Introduction
The placental growth factor (PlGF) is a member of
the vascular endothelial growth factor (VEGF) family of angiogenic
factors that shares with VEGF-A the ability to activate the
transmembrane tyrosine kinase receptor type 1 (VEGFR-1). However,
differently from VEGF-A, which also interacts with VEGFR-2, PlGF
binds exclusively to VEGFR-1. Besides being expressed in
endothelial cells during vessel formation and remodeling, VEGFR-1
is present in monocytes/macrophages and in a variety of human
cancers, where it favors cell migration and survival predicting
poor prognosis and recurrence (1,2).
Notably, in myelomonocytic cells the PlGF/VEGFR-1 pathway plays an
important role in cell recruitment within the tumor mass,
tumor-associated angiogenesis and metastasis (3,4).
Moreover, VEGFR-1 contributes to the differentiation of
CD11b+ myelomonocytic cells from hematopoietic
progenitors (5) and identifies a
monocyte subset with increased chemotactic response towards VEGF-A
and PlGF (6). VEGFR-1 activation by
PlGF is also involved in the downregulation of type 1 T helper
immune responses by modulating the function of dendritic cells
(7) and macrophage polarization
toward a tumor-associated macrophage (TAM) subtype that releases
matrix metalloproteinase-9 (MMP-9) (8). Stimulation of monocytes by PlGF
through activation of VEGFR-1 results in triggering of PI3
kinase/Akt and ERK-1/2 pathways and gene expression induction with
increased production of cytokines (TNF-α and IL-1b) or chemokines
(MCP-1, IL-8 and MIP-1b) (9).
In addition to enhancing tumor aggressiveness, PlGF
and VEGFR-1 overexpression may also contribute to primary or
acquired tumor resistance to current anti-VEGF-A therapies. Indeed,
we observed increased PlGF plasma levels in melanoma patients
treated with the anti-VEGF-A monoclonal antibody (mAb) bevacizumab
compared to healthy donors (10).
One of the mechanisms by which activation of the PlGF/VEGFR-1
pathway may result in failure of anti-VEGF therapies relies on
stimulation of myelomonocytic cell recruitment to the tumor mass
(3,11). We recently demonstrated that in
vivo treatment of melanoma-bearing mice with the anti-VEGFR-1
D16F7 mAb strongly inhibited mobilization of myeloid progenitor
cells from the bone marrow and drastically reduced
monocyte/macrophage infiltration at the tumor border in melanoma
nodules (11). The anti-VEGFR-1 mAb
also exerted inhibitory effects on migration and/or extracellular
matrix (ECM) invasion by endothelial cells as well as cancer cells
(melanoma and glioblastoma) in response to VEGF-A and PlGF
(11–13). The D16F7 mAb has a novel mechanism
of action since it hampers VEGFR-1 activation without preventing
ligand binding.
Poly(ADP-ribose) polymerase (PARP)-1 is the most
abundant isoform of an enzyme family capable of synthesizing
ADP-ribose polymers (PAR) that are transferred to PARP-1 itself and
to a number of target proteins. Thus, PARylation represents a
transient post-translational modification of proteins that is
involved in the regulation of various cellular functions, including
DNA repair and maintenance of genomic integrity, gene transcription
and cell death (reviewed in refs. 14,15).
Moreover, PARP-1 plays an important role in inflammation, either
because it acts as a transcriptional regulator capable of
modulating the expression of pro-inflammatory genes or because,
when overactivated, it leads to NAD+ and ATP depletion
with consequent necrosis that initiates the inflammatory process
(reviewed in ref. 14). In this
regard, PARP inhibition exerts protective effects blocking the
pro-inflammatory activity of PARP-1. Indeed, treatment with PARP
inhibitors (PARPi) of macrophages diminished the production of
inflammatory mediators (16) and
decreased monocytes adhesion and migration across the blood-brain
barrier (BBB) in in vitro models by reducing the activation
of specific integrins (17).
PARPi have been largely investigated for cancer
treatment in combination with chemo- or radiotherapy and as
monotherapy in the case of tumors deficient in homologous
recombination DNA repair (18).
Among the multiple PARPi in clinical development, olaparib,
rucaparib and niraparib have been recently approved. In particular,
olaparib is the first orally bioavailable agent to receive approval
by both FDA and EMA as maintenance monotherapy of patients with
platinum-sensitive relapsed BRCA-mutated ovarian cancer and by FDA
for BRCA-mutated/HER2-negative metastatic breast cancer (19,20).
Moreover, it is currently being studied in a number of clinical
trials for a variety of solid tumors (www.ClinicalTrials.gov). We recently demonstrated that
olaparib exerts cytotoxic effects against acute myeloid leukemia
blasts, while it does not affect the viability of bone marrow
CD34+ enriched peripheral blood cells obtained from
healthy donors (21).
In the present study, we analyzed the effect of the
PARPi olaparib on activation of human myelomonocytic cells by PlGF
and found that olaparib and D16F7 similarly inhibited PlGF-induced
chemotaxis and ECM invasion in a dose-dependent manner. Results
demonstrate that inhibition of monocyte activation mediated by PlGF
may contribute to the antitumor activity of PARPi. Moreover, these
data are expected to be relevant for designing new therapeutic
strategies for neoplastic and inflammatory disorders where PlGF has
been demonstrated to play an important role.
Materials and methods
Cell cultures, human monocyte
isolation and drug treatment
The human promyelocytic HL-60 cell line was obtained
from the American Type Culture Collection (ATCC; Manassas, VA,
USA). Cells were cultured in RPMI-1640 medium (Sigma-Aldrich, St.
Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS;
Sigma-Aldrich), 2 mM L-glutamine, 100 U/ml penicillin and 100 µg/ml
streptomycin sulfate, at 37°C in a 5% CO2 humidified
atmosphere. HL-60 cells were authenticated by STR profiling (BMR
Genomics, Padova, Italy).
For differentiation towards monocytic/macrophagic
cells, the HL-60 cell line was treated with 10 ng/ml phorbol
12-myristate 13-acetate (PMA; Sigma-Aldrich) for 24 h.
Peripheral blood mononuclear cells were separated by
Ficoll-Hypaque density centrifugation of buffy coats obtained from
adult healthy donors. Monocytes were isolated from peripheral blood
mononuclear cells by plastic adherence for 2 h at 37°C in a 5%
CO2 humidified atmosphere. After removal of non-adherent
cells by repeated washing with serum-free Dulbecco's modified
Eagle's medium (DMEM; Sigma-Aldrich), adherent monocytes were
collected by gentle scraping with a plastic scraper.
For the treatment of differentiated HL-60 cells or
monocytes with olaparib, cells were exposed to the indicated
concentrations of the PARPi for 2 h at 37°C. The stock solution of
olaparib (40 mM; Selleckchem, Munich, Germany), was prepared by
dissolving the drug in dimethyl sulfoxide (DMSO; Sigma-Aldrich).
Control cells were always exposed to DMSO at a concentration equal
to that of the drug-treated cells. For the analysis of the
influence of olaparib on maximally stimulated PARP activity,
differentiated HL-60 cells were incubated with 50 µM
H2O2 for 15 min after treatment with the
PARPi.
D16F7 stock solution was prepared in
phosphate-buffered saline (PBS) and in vitro treatment was
performed by incubating the cells in a rotating wheel for 30 min at
room temperature. The generation of the anti-VEGFR-1 D16F7 mAb was
previously described (11).
Analysis of VEGFR-1 transcript
Quantification of the membrane VEGFR-1 transcript
was performed by real-time quantitative reverse
transcription-polymerase chain reaction (qRT-PCR) according to the
dual-labeled fluorigenic probe method and using an ABI Prism 7000
sequence detector (PerkinElmer, Groningen, The Netherlands), as
previously described (22).
Expression levels were calculated by the relative standard curve
method. Primers used were as follows: VEGFR-1, forward
5′-ACCGAATGCCACCTCCATG-3′ and reverse 5′-AGGCCTTGGGTTTGCTGTC-3′.
