Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a lethal
malignancy whose overall five-year survival rate remains at less
than 7%, and PDAC remains the fourth highest cause of
cancer-related mortality worldwide (1). One of the most conspicuous
histopathologic features of PDAC is a highly desmoplastic reaction,
which is characterized by activated pancreatic stellate cells
(PSCs) and a major accumulation of extracellular matrix (ECM)
(2,3). Accumulating evidence indicates that
the desmoplastic stroma enhances stiffness, hyaluronic acid content
and intratumoral hydrostatic pressures, and produces solid-stress
pressure derived from solid tissue components, which may result in
the compression of tumor vessels, thus causing heterogeneous tumor
perfusion in pancreatic cancer (4,5).
Consequently, this desmoplastic stroma is believed to contribute to
hypoxia and to hinder the effective intratumoral drug delivery and
therapeutic efficacy, leading to poorer treatment outcomes
(6).
PSCs are nestin-positive and lipid-storing cells in
the pancreas that have been shown to have a profound role in normal
ECM turnover (7,8). In healthy states, non-activated PSCs
contain abundant cytoplasmic lipid droplets, which are rich in
vitamin A, and produce very low levels of ECM (9). When stimulated by inflammation or
injury of the pancreas, quiescent PSCs, which contain abundant
lipid droplets, trans-differentiate into ‘activated’
myofibroblast-like cells characterized by an increased expression
of α-smooth muscle actin (α-SMA) as well as by the enhanced
synthesis of matrix components, such as fibronectins and collagens.
PSCs are also thought to play a vital role in the pathobiology of
pancreatic cancer. In PDAC, a reciprocal supportive role between
pancreatic cancer cells and activated PSCs is increasingly being
investigated: Pancreatic cancer cells secrete pro-mitogenic and
pro-fibrogenic factors, including sonic hedgehog (SHH),
transforming growth factor-β (TGF-β), and platelet-derived growth
factor (PDGF), which promote PSC activation (9,10).
Reciprocally, activated PSCs facilitate PDAC progression by
providing materials and energy, thereby creating a favorable
microenvironment for pancreatic cancer cells (11). The above mentioned findings
indicated that PSCs, the predominant fibroblastic cell type in the
PDAC microenvironment, may be a potential therapeutic target.
However, mechanisms to target PSCs that can reverse their
activation and block their reciprocal role in pancreatic cancer
remain poorly understood. Prostaglandin-endoperoxide synthase,
generally known as cyclooxygenase (COX), is the rate-limiting
enzyme that catalyzes the conversion of arachidonic acid (AA) to
prostaglandin E2, which occurs via microsomal PGE2 synthases
(12). Cyclooxygenase has three
isoforms known as COX-1, COX-2 and COX-3 (a splice variant of
COX-1). Previous studies have proven that COX-2 is highly expressed
in chronic pancreatitis, pancreatic intraepithelial neoplasia
(PanIN) and pancreatic adenocarcinoma (13–18).
In a transgenic mouse model of PDAC, it has been demonstrated that
the inflammation induced by a high-fat diet promotes the expression
of COX-2, elevates the activity of Kras and leads to the
development of fibrosis during the progression from PanIN to PDAC
(19). Immortalized human
pancreatic stellate cells (HPSCs) express COX-2 as well and
synthesize PGE2. Through its EP4 receptor, PGE2 stimulates the
proliferation and migration of HPSCs and enhances the synthesis of
ECM and MMP genes (20). Indometacin, a well-known
anti-inflammatory drug and a non-selective inhibitor of
cyclooxygenase-2 (COX-2), has previously been shown to have
anticancer activities against many types of neoplastic diseases
(21–24). The mechanism by which these
anticancer activities occur may be through suppression of cell
growth and induction of apoptosis (16). Our previous findings also showed
that indometacin can reverse the invasion and proliferation that is
induced by high levels of glucose though the regulation of
E-cadherin in pancreatic cancer cells (25). However, despite the strong
correlation between the expression of COX-2 and pancreatic
fibrosis, the mechanism involved in the inhibitory role of
indometacin in COX-2-mediated effects on PSCs remains unclear.
In the present study, we hypothesized that
indometacin, as an anti-inflammatory drug, can inhibit PSC
proliferation and migration and suppress its activation through the
inhibition of COX-2, thus potentially blocking bidirectional
interaction between PDAC cells and PSCs. To test this hypothesis in
the present study, we used human PSCs obtained and purified from
the surgically resected tumor tissue of patients suffering from
pancreatic cancer, and we determined the effect of indometacin on
the proliferation, migration and activation of human PSCs, in
addition to the underlying mechanism.
Materials and methods
Ethics approval and patient
consent
All the experimental protocols were authorized by
the Ethics Committee of the First Affiliated Hospital of Medical
College, Xi'an Jiaotong University (Xi'an, China). The protocols
also complied with the Declaration of Helsinki. Written informed
consent was obtained from all the study participants. Only adults
were included in this study.
