Introduction
Pancreatic cancer is the fourth leading cause of
cancer-related deaths in Japan and its overall 5-year survival rate
remains very low (less than 10%) (1). Although surgical resection provides
complete cure for patients with pancreatic cancer, over 80% of
cases are judged to be inoperable at the time of initial diagnosis,
due to advanced disease or distant metastasis, and thus subjected
to chemotherapy and/or radiotherapy (2). The DNA damaging anticancer drug,
gemcitabine (GEM) is a first-line chemotherapy agent for patients.
However, most of them do not respond well to GEM treatment,
followed by subsequent disease progression (3,4). To
overcome this serious issue, a variety of combinations of GEM with
other anticancer drugs such as Nab-paclitaxel (Nab-P) and
5-fluorouracil (5-FU) have been assessed. Unfortunately, these
combinations have resulted in limited clinical benefits for
patients compared to patients treated with GEM alone (5). Since the vast majority of cases carry
loss of function mutations in tumor suppressor gene p53, as
well as CDKN2A and gain of function (GOF) mutations in
proto-oncogene K-Ras (6), it
is probable that these mutations may contribute to the acquisition
of resistance of pancreatic cancer tissues to GEM. Considering
this, the understanding of the precise molecular basis behind the
GEM-resistant property of pancreatic cancer is urgently
required.
RUNX2 (runt-related transcription factor 2) has been
widely accepted to be one of the master regulators of osteoblast
differentiation and bone formation. Previous studies have revealed
that RUNX2-deficient mice lack a mineralized skeleton and
its forced expression potentiates the osteoblast transcription
program (7,8). As expected, RUNX2 transactivates its
multiple downstream target gene promoters implicated in
osteogenesis such as osteocalcin (OCN), type I collagen,
osteopontin (OPN) and collagenase 3 (9). In addition to osteogenesis, a growing
body of evidence indicates that RUNX2 has a pro-oncogenic function.
For example, RUNX2 has been demonstrated to be aberrantly
overexpressed in numerous cancer tissues compared to their
corresponding normal ones (10).
Consistent with these observations, forced expression and depletion
of RUNX2 promoted and suppressed malignant phenotypes of cancer
cells of various origins, respectively (10,11).
Recently, we have demonstrated that RUNX2
gene silencing significantly increased chemosensitivity of human
osteosarcoma and pancreatic cancer cells through the augmentation
of the tumor suppressor p53 family-dependent cell death pathway
(12–15). Tumor suppressor p53 family is
composed of three members including p53, TAp73 and TAp63 (16,17).
According to our observations, RUNX2 knockdown markedly
increased GEM sensitivity of p53-mutated pancreatic cancer
cells (14,15). Since it has been well known that
mutant p53 with an extended half-life participates in the
acquisition/maintenance of chemoresistant properties of aggressive
cancers (16,18), it is indicative that depletion of
RUNX2 may override at least in part, the negative effect of
mutant p53 on their chemosensitivity. To further validate our prior
results, we sought to address these issues under the
anchorage-independent growth of pancreatic cancer cells (spheres)
instead of the conventional two-dimensional (2D) monolayer culture
conditions (19). It has been
considered that multicellular spheres mimic the in vivo
physiological microenvironments of cancer tissues (20).
In the present study, we took advantage of the
clinically-relevant in vitro sphere culture system and
demonstrated that RUNX2 gene silencing increased the GEM
sensitivity of p53-mutated pancreatic cancer MiaPaCa-2
spheres through the stimulation of TAp63-dependent cell death
pathway.
Materials and methods
Cells and cell culture
Human pancreatic cancer-derived SW1990 and MiaPaCa-2
cells obtained from The American Type Culture Collection (Manassas,
VA, USA) were maintained in Dulbecco's modified Eagle's medium
(DMEM; Wako Pure Chemical Industries, Ltd., Osaka, Japan)
supplemented with heat-inactivated 10% fetal bovine serum (FBS;
Invitrogen; Thermo Fisher Scientific, Inc., Carlsbad, CA, USA) and
50 units/ml of penicillin/streptomycin. The cells were cultured in
incubators with a humidified atmosphere of 5% CO2 and
95% air, at 37°C.
Sphere formation
Cells were suspended in serum-free sphere medium
DMEM/F12 (Wako Pure Chemical Industries, Ltd., Osaka, Japan)
supplemented with B27 (Bay Bioscience, Co., Ltd., Tokyo, Japan), 25
ng/ml of basic FGF (bFGF; Miltenyi Biotec, Tokyo, Japan) and 20
ng/ml of EGF (Miltenyi Biotec) and seeded in 6-well plates at a
density of 5×105 cells/well. At the indicated
time-points after seeding (day 0, day 1, day 2 and day 3),
representative images were captured.
siRNA-mediated knockdown
For siRNA-mediated silencing of RUNX2, MiaPaCa-2
spheres were transfected with control siRNA (Santa Cruz
Biotechnology, Santa Cruz, CA, USA) or with siRNA targeting RUNX2
(Dharmacon; Ge Healthcare, Lafayette, CO, USA) using Lipofectamine
2000 transfection reagent according to the manufacturer's
instructions (Invitrogen; Thermo Fisher Scientific, Inc.). The
final concentration of each siRNA was 10 nM. RUNX2 gene silencing
was evaluated by immunoblotting and RT-PCR.
Immunoblotting
Cells were lysed in 1X Laemmli buffer supplemented
with 0.1 M DTT and the commercial protease inhibitor mixture
(product no. P8340; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany).
Equal amounts of cell lysates (30 µg of protein) were analyzed by
10% SDS-polyacrylamide gel electrophoresis, transferred onto
membrane filter (Immobilon; Merck Millipore, Amsterdam, The
Netherlands), and incubated with Tris-buffered saline containing
0.1% Tween-20 (TBS-T) plus 5% of non-fat dry milk at 4°C overnight.
