol/l, respectively (Fig. 1A and B). Then, we determined the
growth rate of each cell line with or without fatostatin treatment
using MTT assays. We observed that the growth rates of the Ishikawa
and HEC-1A cells were notably reduced by fatostatin in a dose- and
time-dependent manner (Fig. 1C and
D). Furthermore, we examined the effect of fatostatin on EC
cell colony formation ability. Following 2 weeks of culture,
fatostatin significantly inhibited the number and size of the
colonies formed in the Ishikawa and HEC-1A cells in a
dose-dependent manner (Fig. 1E and
F). Collectively, these results revealed that fatostatin
inhibits EC cell viability and colony formation.
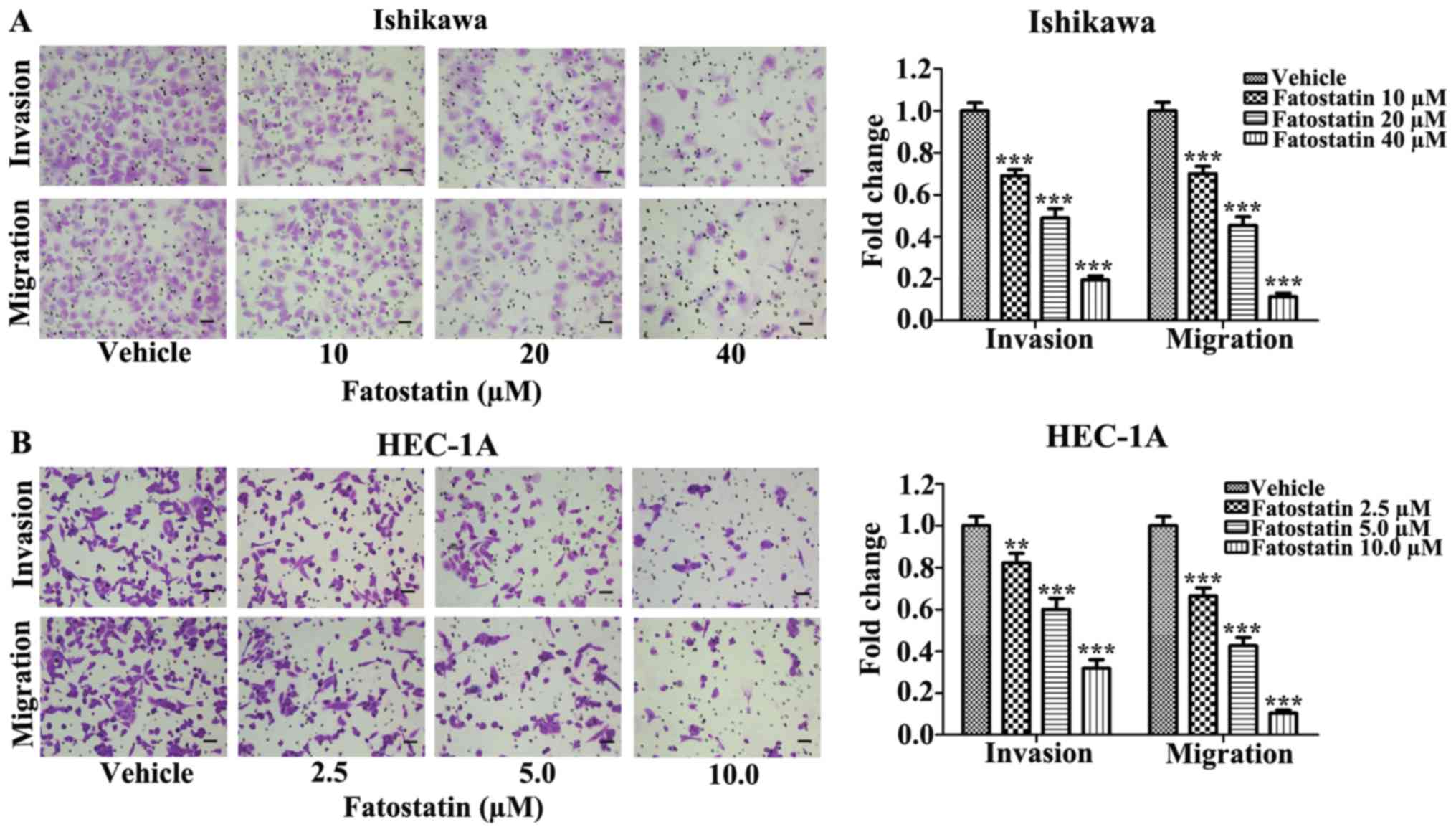
Fatostatin decreases the invasive and
migratory capacities of EC cells
Since invasion and migration are two essential steps
for malignant progression and metastasis, we examined the effects
of fatostatin on the invasive and migratory capacities of EC cells.
We treated Ishikawa and HEC-1A cells with fatostatin at different
concentrations for 48 h and used Boyden chambers to assess cell
invasion and migration, as described in Materials and methods. We
found that the number of cells invading through the membrane and
the number of migrating cells were significantly decreased by
fatostatin compared with those in the vehicle-treated group
(Fig. 2), suggesting that
fatostatin could effectively and dose-dependently inhibit the
invasive and migratory capacities of both cell lines.
