Introduction
Human end-binding protein 1 (EB1), a member of the
plus-end-tracking protein family that was originally reported in
colorectal cancer (1), regulates
microtubule dynamics by promoting microtubule growth and
suppressing catastrophes (2).
Subsequently, EB1 overexpression has been reported in gastric and
hepatic cancers according to proteomic analysis (3,4) and in
oral cancer based on liquid chromatography-mass spectrometry
(5). These observations indicated
that EB1 can serve as a biomarker for tumor progression. The
oncogenic function of EB1 has been experimentally observed in
esophageal, breast and glioblastoma cancer cells. EB1 promotes
tumor cell proliferation by activating Wnt signaling (6), which indicates that it is a marker of
poor prognosis. In addition, EB1 regulates the migration of B16F1
melanoma cells by modulating the balance between lamellipodia and
filopodia protrusions (7).
Previously, we reported that EB1 was a biomarker of radioresistance
in non-small cell lung carcinoma (NSCLC) (8). Although several studies elucidating
EB1 function have been performed using different human tumor
tissues, the precise role of EB1 and its molecular mechanism of
action related to cytotoxicity in cancer remains to be
elucidated.
Cyclooxygenase-2 (COX-2) is a rate-limiting enzyme
in arachidonic acid metabolism and is responsible for regulating
inflammation, pain and angiogenesis (9). The first genetic evidence for the
association between COX-2 and carcinogenesis was obtained for
colorectal cancer and the potential chemotherapeutic role of COX-2
inhibitors has been demonstrated in numerous types of cancer
(10,11). Conversely, COX-2 overexpression
inhibited the cell cycle and tumor progression in osteosarcoma
cells (12). Although our previous
study revealed that EB1 knockdown promoted reactive oxygen species
(ROS)-induced apoptosis in NSCLC cells via nuclear factor-κB
(NF-κB) activation (8), the
correlation between COX-2 and NSCLC cell death and the association
between COX-2 regulation and EB1 remain unclear.
Therefore, in the present study, we aimed to
elucidate the mechanisms regulating ROS production and COX-2
expression in EB1-knockdown lung cancer cells. In the present study
we provided further molecular evidence supporting the potential
application of EB1 as a novel target related to radioresistance in
lung cancer gene therapy.
Materials and methods
Cell culture and treatment
Human A549 lung cancer and SiHa and HeLa cervical
cancer cell lines were purchased from the American Type Culture
Collection (ATCC; Manassas, VA, USA). A549 cells were cultured in
RPMI-1640 medium and SiHa and HeLa cells were cultured in
Dulbecco's modified Eagle's medium (DMEM; Gibco® Life
Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine
serum (FBS; Gibco®, Life Technologies). The cells were
irradiated using a 137Cs source (Atomic Energy of
Canada, Ltd., Mississauga, ON, Canada) at a dose rate 3.81 Gy/min
and treated with 10 mM N-acetyl cysteine (NAC; Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) to scavenge ROS, 1 µM BAY 11-7082 (BAY)
to inhibit NF-κB activity and 20 µM SB203580 (SB) (both from
Calbiochem; Merck KGaA), to inhibit p38 kinase, or 10 µM
cycloheximide (CHX; Sigma-Aldrich; Merck KGaA) to block de
novo protein synthesis.
Cell death assay
Cell death was assessed as previously described
(8). Apoptotic death was also
determined by alterations in cellular morphology.
Quantitative reverse
transcription-polymerase chain reaction (qRT-PCR)
Transcripts were quantified by qRT-PCR as previously
described (13) using the following
primer pairs: EB1 (333-bp product) 5′-CTGCGTATTGTCAGTTTATG-3′
(sense) and 5′-GAGGTTTCTTCGGTTTATTC-3′ (antisense); COX-2 (580-bp
product), 5′-CTGGCGCTCAGCCATACAGC-3′ (sense) and
5′-GGCCCTCGCTTATGATCTGTC-3′ (antisense); and
glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 305-bp product),
5′-CATCTCTGCCCCCTCTGCTGA-3′ (sense) and 5′-GGATGACCTTGCCCACAGCCT-3′
(antisense).
Oncomine data mining
Oncomine (Life Technologies; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) was used for data analysis and
visualization as previously described (14). The expression of EB1 was compared
between lung cancer and normal lung tissue extracts.
Immunohistochemistry
Human tissue microarrays were purchased from
SuperBioChips (cat. no. CC5; Seoul, Korea) and immunohistochemistry
was performed using an anti-EB1 mouse monoclonal antibody
(dilution, 1:250; cat. no. sc-47704; Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA) as previously described (15). Immunostaining was performed using
the avidin-biotin-peroxidase method according to the manufacturer's
instructions (Vector Laboratories, Burlingame, CA, USA). Staining
intensity was scored as follows: 0, no visible staining; 1+, weak
staining; 2+, moderate staining; 3+, strong staining; and 4+, very
strong staining.
Knockdown of proteins by small
interference RNA (siRNA)
The following human-specific siRNAs synthesized
following the manufacturer's instructions (Genolution, Inc., Seoul,
Korea) were used: siEB1, 5′-UUGCCUUGAAGAAAGUGAAUU-3′ (sense) and
5′-UUCACUUUCUUCAAGGCAAUU-3′ (antisense); sip38,
5′-GAAGCUCUCCAGACCAUUUUU-3′ (sense) and 5′-AAAUGGUCUGGAGAGCUUCUU-3′
(antisense); and siCOX-2, 5′-AACUGCUCAACACCGGAAUUUUUUU-3′ (sense)
and 5′-AAAAAUUCCGGUGUUGAGCAGUUUU-3′ (antisense). A scrambled siRNA
that exhibited no significant homology to known gene sequences was
used as a negative control. The cells were transfected with 30 nM
siRNA in medium, as previously described (13).