The membranes were probed with mouse monoclonal anti-p53 (DO-1;
1:4,000; cat. no. sc126; Santa Cruz Biotechnology) which recognizes
both wild-type and mutant p53, rabbit polyclonal anti-TAp73
(1:1,000; cat. no. GTX-109045; GeneTex, Inc., Irvine, CA, USA),
rabbit polyclonal anti-TAp63 (1:1,000; cat. no. GTX-102425;
GeneTex), rabbit monoclonal anti-RUNX2 (1:1,000; cat. no. 8486;
Cell Signaling Technology, Beverley, CA, USA), rabbit polyclonal
anti-E2F-1 (1:1,000; cat. no. 3742; Cell Signaling Technology),
rabbit polyclonal anti-PARP (1:1,000; cat. no. 9542; Cell Signaling
Technology), mouse monoclonal anti-Itch (1:2,000; cat. no. 611199;
BD Transduction Laboratories, Lexington, KY, USA), mouse monoclonal
anti-Lamin B (1:2,000; cat. no. NA12; 101-B7; Calbiochem; Merck
KGaA, St. Louis, MO, USA), mouse monoclonal anti-γH2AX (2F3;
1:2,000; cat. no. 613401; BioLegend, San Diego, CA, USA) or with
mouse monoclonal anti-actin antibody (C-2; 1:2,000; cat. no.
sc-8432; Santa Cruz Biotechnology) at room temperature for 1 h.
Actin was used as a loading control. After washing in TBS-T, the
membranes were incubated with horseradish peroxidase-conjugated
goat anti-mouse (cat. no. 31430) or anti-rabbit IgG (cat. no.
31466) (1:4,000; Invitrogen; Thermo Fisher Scientific, Inc.) at
room temperature for 1 h. Immunoreactive signals were detected with
an enhanced chemiluminescence detection system (ECL; Ge Healthcare
Life Science, Piscataway, NJ, USA).
RNA preparation and RT-PCR
Total RNA was extracted from the indicated cells
using RNeasy Mini kit following the manufacturer's instructions
(Qiagen, Valencia, CA, USA). One microgram of total RNA was
reverse-transcribed using SuperSprict VILO cDNA Synthesis system
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. The resultant cDNA was subsequently used
as a template for PCR-based amplification with gene-specific primer
sets. Gene expression was normalized relative to that of the
housekeeping gene GAPDH. The oligonucleotide primers used for PCR
were as follows: p53 forward, 5′-CTGCCCTCAACAAGATGTTTTG-3′ and
reverse, 5′-CTATCTGAGCAGCGCTCATGG-3′; TAp63 forward,
5′-GACCTGAGTGACCCCATGTG-3′ and reverse, 5′-CGGGTGATGGAGAGAGAGCA-3′;
TAp73 forward, 5′-TCTGGAACCAGACAGCACCT-3′ and reverse,
5′-GTGCTGGACTGCTGGAAAGT-3′; RUNX2 forward,
5′-TCTGGCCTTCCACTCTCAGT-3′ and reverse, 5′-GACTGGCGGGGTGTAAGTAA-3′;
p21WAF1 forward, 5′-ATGAAATTCACCCCCTTTCC-3′ and reverse,
5′-CCCTAGGCTGTGCTCACTTC-3′; NOXA forward,
5′-CTGGAAGTCGAGTGTGCTACT-3′ and reverse
5′-TCAGGTTCCTGAGCAGAAGAG-3′; BAX forward, 5′-AGAGGATGATTGCCGCCGT-3′
and reverse, 5′-CAACCACCCTGGTCTTGGAT-3′; PUMA forward,
5′-GCCCAGACTGTGAATCCTGT-3′ and reverse, 5′-TCCTCCCTCTTCCGAGATTT-3′;
Itch forward, 5′-ACCTGGATGGGAGAAGAGAA-3′ and reverse,
5′-TGTGCGGGGATCTATATAGG-3′; GAPDH forward,
5′-ACCTGACCTGCCGTCTAGAA-3′ and reverse, 5′-TCCACCACCCTGTTGCTGTA-3′.
PCR products were analyzed by 1.5% agarose gel electrophoresis and
visualized using ethidium bromide staining (Wako Pure Chemical
Industries, Ltd.).
WST cell survival assay
Cell viability was determined by standard WST cell
survival assay. In brief, 5×103 cells were seeded in
triplicate in 96-well plates and allowed to attach overnight. Cells
were then treated with or without the indicated concentrations of
GEM. Forty-eight hours after GEM exposure, their proliferation was
assesed by Cell Counting Kit-8 (CCK-8) reagent (Dojindo Molecular
Technologies, Kumamoto, Japan) following the manufacturer's
instructions.
Trypan blue dye exclusion assay
Cells were exposed to the indicated concentrations
of GEM. Forty-eight hours after treatment, floating and adherent
cells were collected and incubated with 0.4% trypan blue solution
(Bio-Rad Laboratories, Hercules, CA, USA) at room temperature for 3
min. The reaction mixtures were then subjected to a TC20 automated
cell counter (Bio-Rad Laboratories). Trypan blue-positive and
-negative cells were considered to be dead and viable cells,
respectively.
Flow cytometric analysis
Forty-eight hours after GEM exposure, floating and
attached cells were harvested, washed in 1X phosphate-buffered
saline (PBS) and fixed in ice-cold 70% ethanol. After fixation,
cells were treated with 1 mg/ml of propidium iodide (PI) and 1
µg/ml of RNase A at 37°C for 30 min in the dark. The cells were
then analyzed by flow cytometry (FACSCalibur; BD Biosciences,
Franklin Lakes, NJ, USA).
Statistical analysis
The results were presented as the mean ± SD of three
independent experiments. One-way ANOVA tests were performed to
determine the statistical significance of difference among the
control and treated groups using Ekuseru-Toukei 2010 software
(Social Survey Research Information Co., Ltd., Tokyo, Japan).
P<0.05 was considered to indicate a statistically significant
difference.