For each sample, the level of VEGFR-1 transcript was normalized to
that of 18S RNA (TaqMan® Gene Expression Assay; Applied
Biosystems, Foster City, CA, USA) and compared to the
VEGFR-1-negative M14 melanoma cell line, to which the arbitrary
value of 1 was assigned.
Western blot analysis
Proteins were run using 10% SDS-polyacrylamide gels
and transferred to supported nitrocellulose membranes by standard
techniques. Immunodetection was performed using the following
primary antibodies: Anti-PAR mouse mAb (1:1,000; cat. no.
4335-MC-100; Trevigen, Gaithersburg, MD, USA), rabbit
anti-E-cadherin and anti-β-catenin mAbs (1:1,000; cat. no. 3195P
and 8480P, respectively; Cell Signaling Technology, Danvers, MA,
USA) or rabbit polyclonal anti-β-actin (1:10,000; cat. no. A2066;
Sigma Aldrich) antibodies. Anti-mouse or anti-rabbit
IgG/horseradish peroxidase secondary antibodies (1:1,000; cat. no.
170-6516 and 170-6515, respectively; Bio-Rad Laboratories,
Hercules, CA, USA) and ECL Western Blotting detection reagents from
GE Healthcare (cat. no. RPN2106; Milan, Italy), were used to
identify the proteins of interest.
Chemotaxis and ECM invasion
assays
In vitro migration assay was performed using Boyden
chambers equipped with 8-µm pore diameter polycarbonate filters
(Nuclepore; Whatman Inc., Clifton, NJ, USA) (for differentiated
HL-60 cells), as previously described (23), or Corning HTS
Transwell®-96 permeable support plates (Sigma-Aldrich)
with 5.0-µm pore polycarbonate membranes (for monocytes), coated
with 5 µg/ml gelatin (Sigma-Aldrich). Vehicle, olaparib and/or
D16F7 pre-treated cells were loaded in the upper compartment of
Boyden chambers (2×105 HL-60 cells/chamber) or Transwell
plates (1.5×105 monocytes/chamber). Migration assay,
toward serum-free medium [containing 0.1% bovine serum albumin
(BSA) and 1 µg/ml heparin] or serum-free medium containing PlGF (50
ng/ml), present in the lower compartment, was performed in the
absence or in the presence of olaparib and/or D16F7 mAb at the
concentrations and incubation times specified in the Figure
legends. Migrated cells, attached to the lower side of the filters,
were fixed in ethanol, stained with crystal violet and counted in
triplicate samples for a total of 12 high power microscopic fields
(×200 and ×400 magnification for HL-60 cells and monocytes,
respectively).
Invasion assay with differentiated HL-60 cells was
performed in Boyden chambers (2×105 cells/chamber)
equipped with 8-µm pore diameter polycarbonate filters coated with
20 µg of the commercial basement membrane matrix Matrigel (BD
Biosciences, Buccinasco, Italy), as previously described (11).
Cell survival assay
Cell culture viability was analyzed using the
tetrazolium compound MTS
[3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)
2-(4-sulphophenyl)-2H-tetrazolium, inner salt] from Promega
(Madison, WI, USA). Increasing numbers of differentiated HL-60
cells, untreated or pretreated with olaparib for 2 h, were seeded
in sextuplicate into flat-bottom 96-well plates and cultured at
37°C in a 5% CO2 humidified atmosphere. After 24 h, 20
µl of 2 mg/ml MTS solution was added to each well and cells were
incubated at 37°C for 2 h. Absorbance was read at 490 nm (reference
wavelength 655 nm) using a 3550-UV Microplate reader (Bio-Rad
Laboratories).
Analysis of NF-κB activity
NF-κB activity was determined utilizing a NF-κB p65
ELISA-based transcription factor assay kit (TransAM assay; Active
Motif Europe, Rixensart, Belgium). The assay was performed
according to the manufacturer's protocol using 20 µg of whole-cell
extracts. The kit contains a 96-well plate with immobilized
oligonucleotides comprising a NF-κB consensus site
(5′-GGGACTTTCC-3′) to which the p65 active form specifically binds.
The NF-κB detecting antibody recognizes an epitope on p65 that is
accessible only when this protein is activated and bound to its
target DNA. After incubation with a horseradish
peroxidase-conjugated secondary antibody, NF-κB activity was
quantified by a microplate reader 3550-UV (Bio-Rad Laboratories) at
450 nm with a reference wavelength of 655 nm.
Cell adhesion to fibronectin
Cell adhesion was tested by seeding monocytes
(1.5×105 cells/well) into flat-bottom 96-well plates
previously coated with fibronectin (5 µg/m; Sigma-Aldrich) and
blocked with 1% BSA/PBS. Selected wells were coated with 1% BSA/PBS
only to evaluate background cell adhesion. After 2 h of incubation
at 37°C, non-adherent cells were washed out and attached cells were
fixed in ethanol and stained with crystal violet. Adhesion
efficiency was determined by counting, in quadruplicate samples,
the number of adherent cells/microscopic field for a total of 12
high power fields (×200 magnification).
Statistical analyses
For multiple comparisons ANOVA followed by
Bonferroni's post-test was used. Statistical significance was
determined at α=0.05 level. Differences were considered
statistically significant when P<0.05.
Results
The PARPi olaparib inhibits migration
and ECM invasion triggered by PlGF in HL-60 cells differentiated to
monocyte/macrophage-like cells
As a model of myelomonocytic cells, we initially
used the HL-60 cell line induced to differentiate towards the
monocytic/macrophage lineage by treatment with PMA. As indicated by
the results of qRT-PCR analysis (Fig.
1A), differentiation of HL-60 cells was accompanied by the
induction of high VEGFR-1 transcript levels and the presence of
this receptor rendered cells responsive to PlGF, as previously
demonstrated (11). In order to
select the concentration of olaparib capable of abrogating
maximally stimulated PARP activity in this model, differentiated
HL-60 cells were pretreated with graded concentrations of the PARPi
(0.1, 1 and 2 µM, for 2 h), exposed to H2O2
for 15 min and then analyzed by western blotting to detect
PARylated proteins. In fact, the oxidant H2O2
is known to generate DNA strand breaks and to induce PARP-1
overactivation. Results indicated that at a concentration as low as
0.1 µM, olaparib markedly inhibited basal or
H2O2-induced PARP-1 activation and consequent
protein PARylation, which was totally abrogated by 1 and 2 µM
olaparib (Fig. 1B). Notably,
exposure of HL-60 differentiated cells to olaparib (2 µM) markedly
inhibited migration (Fig. 1C) and
ECM invasion triggered by PlGF to background values (Fig. 1D). At this concentration, olaparib
did not significantly affect the viability of differentiated HL-60
cells (data not shown).
