Introduction
Prostate cancer (PCa) is the most frequent male
malignancy and a leading cause of cancer-related death in many
Western countries (1). Disease
progression in the majority of patients with androgen-dependent
prostate cancer can generally be controlled; however, patients with
androgen-independent disease and a metastatic phenotype
consistently present with a poor prognosis (2).
The molecular mechanisms that underlie the
metastatic process and secondary foci specificity are poorly
understood. Mainly studies have focused on proteins that function
in normal epithelial cells to maintain intercellular adhesion
(3). For example, E-cadherin plays
an important role in cell-cell adhesion, and a decrease in
E-cadherin is associated with cancer progression (4,5). A
recent study demonstrated that AQP3, a member of the aquaporins,
colocalizes with E-cadherin in the early stages of cell-cell
contact formation (6). Aquaporins
(AQP) belong to a family of water-transporting proteins, and allow
water, glycerol and small solutes through the cell membrane
(7). AQP0, 1, 2, 4, 5, 6 and 8
allow the permeation of water. In addition, AQP3, 7, 9 and 10
transport glycerol and small solutes (8).
Our previous study demonstrated that AQP3 protein is
intensely expressed on the cell membranes in normal human prostate
cells and tissues, whereas in prostate cancer AQP3 is localized
mainly in the cytoplasm (9).
Moreover, a recent study showed that after disrupting the exocyst
complex, AQP3 distribution to E-cadherin-rich cell-cell contacts
was impaired (6). The exocyst, a
conserved hetero-octameric protein complex, is localized to lateral
membranes and developing apical domains of epithelial cells. In
epithelial cells, the exocyst is redistributed from the plasma to
areas of cadherin adhesion, and serves to transport post-Golgi
transport vesicles to adhesion sites (10). Exocyst distribution and activity are
regulated by small GTPases (11).
Two of these, Arf6 and Ral, are important promoters of tumor growth
and progression (12–14). Among the Ral subfamily, RalA and
RalB are highly related GTPases that communicate with parts of the
exocyst (Sec5 and Exo84) in vitro (13).
In the present study we explored the function of RAS
like proto-oncogene A (RalA) in regulating the localization of AQP3
in androgen-independent prostate cancer. We found that depletion of
RalA led to the redistribution of AQP3 into the plasma membrane.
The tumor-promoting function of RalA in prostate cancer is possibly
mediated by the cAMP signaling pathway in prostate cancer.
Materials and methods
Cell culture and reagents
Human prostate cancer cell line PC-3 was obtained
from The American Type Culture Collection (ATCC; Manassas, VA, USA,
USA). In this study, we used cells in fewer than 6 months after
resuscitation. PC-3 was maintained as monolayer cultures in Gibco
F-12 medium (Thermo Fisher Scientific, Inc., Waltham, MA, USA) with
10% fetal bovine serum (FBS), and incubated at 37°C with 5%
CO2 in a humidified atmosphere.
Lentivirus infection
Lentivirus particles carrying shRNA
(5′-GATCCGACAGGTTTCTGTAGAAGATTCAAGAGATCTTCTACAGAAACCTGTCTTTTT-3′)
against RalA or scrambled shRNA
(5′-GATCCTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAATTTTT-3′)
(Santa Cruz Biotechnology, Santa Cruz, CA, USA) which has no
interference with other cellular mRNA in PC-3 cells were
constructed. According to the manufacturer's instructions (Santa
Cruz Biotechnology) cells were treated. All functional assays were
performed after cell infection.
For DNA transfection, PC-3 cells were grown to
75–80% confluence and then according to the manufacturer's
instructions were transfected with a total of 2 mg of DNA (Life
Technologies, Inc., Gaithersburg, MD, USA). E-cadherin expression
vector and the neomycin resistance selection marker (Invitrogen;
Thermo Fisher Scientific, Inc.) at a ratio of 20:1 were performed.
Selective transfectants were tested for transgene expression.
Western blot analysis
For western blot analysis, in brief, protein was
extracted using RIPA lysis buffer and protease inhibitor (Thermo
Fisher Scientific, Inc.). Thirty micrograms of protein from each
sample were separated on 10% SDS-polyacrylamide gel. GAPDH served
as a loading control. Antibodies used included: AQP3 (1:200; rabbit
polyclonal antibody; cat. no. ab125219) RalA (1:1,000; rabbit
monoclonal antibody; cat. no. ab126627), E-cadherin (1:5,000;
rabbit polyclonal antibody; cat. no. ab40772), cAMP (1:30,000;
rabbit polyclonal antibody; cat. no. ab76238), T-PKA (1:2,000;
rabbit polyclonal antibody; cat. no. ab38949), p-PKA (1:2,500;
rabbit monoclonal; cat. no. ab75991) and GAPDH (1:10,000; mouse
monoclonal antibody; cat. no. ab8245; all from Abcam, Cambridge,
MA, USA).
Cell proliferation assays
For the clonogenic assay, cells were seeded onto
60-mm culture dishes at the density of 1,000 cells/well. After
colonies formed (~10 days), the colonies were fixed, stained
(crystal violet; cat no. KGA229; Nanjing KeyGen Biotech, Co., Ltd.,
Nanjing, China) and were counted [colonies with more than 50 cells
were counted under a microscope at ×200 magnification (Leica
microscope DM4000 B; Leica Microsystems, Wetzlar, Germany)].
For the proliferation assay, cells were seeded onto
96-well plates (5,000 per well) in F-12 with 5% FBS, and cells were
treated according to the manufacturer's instructions (Keygentec,
Taoshan, China). Absorbance was measured at 550 nm using Mikrotek
Laborsysteme (Mikrotek, Overath, Germany).
In regards to the MTS assay, the cells were analyzed
using a CellTiter 96® AQueous One Solution Cell
Proliferation Assay Kit (Promega, Madison, WI, USA) at 490 mm
absorbance according to the manufacturer's instructions. Doubling
times were determined from four replicate samples per point.