Results
p53-mutated pancreatic cancer
MiaPaCa-2 cells but not p53-wild-type pancreatic cancer SW1990
cells, form sphere structure
Considering that ~75% of pancreatic cancer tissues
carry p53 mutations and exhibit a serious anticancer drug
resistance (6), we sought to
compare GEM sensitivity between p53-wild-type and p53-mutated
pancreatic cancer cells under the conventional monolayer culture
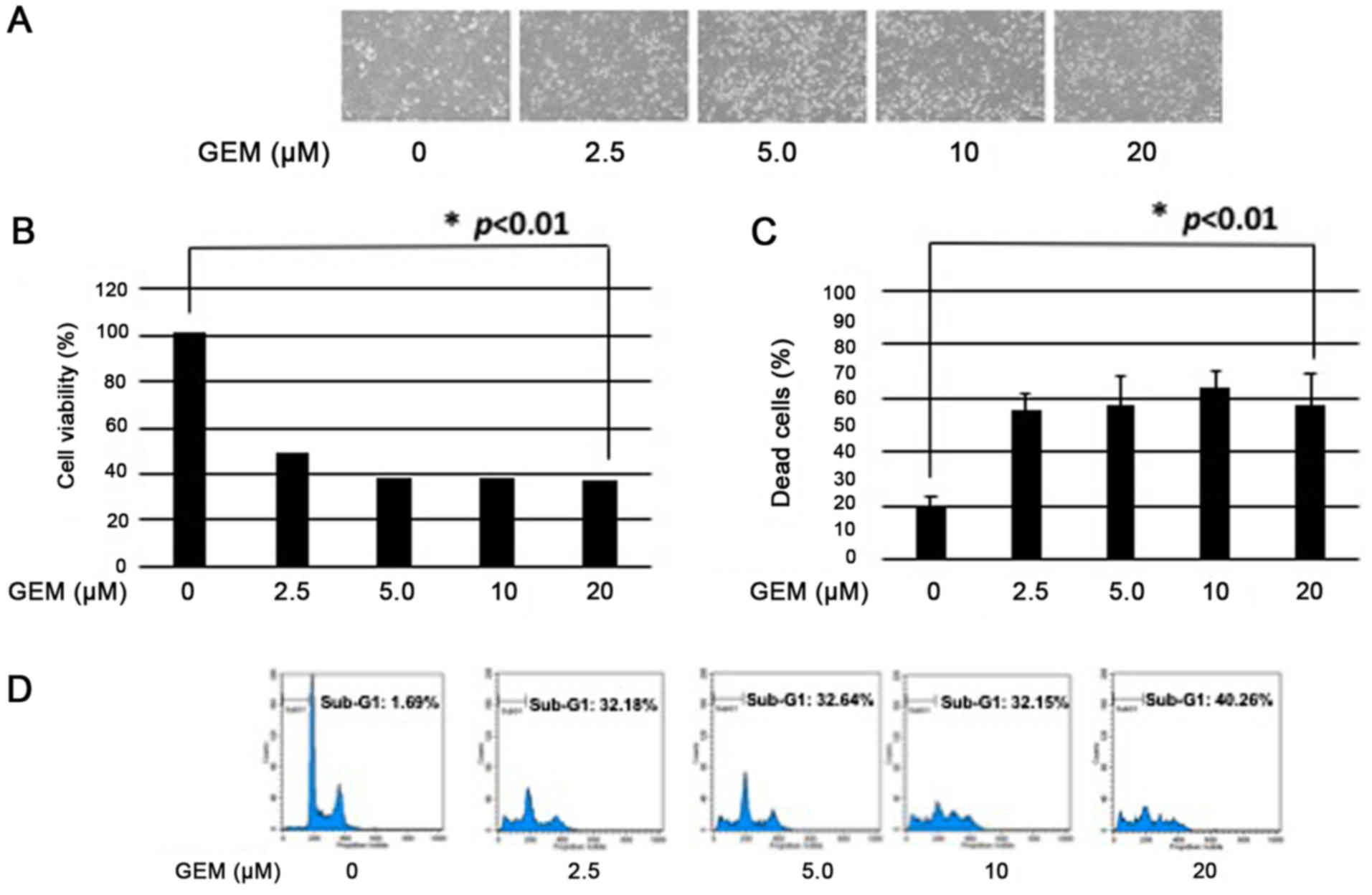
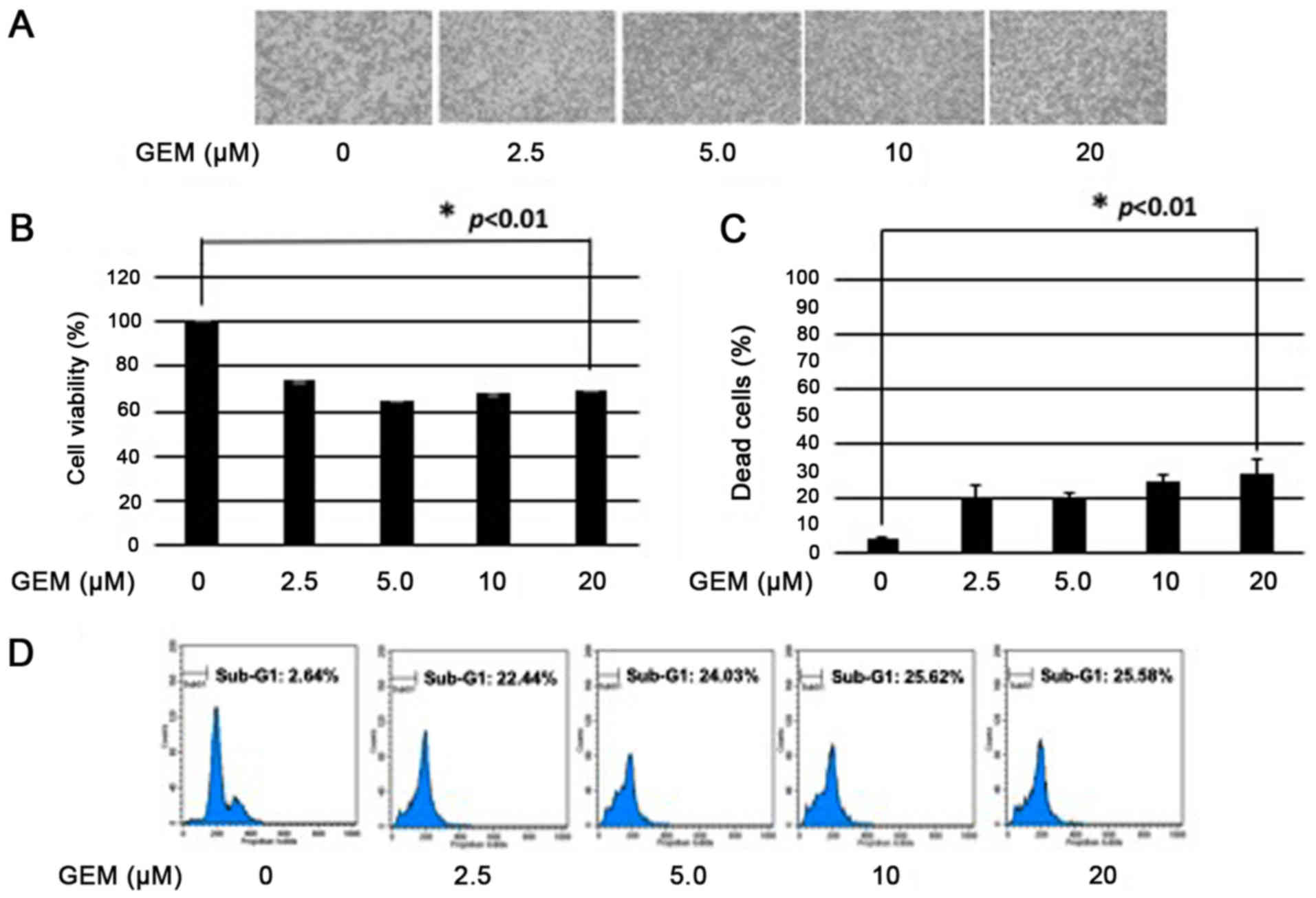
conditions. As clearly displayed in Figs. 1 and 2, p53-mutated MiaPaCa-2 cells poorly
responded to GEM compared to p53-wild-type pancreatic cancer SW1990
cells (P<0.01), indicating that p53 mutation may be involved in
the lower GEM sensitivity of pancreatic cancer cells.