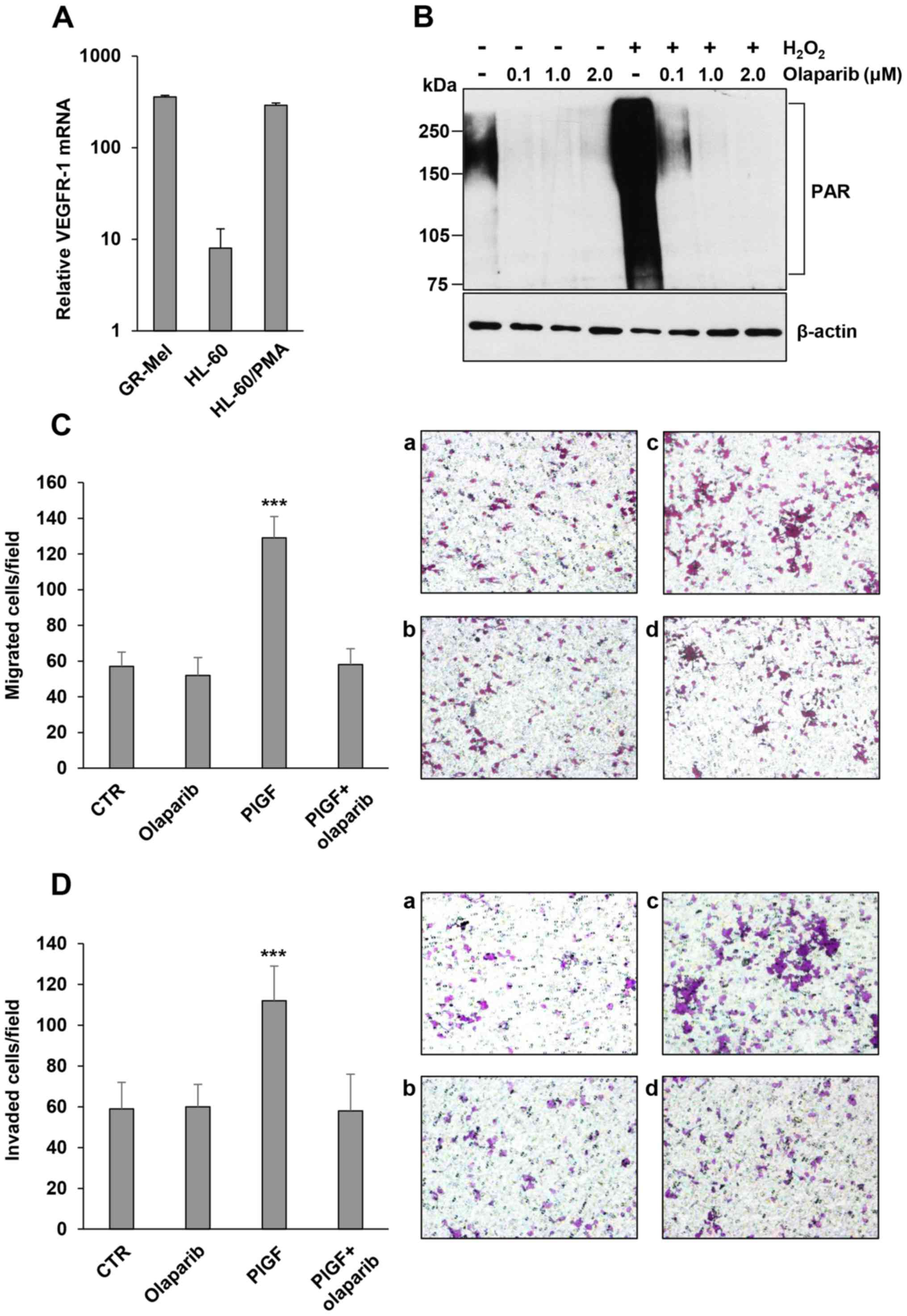 | Figure 1.Influence of the PARPi olaparib on
ECM invasion induced by PlGF in HL-60 cells differentiated in
vitro to the monocyte/macrophage lineage. (A) Analysis of
VEGFR-1 expression. Total RNA, extracted from undifferentiated or
PMA-differentiated HL-60 cells, was analyzed by qRT-PCR analysis.
The human melanoma GR-Mel cells were used as positive control and
the results are in relation to the VEGFR-1-negative M14 melanoma
cell line, to which the arbitrary value of 1 was assigned. (B)
Inhibitory effect of olaparib on protein PARylation in
differentiated HL-60 cells. The analysis of PAR-modified proteins
was performed by western blotting in cells exposed to 50 µM
H2O2 for 15 min after treatment with vehicle
or olaparib (at the indicated µM concentrations for 2 h). (C)
Influence of olaparib on differentiated HL-60 cell migration
induced by PlGF. Migration of differentiated HL-60 cells, vehicle
or olaparib (2 µM for 2 h) pre-treated, in response to serum-free
medium (CTR and olaparib) or 50 ng/ml PlGF was evaluated in Boyden
chambers (4-h incubation) equipped with gelatin-coated filters.
Histogram represents the mean values of the number of migrated
cells/field ± SD of three independent determinations. Results of
statistical analysis using one-way ANOVA followed by Bonferroni's
post-test were as follows: ***P<0.001, PlGF vs. CTR, PlGF vs.
olaparib, PlGF vs. PlGF + olaparib; differences between CTR,
olaparib and PlGF + olaparib were not significant. Representative
images of differentiated HL-60 cell migration are shown
(magnification, ×100): a, untreated non-stimulated control cells
(CTR); b, olaparib-treated non-stimulated cells; c, PlGF-stimulated
cells; d, PlGF + olaparib. (D) Influence of olaparib on ECM
invasion induced by PlGF. Invasion of differentiated HL-60 cells,
vehicle or olaparib (2 µM for 2 h) pre-treated, in response to
serum-free medium (CTR and olaparib) or 50 ng/ml PlGF was evaluated
in Boyden chambers (4-h incubation) equipped with Matrigel-coated
filters. Histogram represents the mean values of the number of
invaded cells/field ± SD of three independent determinations.
Results of statistical analysis using one-way ANOVA followed by
Bonferroni's post-test were as follows: ***P<0.001, PlGF vs.
CTR, PlGF vs. olaparib, PlGF vs. PlGF + olaparib; differences
between CTR, olaparib and PlGF + olaparib were not significant.
Representative images of differentiated HL-60 cell invasion are
shown (magnification, ×100): a, untreated non-stimulated control
cells (CTR); b, olaparib-treated non-stimulated cells; c,
PlGF-stimulated cells; d, PlGF + olaparib. |
Olaparib inhibits PlGF-induced
response similarly to the anti-VEGFR-1 D16F7 mAb
In order to evaluate whether olaparib treatment
might synergize with VEGFR-1 blockade, cells were treated with
olaparib, D16F7 or both agents and then exposed to PlGF. Treatment
with the anti-VEGFR-1 D16F7 mAb, which is known to hamper
PlGF-induced VEGFR-1 activation (11), markedly affected migration of
PMA-differentiated HL-60 cells with an IC50 value of
0.15±0.06 µg/ml (Fig. 2A). Olaparib
inhibited cell migration in a dose-dependent manner (Fig. 2A) with an IC50 of
0.12±0.01 µM. Cells were also treated with olaparib at its
IC50 value in combination with D16F7 at concentrations
encompassing values above and below the mAb IC50 (0.05,
0.1 and 0.2 µg/ml). Results indicated that the PARPi and D16F7 did
not exert synergistic effects (Fig.
2B), suggesting that olaparib might interfere with the same
pathway affected by the anti-VEGFR-1 mAb. Similar results were
obtained by testing ECM invasion induced by PlGF (Fig. 2C). D16F7 was more effective in
inhibiting cell invasiveness as compared to chemotaxis (mAb
IC50: 0.05±0.01 µg/ml). When olaparib and D16F7 were
combined at their IC50 values, the inhibitory effect on
ECM invasion was similar to that obtained with the single agents
(Fig. 2C).