ROS assay
The cells were incubated with 10 nM
2′,7′-dichlorofluorescein diacetate; Molecular Probes, Inc.,
Eugene, OR, USA) for 20 min to detect ROS, as previously described
(8).
Immunofluorescence confocal
microscopy
Immunofluorescence staining for EB1 (Santa Cruz
Biotechnology, Inc.) and COX-2 (Cayman Chemical, Ann Arbor, MI,
USA) was performed as previously described (8). Cell nuclei were identified by staining
with 4,6-diamidino-2-phenylindole (DAPI).
Western blot analysis
Western blot analysis were performed as previously
described (13) using primary
antibodies against the following: EB1 (cat. no. sc-47704), p53
(cat. no. sc-126), Iκ-B (cat. no. sc-371), phospho-ERK (cat. no.
sc-7383) and p38 (cat. no. sc-535) (all from Santa Cruz
Biotechnology, Inc.); cleaved PARP (Asp214, cat. no. 9541),
phospho-p53 (Ser15, cat. no. 9284), phospho-JNK (cat. no. 9251),
JNK (cat. no. 9252), phospho-p38 (cat. no. 9211) and ERK (cat. no.
9102) were obtained from Cell Signaling Technology Inc. (Beverly,
MA, USA) and COX-2 (cat. no. 160106) from Cayman Chemical. β-actin
(A1978; Sigma-Aldrich; Merck KGaA) was used as a loading control.
Blots were reacted with primary antibodies at dilution, 1:1,000 and
secondary antibodies at dilution, 1:5,000.
Statistical analysis
Cell culture experiments were repeated at least
three times. All data are expressed as the mean ± standard
deviation (SD). Statistical differences between groups were
assessed by the Student's t-test and Bonferroni's multiple
comparison test. P<0.05 was considered to indicate a
statistically significant difference.
Results
EB1 protein as a radioresistance and
tumorigenesis biomarker in NSCLC
As displayed in Fig.
1A, treatment with 10 Gy radiation induced cell death in ~63%
H460 vs. 26% A549 lung cancer cells, 65% parental H460 vs. 33%
artificially established R-H460 cells and 66% HeLa vs. 30% SiHa
cervical cancer cells at 72 h post-stimulation. Although the mRNA
level remained unchanged between the parental H460 and R-H460
cells, both EB1 protein and mRNA were highly overexpressed in the
radioresistant A549, R-H460 and SiHa cell lines compared with the
radiosensitive H460 and HeLa cancer cells (Fig. 1B). This observation indicated that
EB1 could serve as a biomarker for radioresistance. qRT-PCR data
revealed that EB1 mRNA levels in A549 and SiHa cells increased by
~2.9- and 2.2-fold compared with that in H460 and HeLa cells,
respectively (Fig. 1C).
Subsequently, the correlation between EB1 expression and lung
cancer severity was examined using the human genetic dataset
analysis tool Oncomine. EB1 mRNA levels were markedly increased in
lung adenocarcinoma tumors (Fig.
1D) compared with normal lung tissues, indicating that EB1 is
also a tumorigenesis factor. Consistent with the Oncomine data,
in vivo evidence based on tissue microarrays for lung cancer
and normal tissues revealed considerably upregulated EB1 expression
in lung cancer tissues compared with normal tissues. Representative
images of lung cancer and normal tissues are shown in Fig. 1E; for the intensity calculation, all
tissue data were analyzed. These results indicated that EB1
induction may play an important role in the acquisition of the
radioresistant phenotype and tumorigenesis in H460 cells.
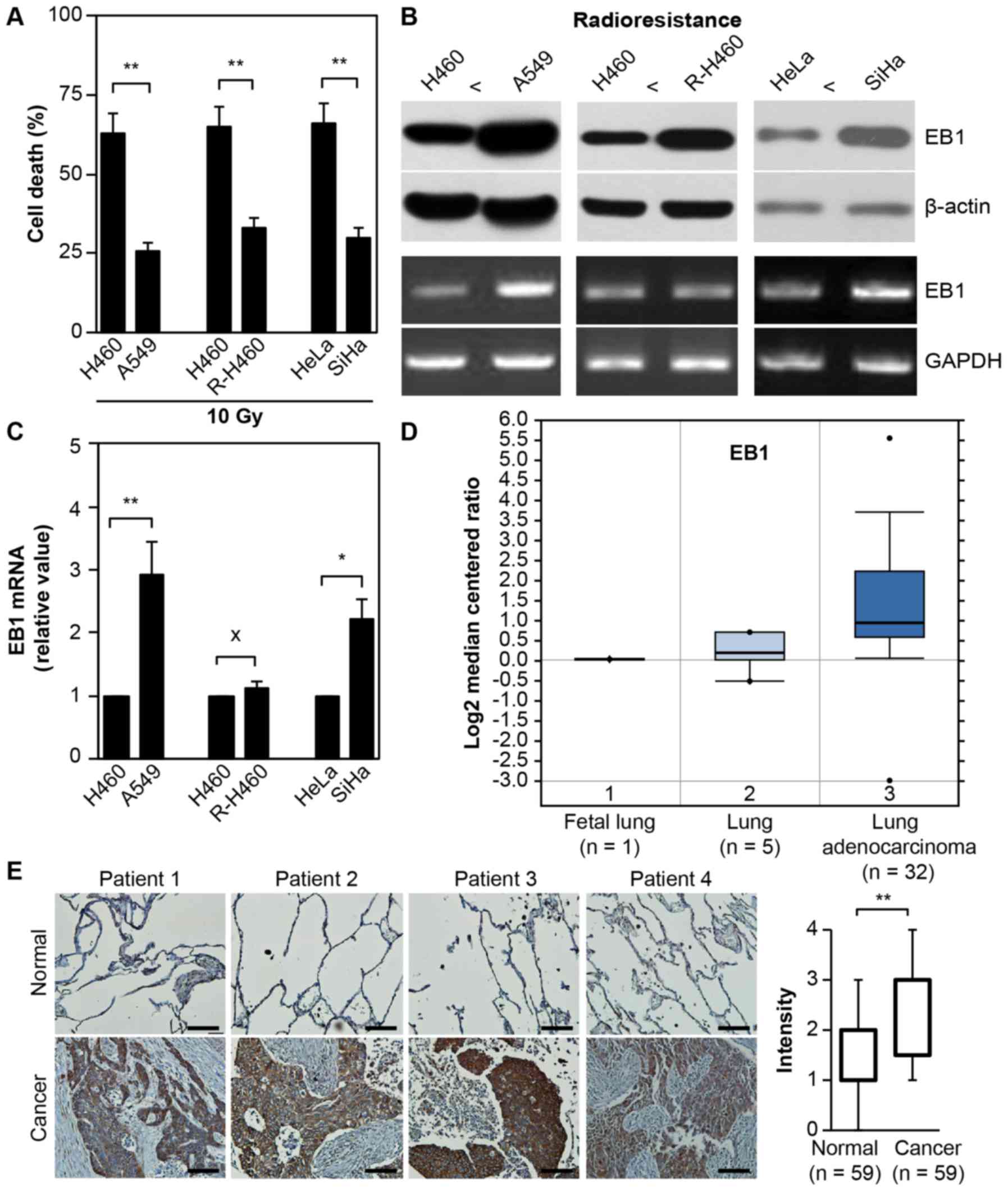 | Figure 1.EB1 protein as a radioresistance and
tumorigenesis biomarker. (A) Lung and cervical cancer cell lines
were treated with 10 Gy radiation for 48 h. Cell death was