Cell cycle distribution assay
Cells were harvested at 48 h and labeled with
propidium iodide (PI) using previously described methods (15). Briefly, cells were resuspended and
fixed, and then PI (0.05 mg/ml; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) was added to the cells and maintained at room
temperature in the dark for 30 min. Cell cycle distribution was
examined using a FACScan instrument equipped with FACStation
running CellQuest software (Becton Dickinson, San Jose, CA,
USA).
Cell apoptosis assay
Apoptosis was assessed by DNA ladder formation,
double stained with Annexin V-fluorescein isothiocyanate (FITC)/PI
and TUNEL assay. For DNA ladder formation assays, tumor cells were
plated in 6-well plates and attached, and then washed twice with
PBS, followed by treatment according to the manufacturer's
instructions (KGA111; Nanjing KeyGen Biotech Co., Ltd.). A NanoDrop
spectrophotometer (Thermo Fisher Scientific) was used to measure
DNA concentrations. Total genomic DNA (5 µg) was separated on a 1%
agarose gel and photographed with a VersaDoc molecular imager
(Bio-Rad Laboratories, Hercules, CA, USA).
For FACS, with or without infection for 48 h, cells
were collected and then double-stained with Annexin V-FITC and PI
(Becton Dickinson). Cells were examined by a FACScan instrument
equipped with FACStation running CellQuest software (Becton
Dickinson).
A TUNEL assay was conducted to detect
apoptosis
The samples were treated with 20 µg/ml
non-deoxyribonuclease (DNase) proteinase K (Merck Drugs &
Biotechnology Inc., Darmstadt, Germany) for 30 min at 37°C to
remove the nuclease, followed by washing with PBS three times for 4
min each. Terminal deoxynucleotidyl transferase (TdT) (F.
Hoffmann-La Roche & Co., Pleasanton, CA, USA) and biotin-dUTP
(F. Hoffmann-La Roche & Co) were added to the samples, and the
mixtures were incubated in the dark at 37°C for 60 min, followed by
PBS washing. Labeling reaction termination solution was added to
the samples, followed by incubation at 37°C for 60 min and washing
with PBS. Streptavidin-HRP enzyme (F. Hoffmann-La Roche & Co.)
(10 µl) and Biotin-dUTP (490 µl) were mixed and added onto the
sections, and the sections were incubated in the dark for 60 min at
37°C. After PBS washing, 3-diaminobenzidine (DAB) solution (F.
Hoffmann-La Roche & Co.) was used to develop the stain. The
samples were counterstained with hematoxylin (Solarbio Science
& Technology Co., Ltd., Beijing, China). The samples were
washed with PBS, dehydrated, mounted in neutral resins and
photographed (fluorescence microscope Leica TCS SP8; Leica
Microsystems).
Wounding healing and invasion
assays
Invasion assays were performed in 24-well BD
BioCoat™ Matrigel Invasion Chambers (BD Biosciences, San Jose, CA,
USA). After incubation, non-invading cells were removed and the
bottom cells were stained with 0.2% crystal violet, and the stained
cells were counted under a microscope (Leica microscope DM4000 B;
Leica Microsystems) at ×200 magnification, and a bar graph was used
to demonstrate the results.
Wound healing assay was performed after transfection
as previously described (16).
Images of the wound area were captured (Leica microscope DM4000 B;
Leica Microsystems) at 0, 24 and 36 h.
Immunofluorescence microscopy
Cells were fixed in 4% paraformaldehyde, and
permeabilized using 0.5% Triton X-100 in PBS and blocked, and then
incubated with primary antibody AQP3 (1:250; rabbit polyclonal
antibody; cat. no. ab125219; Abcam) and then stained with the
secondary antibodies (Rhodamine-labelled secondary antibodies; cat.
no. R-6394; Invitrogen; Thermo Fisher Scientific, Inc.). Nuclei
were counterstained with DAPI (41,6-diamidino-2-phenylindole).
Images were captured on a Leica TCS-SP8 fluorescence microscope
(Leica Microsystems).
Statistical analysis and gene set
enrichment analysis (GSEA)
All data are shown as the mean ± SD. Statistical
analysis was performed with SPSS 17.0 (IBM, Armonk, NY, USA) and
OriginPro 8.0 (Originlab, Northampton, MA, USA). Differences in
mean values between two groups were analyzed by two-tailed
Student's t-test and the mean values of more than two groups were
compared with one-way analysis of variance (ANOVA). A P-value
<0.05 was considered to indicate a statistically significant
result. The prostate cancer dataset was downloaded from the NCBI
Gene Expression Omnibus database, access ID: GSE55945).