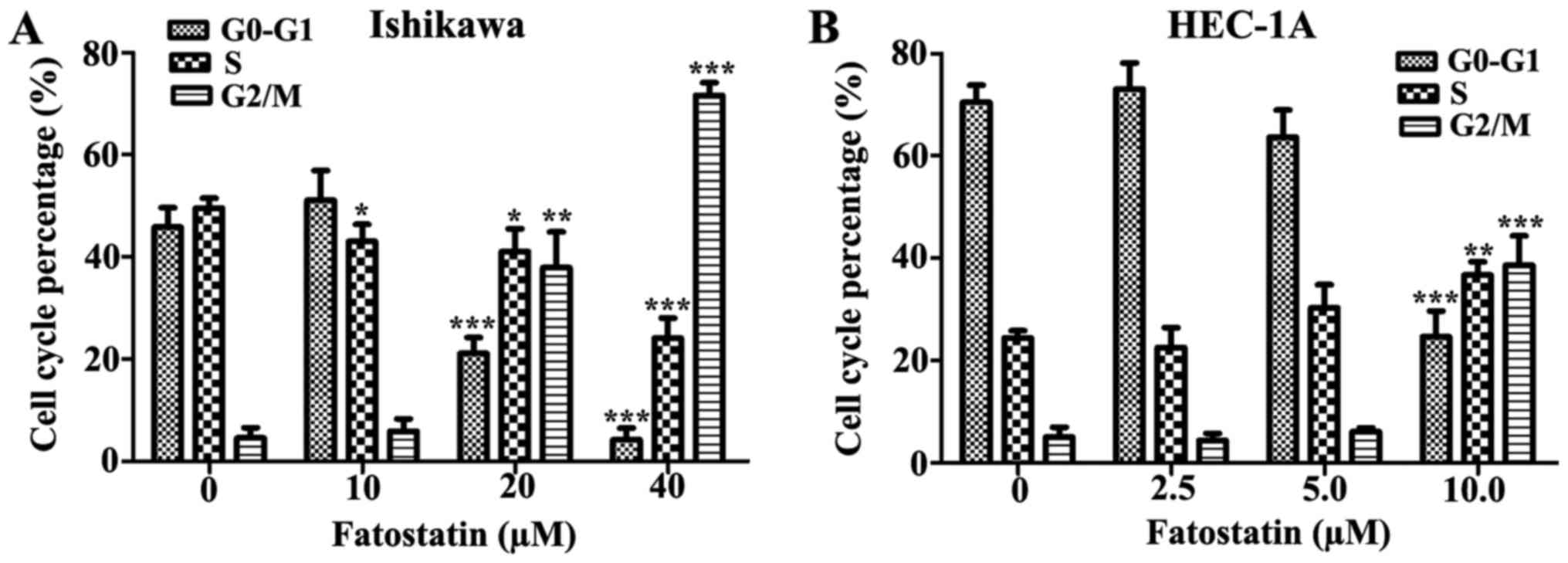
Fatostatin induces G2/M cell cycle
arrest and caspase-dependent apoptosis in EC cells
Cell viability is tightly controlled by the cell
cycle. Therefore, we examined the effects of fatostatin treatment
on cell cycle distribution. Following treatment for 48 h with
either the vehicle control or fatostatin at different
concentrations, the percentage of cells in each cell cycle phase
was determined by PI-staining-based flow cytometry. The data
revealed that fatostatin induced a significant decrease in the
percentage of cells in the G0/G1 phase and a
significant increase in the percentage of cells in the G2/M phase
for both Ishikawa (20 and 40 µM) and HEC-1A (10 µM) cells. In the
Ishikawa cells, the number of cells in the S phase was decreased by
fatostatin treatment, while in the HEC-1A cells, the number of
cells was increased (Fig. 3).
Therefore, fatostatin promoted a significant accumulation of both
Ishikawa and HEC-1A cells in the G2/M phase.
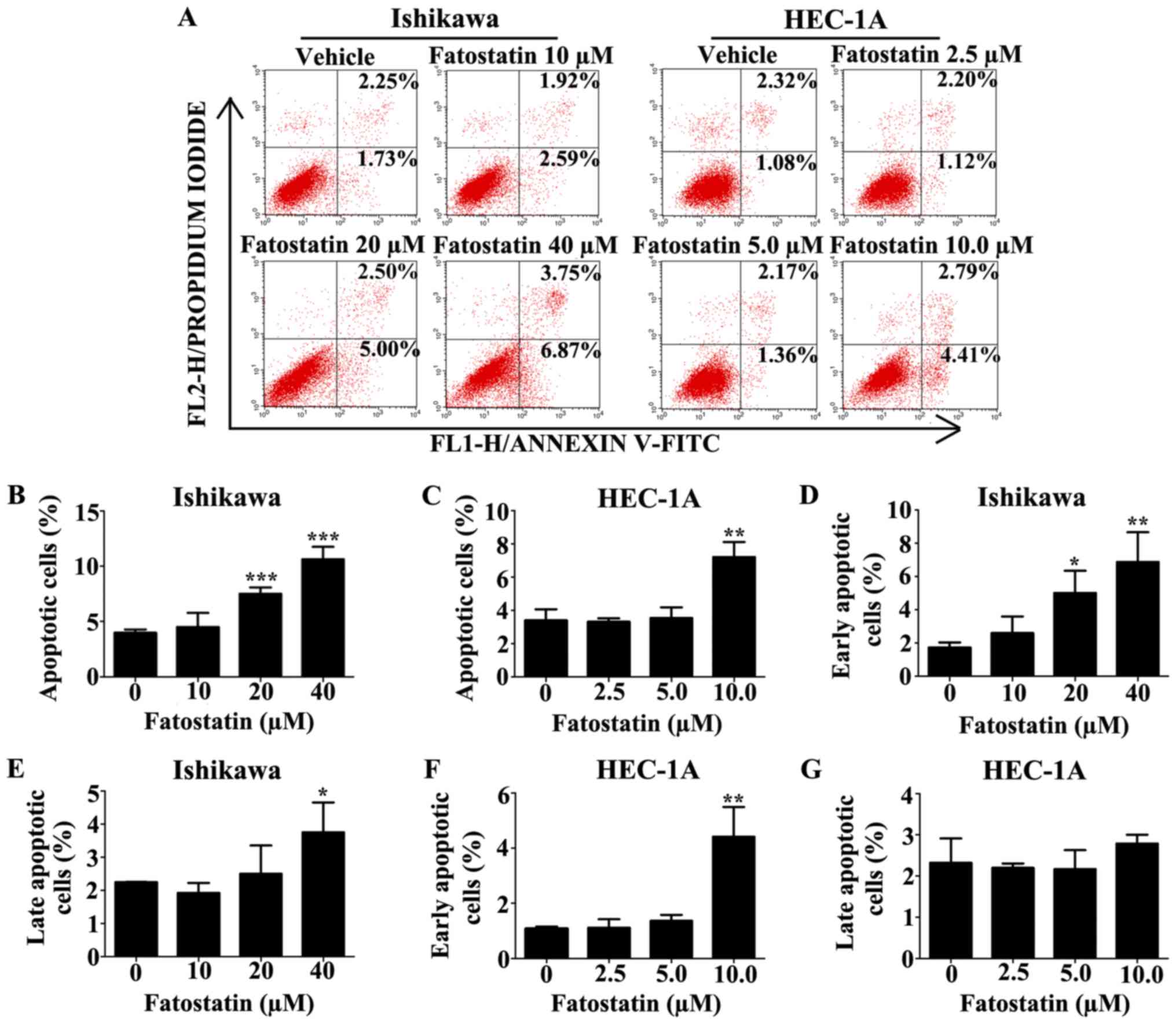
To explore whether fatostatin induced apoptosis, the
level of apoptosis was examined by flow cytometry using the FITC
Annexin V Apoptosis Detection Kit I after treatment with either the
vehicle control or fatostatin at different concentrations for 48 h.