determined by FACS analysis and the data are expressed as the mean
± SD (**P<0.005 compared with radiosensitive control cells). (B
and C) Each cell line was cultured for 36 h without any
stimulation. Protein and transcription levels of EB1 were
determined by western blot analysis (B, top panel) and conventional
PCR (B, bottom panel; loading control, GAPDH), respectively. (C)
The qRT-PCR data are expressed as the mean ± SD (*P<0.05 and
**P<0.005 compared with radiosensitive control cells; X denotes
no significance compared with control cells). (D) Available
datasets in the Oncomine database were queried for EB1 expression
with respect to cancer vs. normal tissue (threshold
P-value=3.87×10-4; fold-change ≥2). (E) Representative microscopic
images of lung cancer tissues and their normal tissue counterparts
stained with an anti-EB1 antibody (left panel, scale bar, 50 µm).
Staining intensity scored as follows in all tissue samples (n=59):
0, no staining; +1, weak; +2, moderate; +3, strong; and +4, very
strong. The data are presented as box and whisker plots.
**P<0.005 compared with the staining intensity of normal
tissues. EB1, end-binding protein 1; FACS, fluorescence-activated
cell sorting; SD, standard deviation; qPCR, quantitative polymerase
chain reaction; GAPDH, glyceraldehyde-3-phosphate
dehydrogenase. |
EB1 depletion promotes p38
kinase-dependent ROS-mediated apoptosis in A549 NSCLC cells
We have previously reported EB1 depletion-mediated
accumulation of ROS in A549 cells, although the upstream activator
of ROS production could not be determined (8). In the present study, we observed that
EB1 depletion in A549 cells markedly induced p38 kinase activity,
but not that of ERK or JNK, without inducing any change in basal
protein levels (Fig. 2A).
Furthermore, as determined by cell staining (Fig. 2B and C, top panel), inhibition of
p38 kinase using a p38-specific siRNA (Fig. 2B) or the chemical inhibitor SB203580
(Fig. 2C) almost completely reduced
ROS levels, which accumulated due to EB1 depletion. In addition,
fluorescence-activated cell sorting (FACS) analysis revealed that
ROS levels were increased by ~1.98-fold in EB1-depleted A549 cells
and ~1.21- and 1.01-fold in p38 siRNA- and SB203580-stimulated A549
cells, respectively, compared with the levels in control cells
(Fig 2B and C, bottom panel).
However, inhibition of ROS generation using a ROS scavenger (NAC)
did not alter p38 kinase activity in EB1-depleted A549 cells
(Fig. 2D), indicating that p38
kinase is involved in ROS production due to siEB1. Subsequently, we
used a p38 loss-of-function approach to examine whether the p38
kinase-induced ROS cascade is critical for siEB1-mediated A549 cell
death. Notably, the reduction in p38 kinase protein due to a
p38-specific siRNA markedly suppressed cleaved PARP levels, which
was significantly induced by an increase in p38 phosphorylation in
EB1-knockdown A549 cells (Fig. 2E).
Consistent with the biochemical results, cell morphology analysis
also revealed a marked restoration of survival in EB1-depleted A549
cells after treatment with both p38 siRNA and SB203580 (Fig. 2F). FACS analysis revealed that cell
death increased by ~3.6-fold in siEB1-treated A549 cells and ~1.9-
and 1.1-fold in p38 siRNA- and SB203580-stimulated A549 cells,
respectively, compared with that in control cells (Fig. 2G). Collectively, our data indicated
that EB1-depleted A549 cell cytotoxicity was attributable to ROS
generation through p38 kinase activation.