Results
Downregulation of expression is
correlated with the subcellular localization of AQP3 in PC-3
cells
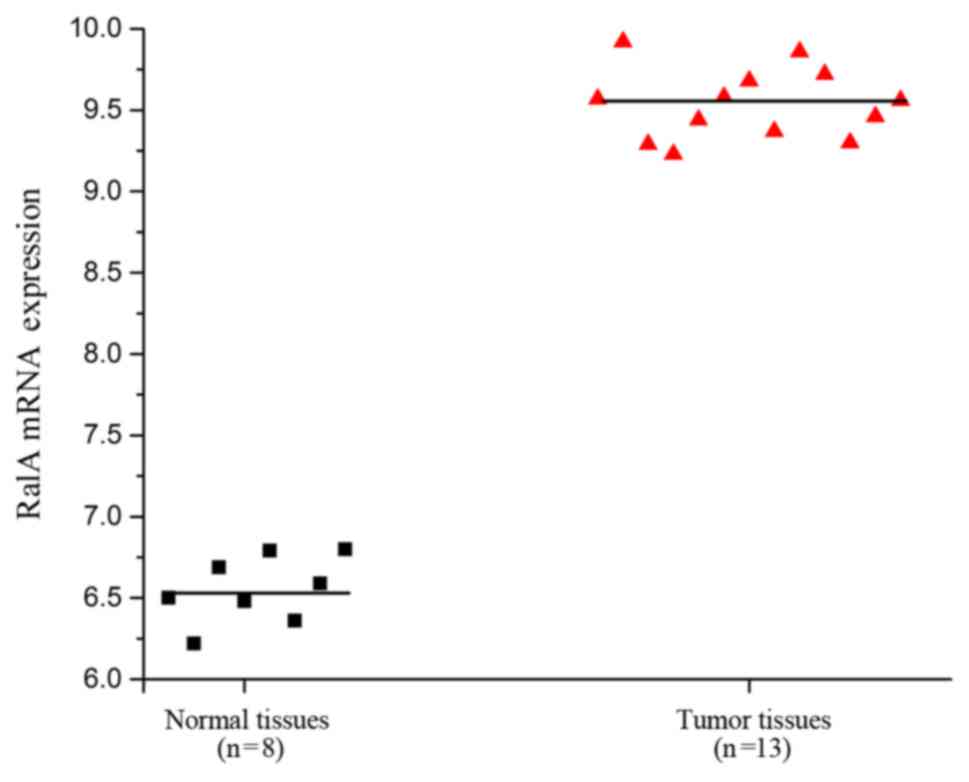
According to the GEO dataset (Access ID: GSE55945),
we found an increase in expression levels of RalA in prostate
cancer tissues compared with level in the adjacent tissues of
patients (Fig. 1, P<0.01). This
result suggests a relationship between RalA mRNA expression and
prostate cancer progression.
To further explore this phenomenon, PC-3 cell
derivatives with stable knockdown of RalA were established.
Moreover, according to Nejsum and Nelson (6), we established PC-3 cell derivatives
with stable overexpression of E-cadherin as a positive control.
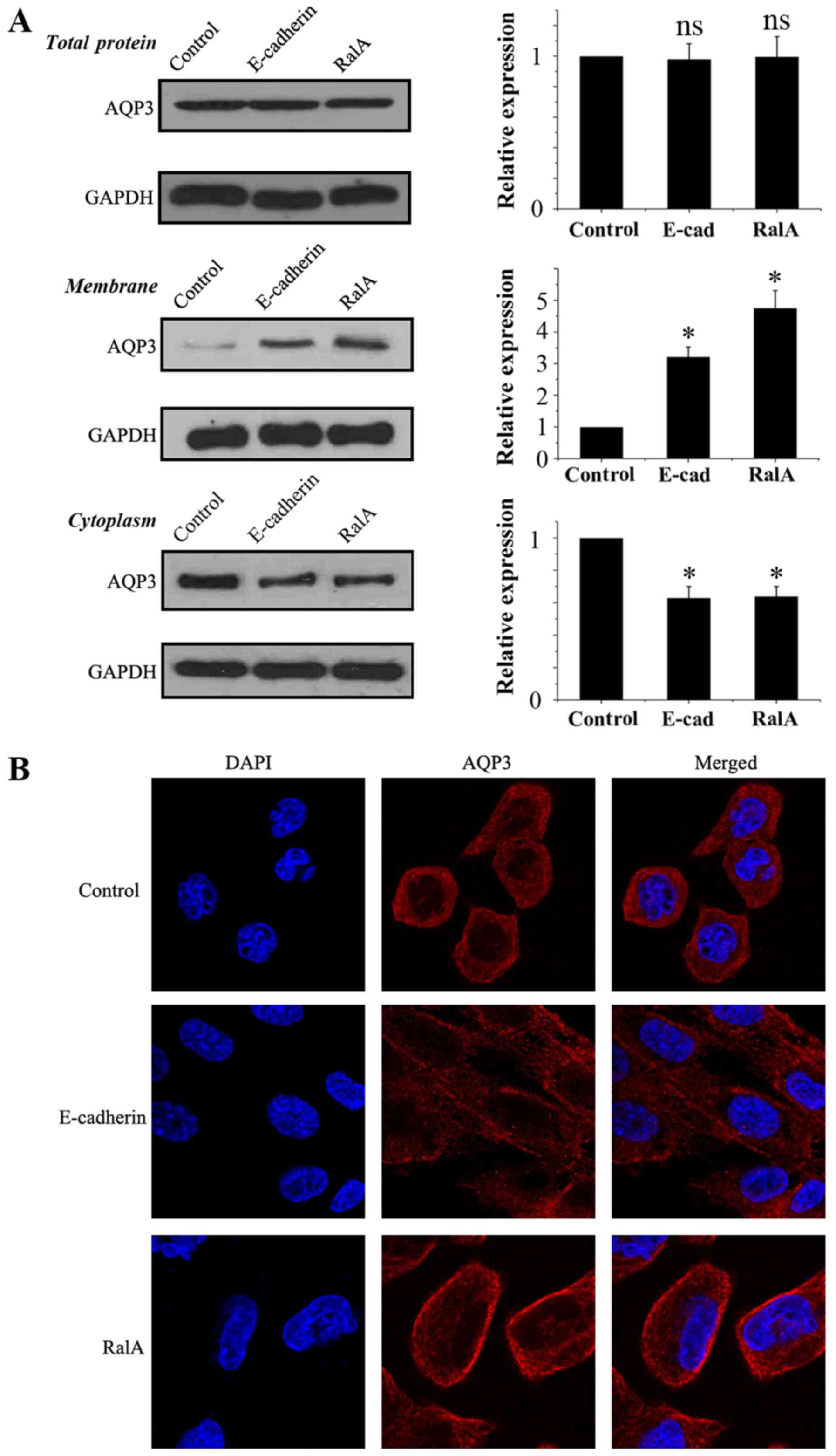
As shown in Fig. 2A,
immunoblot analysis for total AQP3 in the three groups demonstrated
that RalA knockdown and E-cadherin overexpression played no
significantly role in regulating the total protein level of AQP3.