The data revealed that the number of apoptotic cells was notably
increased with fatostatin treatment at higher concentrations
[Ishikawa (20 and 40 µM) and HEC-1A (10 µM)] (Fig. 4A-C). In the Ishikawa cells, the
number of early apoptotic cells was significantly increased by
fatostatin at both treatment concentrations (20 and 40 µM)
(Fig. 4D), while the number of late
apoptotic cells was increased significantly only by the higher
concentration of fatostatin (40 µM) (Fig. 4E). In the HEC-1A cells, the number
of early apoptotic cells was significantly increased with
fatostatin treatment (10 µM) (Fig.
4F), while the number of late apoptotic cells was only mildly
increased, showing no statistical significance (P=0.27) (Fig. 4G). In addition, to further define
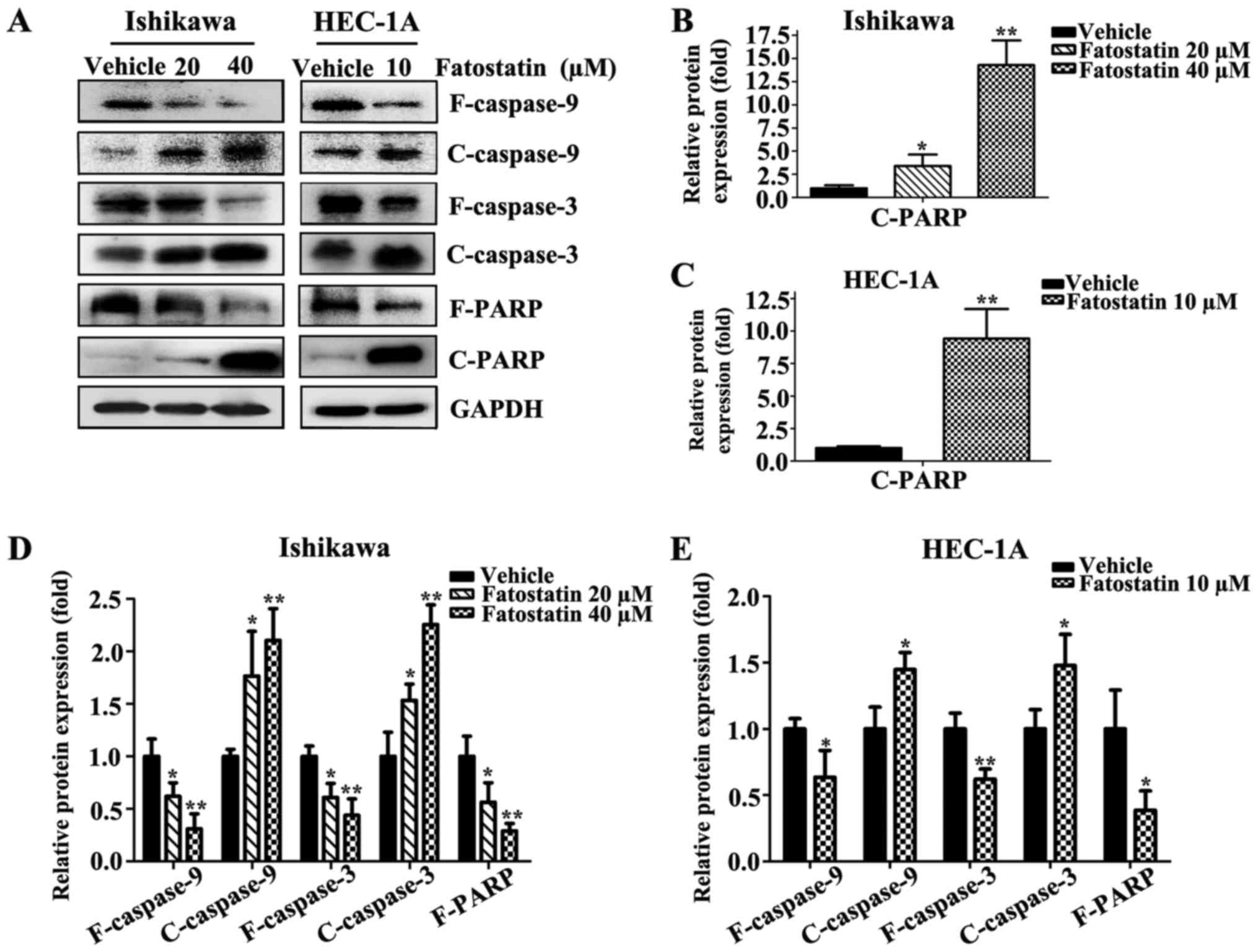
the mechanism underlying fatostatin-induced apoptotic cell death,
we determined the expression levels of caspases by western blot
analysis. Fatostatin decreased the expression of full-length
caspase-9, caspase-3 and PARP and increased the expression of
cleaved caspase-9, caspase-3 and PARP in Ishikawa and HEC-1A cells
(Fig. 5). These results indicated
that fatostatin induced caspase-dependent apoptotic death in EC
cells.