Recently, it has been increasingly recognized that
the response rate of cancer cells to anticancer drugs differs
between two- and three-dimensional (2D and 3D) cancer cell growth
models. Among 3D cultures ranging in complexity from layered
cellular systems to complex multi-cell type spheres, 3D sphere
cultures mimic the architectures and cellular contacts of cells in
cancer tissues (20). These
findings prompted us to evaluate the GEM sensitivity of spheres
generated from MiaPaCa-2 and SW1990 cells, and compare it to that
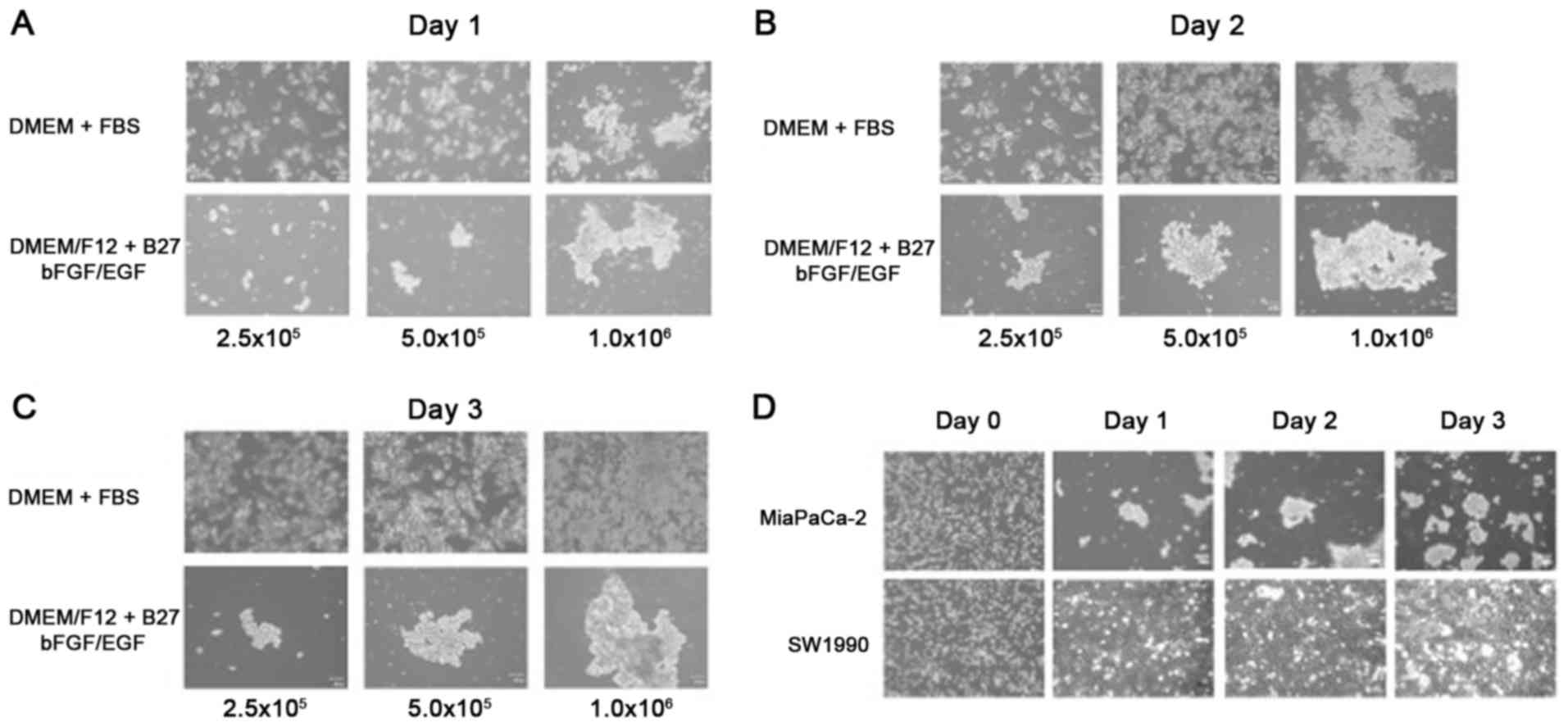
of 2D monolayer cultures. Firstly, we questioned whether MiaPaCa-2
and SW1990 cells could form 3D spheres. The indicated numbers of
MiaPaCa-2 cells were cultured in the standard medium-containing
serum, and then transferred into serum-free sphere-forming medium.