Subcellular fractionation followed by immunoblot analysis showed
the AQP3 band in PC-3 cell plasma and intracellular membranes. RalA
knockdown and E-cadherin overexpression resulted in an increase in
AQP3 in the PC-3 cell membrane and a simultaneous decrease in AQP3
in the plasma. Moreover, confocal immunofluorescence microscopy in
PC-3 cells was in agreement with the quantitative immune blot
analysis (Fig. 2B). These data
suggest that RalA knockdown regulates the subcellular location of
AQP3.
Silencing of RalA inhibits the
proliferation of prostate cancer cells
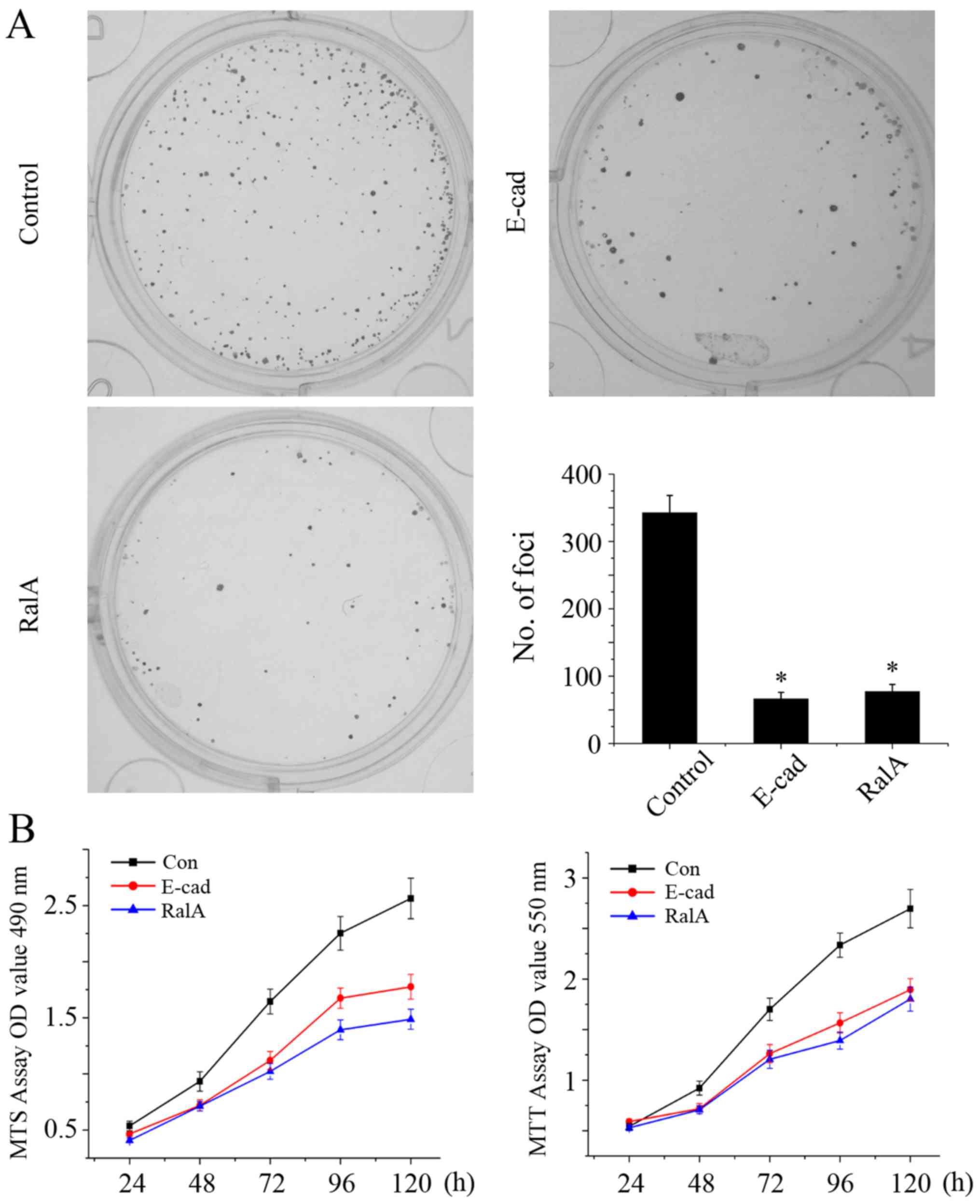
After knockdown of RalA, the number of tumor cells
was significantly less and smaller sizes of cell colonies formed
were noted when compared with the control group (Fig. 3A, P<0.01), Moreover, according to
the MTT and MTS assays, in the absence of RalA, the cell growth
rate was significantly suppressed when compared with corresponding
control (Fig. 3B, left and right
graphs, P<0.01).