Fatostatin significantly inhibits
SREBP metabolic pathways and decreases free fatty acid and total
cholesterol levels in EC cells
Fatostatin is a chemical inhibitor of the SREBP
pathway and directly interacts with SCAP at a distinct domain from
the sterol-binding site and blocks the endoplasmic reticulum (ER)
exit of SCAP and the ER-to-Golgi transport of SREBPs (12,13).
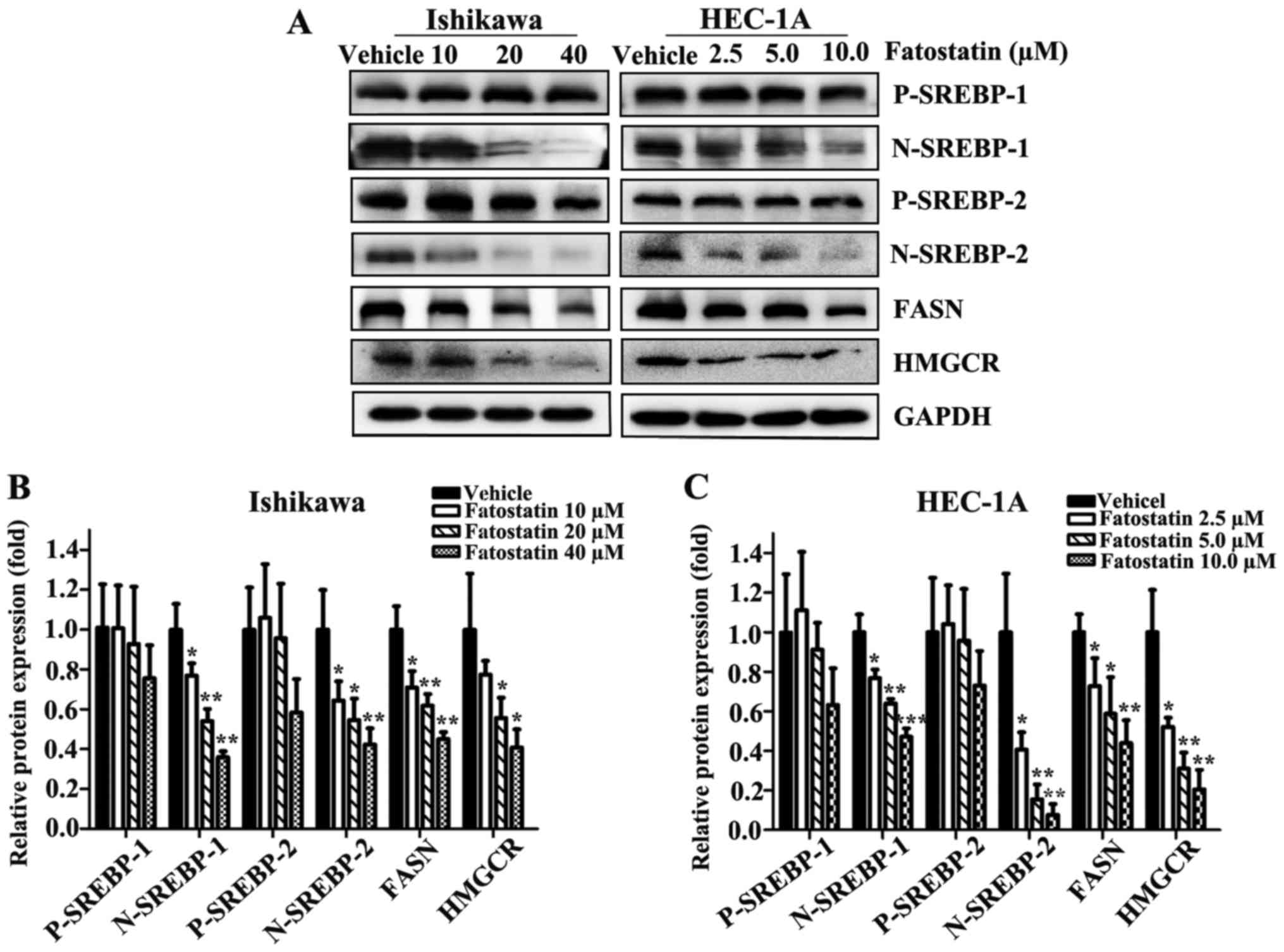
To reveal whether fatostatin suppressed EC through the SREBP
metabolic pathway, we first examined the transcriptional expression
levels of SREBPs. As we hypothesized, following treatment with
fatostatin for 24 h, the expression levels of nuclear SREBP-1 and
SREBP-2 were decreased in a dose-dependent manner in Ishikawa and
HEC-1A cells, while the precursor SREBP-1 and SREBP-2 levels were
mildly changed, showing no statistical significance (Fig. 6A-C). Then, we examined the
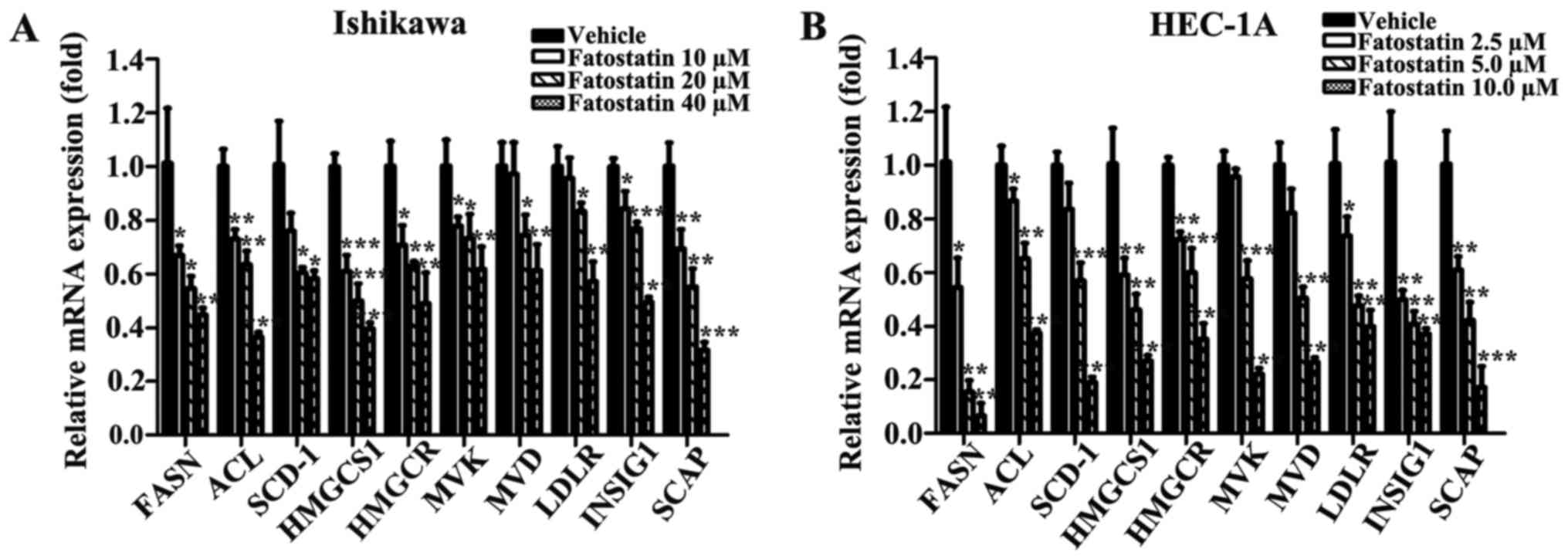
transcriptional expression of the following SREBP-controlled
anabolic genes in Ishikawa and HEC-1A cells: ACL, FASN and SCD-1,
which are involved in lipogenesis; HMGCS1, HMGCR, MVK, MVD and
LDLR, which are involved in cholesterogenesis; and INSIG1 and SCAP,
which are two chaperones. The mRNA expression levels of these genes
were significantly downregulated in the fatostatin-treated cells
compared with those in the vehicle-treated cells (Fig. 7). Similar results were obtained by
western blot analysis. The protein levels of FASN and HMGCR were
decreased by fatostatin in a dose-dependent pattern (Fig. 6A-C). Collectively, these results
indicated that fatostatin significantly inhibited the SREBP
metabolic pathways in EC cells.
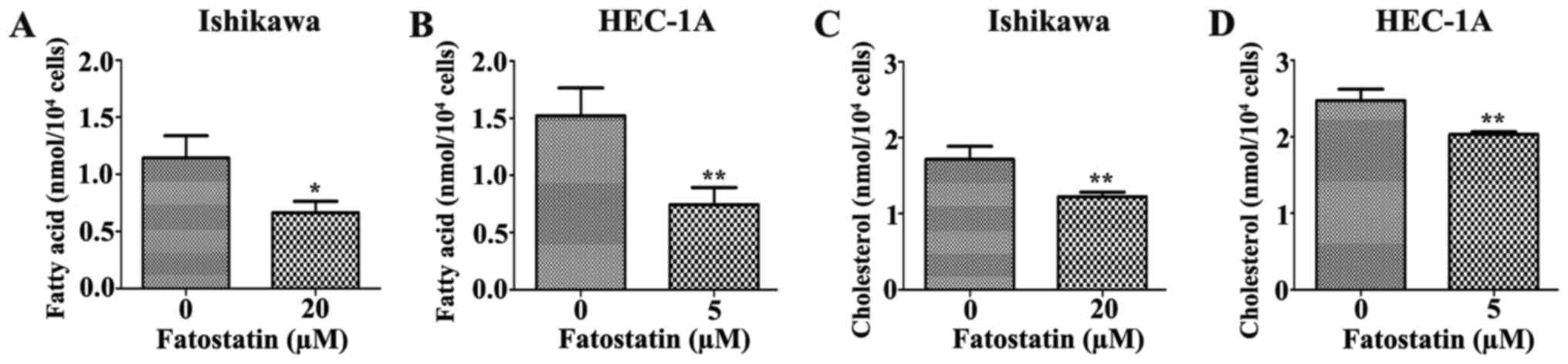
Since fatostatin suppressed several key genes that
were associated with lipogenesis and cholesterogenesis through
SREBPs, we performed quantification assays using a free fatty acid
quantification detection kit and total cholesterol quantification
detection kit to assess the changes in intracellular free fatty
acids and total cholesterol, respectively, that were induced by