Cell culture and reagents
Human PSCs were separated from surgically resected
tumor tissue using the outgrowth method. The tumor tissues were
collected from the Department of Hepatobiliary Surgery at the First
Affiliated Hospital of Xi'an Jiaotong University. The isolated PSCs
were cultured according to methods described in previous studies
(26,27) and grown in F12/DMEM enriched with
10% fetal bovine serum, penicillin G (100 U/ml), and streptomycin
(100 µg/ml) in a humidified atmosphere of 5% CO2 at
37°C. The purity of the PSCs was estimated by morphology, Oil red O
staining of intracellular fat droplets, and immunofluorescence of
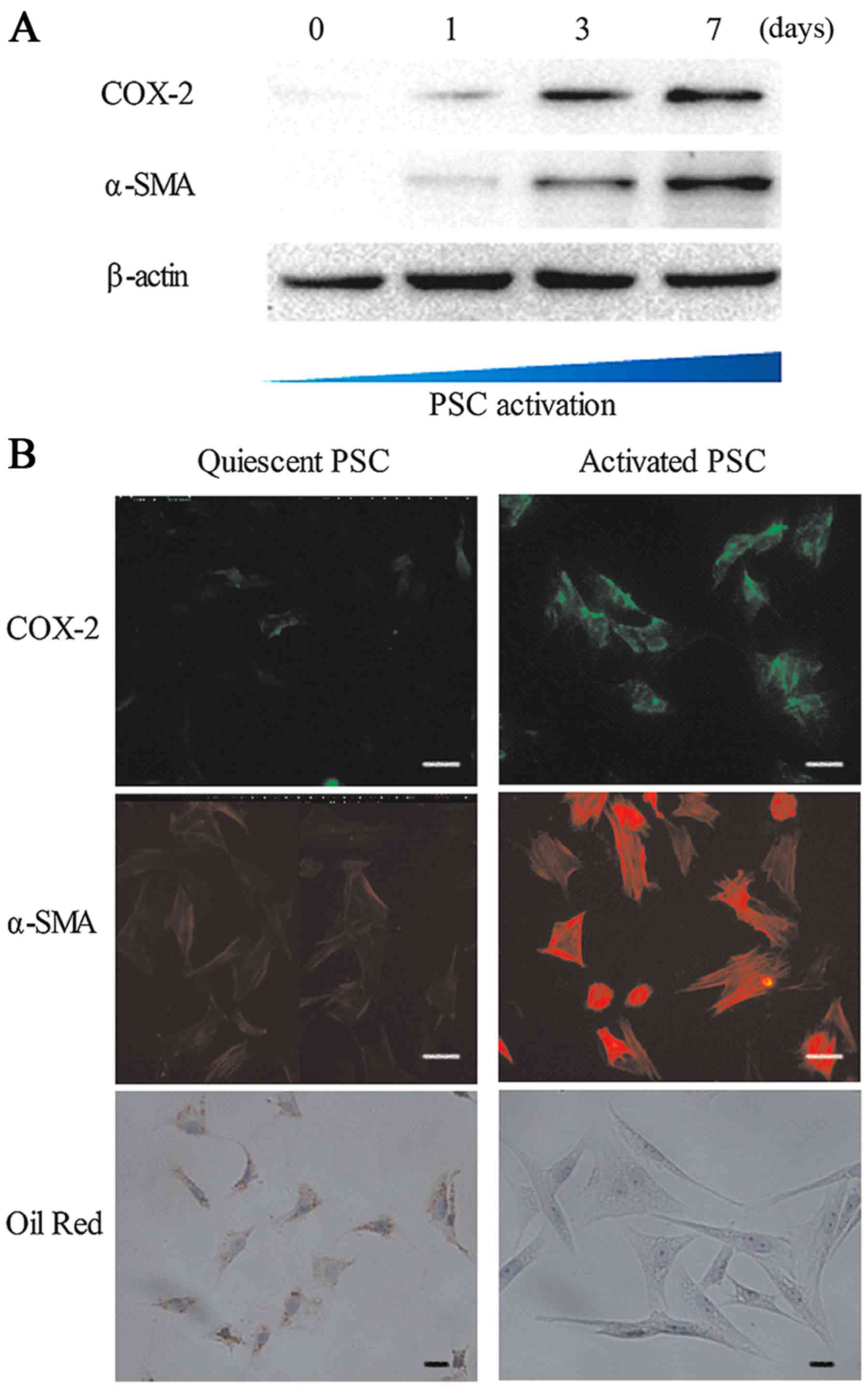
α-smooth muscle actin (Fig. 1).
Cell culture media were obtained from Gibco-BRL (Grand Island, NY,
USA). Indometacin, dimethyl sulfoxide (DMSO), and
3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT)
were obtained from Sigma Chemicals (St. Louis, MO, USA). The stock
solution of indometacin was dissolved in DMSO at 50 g/l. The
antibodies used in this study included anti-COX-2 (Cell Signaling
Technology, Danvers, MA, USA), anti-α-SMA (Sigma, St. Louis, MO,
USA), and anti-β-actin (Santa Cruz Biotechnology, Santa Cruz, CA,
USA).
Cell viability assay
Human PSCs obtained from pancreatic cancer patients
were seeded at 5×103 cells per well into 96-well plates
and treated with indometacin at a concentration gradient (0, 10,
20, 50, 100 and 200 mg/l) for 48 h. Subsequently, the MTT assay was
applied to assess the cell viability, and the absorbance of 490 nm
by a multiwall microplate reader (Bio-Rad, Richmond, CA, USA) was
used for the assessment.
Transwell-migration assay
Assays were performed to assess human PSC migration
by Transwell chambers (Millipore, Billerica, MA, USA) in accordance
with the protocol described previously (28). Briefly, human PSCs were
serum-starved for 6–8 h, and then indometacin was used to pretreat
the cells for 24 h. Subsequently, PSCs (1×105) were
digested, resuspended in serum-free medium and seeded into an upper
chamber of a Transwell plate. The PSCs were given 24 h to migrate
into the lower chamber containing a 10% serum gradient. After 24 h,
the non-migratory cells on the upper side were scraped using a
cotton swab, and the membrane of the chamber was then fixed with 4%
paraformaldehyde and stained with 0.1% crystal violet. The number
of migrated cells on each membrane in 10 random fields was counted,
imaged and recorded at a magnification of ×100. The values reported
here are the averages of triplicate experiments.
Reverse transcriptase-quantitative PCR
(RT-qPCR)
After completion of the specified intervention,
total ribonucleic acid (total RNA) was extracted from these cells
using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to
the manufacturer's protocol. The extracted total RNA was then
reverse-transcribed into cDNA using a PrimeScript RT reagent kit
(Takara Biotechnology, Dalian, China). An iQ5 multicolor real-time
PCR detection system (Bio-Rad, Hercules, CA, USA) and a SYBR-Green
PCR kit (Takara Biotechnology) were used for qPCR according to the
manufacturer's protocols. The following PCR program was used:
Denaturation at 95°C for 30 sec, followed by 40 cycles consisting
of denaturation at 95°C for 5 sec, annealing at 60°C for 30 sec,
and extension at 72°C for 30 sec. The specificity of the amplified
PCR products was evaluated by melting curve analysis, and the
comparative C(q) method, with GAPDH as the normalization control,
was used to assess the expression level of each target gene, as
previously described (29). The
primer sequences used for RT-qPCR were: For COX-2 sense,
5′-TTCAAATGAGATTGTGGGAAAATTGCT-3′ and antisense,
5′-GTGCATCAACACAGGCGCCTCTTC-3′; and for β-actin sense,
5′-ATCGTGCGTGACATTAAGGAGAAG-3′ and antisense,
5′-AGGAAGGCTGGAAGAGTG-3′.