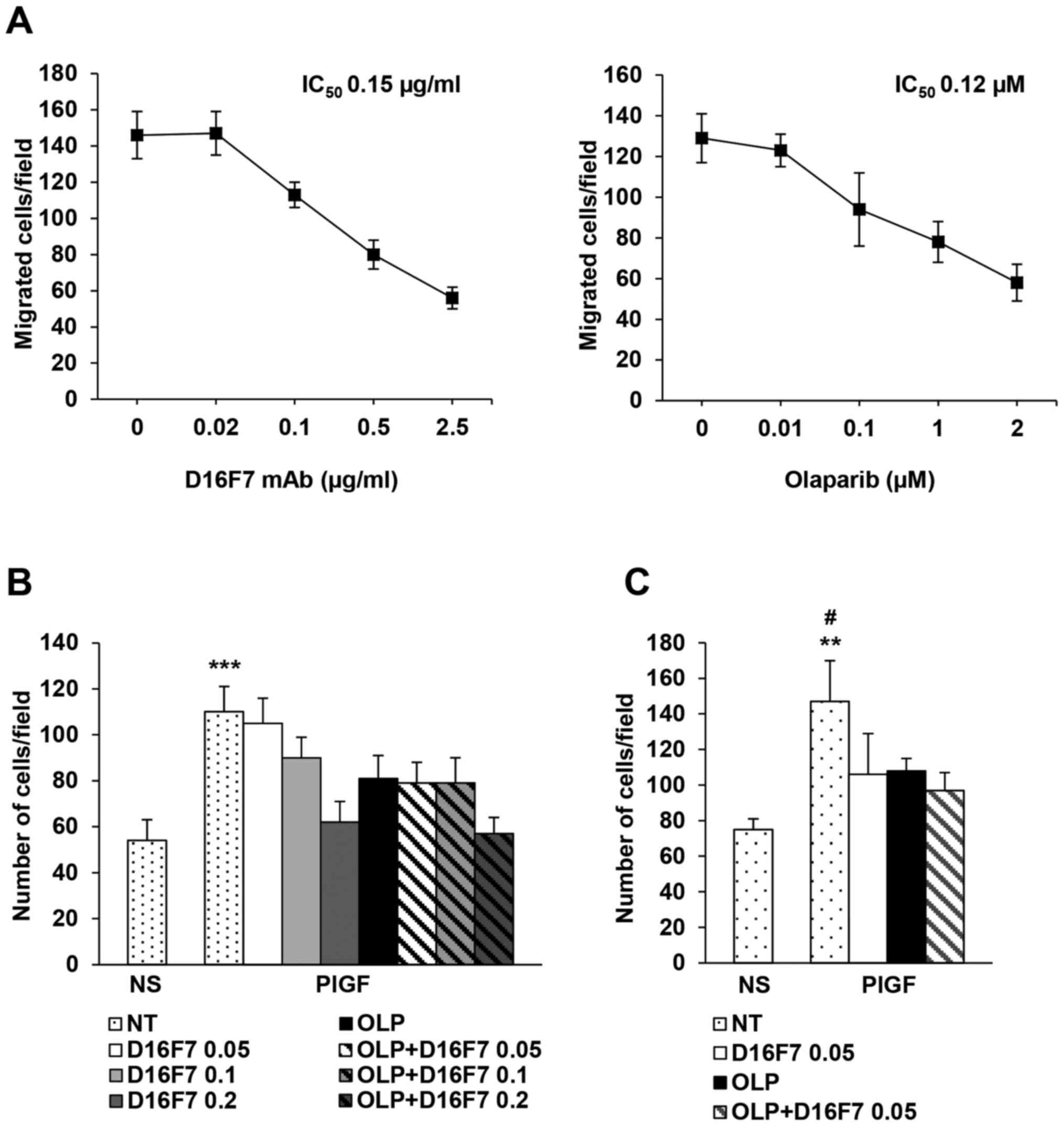 | Figure 2.Analysis of the influence of
olaparib, as a single agent or in combination with the anti-VEGFR-1
D16F7 mAb, on PlGF-induced chemotaxis and ECM invasion of
differentiated HL-60 cells. (A) Dose-dependent inhibitory effect of
D16F7 mAb or olaparib on PlGF-induced chemotaxis. Migration of
differentiated HL-60 cells, vehicle (0 in the x-axis) and D16F7 mAb
or olaparib pre-treated (at the indicated concentrations), for 30
min or 2 h, respectively, in response to PlGF (50 ng/ml) was
evaluated in Boyden chambers equipped with gelatin-coated filters.
(B) Influence of olaparib and D16F7 mAb combined treatment on
differentiated HL-60 cell migration in response to PlGF. Migration
assay was performed using vehicle (not treated, NT) or D16F7
pre-treated cells in the absence or in the presence of a fixed
concentration of olaparib (OLP, 0.1 µM). Results of statistical
analysis using one-way ANOVA followed by Bonferroni's post-test
were as follows: ***P<0.001, PlGF vs. all groups (except PlGF +
0.05 µg/ml D16F7). Differences between PlGF + 0.1 µg/ml D16F7 and
PlGF + 0.1 µg/ml D16F7 + olaparib and differences between PlGF +
0.2 µg/ml D16F7 and PlGF + 0.2 µg/ml D16F7 + olaparib were not
significant. NS, non-stimulated cells. (C) Influence of olaparib
and D16F7 mAb combined treatment on ECM invasion by differentiated
HL-60 cells in response to PlGF. Invasion assay was performed using
vehicle (not treated, NT) or D16F7 pre-treated cells (0.05 µg/ml,
i.e., mAb IC50 on ECM invasion) in the absence or in the
presence of a fixed concentration of olaparib (OLP, 0.1 µM).
Results of statistical analysis using one-way ANOVA followed by
Bonferroni's post-test were as follows: **P<0.01, PlGF vs.
non-stimulated cells (NS); #P<0.05, PLGF vs. all the
other groups. Differences between PlGF + 0.05 µg/ml D16F7, PlGF +
olaparib and PlGF + 0.05 µg/ml D16F7 + olaparib were not
significant. Histograms represent the mean values of the number of
migrated cells/field ± SD of three independent determinations. |
In order to evaluate whether the inhibitory effect
of olaparib on PlGF-induced ECM invasion was due to modulation of
epithelial to mesenchymal transition (EMT) markers, we tested the
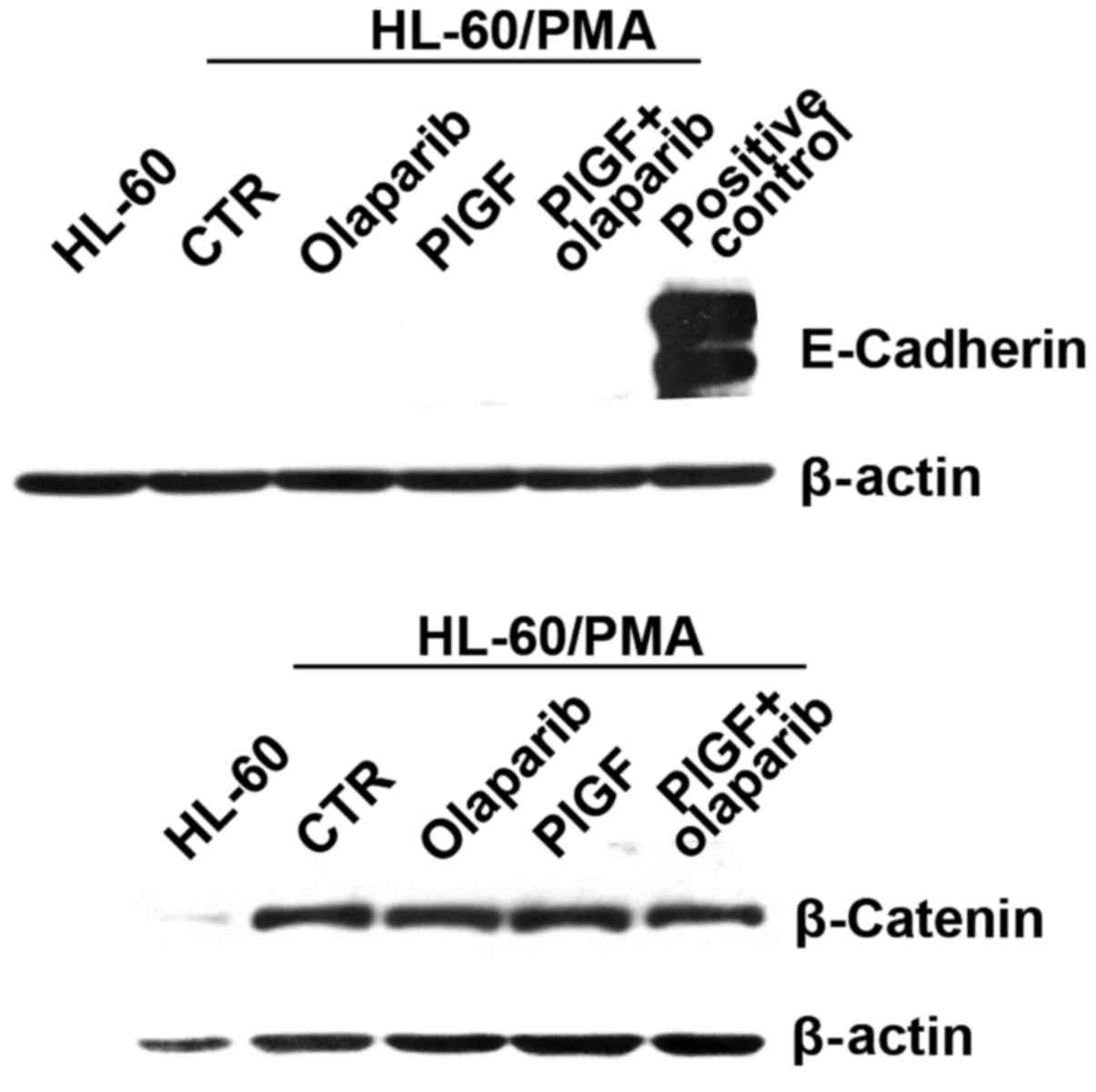
expression of E-cadherin and β-catenin in differentiated HL-60
cells exposed to PlGF and olaparib by western blot analysis. The
results showed that cells did not express E-cadherin, while they
expressed high levels of β-catenin, which is compatible with a
mesenchymal phenotype. However, no modulation of protein expression
was observed in response to the different treatments (Fig. 3).