At the indicated time-points following incubation in the
sphere-forming medium, representative images were captured. As
displayed in Fig. 3A-C, MiaPaCa-2
cells (5×105 and 1×106 cells/plate) gave rise
to obvious multi-cellular structures under our experimental
conditions. In contrast to MiaPaCa-2, SW1990 cells did not form
sphere structures until 3 days of incubation (Fig. 3D), indicating that p53 status
may be involved in sphere formation of pancreatic cancer cells.
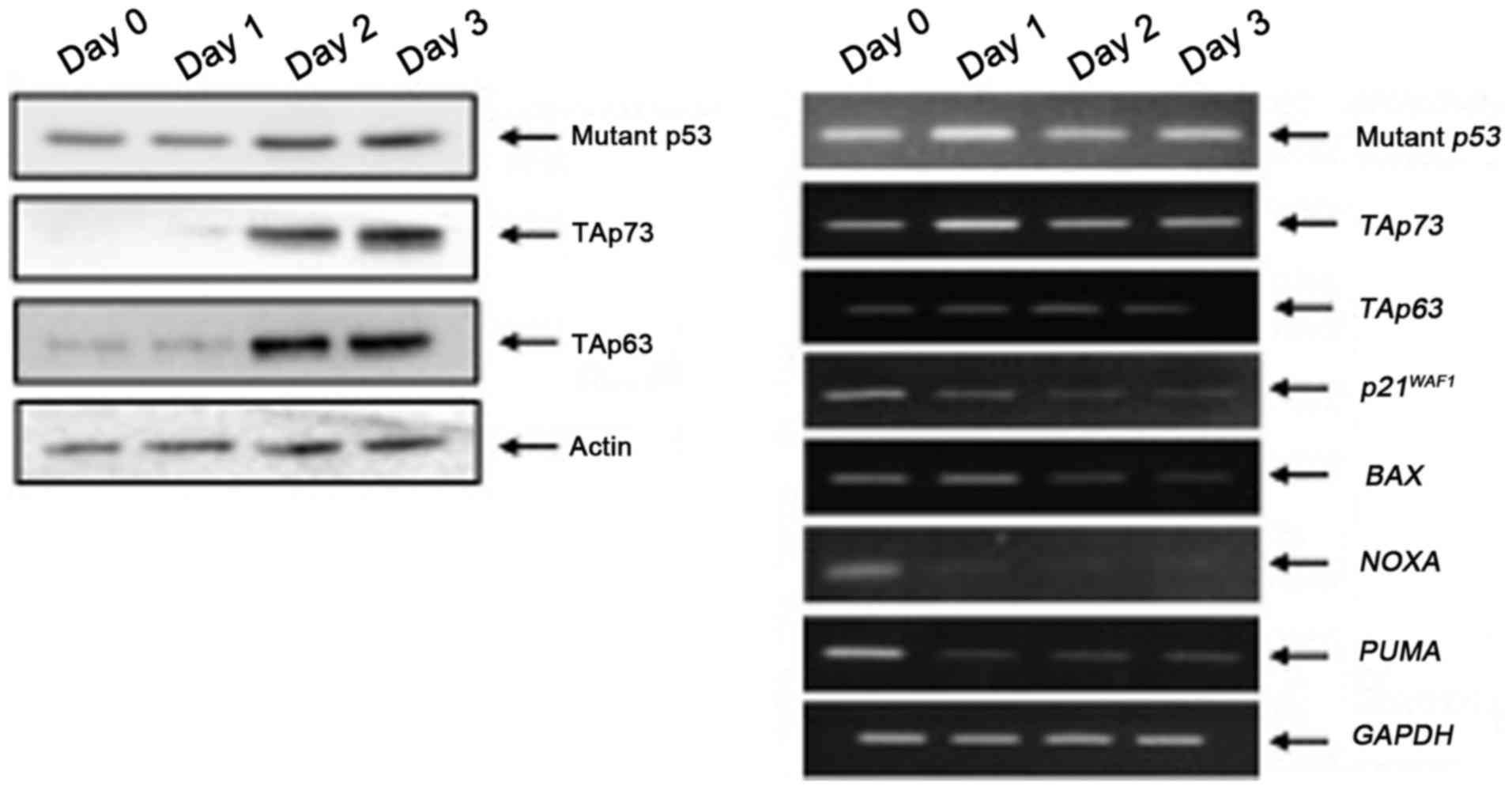
Under the same experimental conditions, cell lysates
and total RNA were prepared from MiaPaCa-2 spheres and analyzed by
immunoblotting and RT-PCR, respectively. As displayed in Fig. 4, the expression levels of mutant
p53, TAp73 and TAp63 proteins were elevated during the sphere
formation. RT-PCR analysis revealed that p53 family target gene
transcription was markedly reduced in MiaPaCa-2 spheres compared to
that of the adherent MiaPaCa-2 cells (day 0), indicating that
mutant p53 prohibited the transcriptional activity of
TAp73/TAp63.
MiaPaCa-2 spheres respond poorly to
GEM
Since it has been demonstrated that spheres often
display the lower sensitivity to anticancer drugs (21,22),
we thus examined the cytotoxic effect of GEM on MiaPaCa-2 spheres.