48-h treatments with fatostatin. As we hypothesized, the level of
intracellular free fatty acids was significantly decreased by
fatostatin in Ishikawa and HEC-1A cells compared with that in the
vehicle-treated cells (Fig. 8A and
B). Similar results were observed for the total cholesterol
levels. Fatostatin significantly decreased the level of
intracellular total cholesterol in Ishikawa and HEC-1A cells
compared with that in the vehicle-treated cells (Fig. 8C and D). Therefore, these results
indicated that fatostatin significantly decreased the levels of
intracellular free fatty acid and total cholesterol through
inhibition of SREBPs in EC cells.
Discussion
Accumulating evidence has shown that tumor cells
reprogram their metabolic pathways to sustain higher proliferative
rates, enhance tumor growth and resist cell death signals (17,18).
In cancer cells, lipid metabolism is increased to meet high
metabolic demands (18,19). Disordered lipid metabolism
contributes to different aspects of tumorigenesis. In EC, lipid
metabolism is the most upregulated metabolic pathway and impacts
the outcome of treatment and/or disease progression in patients
with type I EC (20). SREBPs are
central regulators of lipid homeostasis and function by
transcriptionally activating genes that are involved in fatty acid
and cholesterol homeostasis (5).
Several studies have reported that SREBPs function as oncogenes in
various malignant tumors and that they can promote tumor
progression by regulating lipogenesis (6–9). Li
et al demonstrated that the expression of SREBP-1 was
significantly elevated in EC compared with that in healthy
endometrium, that the expression levels were positively correlated
with cancer progression, and that knockdown of SREBP-1 expression
in EC cells suppressed cell proliferation, reduced clonogenic
capacity and induced apoptosis in vitro and in vivo
(10). Thus, SREBP-1 functions as
an oncogene in EC and can promote EC progression by regulating
lipid metabolism, suggesting SREBP-1 as a novel therapeutic target
for EC treatment.