Olaparib inhibits PARP activity and
PlGF-induced chemotaxis of freshly isolated human monocytes
Primary human monocytes were isolated from buffy
coats obtained from 6 healthy donors and analyzed for the presence
of the VEGFR-1 transcript. Results of qRT-PCR indicated that
VEGFR-1 was detected in all monocyte samples tested even though at
different levels (Fig. 4A).
Analysis by western blotting showed that treatment with 2 µM
olaparib for 2 h markedly inhibited PARylation of cellular proteins
in a monocyte preparation that presented high basal levels of PARP
activity (Fig. 4B). The same
olaparib concentration abrogated migration in response to PlGF of
all monocyte preparations tested with similar efficacy compared to
the anti-VEGFR-1 D16F7 mAb (Fig. 4C and
D).
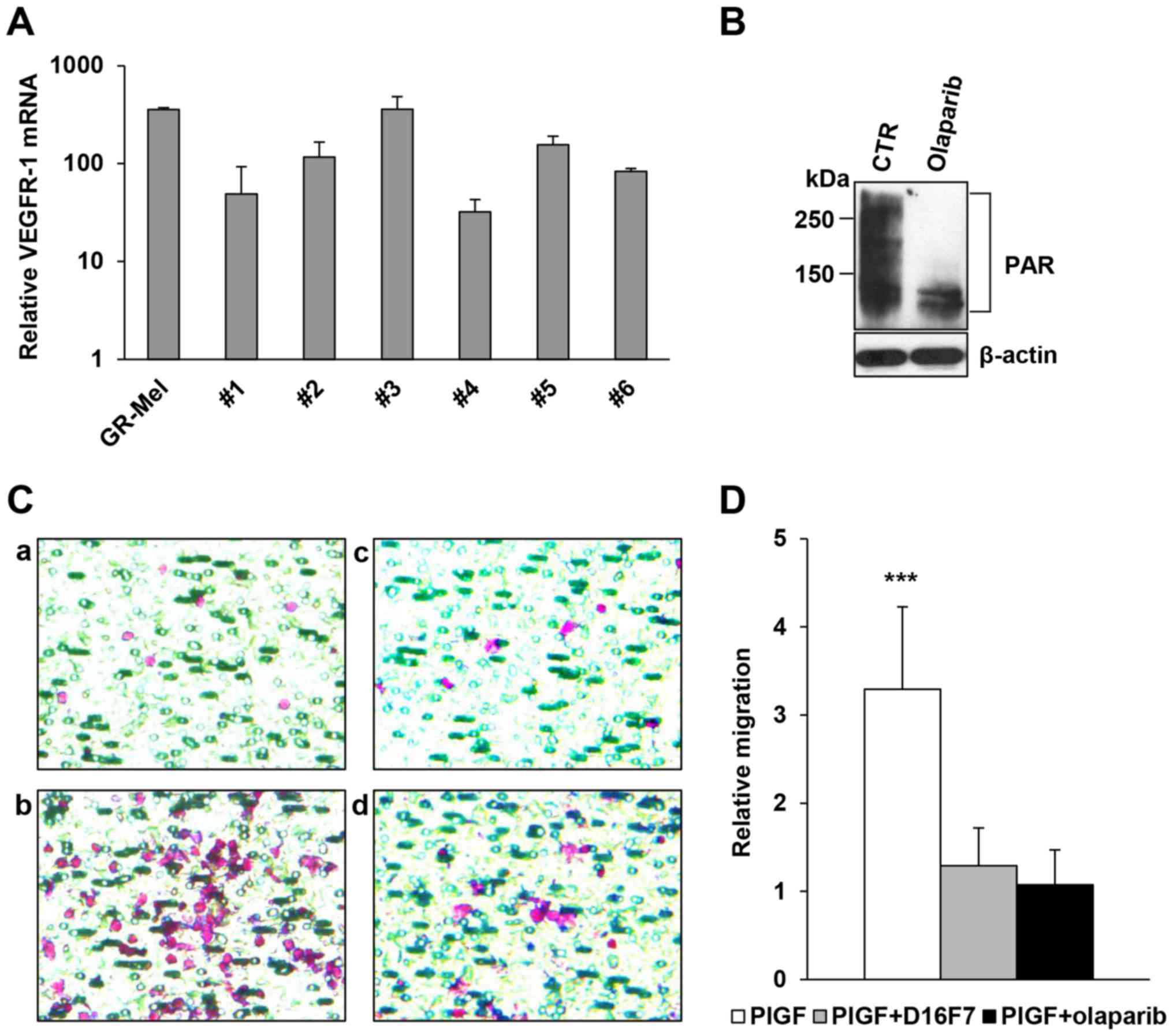 | Figure 4.Inhibitory activity of olaparib and
the anti-VEGFR-1 D16F7 mAb on the chemotactic response of human
monocytes to PlGF. (A) Analysis of VEGFR-1 expression. Total RNA
was extracted from freshly isolated human monocytes obtained from 6
healthy donors and analyzed by qRT-PCR. The human melanoma GR-Mel
cells were used as positive control and results were in relation to
the VEGFR-1-negative M14 melanoma cell line, to which the arbitrary
value of 1 was assigned. (B) Inhibitory effect of olaparib on
protein PARylation in human monocytes. The analysis of PAR-modified
proteins was performed by western blotting in vehicle or olaparib
pre-treated (2 µM for 2 h) monocytes. (C) Influence of olaparib and
D16F7 mAb, as single agents or in combination, on PlGF-induced
monocyte chemotaxis. Monocytes were exposed to 2 µM olaparib for 2
h and/or to 5 µg/ml D16F7 for 30 min and cell migration was
analyzed in Corning HTS Transwell®-96 permeable support
plates equipped with gelatin-coated filters (1.5×105
cells/well, 2-h incubation). Representative images of monocyte
migration are shown (magnification, ×400); a, untreated
non-stimulated control; b, PlGF stimulated cells; c, PlGF + D16F7
mAb; d, PlGF + olaparib. (D) Results of monocyte chemotaxis
analysis are expressed as the ratio between the number of migrated
monocytes stimulated with PlGF and that of non-stimulated,
untreated control cells. Data are the mean values obtained from 6
different monocyte preparations. Results of statistical analysis
using one-way ANOVA followed by Bonferroni's post-test were as
follows: ***P<0.001, PlGF vs. PlGF + D16F7 and PlGF vs. PlGF +
olaparib; differences between PlGF + D16F7 and PlGF + olaparib were
not significant. |
Influence of olaparib on PlGF-induced
signaling pathways in monocytes
It has been demonstrated that PlGF acts as survival
factor for tumor cells by upregulating nuclear factor-κB (NF-κB)
activity (24). Therefore, we
investigated whether PlGF-mediated stimulation of monocytes might
result in NF-κB induction and whether olaparib modulates NF-κB
activation. Results indicated that PlGF caused a significant
increase in NF-κB activity that was not affected by pre-treatment
with olaparib (Fig. 5A).