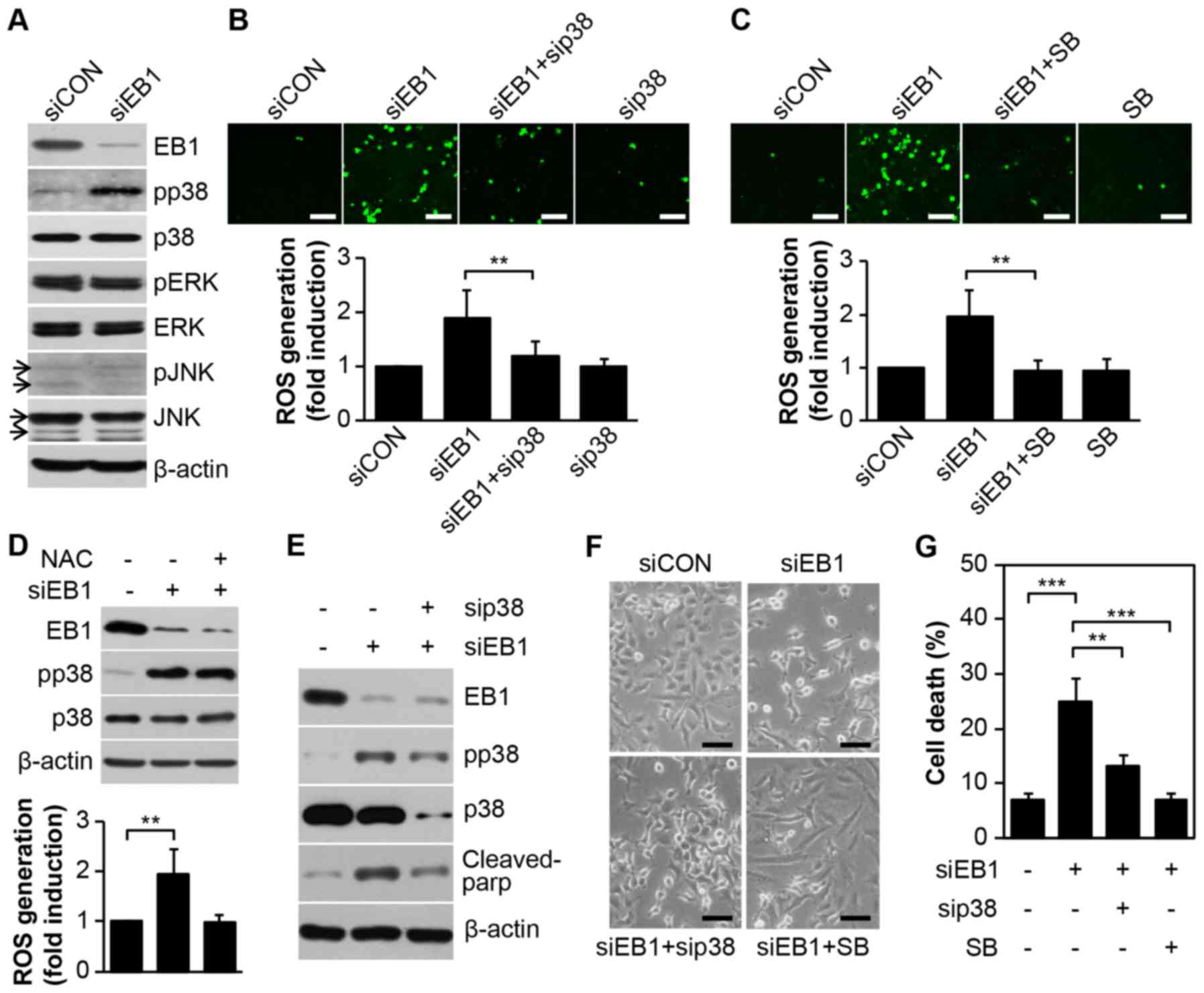 | Figure 2.EB1 depletion promotes p38 kinase
activation-dependent ROS generation to induce A549 cancer cell
death. (A) A549 cells were transfected with 30 nM control siRNA
(siCON) or EB1 siRNA (siEB1) for 36 h. Protein and phosphorylation
levels of EB1 and MAPKs were determined by western blot analysis.
(B) A549 cells were transfected with 30 nM siCON, EB1 siRNA and/or
p38 siRNA (sip38) for 36 h. Intracellular ROS levels were assessed
by confocal microscopy (top panel, scale bar, 0.1 mm) and
determined by FACS analysis. The data are expressed as the mean ±
SD (**P<0.005 compared with siEB1-transfected cells, bottom
graph). (C) A549 cells were transfected with 30 nM siCON or EB1
siRNA for 36 h in the absence or presence of 20 µM SB203580 (SB).
Intracellular ROS levels were determined as noted in B. (D) A549
cells were transfected as described in A in the absence or presence
of 10 mM NAC. Protein and phosphorylation levels of EB1 and p38
kinase were determined by western blot analysis (top panel). ROS
production was assessed by FACS analysis. The data are expressed as
the mean ± SD (**P<0.005 compared with siEB1 alone-transfected
cells, bottom graph). (E-G) A549 cells were transfected as
mentioned in B and C. Protein levels of EB1, p38 and cleaved PARP
and phosphorylation level of p38 were determined by (E) western
blot analysis. Morphological changes were observed by (F) light
microscopy (scale bar, 0.1 mm) and cell death was determined by (G)
FACS analysis. The data are expressed as the mean ± SD
(**P<0.005 compared with siEB1-transfected cells) (G). EB1,
end-binding protein 1; MAPK, mitogen-activated protein kinase; ROS,
reactive oxygen species; FACS, fluorescence-activated cell sorting;
SD, standard deviation; PARP, poly(ADP-ribose) polymerase. |
Regulation of COX-2 expression is p38
kinase dependent but not NF-κB dependent in EB1-depleted A549 NSCLC
cells
Given that the COX-2 enzyme plays critical roles in
numerous biological processes, including inflammation, cancer cell
death and development (16,17), we examined the correlation between
COX-2 and EB1 knockdown-induced cytotoxicity in A549 cells. As
displayed in Fig. 3A, silencing of
EB1 in A549 cells markedly induced the expression of COX-2 protein
(top panel) and transcript (middle panel), as determined by western
blot analysis and conventional PCR analyses, respectively. qRT-PCR
revealed that EB1 and COX-2 mRNA levels were reduced by ~80% and
increased by ~2.21-fold, respectively, in EB1-depleted A549 cells
compared with control cells (Fig.