SREBPs are membrane-bound, basic
helix-loop-helix-leucine zipper (HLH-LZ) transcription factors
(5). SREBPs are subject to complex
post-translational regulation and SCAP is a critical regulator of
this process (21). Precursor
SREBPs, which associate with two chaperone proteins, namely, INSIG
and SCAP, are embedded in the ER. SCAP and INSIG bind to ER
membrane-associated cholesterol or oxysterols molecules via
sterol-sensing domains that are sensitive to ER membrane sterol
levels. When ER membrane sterol levels decrease, INSIG and SCAP
undergo conformational changes, and the SCAP/SREBP complex is
released from INSIG (22). Then,
the SREBP/SCAP complex is escorted from the ER to the Golgi by
binding to Sec24, which is a component of the Sar1/Sec23/Sec24
complex of the COPII protein coat (22,23).
In the Golgi, SREBPs are released from SCAP and cleaved via site-1
and site-2 proteases to generate nuclear SREBPs. Subsequently,
nuclear SREBPs activate target genes by binding to sterol response
elements and maintain fatty acid and cholesterol homeostasis
(21).
Fatostatin is a chemical inhibitor of the SREBP
pathway. Fatostatin interacts directly with SCAP at a distinct
domain from the sterol-binding site and blocks the ER exit of SCAP
and the ER-to-Golgi transport of SREBPs (12,13).
Thus, fatostatin can decrease the expression levels of nuclear
SREBPs and their downstream genes and subsequently decrease lipid
metabolism. As was aforementioned, Li et al investigated the
role of SREBP-1 in EC and demonstrated that SREBP-1 was essential
for EC cell growth both in vitro and in vivo as
determined by immunohistochemistry staining, cell transfection and
transduction and subcutaneous tumor implantation. Their results
revealed that SREBP-1 functions as an oncogene in EC and may be a
novel therapeutic target for EC treatment (10). Therefore, blocking SREBP-regulated
metabolic pathways via pharmacological intervention may be a novel
therapeutic approach for treating EC. However, they did not perform
any further studies on this topic. In the present study, we
speculated that fatostatin, which is a chemical inhibitor of the
SREBP pathway and can block SREBP-regulated metabolic pathways, may
be a novel therapeutic approach for EC treatment, and aimed to
investigate the antitumor effects of fatostatin against EC.
Studies have demonstrated that fatostatin promotes a
significant reduction in nuclear SREBPs and their downstream genes
in prostate cancer and pancreatic cancer cells (14–16).
Ankur et al determined that the anticancer properties of
fatostatin were not only due to its inhibition of SREBPs and
effects on lipid metabolism but also attributed to its inhibition
of cell division (24). Li et
al determined that, in prostate cancer cells, fatostatin
inhibited cell proliferation, invasion and migration, promoted G2/M
cell cycle arrest and induced caspase-mediated apoptosis. The
authors also demonstrated that, in prostate cancer cells,
fatostatin suppressed SREBP processing, SREBP transcriptional
activity, several key enzymes for lipogenesis and
cholesterogenesis, and fatty acid and cholesterol levels (14,15).
Siqingaowa et al demonstrated that fatostatin decreased
pancreatic cancer cell viability and proliferation (16). In our present study of EC cells,
fatostatin significantly decreased the expression of nuclear SREBPs
and their downstream genes and reduced free fatty acid and total
cholesterol levels. In addition, fatostatin inhibited EC cell
viability and colony formation, decreased the invasive and
migratory capacities of EC cells, induced G2/M EC cell cycle arrest
and promoted caspase-mediated apoptosis in EC cells. Therefore, our
study revealed that fatostatin exhibited an antitumor effect by
blocking SREBP-regulated lipid metabolic pathways in EC.
Lipid homeostasis is important for maintaining
cellular structure and normal function. Fatty acids play essential
roles in multiple cellular processes (25), are essential constituents of all
biological membrane lipids, and are important substrates for energy
storage and metabolism (25,26).
FASN, ACL and SCD-1 are three rate-limiting enzymes involved in the
biosynthesis of long-chain fatty acids (14). SREBP-1 functions as the key
regulator of fatty acid metabolism by transcriptionally regulating
the expression of these three lipogenic genes (5,14).
Studies have demonstrated that SREBP-1 and FASN play crucial roles
in the processes of EC oncogenesis and progression (10,27).
ACL expression and activity are markedly increased in cancer cells,
including EC cells, and ACL inhibition can suppress tumor cell
growth (20,28). Evidence has shown that SCD-1 can
enhance tumorigenesis by accelerating the cancer cell proliferation
rate, increasing cell invasiveness, and enhancing cell survival
(29). Our results revealed that
fatostatin markedly suppressed the expression of FASN, ACL and
SCD-1 and significantly reduced the levels of intracellular fatty
acids in EC cells. The inhibitory mechanism of fatostatin could be
attributed to decreasing SREBP-1 transcriptional activity, since
SREBP-1 has been demonstrated to transcriptionally regulate the
expression of these three lipogenic genes and fatostatin is a
chemical inhibitor of the SREBP pathway. These data revealed that
fatostatin decreased the expression of cancer-associated lipogenic
genes and inhibited EC growth, oncogenesis and progression in
vitro by interrupting SREBP-1-regulated fatty acid
metabolism.
Cholesterol is a major component of lipid rafts,
which exhibit a special structure on the cellular membrane and
consist of cholesterol and sphingolipids (30). Several kinase receptors and
signaling molecules, such as EGFR and AK, are located in lipid
rafts (31,32). Therefore, the maintenance of the
intracellular cholesterol level is important for maintaining
lipid-raft-mediated survival signaling pathways, cell morphology
and cell function (30). Studies
have suggested that SREBP-2 is a major regulator of cholesterol
metabolism and that SREBP-2 upregulates several important
cholesterogenic genes, such as HMGCS1, HMGCR and LDLR (5,14).