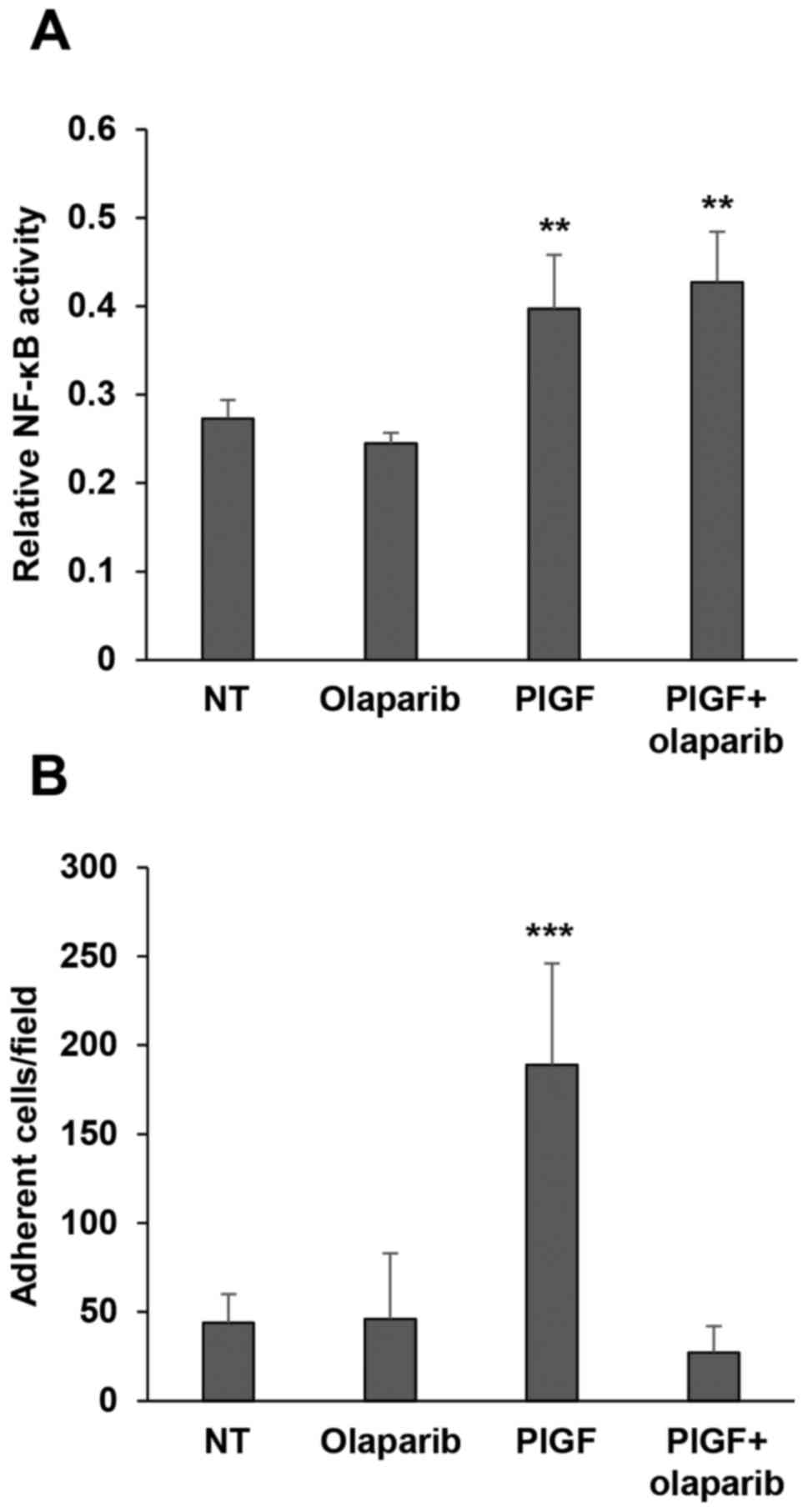 | Figure 5.Olaparib inhibits PlGF-induced
monocyte adhesion to fibronectin while it does not affect NF-κB
activation by the angiogenic factor. (A) Influence of olaparib on
NF-κB activation in response to PlGF. Monocytes, either vehicle
(not treated, NT) or olaparib pre-treated (2 µM for 2 h), were
incubated in the presence of 50 ng/ml PlGF for 20 h. Whole-cell
extracts were then analyzed for NF-κB activity by a quantitative
Trans-AM kit, as described in Materials and methods. Data are
representative of one out of three independent experiments with
similar results and are the mean values from three independent
determinations ± SD. Results of statistical analysis using one-way
ANOVA followed by Bonferroni's post-test were as follows:
**P<0.01, PlGF vs. NT, PlGF vs. olaparib; **P<0.01, PlGF +
olaparib vs. NT and PlGF + olaparib vs. olaparib; differences
between PlGF and PlGF + olaparib were not significant. (B)
Influence of olaparib on monocyte adhesion to fibronectin. The
ability of control or PlGF-stimulated monocytes (100 ng/ml) to
adhere to fibronectin was evaluated, as described in Materials and
methods, in vehicle (not treated, NT) or olaparib (2 µM for 2 h)
pre-treated cells. Histogram represents the mean values of the
number of adherent cells/field ± SD. Results of statistical
analysis using one-way ANOVA followed by Bonferroni's post-test
were as follows: ***P<0.001, PlGF vs. NT, PlGF vs. olaparib and
PlGF vs. PlGF + olaparib; differences between NT, olaparib and PlGF
+ olaparib were not significant. |
In order to shed light on the mechanism of the
olaparib-mediated inhibition of monocyte chemotaxis and
invasiveness stimulated by PlGF and based on PARPi ability to
modulate integrin expression in leukocytes (17), we evaluated the influence of
pre-treatment with olaparib on monocyte adhesion to fibronectin, a
process that requires integrin activation (25). Analysis of cell adhesion to
fibronectin revealed that PlGF strongly induced monocytes to adhere
to this ECM component and that olaparib hampered this effect
(Fig. 5B). These data strongly
suggested that inhibition of integrin activation by olaparib may
contribute to the observed effects on the motility of
PlGF-stimulated monocytes.
Discussion
In the present study, we demonstrated for the first
time that the PARPi olaparib hampers PlGF-driven stimulation of
myelomonocytic cells. This effect is due, at least in part, to
inhibition of integrin activation that seems to be required for
monocytic cell ability to migrate and invade the ECM in response to
VEGFR-1 activation by PlGF. The inhibitory activity of olaparib on
monocytic cells is indeed comparable to that exerted by the
recently described anti-VEGFR-1 D16F7 mAb (11).
Besides its role as an angiogenic factor in
tumor-associated vascularization, PlGF has been shown to influence
the aggressiveness of tumor cells from different tissue origin.
Actually, PlGF is upregulated in many human cancer types, where its
expression directly correlates with tumor stage, metastasis or
recurrence, and inversely correlates with survival (24,26).
In addition, PlGF plasma levels are frequently high in patients
treated with anti-VEGF agents (24,26),
suggesting an involvement in innate and/or acquired resistance
mechanisms to these therapies (27–29).
Indeed, PlGF can directly affect tumor cells increasing or inducing
migration and ECM invasion as demonstrated in colorectal,
pancreatic or breast carcinomas and melanoma (11,30–32).
Furthermore, the responsiveness to PlGF of cancer cell lines
requires the expression of VEGFR-1 (33).
PlGF secreted by tumor cells also contributes to the
recruitment of monocytes/macrophages into the tumor mass (i.e.,
TAMs) and is involved in TAM polarization to a
pro-angiogenic/immune-suppressive M2-like phenotype (34). M2-like TAMs secrete a number of
growth factors and proteases that promote angiogenesis and ECM
remodeling, and suppress antitumor immune responses thereby
stimulating tumor growth, invasion and metastasis (35).