2A, bottom panel). Although no significant alteration in
nuclear COX-2 distribution dynamics was observed in A549 cells
following EB1 knockdown (Fig. 3B,
top panel), COX-2 expression surprisingly increased by ~3.02-fold
compared with control cells. This result was consistent with the
results of immunofluorescence confocal microscopy (Fig. 3B, bottom panel). However, in A549
cells, selective inhibition of p38 kinase with SB203580 decreased
COX-2 protein (Fig. 3C, top panel)
and transcription levels mediated by EB1 depletion (Fig. 3C, middle panel). qRT-PCR results
also demonstrated similar levels of COX-2 mRNA in EB1-depleted
SB203580-treated A549 cells and in unstimulated control cells
(Fig. 3C, bottom panel).
Additionally, inhibition of p38 kinase protein expression using a
p38-specific siRNA considerably suppressed EB1 depletion-induced
COX-2 expression in A549 cells (Fig.
3D), indicating that p38 kinase is an upstream regulator of the
expression of COX-2. The suppression of ROS in EB1-depleted A549
cells using NAC resulted in COX-2 expression patterns that were
similar to those observed after p38 kinase inhibition (Fig. 3E), indicating that the p38-ROS
signaling axis is a key player in COX-2 regulation. Although the
COX-2 gene is a target of the transcription factor NF-κB, we
observed that NF-κB inhibition using BAY 11-7082 (BAY) in
EB1-knockdown A549 cells did not alter the high levels of COX-2
expression (Fig. 3F). This finding
indicated that p38 kinase-dependent and NF-κB-independent COX-2
regulation is associated with EB1-mediated signaling.
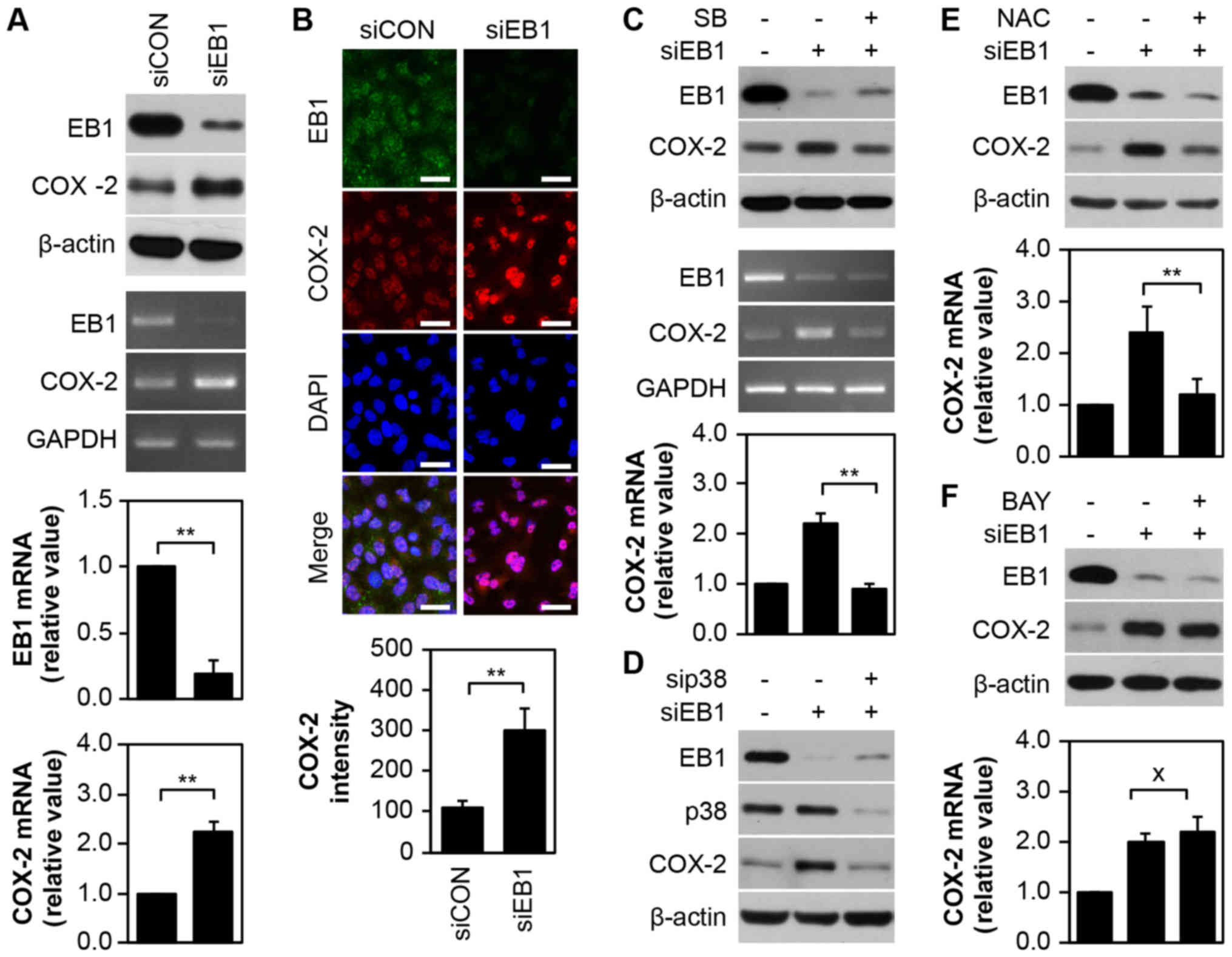 | Figure 3.EB1 depletion upregulates the
expression of COX-2 via p38-ROS signaling activation, but not the
NF-κB pathway in A549 cancer cells. (A and B) A549 cells were
transfected with 30 nM siCON or siEB1 for 36 h. Protein and
transcription levels of EB1 and COX-2 were determined by western
blot analysis (A, top panel) and conventional PCR (A, middle panel;
loading control: GAPDH), respectively. The qRT-PCR (A, bottom
panel) data are expressed as the mean ± SD (**P<0.005 compared
with control cells). (B, top panel) EB1 and COX-2 localization was
visualized by confocal microscopy and representative images are
shown (scale bar, 0.1 mm). (B, bottom panel). Quantification of the
fluorescent area of the cells is expressed as the mean ± SD
(**P<0.005 compared with control cells). (C) A549 cells were
transfected with 30 nM siCON or siEB1 for 36 h in the absence or
presence of 20 µM SB203580. Protein and transcription levels of EB1
and COX-2 were determined by western blot analysis (top panel) and
conventional PCR (middle panel, loading control: GAPDH),
respectively. The qRT-PCR data (bottom panel) are expressed as the
mean ± SD (**P<0.005 compared with siEB1 alone-transfected