Knockdown of RalA enhances the
apoptosis of prostate cancer cells
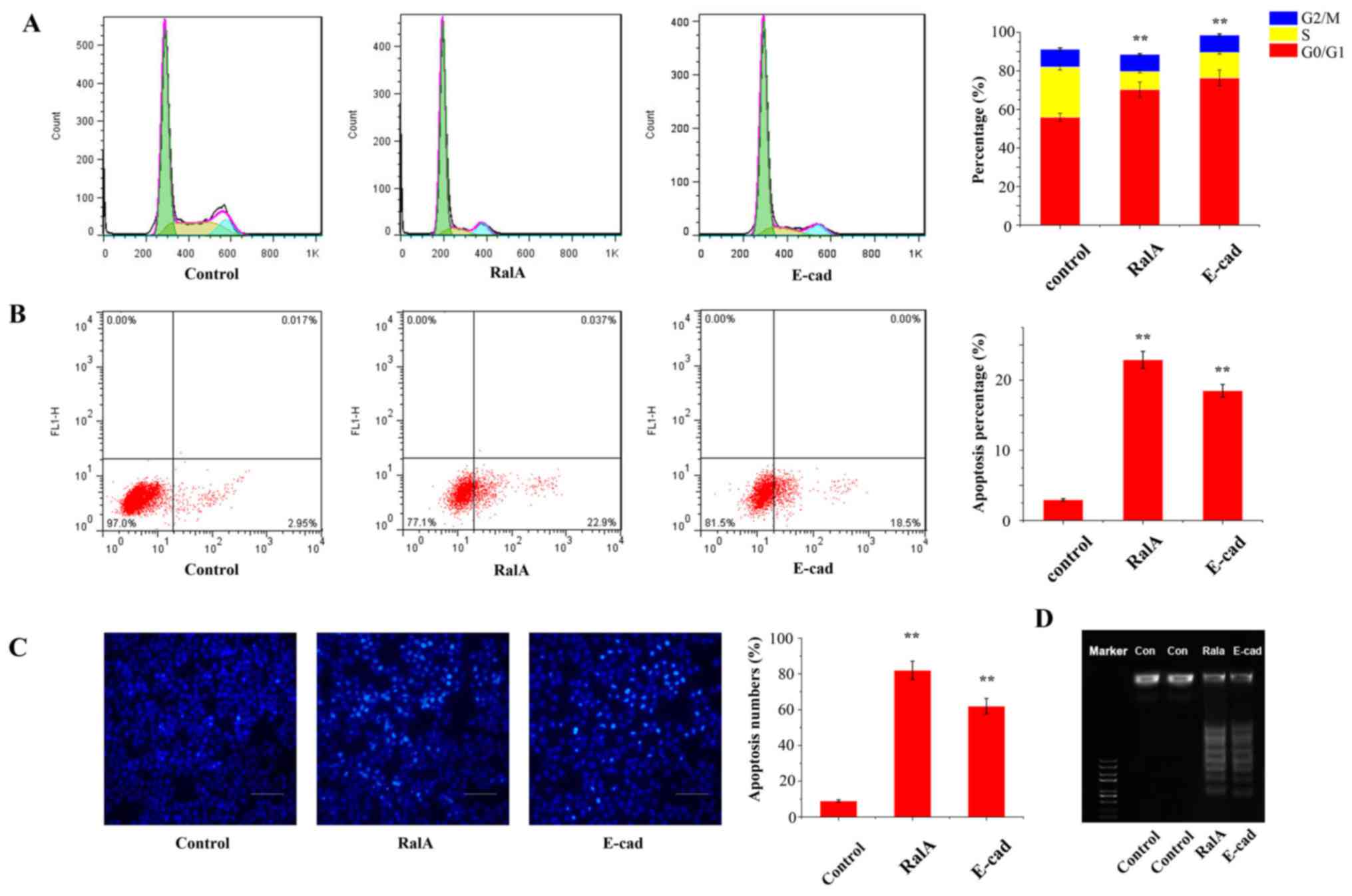
We next aimed to explore how RalA affects cell cycle
progression. Flow cytometric analysis revealed that the percentage
of PC-3 cells in the G0/G1 phase with viral transfection was
markedly increased by 21.2% (Fig.
4A, P<0.05), and the percentage of S phase cells was
significantly decreased, compared with the control cells. These
results indicated that RalA enhanced G1/S cell cycle transition in
prostate cancer cells.
We further determined the apoptotic rate after
knockdown of RalA in the PC-3 cells by Annexin V-FITC/PI staining
assay. As shown in Fig. 4B, flow
cytometric analysis revealed that the silencing of RalA and
overexpression of E-cadherin in PC-3 cells markedly enhanced cell
death compared with that noted in the control group (RalA,
22.5±1.31% vs. E-cadherin, 17.9±1.26% vs. control, 3.2±0.33%;
P<0.01).
In the TUNEL assay, we showed that AQP3
redistribution significantly induced cell apoptosis compared with
the control (Fig. 4C; RalA,
78.9±4.5 cells/field vs. E-cadherin, 59.7±3.8 cells/field vs.
control, 9.2±1.7 cells/field; P< 0.01).
Moreover, in the DNA Ladder assay, the treatment
groups formed a ladder-like trend which suggested that AQP3
redistribution in PC-3 cells markedly enhanced cell death compared
with that noted in the control group (Fig. 4D).
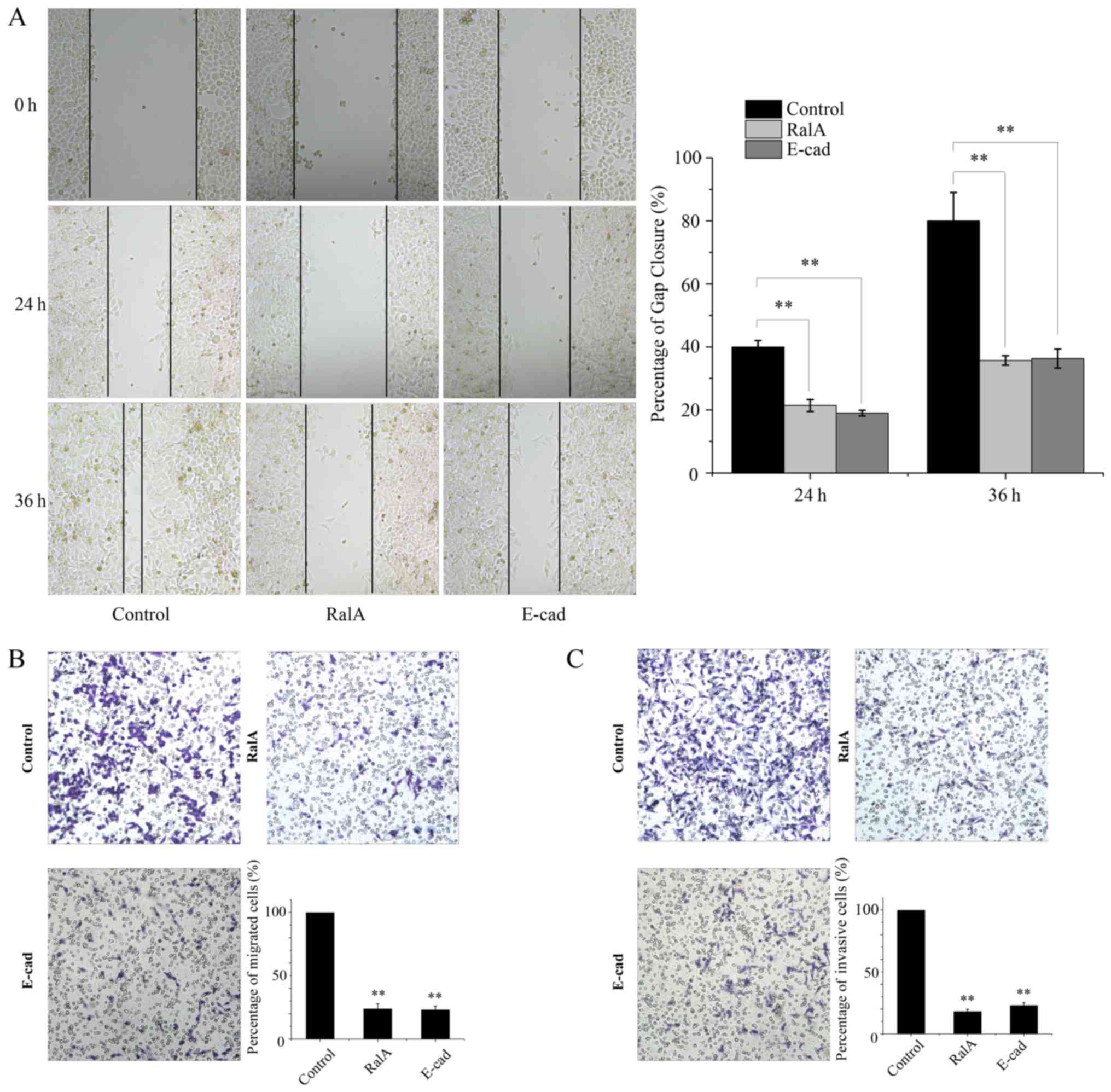
Silencing of RalA suppresses the
migration and invasion of prostate cancer cells
We then investigated the function of RalA in the
migration and invasion in the treated tumor cells. To this end, we
conducted wound-healing and Transwell chamber assays. The results