Western blot analysis
Total protein was extracted from 1×106
PSCs grown under the above experimental conditions using RIPA lysis
buffer (Beyotime, Guangzhou, China). The BCA protein assay kit
(Pierce Biotechnology, Rockford, IL, USA) was used to test the
concentration of the proteins based on the manufacturer's
instructions. The details of the western blot assay were previously
described (30). The expression of
specific proteins was determined using a peroxidase reaction to
visualize the chemiluminescence of immunoreactive bands, and the
images of the bands were recorded by the ChemiDoc XRS imaging
system (Bio-Rad). Quantity One image software (version 4.6;
Bio-Rad) was used for the densitometric analysis of each band, and
β-actin was used as the internal loading control.
Immunofluorescence microscopy
After a specific intervention was completed, PSCs
were washed with phosphate-buffered saline (PBS) and fixed in 4%
paraformaldehyde for 30 min at room temperature, washed with PBS
again, and permeabilized in 0.5% Triton X-100 (diluted in PBS) for
10 min. The cells on the slides were subsequently blocked with 5%
BSA for 1 h at room temperature, and then incubated with the
primary antibody [COX-2 (D5H5); Cell Signaling Technology, Danvers,
MA, USA, rabbit monoclonal antibody, dilution at 1:100, cat. no.
12282; α-SMA; Sigma, St. Louis, MO, USA; mouse monoclonal antibody,
dilution at 1:200, cat. no. A5228] at 4°C overnight. Following
incubation with the secondary antibody [Alexa Fluor 488-conjugated
goat anti-rabbit IgG (green), dilution at 1:200; Alexa Fluor
488-conjugated goat anti-mouse IgG (green), dilution at 1:200;
Alexa Fluor 594-conjugated goat anti-mouse IgG (red), dilution at
1:200] from Jackson Immunoresearch Laboratories (West Grove, PA,
USA) for 1 h at room temperature, the cell nuclei were stained with
4′-6-diamidino-2-phenylindole (DAPI) and then sealed on the glass
slides. A Zeiss Instruments confocal microscope (Carl Zeiss
Microscopy; LLC, Thornwood, NY, USA) was used to capture images and
record the cells on the slides using appropriate excitation and
emission spectra at a magnification of ×400.
Oil red O staining
Oil red O staining was applied to visualize
intracellular lipid content in PSCs. Briefly, PSCs on the slides
were washed with phosphate-buffered saline (PBS) and fixed in 4%
paraformaldehyde for 1 h at room temperature. After washing the
PSCs with isopropanol, pre-warmed 0.25% oil red O working solution
was used to stain intracellular lipid content for 15 min in a 60°C
oven. After being washed with PBS twice, the cells were re-stained
with hematoxylin for 15 sec and sealed with glycerin on glass
slides. Finally, a light microscope (Nikon Eclipse Ti-S; Nikon
Corporation, Tokyo, Japan) at a magnification of ×200 was used to
photograph the cells stained with oil red O.
Statistical analysis
The data shown in this study were representative of
three independent experiments, for each experiment, at least 3
samples were used in each treatment group. Data are presented as
the means ± standard deviation. Differences between groups were
evaluated using one-way analysis of variance (ANOVA) and post hoc
analysis of significant effects was performed using Dunnett's test.
P<0.05 was considered statistically significant.
Results
COX-2 levels are elevated during PSC
activation when cultured in vitro
We extracted protein from PSCs at various stages
(after 0, 1, 3 or 7 days of culture) of activation for western blot
analysis. As shown in Fig. 1A,
during the process of activation of PSCs that were cultured in
vitro, the expression of COX-2 and α-SMA simultaneously
increased with the increasing culture time, while the cytoplasmic
lipid droplets in PSCs were substantially decreased, as determined
by oil red O staining (Fig. 1B).
The immunofluorescence staining in Fig.
1B revealed cytoplasmic staining for COX-2 in cells that were
also αSMA-positive. The fluorescence intensity of COX-2 and α-SMA
in activated PSCs was higher than that in quiescent PSCs. These
results showed that COX-2 levels are elevated during PSC activation
when cultured in vitro.
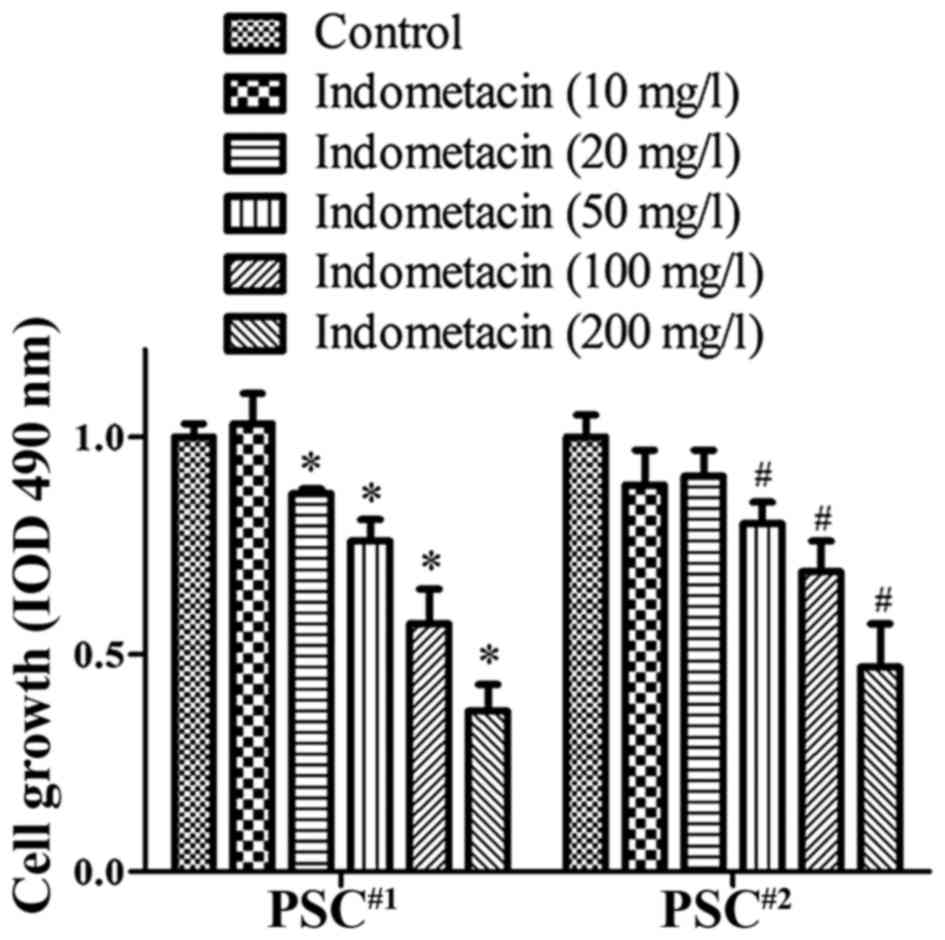
Indometacin restrains the
proliferation of human PSCs
To explore the effect of indometacin on PSCs, we
first investigated the effect of indometacin on the proliferation
of these cells using an MTT assay. PSCs were taken from patients
with pancreatic cancer were treated with a series of gradually
increasing concentrations of indometacin (0 as a control, then 10,
20, 50, 100, or 200 mg/l) for 48 h. Our results showed that
indometacin decreased the growth rate of human PSCs in a
dose-dependent manner (Fig. 2). The
statistical analysis showed that indometacin started to inhibit the
proliferation of all human PSCs at a concentration of 50 mg/l, and
the cell growth rate was significantly decreased at a concentration
of 200 mg/l. According to the data from the MTT assays, we chose
the concentrations of 50 and 100 mg/l for further experiments.