Although the importance of SREBP-2 in cancer cells and oncogenesis
remains relatively unexplored, unlike SREBP-1, a combination
knockdown of SREBP-2 and SREBP-1 in cancer cell lines causes EC
stress and induces apoptosis in lipoprotein-depleted conditions,
and the simultaneous targeting of HMGCR and SREBP-2 is a promising
novel antitumor therapeutic strategy (33). HMGCR is the action target of statins
and is used to treat high cholesterol levels. Multiple studies have
demonstrated that the use of statins is associated with decreased
incidences of cancer and decreased deaths from cancers of the
breast, colon, pancreas, gastrointestinal tract, liver,
endometrium, and ovaries. Statin treatment has the ability to
inhibit various cancer processes, including tumorigenesis, growth,
angiogenesis, and metastasis (34).
In addition, statins have anti-proliferative and anti-metastatic
effects on EC cell lines in vitro (33,35).
These data demonstrated that HMGCR inhibition has antitumor
effects. HMGCS1 is upstream of HMGCR and synthesizes HMG-CoA, the
substrate of HMGCR. Analysis of cancer genomic datasets using
cBioPortal revealed that HMGCS1 can be amplified in various
cancers. HMGCS1 and SREBP-2 knockdown, together with
statin-mediated HMGCR inhibition, can robustly enhance tumor cell
apoptosis (33). In the present
study, we demonstrated that fatostatin markedly decreased the
expression levels of these cholesterogenic genes, including HMGCR
and HMGCS1, and significantly decreased the total intracellular
cholesterol level in EC cells, which is important for maintaining
lipid-raft-mediated survival signaling pathways, cell morphology
and cell function. The inhibitory mechanism of fatostatin could be
attributed to decreasing SREBP-2 transcriptional activity, since
SREBP-2 has been demonstrated to transcriptionally regulate the
expression of these cholesterogenic genes and fatostatin is a
chemical inhibitor of the SREBP pathway. These data revealed that
fatostatin decreased the expression of cholesterogenic genes and
exhibited an antitumor effect against EC in vitro by
interrupting the SREBP-2-regulated cholesterogenic pathway.
Apoptosis is a regulated cellular suicide mechanism,
with characteristics including nuclear condensation, cell
shrinkage, membrane blebbing, and DNA fragmentation (36). The caspase family consists of the
central regulators of apoptosis and contains two types of caspase
enzymes, namely, initiator and executioner caspases. Initiator
caspases (including caspase-2, caspase-8, caspase-9, caspase-10,
caspase-11, and caspase-12) are closely correlated to pro-apoptotic
signals. Once activated, full-length caspases are cleaved to form
cleaved-caspases and activate downstream effector caspases
(including caspase-3, caspase-6, and caspase-7), which in turn
induce apoptosis (37). Studies
have demonstrated that PARP is an intracellular ‘death substrate’
and that cleavage of PARP by caspases is likely a prerequisite for
the induction of apoptosis in various cells (38). In our present study, we demonstrated
that fatostatin decreased the expression levels of full-length
caspase-9, caspase-3 and PARP, increased the expression levels of
cleaved caspase-9, cleaved caspase-3 and cleaved PARP in EC cells,
and increased EC cell apoptosis. These data revealed that
fatostatin induced caspase-dependent apoptosis in EC cells. Since
the potential signaling pathways between fatostatin and the caspase
family in EC and the potential underlying molecular mechanisms by
which fatostatin decreases the invasive and migratory capacities of
EC cells are not fully understood and no in vivo tests were
performed, more studies are warranted.
In summary, in the present study we revealed the
potential underlying molecular mechanisms by which fatostatin
suppressed EC growth and tumorigenesis. We provided evidence that
fatostatin inhibits cell viability and colony formation, decreases
the invasive and migratory capacities of EC cells and enhances EC
cell apoptosis by blocking SREBP-regulated metabolic pathways. In
brief, we demonstrated that fatostatin displayed high antitumor
effects against EC in vitro by blocking SREBP-regulated
metabolic pathways. Although several underlying mechanisms require
further investigation, our findings revealed that fatostatin can be
a novel therapeutic strategy for the treatment of EC.
Acknowledgements
This study was supported by a grant from the
National Natural Science Foundation of China (no. 81372808).
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Suri V and Arora A: Management of
endometrial cancer: A review. Rev Recent Clin Trials. 10:309–316.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gunderson CC, Fader AN, Carson KA and
Bristow RE: Oncologic and reproductive outcomes with progestin
therapy in women with endometrial hyperplasia and grade 1
adenocarcinoma: A systematic review. Gynecol Oncol. 125:477–482.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hahn HS, Yoon SG, Hong JS, Hong SR, Park
SJ, Lim JY, Kwon YS, Lee IH, Lim KT, Lee KH, et al: Conservative
treatment with progestin and pregnancy outcomes in endometrial
cancer. Int J Gynecol Cancer. 19:1068–1073. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Horton JD, Goldstein JL and Brown MS:
SREBPs: Activators of the complete program of cholesterol and fatty
acid synthesis in the liver. J Clin Invest. 109:1125–1131. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li JN, Mahmoud MA, Han WF, Ripple M and
Pizer ES: Sterol regulatory element-binding protein-1 participates
in the regulation of fatty acid synthase expression in colorectal
neoplasia. Exp Cell Res. 261:159–165. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Swinnen JV: Increased lipogenesis in
steroid-responsive cancer cells: Mechanisms of regulation, role in
cancer cell biology and perspectives on clinical applications. Verh
K Acad Geneeskd Belg. 63:321–333. 2001.PubMed/NCBI
|
|
8
|
Yang Yu, Morin PJ, Han WF, Chen T, Bornman
DM, Gabrielson EW and Pizer ES: Regulation of fatty acid synthase
expression in breast cancer by sterol regulatory element binding
protein-1c. Exp Cell Res. 282:132–137. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yahagi N, Shimano H, Hasegawa K, Ohashi K,
Matsuzaka T, Najima Y, Sekiya M, Tomita S, Okazaki H, Tamura Y, et
al: Co-ordinate activation of lipogenic enzymes in hepatocellular
carcinoma. Eur J Cancer. 41:1316–1322. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li W, Tai Y, Zhou J, Gu W, Bai Z, Zhou T,
Zhong Z, McCue PA, Sang N, Ji JY, et al: Repression of endometrial
tumor growth by targeting SREBP1 and lipogenesis. Cell Cycle.