cells). (D) A549 cells were transfected with 30 nM siCON, siEB1
and/or sip38 for 36 h. EB1, p38 and COX-2 protein levels were
determined by western blot analysis. (E and F) A549 cells were
transfected with 30 nM siCON or siEB1 for 36 h in the absence or
presence of 10 mM NAC (E) or 2 µM Bay (F). EB1 and COX-2 protein
levels were determined by western blot analysis (top panel) and
COX-2 transcription levels were determined by qRT-PCR (bottom
panel). The data are expressed as the mean ± SD (*P<0.05
compared with siEB1 alone-transfected cells; × denotes no
significance compared with siEB1-transfected cells). EB1,
end-binding protein 1; COX-2, cyclooxygenase-2; ROS, reactive
oxygen species; NF-κB nuclear factor-κB; GAPDH,
glyceraldehyde-3-phosphate dehydrogenase; GAPDH,
glyceraldehyde-3-phosphate dehydrogenase; qPCR, quantitative
polymerase chain reaction; siCON, control siRNA; siEB1, EB1
siRNA. |
COX-2 plays a critical role in EB1
knockdown-mediated A549 NSCLC cell death
Consistent with the results demonstrating
overexpression of the COX-2 protein during EB1 depletion-induced
A549 cell death (Fig. 4A, left
panel), other radioresistant cell lines, such as SiHa cervical
cancer cells, also exhibited induction of COX-2 protein expression
under the same conditions. In addition, two important apoptotic
markers, cleaved PARP and activated caspase-3, were also increased
(Fig. 4A) in EB1 depleted A549 and
SiHa cell lines. This finding indicated the potential association
between the overexpression of COX-2 and EB1 knockdown-mediated cell
death. Furthermore, COX-2 levels could not be detected in control
cells and were significantly stable (half-life, >9 h) in
EB1-knockdown A549 cells treated with the translation inhibitor
CHX, demonstrating that EB1 was involved in regulating COX-2
protein stability (data not shown). Finally, to establish the
primary role of COX-2 in EB1 knockdown-mediated cell death, A549
cells were transfected with a COX-2-specific siRNA alone or in
combination with EB1-specific siRNA. Notably, EB1 knockdown-induced
A549 cell death was markedly reduced by direct inhibition of COX-2
expression, as determined by western blot analysis for two
apoptotic markers (Fig. 4B). Data
on changes in cell morphology also corroborated the above mentioned
biochemical results (Fig. 4C). FACS
analysis also demonstrated that cell death was increased to ~28.6%
in siEB1-treated A549 cells and reduced to ~9.2% in cells also
transfected with COX-2 siRNA compared with ~8% in control cells
(Fig. 4D). Collectively, our
results indicated that COX-2 is essential for EB1-mediated
regulation of the cell fate in different cell lines, including
radioresistant A549 NSCLC cells. However, this observation may not
be limited to lung cancer cells.