To this end, MiaPaCa-2 cells were cultured in sphere-forming medium
and then exposed to different concentrations of GEM up to 10 µM. At
the indicated time-points after treatment, the cells were analyzed
by trypan blue dye exclusion assay. Although MiaPaCa-2 spheres
underwent cell death following GEM exposure, there were no marked
differences in dose response among MiaPaCa-2 spheres at a dose
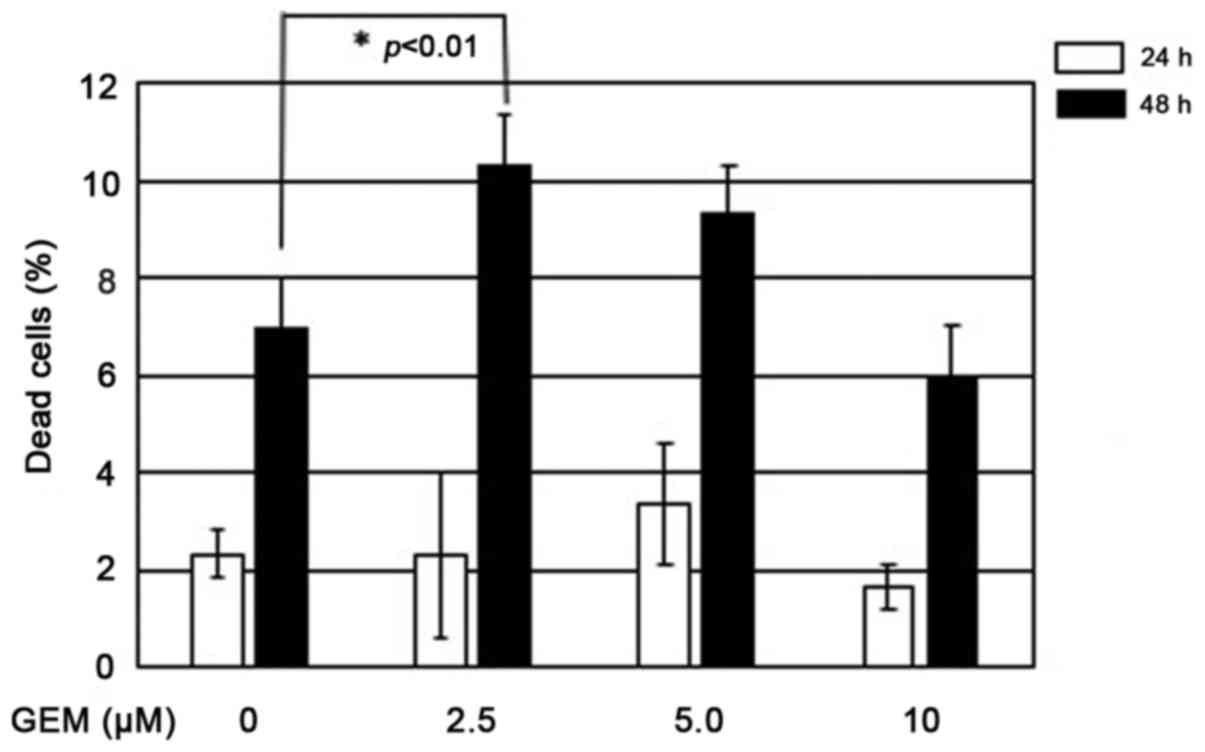
range of 2.5–10 µM of GEM (Fig. 5).
Therefore, for further experiments, we treated MiaPaCa-2 spheres
with 2.5 µM of GEM for 48 h. Under these experimental conditions,
representative images were captured and the spheres were
subsequently subjected to FACS analysis. As displayed in Fig. 6A, GEM treatment caused at most
2-fold increase in the number of cells with sub-G1 DNA
content relative to the untreated ones, which was almost in
accordance with the results obtained from the trypan blue dye
exclusion assay.
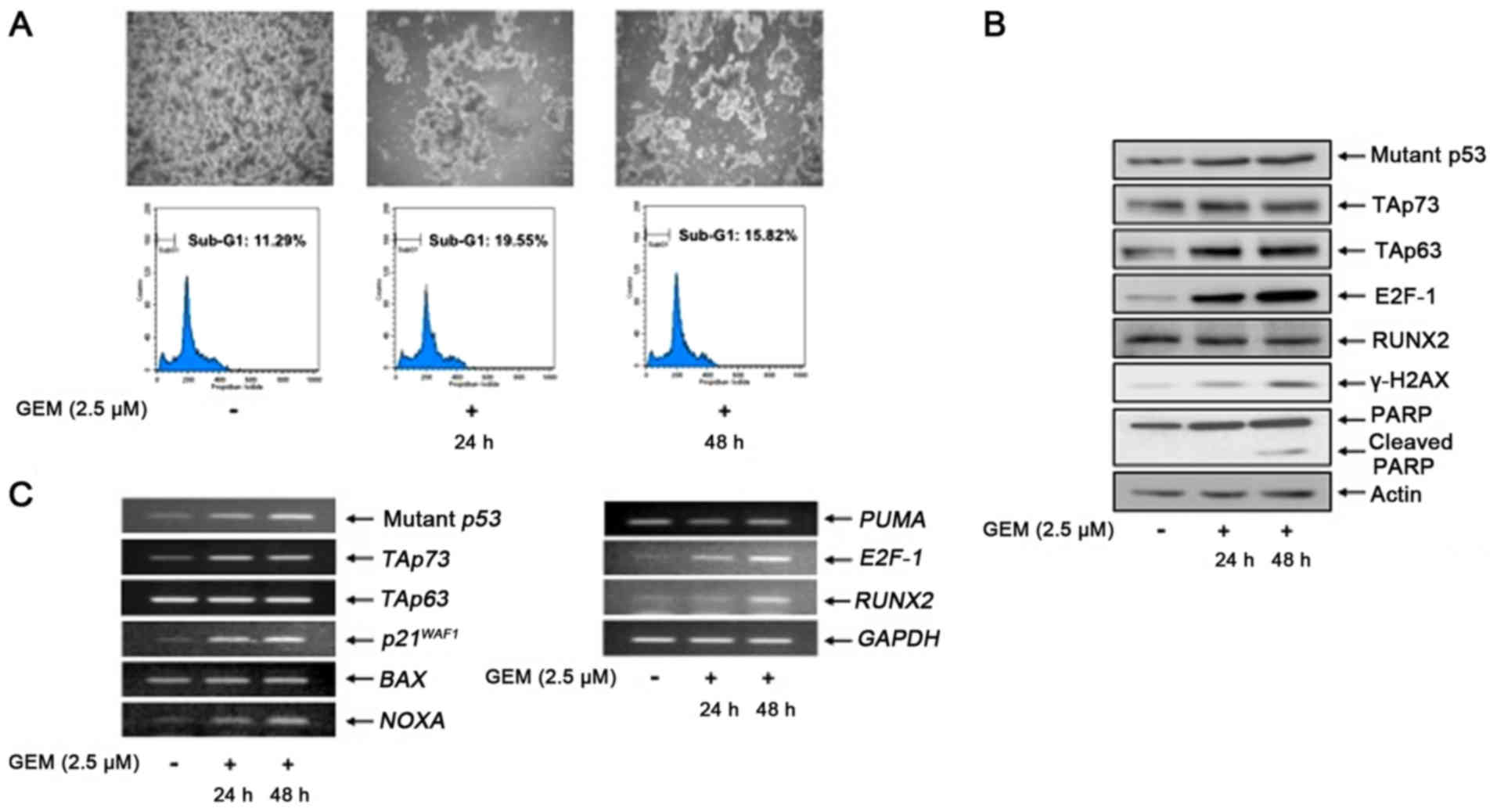
To gain an insight into understanding the molecular
basis behind the poor response of MiaPaCa-2 spheres to GEM, the
expression patterns of p53 family members and their target genes
were examined. As displayed in Fig.