11:2348–2358. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Eberhard Y, Gronda M, Hurren R, Datti A,
Maclean N, Ketela T, Moffat J, Wraba JL and Schimmer AD: Inhibition
of SREBP1 sensitizes cells to death ligands. Oncotarget. 2:186–196.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kamisuki S, Mao Q, Abu-Elheiga L, Gu Z,
Kugimiya A, Kwon Y, Shinohara T, Kawazoe Y, Sato S, Asakura K, et
al: A small molecule that blocks fat synthesis by inhibiting the
activation of SREBP. Chem Biol. 16:882–892. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shao W, Machamer CE and Espenshade PJ:
Fatostatin blocks ER exit of SCAP but inhibits cell growth in a
SCAP-independent manner. J Lipid Res. 57:1564–73. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li X, Chen YT, Hu P and Huang WC:
Fatostatin displays high anti-tumor activity in prostate cancer by
blocking SREBP-regulated metabolic pathways and androgen receptor
signaling. Mol Cancer Ther. 13:855–866. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li X, Wu JB, Chung LW and Huang WC:
Anti-cancer efficacy of SREBP inhibitor, alone or in combination
with docetaxel, in prostate cancer harboring p53 mutations.
Oncotarget. 6:41018–41032. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Siqingaowa, Sekar S, Gopalakrishnan V and
Taghibiglou C: Sterol regulatory element-binding protein 1
inhibitors decrease pancreatic cancer cell viability and
proliferation. Biochem Biophys Res Commun. 488:136–140. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tennant DA, Durán RV and Gottlieb E:
Targeting metabolic transformation for cancer therapy. Nat Rev
Cancer. 10:267–277. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ward PS and Thompson CB: Metabolic
reprogramming: A cancer hallmark even warburg did not anticipate.
Cancer Cell. 21:297–308. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Deberardinis RJ, Sayed N, Ditsworth D and
Thompson CB: Brick by brick: Metabolism and tumor cell growth. Curr
Opin Genet Dev. 18:54–61. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Byrne FL, Poon IK, Modesitt SC, Tomsig JL,
Chow JD, Healy ME, Baker WD, Atkins KA, Lancaster JM, Marchion DC,
et al: Metabolic vulnerabilities in endometrial cancer. Cancer Res.
74:5832–5845. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Williams KJ, Argus JP, Zhu Y, Wilks MQ,
Marbois BN, York AG, Kidani Y, Pourzia AL, Akhavan D, Lisiero DN,
et al: An essential requirement for the SCAP/SREBP signaling axis
to protect cancer cells from lipotoxicity. Cancer Res.
73:2850–2862. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Radhakrishnan A, Goldstein JL, McDonald JG
and Brown MS: Switch-like control of SREBP-2 transport triggered by
small changes in ER cholesterol: A delicate balance. Cell Metab.
8:512–521. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Goldstein JL, DeBose-Boyd RA and Brown MS:
Protein sensors for membrane sterols. Cell. 124:35–46. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gholkar AA, Cheung K, Williams KJ, Lo YC,
Hamideh SA, Nnebe C, Khuu C, Bensinger SJ and Torres JZ: Fatostatin
inhibits cancer cell proliferation by affecting mitotic microtubule
spindle assembly and cell division. J Biol Chem. 291:17001–17008.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Johannes VS, Koen B and Guido V: Increased
lipogenesis in cancer cells: New players, novel targets. Curr Opin
Clin Nutr Metab Care. 9:358–365. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Menendez JA and Lupu R: Fatty acid
synthase and the lipogenic phenotype in cancer pathogenesis. Nat
Rev Cancer. 7:763–777. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Qiu C, Dongol S, Lv QT, Gao X and Jiang J:
Sterol regulatory element-binding protein-1/fatty acid synthase
involvement in proliferation inhibition and apoptosis promotion
induced by progesterone in endometrial cancer. Int J Gynecol
Cancer. 23:1629–1634. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hatzivassiliou G, Zhao F, Bauer DE,
Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA and
Thompson CB: ATP citrate lyase inhibition can suppress tumor cell
growth. Cancer Cell. 8:311–321. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Igal RA: Stearoyl-CoA desaturase-1: A
novel key player in the mechanisms of cell proliferation,
programmed cell death and transformation to cancer. Carcinogenesis.
31:1509–1515. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guo D, Bell EH and Chakravarti A: Lipid
metabolism emerges as a promising target for malignant glioma
therapy. CNS Oncol. 2:289–299. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Freeman MR, Cinar B, Kim J, Mukhopadhyay
NK, Di Vizio D, Adam RM and Solomon KR: Transit of hormonal and EGF
receptor-dependent signals through cholesterol-rich membranes.
Steroids. 72:210–217. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lingwood D and Simons K: Lipid rafts as a
membrane-organizing principle. Science. 327:46–50. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pandyra AA, Mullen PJ, Goard CA, Ericson
E, Sharma P, Kalkat M, Yu R, Pong JT, Brown KR, Hart T, et al:
Genome-wide RNAi analysis reveals that simultaneous inhibition of
specific mevalonate pathway genes potentiates tumor cell death.
Oncotarget. 6:26909–26921. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wiemer AJ, Hohl RJ and Wiemer DF: The
intermediate enzymes of isoprenoid metabolism as anticancer
targets. Anticancer Agents Med Chem. 9:526–542. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nevadunsky NS, Van Arsdale A, Strickler
HD, Spoozak LA, Moadel A, Kaur G, Girda E, Goldberg GL and Einstein
MH: Association between statin use and endometrial cancer survival.
Obstet Gynecol. 126:144–150. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Indran IR, Tufo G, Pervaiz S and Brenner
C: Recent advances in apoptosis, mitochondria and drug resistance
in cancer cells. Biochim Biophys Acta. 1807:735–745. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Vyas VK, Chintha C and Pandya MR: Biology
and medicinal chemistry approaches towards various apoptosis
inducers. Anticancer Agents Med Chem. 13:433–455. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Soldatenkov VA and Potaman VN: DNA-binding
properties of poly(ADP-ribose) polymerase: A target for anticancer
therapy. Curr Drug Targets. 5:357–365. 2004. View Article : Google Scholar : PubMed/NCBI
|