In this context, the inhibitory effects of olaparib
on monocytic cell activation by PlGF suggests a role for this PARPi
in pathological states where PlGF is overexpressed. Due to their
ability of inhibiting DNA repair, PARPi were initially developed as
radio- and chemosensitizers and thereafter approved as a novel
class of anticancer drugs to be used in monotherapy for homologous
recombination defective tumors. Notably, these compounds have been
recently proposed as potentially effective therapeutic agents for a
variety of non-oncological disorders in which DNA-damage-dependent
and -independent mechanisms of PARP activation may play a
pathophysiological role (14).
Therefore, olaparib treatment might be of benefit also for other
non-cancerous diseases that are associated with monocyte activation
in response to PlGF through inhibition of signal transduction
mechanisms and independently on its effects on DNA repair.
Regarding the mechanisms by which olaparib modulates
the monocytic cell response to PlGF, we found that the PARPi, at
concentrations below the plasma peak values detected in treated
cancer patients (36–38) and that totally abrogate cellular
PARP activity, hampered PlGF-induced monocyte adhesion to
fibronectin, while it did not affect NF-κB activation in response
to this angiogenic factor.
The transcription factor NF-κB plays a key role in
the regulation of cell proliferation, inflammation, angiogenesis
and suppression of apoptosis, and, when constitutively activated,
may be critical in the development of drug resistance in tumor
cells (reviewed in ref. 39). PlGF
significantly increases tumor cell resistance to chemotherapy and
this effect is associated with activation of NF-κB signaling
pathways (24). Our results showed
that PlGF induces NF-κB activation also in human monocytes.
However, monocyte treatment with olaparib did not prevent
PlGF-induced NF-κB upregulation, suggesting that the inhibitory
effect of olaparib on chemotaxis might involve alternative
mechanisms. Nevertheless, it cannot be excluded that olaparib might
affect NF-κB translocation to the nucleus potentially promoted by
PlGF and further studies are in progress to clarify this issue.
On the other hand, we found that PlGF markedly
stimulated monocyte adhesion to the ECM component fibronectin,
which suggests that integrin activation may be crucial for the
promotion of growth factor-induced cell motility. Adhesion of
monocytes to ECM (through fibronectin) or to activated endothelial
cells (through the adhesion molecule VCAM-1) is regulated by
integrin β1 conformational changes (40). In fact, integrin must switch from an
inactive (closed/non-adherent) to an active (open/adherent) form.
This inside-out change exposes the integrin binding site and is
regulated by stimulation of G protein coupled receptors via
intracellular signals (40).
Notably, PI3K has a crucial role in the regulation of monocyte
integrin activation (41) and
migration (42). Indeed,
PlGF-mediated stimulation of VEGFR-1 in primary monocytes results
in the phosphorylation of PI3K, ERK1/2, p38 and Akt (Ser473)
kinases, PI3K being a central regulator of PlGF-induced human
monocytes chemotaxis (42). To this
regard, it has been reported that several PARPi down-modulate
monocyte adhesion to microvascular endothelial cells by preventing
the conformational activation of integrin β1 (16), which is required for inflammatory
cell mobility. Consistently, we observed that olaparib
pre-treatment abrogated the stimulating effect of PlGF on monocytic
cell adhesion to fibronectin. Although results on olaparib ability
to inhibit chemotaxis and ECM invasion activity were obtained using
several human monocyte preparations, only one myelomonocytic cell
line was tested in the present study. A greater number of cell
lines would be required to strengthen our conclusion.
It could be, therefore, hypothesized that activation
of VEGFR-1 by PlGF in monocytic cells might trigger signal
transduction pathways that result in two different effects:
proliferation, suppression of apoptosis and survival, which depend
on NF-κB activity; cell adhesion and migration, which depend on
integrin activation. Our results using the PARPi olaparib are in
agreement with this hypothesis. Actually, the data herein described
suggest that olaparib interferes with a specific signaling pathway
triggered by PlGF through VEGFR-1, which involves specific integrin
activation, and that inhibition of PlGF-induced monocyte activation
may contribute to PARPi antitumor activity.
Acknowledgements
Not applicable.
Glossary
Abbreviations
Abbreviations:
|
PlGF
|
placental growth factor
|
|
VEGFR
|
vascular endothelial growth factor
receptor
|
|
PARP
|
poly(ADP-ribose) polymerase
|
|
PARPi
|
PARP inhibitor
|
References
|
1
|
Clauss M, Weich H, Breier G, Knies U,
Röckl W, Waltenberger J and Risau W: The vascular endothelial
growth factor receptor Flt-1 mediates biological activities.
Implications for a functional role of placenta growth factor in
monocyte activation and chemotaxis. J Biol Chem. 271:17629–17634.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kim KJ, Cho CS and Kim WU: Role of
placenta growth factor in cancer and inflammation. Exp Mol Med.
44:10–19. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ding Y, Huang Y, Song N, Gao X, Yuan S,
Wang X, Cai H, Fu Y and Luo Y: NFAT1 mediates placental growth
factor-induced myelomonocytic cell recruitment via the induction of
TNF-alpha. J Immunol. 184:2593–2601. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kerber M, Reiss Y, Wickersheim A, Jugold
M, Kiessling F, Heil M, Tchaikovski V, Waltenberger J, Shibuya M,
Plate KH and Machein MR: Flt-1 signaling in macrophages promotes
glioma growth in vivo. Cancer Res. 68:7342–7351. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Laurent J, Hull EF, Touvrey C, Kuonen F,
Lan Q, Lorusso G, Doucey MA, Ciarloni L, Imaizumi N, Alghisi GC, et
al: Proangiogenic factor PlGF programs CD11b+
myelomonocytes in breast cancer during differentiation of their
hematopoietic progenitors. Cancer Res. 71:3781–3791. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Czepluch FS, Olieslagers S, van Hulten R,
Vöö SA and Waltenberger J: VEGF-A-induced chemotaxis of CD16+
monocytes is decreased secondary to lower VEGFR-1 expression.
Atherosclerosis. 215:331–338. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lin YL, Liang YC and Chiang BL: Placental
growth factor down-regulates type 1 T helper immune response by
modulating the function of dendritic cells. J Leukoc Biol.
82:1473–1480. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou X and Qi Y: Larynx carcinoma
regulates tumor-associated macrophages through PLGF signaling. Sci
Rep. 5:100712015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Selvaraj SK, Giri RK, Perelman N, Johnson
C, Malik P and Kalra VK: Mechanism of monocyte activation and
expression of proinflammatory cytochemokines by placenta growth
factor. Blood. 102:1515–1524. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pagani E, Ruffini F, Cappellini Antonini
GC, Scoppola A, Fortes C, Marchetti P, Graziani G, D'Atri S and
Lacal PM: Placenta growth factor and neuropilin-1 collaborate in
promoting melanoma aggressiveness. Int J Oncol. 48:1581–1589. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Graziani G, Ruffini F, Tentori L, Scimeca
M, Dorio AS, Atzori MG, Failla CM, Morea V, Bonanno E, D'Atri S and
Lacal PM: Antitumor activity of a novel anti-vascular endothelial
growth factor receptor-1 monoclonal antibody that does not
interfere with ligand binding. Oncotarget. 7:72868–72885. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Atzori MG, Tentori L, Ruffini F, Ceci C,
Lisi L, Bonanno E, Scimeca M, Eskilsson E, Daubon T, Miletic H, et
al: The anti-vascular endothelial growth factor receptor-1
monoclonal antibody D16F7 inhibits invasiveness of human
glioblastoma and glioblastoma stem cells. J Exp Clin Cancer Res.