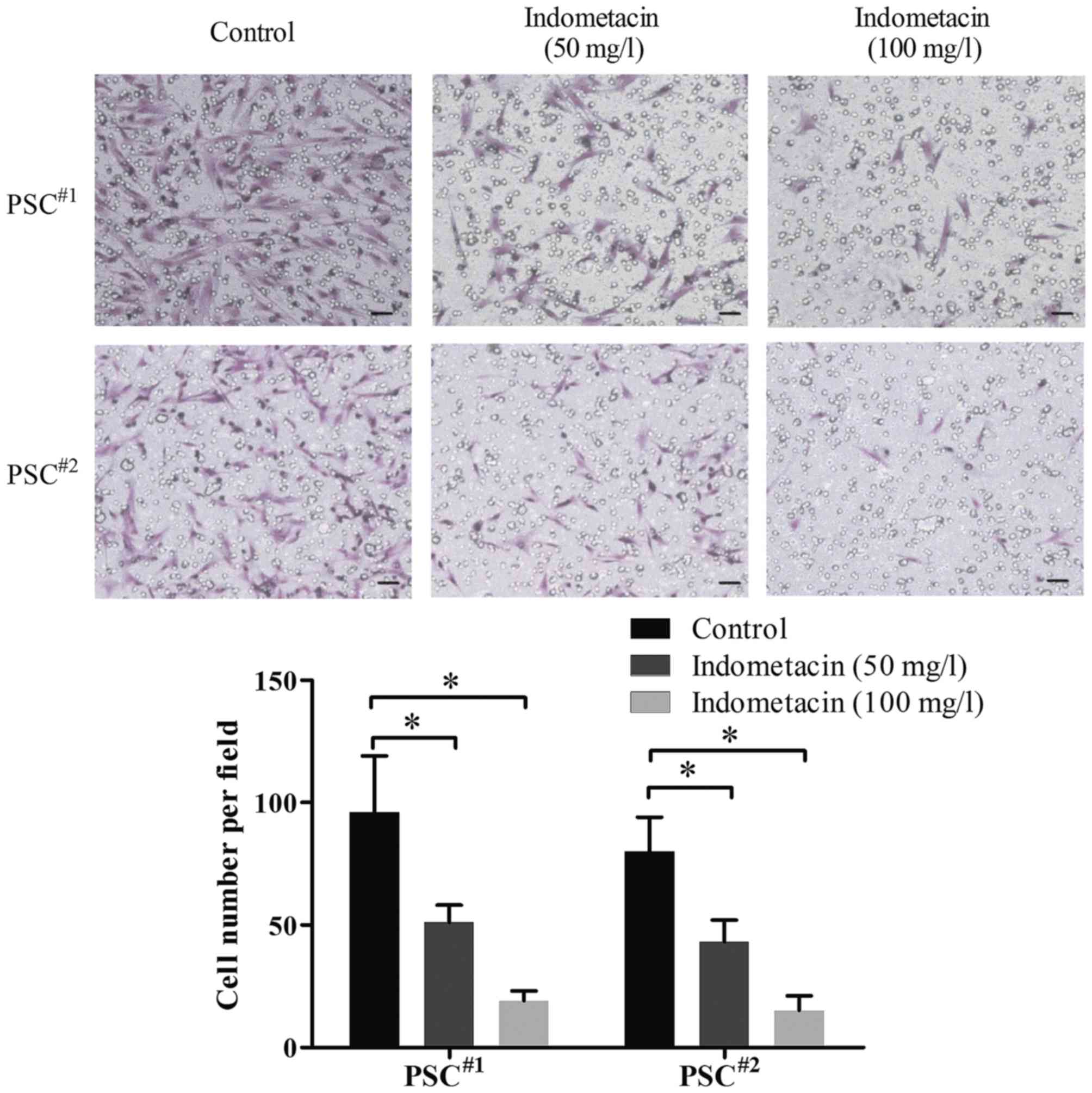
Indometacin inhibits the migration of
human PSCs
Next, we studied the effect of indometacin on the
migration of human PSCs using Transwell-migration assays. As shown
in Fig. 3, treatment with 50 mg/l
indometacin markedly decreased the migration ability of human PSCs
compared to the untreated cells in the control group, and almost no
cells migrated with an indometacin concentration of 100 mg/l. The
statistical analysis showed that there was a significant difference
in the migration ability between the untreated control cells and
PSCs treated with indometacin. These results suggested that
indometacin inhibits the migration capacity of human PSCs obtained
from pancreatic cancer patients in vitro.