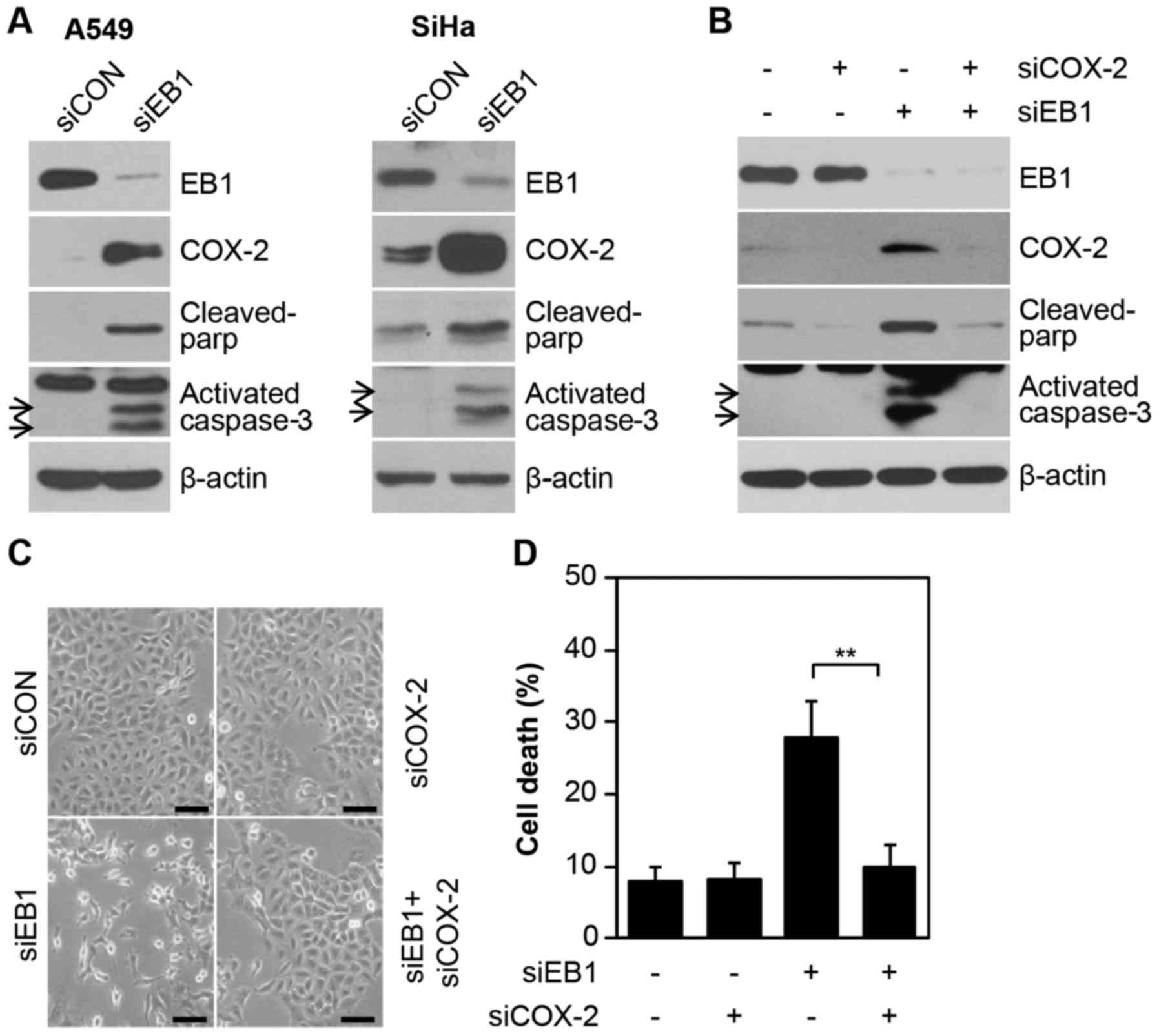 | Figure 4.COX-2 induced by EB1 knockdown plays a
critical role in A549 cancer cell death. (A) A549 (left panel) and
SiHa (right panel) cells were transfected with 30 nM siCON or siEB1
for 36 h. EB1, COX-2, cleaved PAR and activated caspase-3 protein
levels were determined by western blot analysis. (B-D) A549 cells
were transfected with 30 nM siCON or siEB1 and/or COX-2 siRNA
(siCOX-2) for 48 h. EB1, COX-2, cleaved PARP and activated
caspase-3 protein levels were determined by western blot analysis.
(B) Morphological changes were observed by (C) light microscopy
(scale bar, 0.1 mm) and cell death was determined by (D) FACS
analysis. The data are expressed as the mean ± SD (**P<0.005
compared with siEB1-transfected cells). EB1, end-binding protein 1;
COX-2, cyclooxygenase-2; siCON, control siRNA; siEB1, EB1 siRNA;
PARP, poly(ADP-ribose) polymerase. |
Discussion
In the present study, we aimed to identify novel
strategies for improving the response ratio of radiotherapy and to
determine radioresistance factors in lung cancer cells. We have
previously reported (8) that EB1
was involved in regulating tumor cell death leading to
radioresistance and that EB1 knockdown promoted apoptosis in lung
cancer cells by increasing expression of the pro-apoptotic protein
Bax via ROS-dependent NF-κB activation (8). However, the molecular mechanisms and
detailed pathways underlying radiation-induced resistance in lung
cancer cells has remained a topic of further investigation. We
performed the present study to explore the molecular mechanisms
underlying EB1-mediated inhibition of tumor cell cytotoxicity
following exposure to radiation. We observed that the mechanisms
underlying this effect involved EB1 depletion, which promoted p38
kinase-dependent ROS-mediated apoptosis in A549 cells. This
p38-dependent apoptosis was critical for upregulating COX-2.
Given that MAPKs are central mediators of cell death
and survival pathways and are implicated in the response of tumor
cells to diverse antitumor signals, including radiation, we first
investigated the MAPK signaling pathway (18). EB1 silencing in A549 cells
significantly induced p38 phosphorylation. However, ERK and JNK
were not activated. Previously, our group demonstrated that
downregulation of EB1 is related to NF-κB-dependent signaling
cascades. In the present study, NF-κB, another known
transcriptional regulator of COX-2, was not found to be involved in
the increase of COX-2 via the EB1-p38-COX-2 signaling axis. Several
studies have suggested the involvement of COX-2 in tumor
progression and carcinogenesis and different protein
kinase-mediated pathways regulate COX-2 expression in response to
different cellular stress signals (16). Although COX-2 modulates cell
proliferation or apoptosis in several solid tumors, its functions
appear to be controversial (17).
Thus, COX-2 modulation is a promising field investigated by
numerous research groups. Our results indicated that COX-2 is
essential for EB1 knockdown-mediated A549 cell death. We
demonstrated that the upregulation of COX-2 in EB1-depleted cells
required the activation of the p38 MAPK pathway and led to
apoptosis.
In conclusion, we revealed in the present study that
EB1 knockdown in radioresistant lung cancer cells (A549) relied on
the signaling pathway leading to p38-dependent COX-2 upregulation,
which ultimately induced ROS-mediated apoptosis. Our results also
indicated that knockdown of EB1 may effectively improve the
therapeutic effects of RT in patients with radioresistant lung
cancer. Therefore, a better understanding of the potential
molecular mechanisms underlying the radioresistance effect of EB1
is of immense importance in lung cancer research and radiation
therapy. Collectively, our results indicated that EB1 could be used
as a biomarker for RT.