showed that RalA knockdown decreased the motility of tumor cells
(Fig. 5A). Furthermore, the RalA
knockdown group showed suppressed invasion in the Matrigel-coated
Transwells when compared with the control group (Fig. 5B).
Ral GTPases stimulate GTP exchange on two
substrates, RalA and RalB (17).
Moreover, Shipitsin et al indicated that RalA enhances the
delivery of vesicles containing E-cadherin between the trans-Golgi
and the basolateral plasma membrane (18). In addition, Nejsum and Nelson
concluded that basolateral membrane AQP3 is localized directly to
initial areas of E-cadherin-mediated cell-cell adhesion (6). Therefore, we investigated that the
presence of RalA components has an important effect on AQP3. To
further explore the mechanism by which the signaling pathway is
involved in regulating AQP3 via RalA, we focused on the PKA-cAMP
signaling pathway which was found to regulate AQP8 subcellular
location in rat hepatocytes (19).
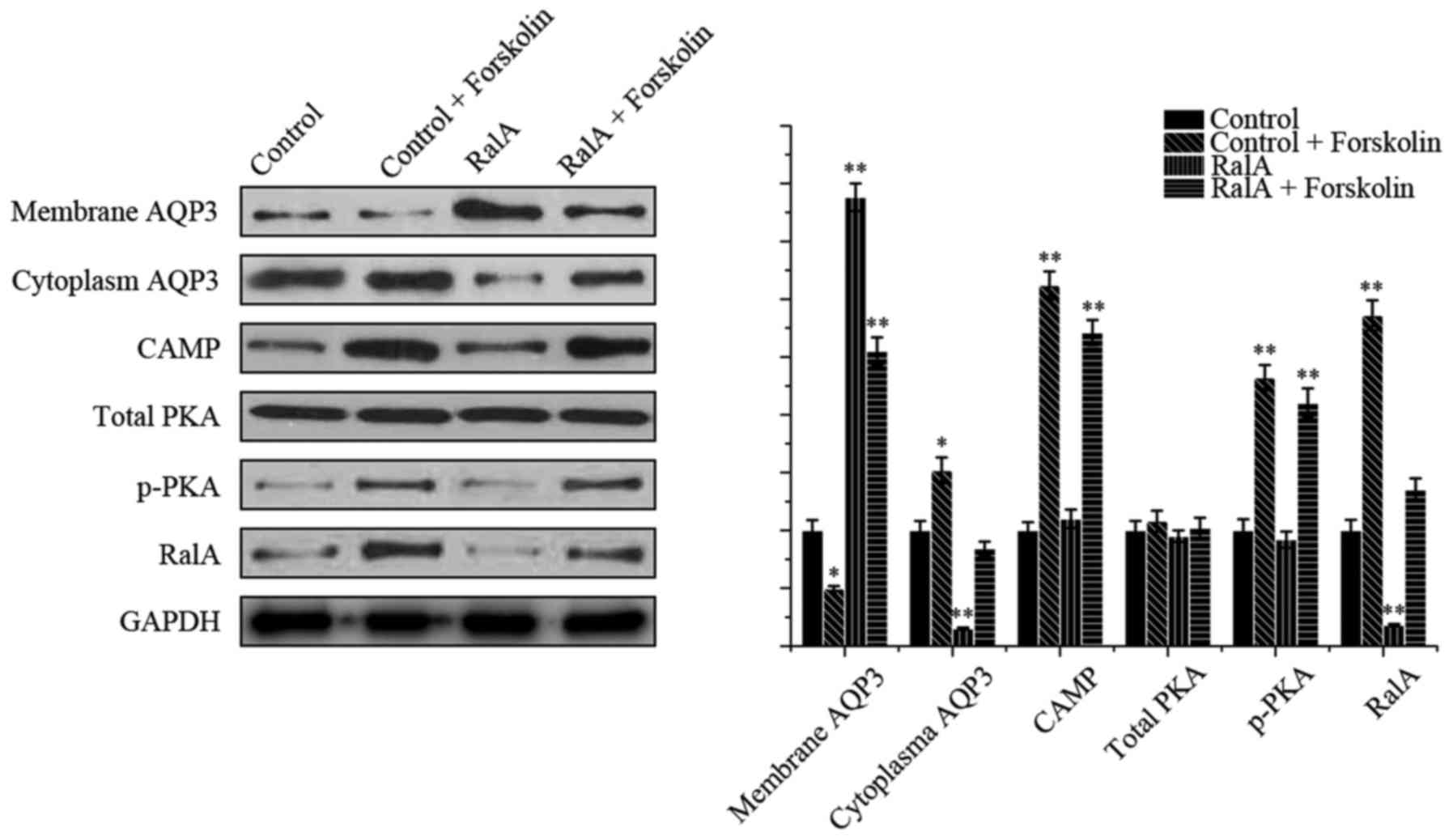
Forskolin, a direct activator of adenylyl cyclases,
was added to test the signaling pathway, The localization of AQP3
and the expression of pathway-associated protein were evaluated by
western blot analysis (Fig. 6). As
expected, the presence of forskolin (10 µM) resulted in less
membranous and more cytoplasmatic distribution of AQP3 in the PC-3
cells, compared with the control group. In the shRNA-RalA group,
treatment with forskolin redistributed AQP3. In addition, both
p-PKA and RalA levels were increased compared with the control
group, which indicated that a cAMP-PKA dependent pathway activated
RalA and promoted AQP3 distribution.