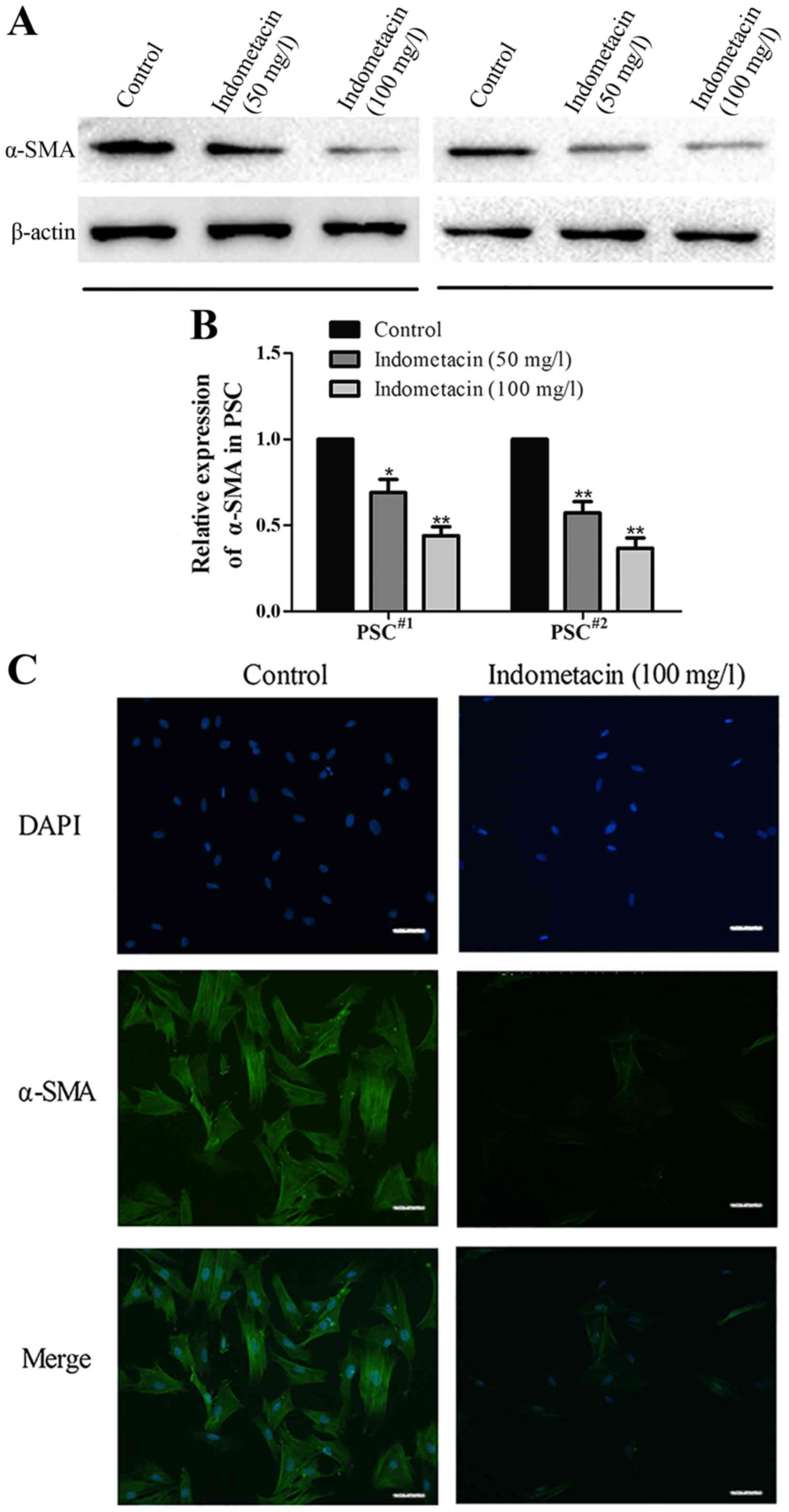
Indometacin inhibits human PSCs
activation
To determine the effect of indometacin on PSC
activation, we treated human PSCs with indometacin in vitro.
The serum-free starvation-synchronized PSCs were treated with
indometacin (50 or 100 mg/l) for 24 h. Subsequently, PSC activation
was verified by the detection of α-SMA level using western blot
analysis and immunofluorescence staining. As shown in Fig. 4A and B, the western blot results
indicated that indometacin decreased the expression of α-SMA in a
dose-dependent manner. The α-SMA level was significantly lower in
cells treated with 100 mg/l indometacin than in untreated cells in
the control group. Similarly, immunofluorescence labeling showed
that treatment with 100 mg/l indometacin markedly decreased the
α-SMA level (Fig. 4C).
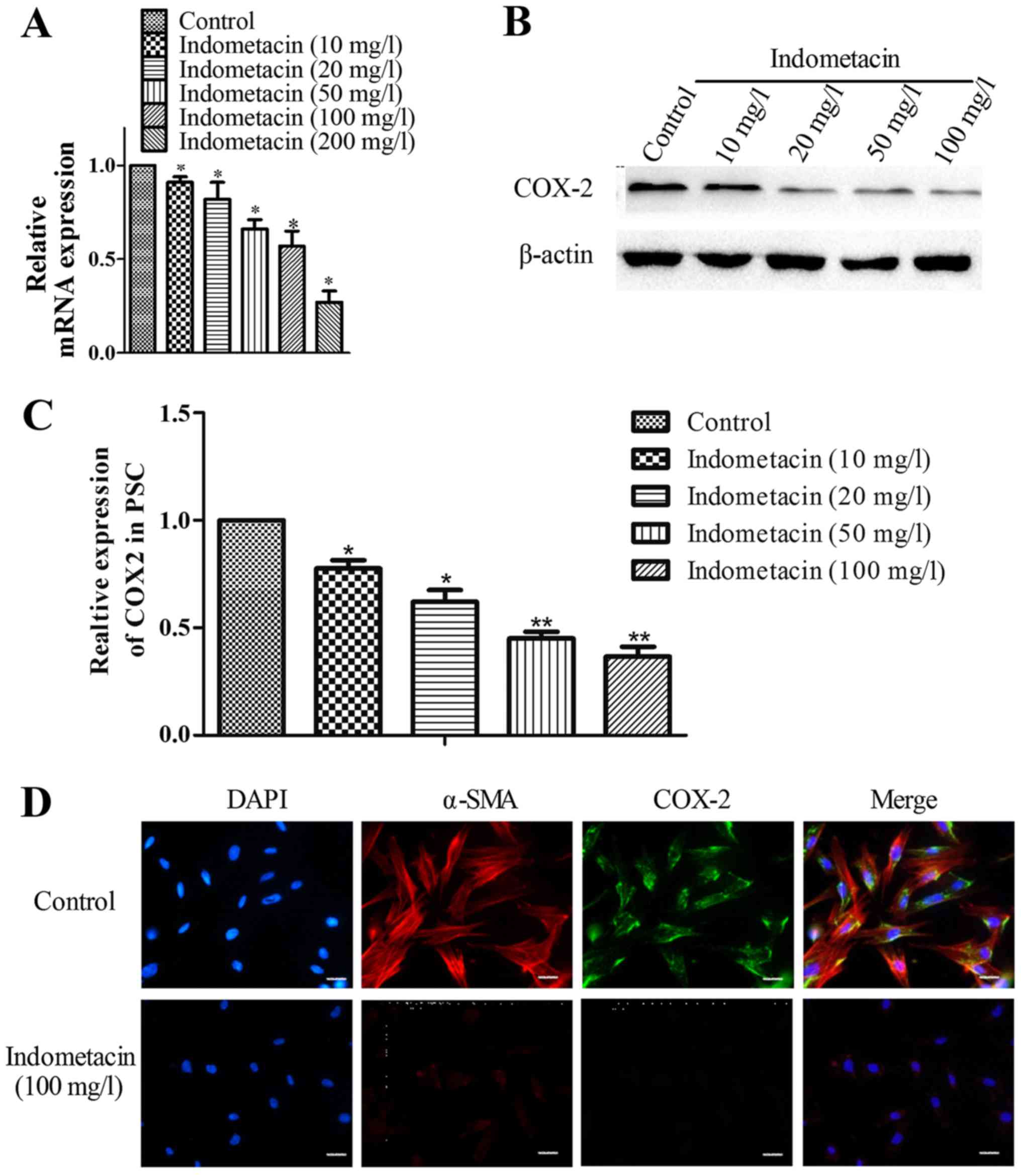
Indometacin suppresses PSC activation
through the downregulation of COX-2 expression
To determine whether indometacin affected PSC
activation via inhibition of the expression of COX-2, human PSCs
were treated with indometacin (at concentrations of 0, 10, 20, 50,
100 or 200 mg/l) for 24 h. Total cell RNA was extracted to evaluate
the mRNA levels of COX-2 by RT-qPCR. Similarly, the cells were
treated with indometacin (at concentrations of 0, 10, 20, 50 or 100
mg/l) for 48 h, and COX-2 protein expression was determined by
western blot analysis in human PSCs. The findings are shown in
Fig. 5A-C. Indometacin treatment
decreased COX-2 expression at the mRNA and protein levels in a
dose-dependent manner. To further investigate indometacin
suppression of PSC activation via the inhibition of COX-2,
immunofluorescent double staining was used to synchronously
visualize COX-2 expression and PSC activation. As shown in Fig. 5D, the immunofluorescence results
show that the level of PSC activation and the expression of COX-2
were simultaneously decreased at an indometacin concentration of
100 mg/l. These results demonstrated that indometacin suppresses
PSC activation to some extent through the downregulation of COX-2
expression.
Discussion
As Dvorak noted, cancers are similar to wounds that
do not heal (2), and accumulating
evidence has shown that a reactive stroma microenvironment exerts a
supportive role on PDAC progression (31,32).
PSCs are the main components of this reactive stroma. In the
initiation and development of pancreatic cancer, stellate cells in
a quiescent state undergo phenotypic changes that
transdifferentiate them from vitamin-A-rich cells into ‘activated’
PSCs that overexpress α-SMA and that synthesize large amounts of
extracellular matrix components, such as fibronectin and collagens;
these components form a stroma-rich and hypoxic microenvironment
(6). Then, intratumoral hypoxia
fuels tumor progression by inducing angiogenesis, genomic