Acknowledgments
Not applicable.
Funding
The present study was supported by a grant from the
Korea Institute of Radiological and Medical Sciences (KIRAMS)
funded by the Ministry of Science and ICT (MSIT), Republic of Korea
(50531–2018).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
SGH, EHK conceived the idea, designed the experiment
setup and wrote the manuscript. JHB, JHY and EHH performed the
experiments. SGH, EHK analysed the data. JYS, HDU, JKP, ICP, JSK
and CWL discussed the data. All authors read and approved the
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Orimo T, Ojima H, Hiraoka N, Saito S,
Kosuge T, Kakisaka T, Yokoo H, Nakanishi K, Kamiyama T, Todo S, et
al: Proteomic profiling reveals the prognostic value of adenomatous
polyposis coli-end-binding protein 1 in hepatocellular carcinoma.
Hepatology. 48:1851–1863. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kumar M, Mehra S, Thakar A, Shukla NK,
Roychoudhary A, Sharma MC, Ralhan R and Chauhan SS: End Binding 1
(EB1) overexpression in oral lesions and cancer: A biomarker of
tumor progression and poor prognosis. Clin Chim Acta. 459:45–52.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang Y, Zhou X, Zhu H, Liu S, Zhou C,
Zhang G, Xue L, Lu N, Quan L, Bai J, et al: Overexpression of EB1
in human esophageal squamous cell carcinoma (ESCC) may promote
cellular growth by activating beta-catenin/TCF pathway. Oncogene.
24:6637–6645. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dong X, Liu F, Sun L, Liu M, Li D, Su D,
Zhu Z, Dong JT, Fu L and Zhou J: Oncogenic function of microtubule
end-binding protein 1 in breast cancer. J Pathol. 220:361–369.
2010.PubMed/NCBI
|
|
5
|
Berges R, Baeza-Kallee N, Tabouret E,
Chinot O, Petit M, Kruczynski A, Figarella-Branger D, Honore S and
Braguer D: End-binding 1 protein overexpression correlates with
glioblastoma progression and sensitizes to Vinca-alkaloids in vitro
and in vivo. Oncotarget. 5:12769–12787. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nishigaki R, Osaki M, Hiratsuka M, Toda T,
Murakami K, Jeang KT, Ito H, Inoue T and Oshimura M: Proteomic
identification of differentially-expressed genes in human gastric
carcinomas. Proteomics. 5:3205–3213. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schober JM, Cain JM, Komarova YA and
Borisy GG: Migration and actin protrusion in melanoma cells are
regulated by EB1 protein. Cancer Lett. 284:30–36. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim M-J, Yun HS, Hong E-H, Lee SJ, Baek
JH, Lee CW, Yim JH, Kim JS, Park JK, Um HD, et al: Depletion of
end-binding protein 1 (EB1) promotes apoptosis of human
non-small-cell lung cancer cells via reactive oxygen species and
Bax-mediated mitochondrial dysfunction. Cancer Lett. 339:15–24.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Smith WL, DeWitt DL and Garavito RM:
Cyclooxygenases: Structural, cellular, and molecular biology. Annu
Rev Biochem. 69:145–182. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Oshima M, Dinchuk JE, Kargman SL, Oshima
H, Hancock B, Kwong E, Trzaskos JM, Evans JF and Taketo MM:
Suppression of intestinal polyposis in Apc delta716 knockout mice
by inhibition of cyclooxygenase 2 (COX-2). Cell. 87:803–809. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Howe LR and Dannenberg AJ: A role for
cyclooxygenase-2 inhibitors in the prevention and treatment of
cancer. Semin Oncol. 29:111–119. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu Z, Choudhary S, Voznesensky O, Mehrotra
M, Woodard M, Hansen M, Herschman H and Pilbeam C: Overexpression
of COX-2 in human osteosarcoma cells decreases proliferation and
increases apoptosis. Cancer Res. 66:6657–6664. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim JS, Chang JW, Yun HS, Yang KM, Hong
EH, Kim DH, Um HD, Lee KH, Lee SJ and Hwang SG: Chloride
intracellular channel 1 identified using proteomic analysis plays
an important role in the radiosensitivity of HEp-2 cells via
reactive oxygen species production. Proteomics. 10:2589–2604. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yun HS, Baek J-H, Yim J-H, Lee SJ, Lee CW,
Song JY, Um HD, Park JK, Park IC and Hwang SG: Knockdown of
hepatoma-derived growth factor-related protein-3 induces apoptosis
of H1299 cells via ROS-dependent and p53-independent NF-κB
activation. Biochem Biophys Res Commun. 449:471–476. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim J-S, Kim EJ, Oh JS, Park I-C and Hwang
S-G: CIP2A modulates cell-cycle progression in human cancer cells
by regulating the stability and activity of Plk1. Cancer Res.
73:6667–6678. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Williams CS, Mann M and DuBois RN: The
role of cyclooxygenases in inflammation, cancer, and development.
Oncogene. 18:7908–7916. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sobolewski C, Cerella C, Dicato M,
Ghibelli L and Diederich M: The role of cyclooxygenase-2 in cell
proliferation and cell death in human malignancies. Int J Cell Biol
2010. 2151582010.doi: 10.1155/2010/215158.
|
|
18
|
Munshi A and Ramesh R: Mitogen-activated
protein kinases and their role in radiation response. Genes Cancer.
4:401–408. 2013. View Article : Google Scholar : PubMed/NCBI
|