36:1062017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Atzori MG, Tentori L, Ruffini F, Ceci C,
Bonanno E, Scimeca M, Lacal PM and Graziani G: The anti-vascular
endothelial growth factor receptor-1 monoclonal antibody D16F7
inhibits glioma growth and angiogenesis in vivo. J Pharmacol Exp
Ther. 364:77–86. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Berger NA, Besson VC, Boulares AH, Bürkle
A, Chiarugi A, Clark RS, Curtin NJ, Cuzzocrea S, Dawson TM, Dawson
VL, et al: Opportunities for the repurposing of PARP inhibitors for
the therapy of non-oncological diseases. Br J Pharmacol.
175:192–222. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Martin-Hernandez K, Rodriguez-Vargas JM,
Schreiber V and Dantzer F: Expanding functions of ADP-ribosylation
in the maintenance of genome integrity. Semin Cell Dev Biol.
63:92–101. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bai P and Virág L: Role of
poly(ADP-ribose) polymerases in the regulation of inflammatory
processes. FEBS Lett. 586:3771–3777. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rom S, Zuluaga-Ramirez V, Reichenbach NL,
Dykstra H, Gajghate S, Pacher P and Persidsky Y: PARP inhibition in
leukocytes diminishes inflammation via effects on
integrins/cytoskeleton and protects the blood-brain barrier. J
Neuroinflammation. 13:2542016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shah GM, Robu M, Purohit NK, Rajawat J,
Tentori L and Graziani G: PARP inhibitors in cancer therapy: Magic
bullets but moving targets. Front Oncol. 3:2792013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pujade-Lauraine E, Ledermann JA, Selle F,
Gebski V, Penson RT, Oza AM, Korach J, Huzarski T, Poveda A,
Pignata S, et al: Olaparib tablets as maintenance therapy in
patients with platinum-sensitive, relapsed ovarian cancer and a
BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind,
randomised, placebo-controlled, phase 3 trial. Lancet Oncol.
18:1274–1284. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Robson M, Im SA, Senkus E, Xu B, Domchek
SM, Masuda N, Delaloge S, Li W, Tung N, Armstrong A, et al:
Olaparib for metastatic breast cancer in patients with a Germline
BRCA mutation. N Engl J Med. 377:523–533. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Faraoni I, Compagnone M, Lavorgna S,
Angelini DF, Cencioni MT, Piras E, Panetta P, Ottone T, Dolci S,
Venditti A, et al: BRCA1, PARP1 and γH2AX in acute myeloid
leukemia: Role as biomarkers of response to the PARP inhibitor
olaparib. Biochim Biophys Acta. 1852:462–472. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ruffini F, Failla CM, Orecchia A, Bani MR,
Dorio AS, Fortes C, Zambruno G, Graziani G, Giavazzi R, D'Atri S,
et al: Expression of the soluble vascular endothelial growth factor
receptor-1 in cutaneous melanoma: Role in tumour progression. Br J
Dermatol. 164:1061–1070. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lacal PM, Morea V, Ruffini F, Orecchia A,
Dorio AS, Failla CM, Soro S, Tentori L, Zambruno G, Graziani G, et
al: Inhibition of endothelial cell migration and angiogenesis by a
vascular endothelial growth factor receptor-1 derived peptide. Eur
J Cancer. 44:1914–1921. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Levati L, Ruffini F, Muzi A, Umezawa K,
Graziani G, D'Atri S and Lacal PM: Placenta growth factor induces
melanoma resistance to temozolomide through a mechanism that
involves the activation of the transcription factor NF-κB. Int J
Oncol. 38:241–247. 2011.PubMed/NCBI
|
|
25
|
White ES, Livant DL, Markwart S and
Arenberg DA: Monocyte-fibronectin interactions, via alpha(5)beta(1)
integrin, induce expression of CXC chemokine-dependent angiogenic
activity. J Immunol. 167:5362–5366. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dewerchin M and Carmeliet P: Placental
growth factor in cancer. Expert Opin Ther Targets. 18:1339–1354.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jain RK, Duda DG, Willett CG, Sahani DV,
Zhu AX, Loeffler JS, Batchelor TT and Sorensen AG: Biomarkers of
response and resistance to antiangiogenic therapy. Nat Rev Clin
Oncol. 6:327–338. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bagley RG, Ren Y, Weber W, Yao M,
Kurtzberg L, Pinckney J, Bangari D, Nguyen C, Brondyk W, Kaplan J
and Teicher BA: Placental growth factor upregulation is a host
response to antiangiogenic therapy. Clin Cancer Res. 17:976–988.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Carmeliet P and Jain RK: Molecular
mechanisms and clinical applications of angiogenesis. Nature.
473:298–307. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fischer C, Jonckx B, Mazzone M, Zacchigna
S, Loges S, Pattarini L, Chorianopoulos E, Liesenborghs L, Koch M,
De Mol M, et al: Anti-PlGF inhibits growth of
VEGF(R)-inhibitor-resistant tumors without affecting healthy
vessels. Cell. 131:463–475. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Coenegrachts L, Maes C, Torrekens S, Van
Looveren R, Mazzone M, Guise TA, Bouillon R, Stassen JM, Carmeliet
P and Carmeliet G: Anti-placental growth factor reduces bone
metastasis by blocking tumor cell engraftment and osteoclast
differentiation. Cancer Res. 70:6537–6547. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wei SC, Tsao PN, Weng MT, Cao Z and Wong
JM: Flt-1 in colorectal cancer cells is required for the tumor
invasive effect of placental growth factor through a p38-MMP9
pathway. J Biomed Sci. 20:392013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yao J, Wu X, Zhuang G, Kasman IM, Vogt T,
Phan V, Shibuya M, Ferrara N and Bais C: Expression of a functional
VEGFR-1 in tumor cells is a major determinant of anti-PlGF
antibodies efficacy. Proc Natl Acad Sci USA. 108:11590–11595. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rolny C, Mazzone M, Tugues S, Laoui D,
Johansson I, Coulon C, Squadrito ML, Segura I, Li X, Knevels E, et
al: HRG inhibits tumor growth and metastasis by inducing macrophage
polarization and vessel normalization through downregulation of
PlGF. Cancer Cell. 19:31–44. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Galdiero MR, Garlanda C, Jaillon S, Marone
G and Mantovani A: Tumor associated macrophages and neutrophils in
tumor progression. J Cell Physiol. 228:1404–1412. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fong PC, Boss DS, Yap TA, Tutt A, Wu P,
Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, et
al: Inhibition of poly(ADP-ribose) polymerase in tumors from
BRCA mutation carriers. N Engl J Med. 361:123–134. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Roth J, Peer CJ, Mannargudi B, Swaisland
H, Lee JM, Kohn EC and Figg WD: A sensitive and robust ultra HPLC
assay with tandem mass spectrometric detection for the quantitation
of the PARP inhibitor olaparib (AZD2281) in Human plasma for
pharmacokinetic application. Chromatography. 1:82–95. 2014.
View Article : Google Scholar
|
|
38
|
Plummer R, Swaisland H, Leunen K, van
Herpen CM, Jerusalem G, De Grève J, Lolkema MP, Soetekouw P,
Mau-Sørensen M, Nielsen D, et al: Olaparib tablet formulation:
Effect of food on the pharmacokinetics after oral dosing in
patients with advanced solid tumours. Cancer Chemother Pharmacol.
76:723–729. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xia Y, Shen S and Verma IM: NF-κB, an
active player in human cancers. Cancer Immunol Res. 2:823–830.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Askari JA, Buckley PA, Mould AP and
Humphries MJ: Linking integrin conformation to function. J Cell
Sci. 122:165–170. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ferreira AM, Isaacs H, Hayflick JS, Rogers
KA and Sandig M: The p110delta isoform of PI3K differentially
regulates beta1 and beta2 integrin-mediated monocyte adhesion and
spreading and modulates diapedesis. Microcirculation. 13:439–456.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tchaikovski V, Fellbrich G and
Waltenberger J: The molecular basis of VEGFR-1 signal transduction
pathways in primary human monocytes. Arterioscler Thromb Vasc Biol.
28:322–328. 2008. View Article : Google Scholar : PubMed/NCBI
|