instability, a switch to anaerobic metabolism, induction of a
cancer ‘stem cell’ phenotype, EMT, metastasis, inflammation,
fibrosis, immunosuppression, and resistance to apoptosis/autophagy
and chemotherapy (33). Previous
findings have shown that eradicating tumor stroma by targeted
deletion leads to undifferentiated, aggressive pancreatic cancer,
suggesting that tumor-associated fibroblasts may play an inhibitory
role rather than a tumor-promoting role (28,30). A
possible reason for this phenomenon is that these cells likely
exist to eliminate some key stromal components that are necessary
for maintaining tissue homeostasis. However, the study of Sherman
et al offers a new strategy to cure pancreatic cancer
(34). Sherman et al
(34) revealed that the vitamin D
receptor (VDR) plays an important role in the activation of
pancreatic stellate cells and that VDR activation by VDR ligand
calcipotriol leads to stromal reprogramming, which includes
reversing reactive stroma back to a quiescent state, thus reducing
fibrosis-related inflammatory markers, decreasing energy supply for
the tumor, restoring normal function, promoting chemotherapeutic
efficacy and enhancing potential immune responses to pancreatic
cancer. All-trans retinoic acid (ATRA) can also reverse activated
PSCs into a resting state, thereby inhibiting the proliferation of
cancer cells and β-catenin nuclear translocation, increasing tumor
cell apoptosis and changing tumor morphology (35). As such, reversing activated PSCs
back to the quiescent, vitamin-A-rich and lipid droplet-positive
stromal cells that exist in healthy tissue is a new paradigm for
treating pancreatic cancer.
Overexpression of COX-2 has been observed in 46 to
70% of pancreatic tumors (13,36,37)
and is mainly overexpressed in pancreatic carcinoma cells (38). Previous research also suggests that
in chronic pancreatitis, COX-2 is of great significance in the
initiation and promotion of pancreatic stellate cell activation
(39). However, only a few studies
have shown that COX-2 is expressed in pancreatic stellate cells,
and those studies found no detectable COX-2 expression in the
stroma of pancreatic cancer (40,41).
In the present study, double staining immunofluorescence revealed
that the expression of COX-2 and α-SMA in PSCs taken from tumor
tissue was elevated with increasing culture time, which is in
agreement with the findings of Charo et al (20), who showed that PSCs cultured in
vitro express COX-2 and produce PGE2. PGE2, via its EP4
receptor, promotes a series of behavioral cellular changes,
including increased proliferation, migration, and activation of
PSCs and the synthesis of the collagen I matrix. In contrast to the
findings of Charo et al (20), other authors found that PGE2 exerts
inhibitory effects on the proliferation and fibrogenesis of PSCs
through its receptor EP2, a process that is mediated by the cAMP
pathway (41).