Discussion
To the best of our knowledge, this is the first
study on the subcellular location of AQP3 protein in prostate
cancer cells. Our data suggest that AQP3 is localized largely in
the cytoplasm and can be targeted to the plasma membrane in
prostate cancer cells via a cAMP/PKA/RalA pathway.
Aquaporins play an important role in transporting
water and small solutes. To date, 13 subtypes (AQP0-AQP12) have
been detected in mammals (7,8).
Expression of AQPs promotes disease progression and can predict the
poor survival of cancer (8).
Increased levels of AQP3 have been detected in squamous cell
carcinoma of the skin, pulmonary adenocarcinoma, gastric cancer and
colorectal adenocarcinoma (20–23).
However, few studies have focused on the subcellular location
change of AQP3 between normal prostate tissues and prostate
cancer.
Nejsum and Nelson demonstrated that AQP3 targets
directly to E-cadherin-mediated cell-cell contacts (6). Moreover, active RalA enhances the rate
of delivery of E-cadherin (18).
Therefore, we focused our efforts on investigating RalA in prostate
cancer.
Ras-like (Ral) GTPases include RalA and RalB, which
both have differential functions (24). In several types of cancers,
increased expression of RalA has been identified (25–27).
Consistent with these studies, according to the GEO dataset (Access
ID: GSE45016), we found increased expression levels of RalA in
prostate cancer tissues. Moreover, when we silenced RalA in PC-3
cells, we found that the intracellular AQP3 was re-localized to the
cell membrane, thereby confirming our hypothesis. In addition, we
knocked down RalA in PC-3 cells and found significantly reduced
colony formation and cell proliferation, as well as suppression of
G1/S cell cycle phase cells and enhanced cell apoptosis. This was
consistent with a study by Ismail et al who found that
cryotherapy also redistributed the localization of AQP3 and
enhanced the sensitivity of prostate cancer cells to cryotherapy
treatment (28). In addition, RalA
knockdown suppressed migration and invasion of tumor cells.
The mechanism underlying the regulation of AQP3
subcellular location in prostate cancer remains unclear. We suggest
that localization of AQP3 expression was regulated by the
cAMP/PKA/RalA signaling pathway. This signaling pathway has been
considered as a regulator of cellular migration, invasion and
metastasis (29,30). Our data showed that enhanced
adenylyl cyclases activation increased RalA expression in
shRNA-RalA cells, indicating an important role for the
cAMP-PKA-RalA pathway in regulating AQP3 subcellular location.
In summary, we found that RalA was overexpressed in
prostate cancer and plays an important role in regulating
localization of AQP3 expression and further enhances the
proliferation, apoptosis and metastasis of prostate cancer cells.
The localization of AQP3 expression may be regulate by these
biological processes through the cAMP/PKA/RalA signaling pathway.
These data provide evidence for targeted therapy.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (nos. 81372761 and
31570953).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ruscetti M, Quach B, Dadashian EL,
Mulholland DJ and Wu H: Tracking and functional characterization of
epithelial-mesenchymal transition and mesenchymal tumor cells
during prostate cancer metastasis. Cancer Res. 75:2749–2759. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Egan A, Dong Y, Zhang H, Qi Y, Balk SP and
Sartor O: Castration-resistant prostate cancer: Adaptive responses
in the androgen axis. Cancer Treat Rev. 40:426–433. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wu YC, Tang SJ, Sun GH and Sun KH: CXCR7
mediates TGFβ1-promoted EMT and tumor-initiating features in lung
cancer. Oncogene. 35:2123–2132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
An HT, Yoo S and Ko J: α-Actinin-4 induces
the epithelial-to-mesenchymal transition and tumorigenesis via
regulation of Snail expression and beta-catenin stabilization in
cervical cancer. Oncogene. 35:5893–5904. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nejsum LN and Nelson WJ: A molecular
mechanism directly linking E-cadherin adhesion to initiation of
epithelial cell surface polarity. J Cell Biol. 178:323–335. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Papadopoulos MC and Saadoun S: Key roles
of aquaporins in tumor biology. Biochim Biophys Acta.