6B, the amount of γH2AX, which is one of the reliable molecular
markers for DNA damage, was increased following GEM exposure in a
dose-dependent manner, indicating that MiaPaCa-2 spheres received
DNA damage. In addition, the proteolytic cleavage of PARP which may
be catalyzed by the activated caspase-3, was detectable at 48 h
after GEM treatment. For p53 family members, mutant p53, TAp73 and
TAp63 proteins were upregulated in response to GEM. Since E2F-1,
which acts as a transcriptional activator of TAp73 (23), was significantly induced following
GEM exposure, it is likely that E2F-1 is responsible for the
GEM-mediated induction of TAp73. Concurrently, RUNX2 remained
basically unchanged regardless of the GEM treatment. RT-PCR
analysis demonstrated that GEM treatment stimulated the
transcription of p53 family members (mutant p53 and
TAp73) as well as E2F-1 (Fig. 6C). By contrast, TAp63 was
unaffected by GEM. For p53 family-target genes, GEM-mediated
transcriptional activation of p21WAF1 and
NOXA was observed, whereas we did not detect a significant
change in the expression levels of BAX and PUMA in
response to GEM. As displayed in Fig.
6B, the amount of RUNX2 protein remained constant in the
presence or absence of GEM, whereas GEM treatment led to a marked
increase in RUNX2 transcription level. At present, we do not
know the molecular mechanism(s) underlying this discrepancy.
Based on these expression analyses, it is probable
that, despite GEM-mediated stimulation of TAp73/TAp63 expression,
poor response of MiaPaCa-2 spheres to GEM may be due to GEM-induced
further accumulation of mutant p53 and/or the constitutive
expression of RUNX2.
Knockdown of RUNX2 increases the GEM
sensitivity of MiaPaCa-2 spheres
Recently, we have revealed that RUNX2 knockdown
sensitizes MiaPaCa-2 cells to GEM under the conventional 2D
monolayer culture conditions (14).
To examine the possible impact of RUNX2 on the GEM sensitivity of
MiaPaCa-2 spheres, siRNA-mediated depletion of RUNX2 was performed.
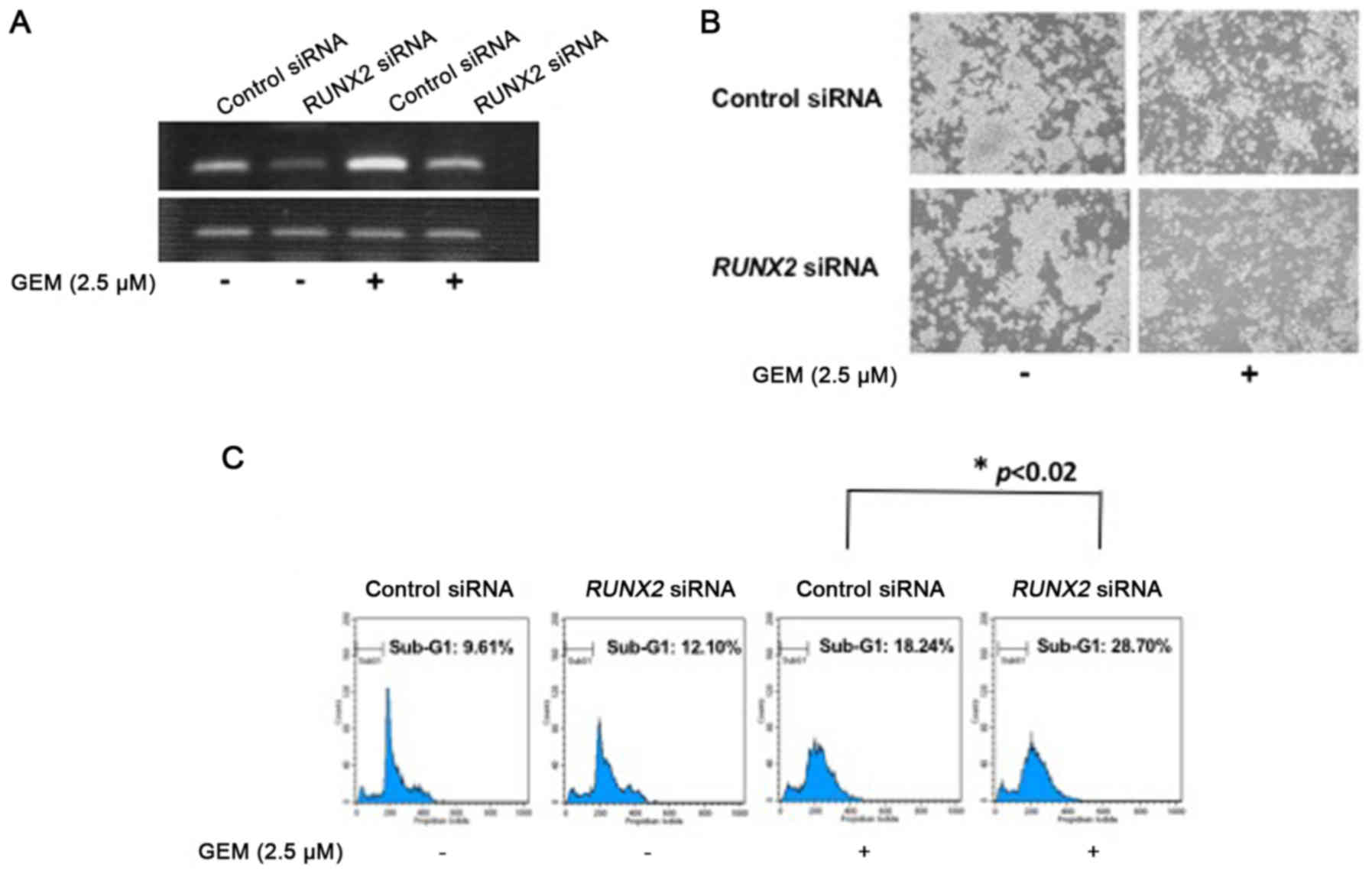
As displayed in Fig. 7A, RUNX2 gene
silencing was successful under our experimental conditions as
verified by RT-PCR analysis. Twenty-four hours after the siRNA
transfection, MiaPaCa-2 spheres were exposed to 2.5 µM of GEM or
left untreated. Forty-eight hours after treatment, representaive
images were captured and spheres were processed for the subsequent
FACS analysis. As clearly displayed in Fig. 7B, GEM-induced decrease in the number
of spheres and disruption of their multi-cellular structures were
augmented by RUNX2 depletion. Furthermore, FACS analysis
demonstrated that RUNX2 knockdown increased the number of cells
with sub-G1 DNA content of MiaPaCa-2 spheres in response
to GEM compared to non-silencing ones exposed to GEM (Fig. 7C). These results indicated that,
similar to 2D monolayer cultures (14), RUNX2 gene silencing increased the
sensitivity of MiaPaCa-2 spheres to GEM.