Drug ‘repurposing’ can be used to identify novel
applications for drugs that have already been approved by the US
FDA for another purpose. As drug repurposing can reduce the length
and cost of research and avoid clinical trials, it is more
affordable and achievable than novel drug discovery (42). Indometacin is a non-steroidal
anti-inflammatory drug (NSAID) and is also a non-selective
inhibitor of COX-2. Recently, treatment with indometacin has been
shown to have effects against PDAC, while its effects on PSCs are
largely unclear (25). Although the
stroma plays an important role in PDAC progression, how stromal
activity and downstream signaling are regulated at a molecular
level has not been completely clarified. Reversing activated PSCs
back to a quiescent state seems to be a promising strategy, and our
study provides evidence that indometacin inhibits PSC activation
and major behavior changes in PSCs. These changes include
suppression of the activation, proliferation and migration of PSCs,
and the underlying mechanism of this process may be through the
inhibition COX-2. However, whether indometacin can reverse
activated PSCs back to a quiescent state as well as the definite
mechanism by which this occurs remains to be confirmed.
Acknowledgements
Not applicable.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dvorak HF: Tumors: Wounds that do not
heal. Similarities between tumor stroma generation and wound
healing. N Engl J Med. 315:1650–1659. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Whatcott CJ, Diep CH, Jiang P, Watanabe A,
LoBello J, Sima C, Hostetter G, Shepard HM, Von Hoff DD and Han H:
Desmoplasia in primary tumors and metastatic lesions of pancreatic
cancer. Clin Cancer Res. 21:3561–3568. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stylianopoulos T, Martin JD, Chauhan VP,
Jain SR, Diop-Frimpong B, Bardeesy N, Smith BL, Ferrone CR,
Hornicek FJ, Boucher Y, et al: Causes, consequences, and remedies
for growth-induced solid stress in murine and human tumors. Proc
Natl Acad Sci USA. 109:15101–15108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chauhan VP, Boucher Y, Ferrone CR, Roberge
S, Martin JD, Stylianopoulos T, Bardeesy N, DePinho RA, Padera TP,
Munn LL and Jain RK: Compression of pancreatic tumor blood vessels
by hyaluronan is caused by solid stress and not interstitial fluid
pressure. Cancer Cell. 26:14–15. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Provenzano PP, Cuevas C, Chang AE, Goel
VK, Von Hoff DD and Hingorani SR: Enzymatic targeting of the stroma
ablates physical barriers to treatment of pancreatic ductal
adenocarcinoma. Cancer Cell. 21:418–429. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Apte MV, Haber PS, Applegate TL, Norton
ID, McCaughan GW, Korsten MA, Pirola RC and Wilson JS: Periacinar
stellate shaped cells in rat pancreas: Identification, isolation,
and culture. Gut. 43:128–133. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Phillips PA, McCarroll JA, Park S, Wu MJ,
Pirola R, Korsten M, Wilson JS and Apte MV: Rat pancreatic stellate
cells secrete matrix metalloproteinases: Implications for
extracellular matrix turnover. Gut. 52:275–282. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Apte MV and Wilson JS: Dangerous liaisons:
Pancreatic stellate cells and pancreatic cancer cells. J
Gastroenterol Hepatol. 27 Suppl 2:S69–S74. 2012. View Article : Google Scholar
|
|
10
|
Bailey JM, Swanson BJ, Hamada T, Eggers
JP, Singh PK, Caffery T, Ouellette MM and Hollingsworth MA: Sonic
hedgehog promotes desmoplasia in pancreatic cancer. Clin Cancer
Res. 14:5995–6004. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hwang RF, Moore T, Arumugam T,
Ramachandran V, Amos KD, Rivera A, Ji B, Evans DB and Logsdon CD:
Cancer-associated stromal fibroblasts promote pancreatic tumor
progression. Cancer Res. 68:918–926. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fosslien E: Molecular pathology of
cyclooxygenase-2 in neoplasia. Ann Clin Lab Sci. 30:3–21.
2000.PubMed/NCBI
|
|
13
|
Schlosser W, Schlosser S, Ramadani M,
Gansauge F, Gansauge S and Beger HG: Cyclooxygenase-2 is
overexpressed in chronic pancreatitis. Pancreas. 25:26–30. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tucker ON, Dannenberg AJ, Yang EK, Zhang
F, Teng L, Daly JM, Soslow RA, Masferrer JL, Woerner BM, Koki AT
and Fahey TJ III: Cyclooxygenase-2 expression is up-regulated in
human pancreatic cancer. Cancer Res. 59:987–990. 1999.PubMed/NCBI
|
|
15
|
Kokawa A, Kondo H, Gotoda T, Ono H, Saito
D, Nakadaira S, Kosuge T and Yoshida S: Increased expression of
cyclooxygenase-2 in human pancreatic neoplasms and potential for
chemoprevention by cyclooxygenase inhibitors. Cancer. 91:333–338.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Koshiba T, Hosotani R, Miyamoto Y, Wada M,
Lee JU, Fujimoto K, Tsuji S, Nakajima S, Doi R and Imamura M:
Immunohistochemical analysis of cyclooxygenase-2 expression in
pancreatic tumors. Int J Pancreatol. 26:69–76. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Merati K, Siadaty Said M, Andea A, Sarkar
F, Ben-Josef E, Mohammad R, Philip P, Shields AF, Vaitkevicius V,
Grignon DJ and Adsay NV: Expression of inflammatory modulator COX-2
in pancreatic ductal adenocarcinoma and its relationship to
pathologic and clinical parameters. Am J Clin Oncol. 24:447–452.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Maitra A, Ashfaq R, Gunn CR, Rahman A, Yeo
CJ, Sohn TA, Cameron JL, Hruban RH and Wilentz RE: Cyclooxygenase 2
expression in pancreatic adenocarcinoma and pancreatic
intraepithelial neoplasia: An immunohistochemical analysis with
automated cellular imaging. Am J Clin Pathol. 118:194–201. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Philip B, Roland CL, Daniluk J, Liu Y,
Chatterjee D, Gomez SB, Ji B, Huang H, Wang H, Fleming JB, et al: A
high-fat diet activates oncogenic Kras and COX2 to induce
development of pancreatic ductal adenocarcinoma in mice.