1848:2576–2583. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ribatti D, Ranieri G, Annese T and Nico B:
Aquaporins in cancer. Biochim Biophys Acta. 1840:1550–1553. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang J, Tanji N, Kikugawa T, Shudou M,
Song X and Yokoyama M: Expression of aquaporin 3 in the human
prostate. Int J Urol. 14:1088–1092. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yeaman C, Grindstaff KK and Nelson WJ:
Mechanism of recruiting Sec6/8 (exocyst) complex to the apical
junctional complex during polarization of epithelial cells. J Cell
Sci. 117:559–570. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Prigent M, Dubois T, Raposo G, Derrien V,
Tenza D, Rossé C, Camonis J and Chavrier P: ARF6 controls
post-endocytic recycling through its downstream exocyst complex
effector. J Cell Biol. 163:1111–1121. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chabu C, Li DM and Xu T: EGFR/ARF6
regulation of Hh signalling stimulates oncogenic Ras tumour
overgrowth. Nat Commun. 8:146882017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pawar A, Meier JA, Dasgupta A, Diwanji N,
Deshpande N, Saxena K, Buwa N, Inchanalkar S, Schwartz MA and
Balasubramanian N: Ral-Arf6 crosstalk regulates Ral dependent
exocyst trafficking and anchorage independent growth signalling.
Cell Signal. 28:1225–1236. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gentry LR, Nishimura A, Cox AD, Martin TD,
Tsygankov D, Nishida M, Elston TC and Der CJ: Divergent roles of
CAAX motif-signaled posttranslational modifications in the
regulation and subcellular localization of Ral GTPases. J Biol
Chem. 290:22851–22861. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Henry MK, Lynch JT, Eapen AK and Quelle
FW: DNA damage-induced cell-cycle arrest of hematopoietic cells is
overridden by activation of the PI-3 kinase/Akt signaling pathway.
Blood. 98:834–841. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen Q, Yang D, Zong H, Zhu L, Wang L,
Wang X, Zhu X, Song X and Wang J: Growth-induced stress enhances
epithelial-mesenchymal transition induced by IL-6 in clear cell
renal cell carcinoma via the Akt/GSK-3β/β-catenin signaling
pathway. Oncogenesis. 6:e3752017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Thomas JC, Cooper JM, Clayton NS, Wang C,
White MA, Abell C, Owen D and Mott HR: Inhibition of Ral GTPases
using a stapled peptide approach. J Biol Chem. 291:18310–18325.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shipitsin M and Feig LA: RalA but not RalB
enhances polarized delivery of membrane proteins to the basolateral
surface of epithelial cells. Mol Cell Biol. 24:5746–5756. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
García F, Kierbel A, Larocca MC, Gradilone
SA, Splinter P, LaRusso NF and Marinelli RA: The water channel
aquaporin-8 is mainly intracellular in rat hepatocytes, and its
plasma membrane insertion is stimulated by cyclic AMP. J Biol Chem.
276:12147–12152. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hua Y, Ma X, Liu X, Yuan X, Qin H and
Zhang X: Identification of the potential biomarkers for the
metastasis of rectal adenocarcinoma. APMIS. 125:93–100. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xia H, Ma YF, Yu CH, Li YJ, Tang J, Li JB,
Zhao YN and Liu Y: Aquaporin 3 knockdown suppresses tumour growth
and angiogenesis in experimental non-small cell lung cancer. Exp
Physiol. 99:974–984. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen J, Wang T, Zhou YC, Gao F, Zhang ZH,
Xu H, Wang SL and Shen LZ: Aquaporin 3 promotes
epithelial-mesenchymal transition in gastric cancer. J Exp Clin
Cancer Res. 33:382014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ishimoto S, Wada K, Usami Y, Tanaka N,
Aikawa T, Okura M, Nakajima A, Kogo M and Kamisaki Y: Differential
expression of aquaporin 5 and aquaporin 3 in squamous cell
carcinoma and adenoid cystic carcinoma. Int J Oncol. 41:67–75.
2012.PubMed/NCBI
|
|
24
|
Hobbs GA, Der CJ and Rossman KL: RAS
isoforms and mutations in cancer at a glance. J Cell Sci.
129:1287–1292. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Padavano J, Henkhaus RS, Chen H, Skovan
BA, Cui H and Ignatenko NA: Mutant K-RAS promotes invasion and
metastasis in pancreatic cancer through GTPase signaling pathways.
Cancer Growth Metastasis. 8 Suppl 1:S95–S113. 2015.
|
|
26
|
Győrffy B, Stelniec-Klotz I, Sigler C,
Kasack K, Redmer T, Qian Y and Schäfer R: Effects of RAL signal
transduction in KRAS- and BRAF-mutated cells and prognostic
potential of the RAL signature in colorectal cancer. Oncotarget.
6:13334–13346. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang K, Terai K, Peng W, Rouyanian A, Liu
J, Roby KF, Wise AL, Ezzeldin M, Larson J, Woo RA, et al: The role
of RalA in biology and therapy of ovarian cancer. Oncotarget.
2013.
|
|
28
|
Ismail M, Bokaee S, Morgan R, Davies J,
Harrington KJ and Pandha H: Inhibition of the aquaporin 3 water
channel increases the sensitivity of prostate cancer cells to
cryotherapy. Br J Cancer. 100:1889–1895. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ezzeldin M, Borrego-Diaz E, Taha M,
Esfandyari T, Wise AL, Peng W, Rouyanian A, Kermani Asvadi A,
Soleimani M, Patrad E, et al: RalA signaling pathway as a
therapeutic target in hepatocellular carcinoma (HCC). Mol Oncol.
8:1043–1053. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yu T, Yang G, Hou Y, Tang X, Wu C, Wu XA,
Guo L, Zhu Q, Luo H, Du YE, et al: Cytoplasmic GPER translocation
in cancer-associated fibroblasts mediates cAMP/PKA/CREB/glycolytic
axis to confer tumor cells with multidrug resistance. Oncogene.
36:2131–2145. 2017. View Article : Google Scholar : PubMed/NCBI
|