Gastroenterology. 145:1449–1458. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Charo C, Holla V, Arumugam T, Hwang R,
Yang P, Dubois RN, Menter DG, Logsdon CD and Ramachandran V:
Prostaglandin E2 regulates pancreatic stellate cell activity via
the EP4 receptor. Pancreas. 42:467–474. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kubatka P, Kalická K, Bojková B, Ahlers I,
Ahlersová E and Péč M: Neoplastic effect of indomethacin in
N-methyl-N-nitrosourea induced mammary carcinogenesis in female
rats. Klin Onkol. 25:359–363. 2012.(In Slovak). PubMed/NCBI
|
|
22
|
Lange A, Gustke H, Glassmeier G, Heine M,
Zangemeister-Wittke U, Schwarz JR, Schumacher U and Lange T:
Neuronal differentiation by indomethacin and IBMX inhibits
proliferation of small cell lung cancer cells in vitro. Lung
Cancer. 74:178–187. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
de Groot DJ, van der Deen M, Le TK,
Regeling A, de Jong S and de Vries EG: Indomethacin induces
apoptosis via a MRP1-dependent mechanism in doxorubicin-resistant
small-cell lung cancer cells overexpressing MRP1. Br J Cancer.
97:1077–1083. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Das A, Banik NL and Ray SK:
Methylprednisolone and indomethacin inhibit oxidative stress
mediated apoptosis in rat C6 glioblastoma cells. Neurochem Res.
32:1849–1856. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Han L, Peng B, Ma Q, Ma J, Li J, Li W,
Duan W, Chen C, Liu J, Xu Q, et al: Indometacin ameliorates high
glucose-induced proliferation and invasion via modulation of
e-cadherin in pancreatic cancer cells. Curr Med Chem. 20:4142–4152.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gao Z, Wang X, Wu K, Zhao Y and Hu G:
Pancreatic stellate cells increase the invasion of human pancreatic
cancer cells through the stromal cell-derived factor-1/CXCR4 axis.
Pancreatology. 10:186–193. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bachem MG, Schneider E, Gross H,
Weidenbach H, Schmid RM, Menke A, Siech M, Beger H, Grünert A and
Adler G: Identification, culture, and characterization of
pancreatic stellate cells in rats and humans. Gastroenterology.
115:421–432. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rhim AD, Oberstein PE, Thomas DH, Mirek
ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP,
Tattersall IW, et al: Stromal elements act to restrain, rather than
support, pancreatic ductal adenocarcinoma. Cancer Cell. 25:735–747.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Özdemir BC, Pentcheva-Hoang T, Carstens
JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C,
Novitskiy SV, et al: Depletion of carcinoma-associated fibroblasts
and fibrosis induces immunosuppression and accelerates pancreas
cancer with reduced survival. Cancer Cell. 25:719–734. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pandol S, Edderkaoui M, Gukovsky I, Lugea
A and Gukovskaya A: Desmoplasia of pancreatic ductal
adenocarcinoma. Clin Gastroenterol Hepatol. 7 11 Suppl:S44–S47.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schober M, Jesenofsky R, Faissner R,
Weidenauer C, Hagmann W, Michl P, Heuchel RL, Haas SL and Löhr JM:
Desmoplasia and chemoresistance in pancreatic cancer. Cancers.
6:2137–2154. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jain RK: Antiangiogenesis strategies
revisited: From starving tumors to alleviating hypoxia. Cancer
Cell. 26:605–622. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sherman MH, Yu RT, Engle DD, Ding N,
Atkins AR, Tiriac H, Collisson EA, Connor F, Van Dyke T, Kozlov S,
et al: Vitamin D receptor-mediated stromal reprogramming suppresses
pancreatitis and enhances pancreatic cancer therapy. Cell.
159:80–93. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Froeling FE, Feig C, Chelala C, Dobson R,
Mein CE, Tuveson DA, Clevers H, Hart IR and Kocher HM: Retinoic
acid-induced pancreatic stellate cell quiescence reduces paracrine
Wnt-β-catenin signaling to slow tumor progression.
Gastroenterology. 141(1486–1497): e1–e14. 2011.
|
|
36
|
Okami J, Yamamoto H, Fujiwara Y, Tsujie M,
Kondo M, Noura S, Oshima S, Nagano H, Dono K, Umeshita K, et al:
Overexpression of cyclooxygenase-2 in carcinoma of the pancreas.
Clin Cancer Res. 5:2018–2024. 1999.PubMed/NCBI
|
|
37
|
Yip-Schneider MT, Barnard DS, Billings SD,
Cheng L, Heilman DK, Lin A, Marshall SJ, Crowell PL, Marshall MS
and Sweeney CJ: Cyclooxygenase-2 expression in human pancreatic
adenocarcinomas. Carcinogenesis. 21:139–146. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Colby JK, Klein RD, McArthur MJ, Conti CJ,
Kiguchi K, Kawamoto T, Riggs PK, Pavone AI, Sawicki J and Fischer
SM: Progressive metaplastic and dysplastic changes in mouse
pancreas induced by cyclooxygenase-2 overexpression. Neoplasia.
10:782–796. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Aoki H, Ohnishi H, Hama K, Shinozaki S,
Kita H, Osawa H, Yamamoto H, Sato K, Tamada K and Sugano K:
Cyclooxygenase-2 is required for activated pancreatic stellate
cells to respond to proinflammatory cytokines. Am J Physiol Cell
Physiol. 292:C259–C268. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gong J, Xie J, Bedolla R, Rivas P,
Chakravarthy D, Freeman JW, Reddick R, Kopetz S, Peterson A, Wang
H, et al: Combined targeting of STAT3/NF-κB/COX-2/EP4 for effective
management of pancreatic cancer. Clin Cancer Res. 20:1259–1273.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Pomianowska E, Sandnes D, Grzyb K,
Schjølberg AR, Aasrum M, Tveteraas IH, Tjomsland V, Christoffersen
T and Gladhaug IP: Inhibitory effects of prostaglandin E2 on
collagen synthesis and cell proliferation in human stellate cells
from pancreatic head adenocarcinoma. BMC Cancer. 14:4132014.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pessetto ZY, Weir SJ, Sethi G, Broward MA
and Godwin AK: Drug repurposing for gastrointestinal stromal tumor.
Mol Cancer Ther. 12:1299–1309. 2013. View Article : Google Scholar : PubMed/NCBI
|