RUNX2 depletion causes further
augmentation of the GEM-mediated accumulation of TAp63 protein in
MiaPaCa-2 spheres
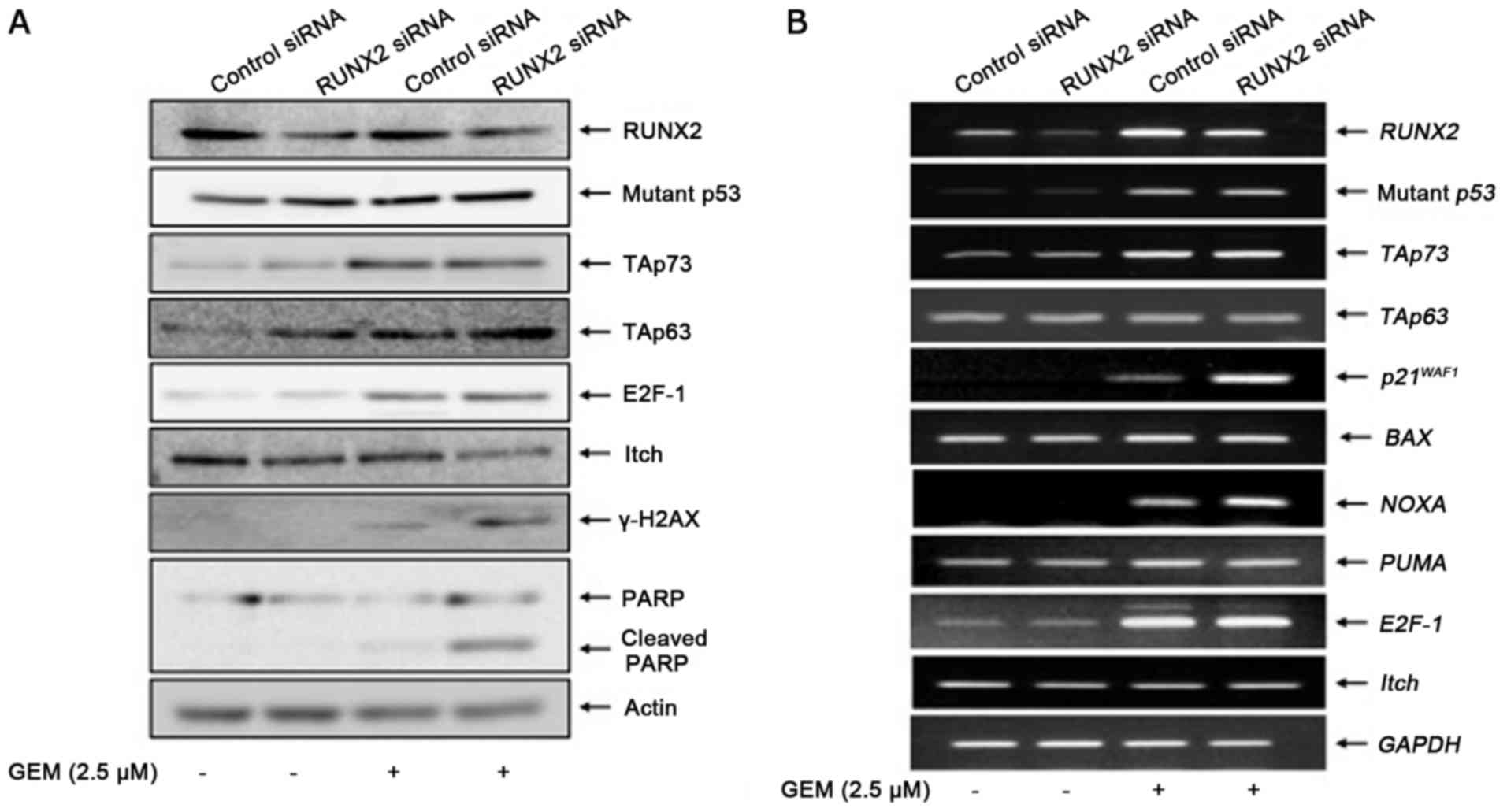
To investigate the molecular mechanism(s) by which
RUNX2 gene silencing increased the GEM sensitivity of MiaPaCa-2
spheres, we examined the expression patterns of the p53 family
members and their target genes in RUNX2-depleted spheres in
response to GEM. In support of the results obtained by FACS
analysis, GEM-mediated proteolytic cleavage of PARP was markedly
stimulated in RUNX2-silencing MiaPaCa-2 spheres (Fig. 8A). Notably, RUNX2 depletion
augmented GEM-induced accumulation of γH2AX, while, RUNX2 gene
silencing had a negligible effect on GEM-mediated induction of
mutant p53, TAp73 and E2F-1 proteins. Unlike TAp73 protein,
GEM-dependent promotion of the TAp63 protein expression was further
stimulated by RUNX2 knockdown. In accordance with these results,
Itch which acts as an E3 ubiquitin protein ligase for TAp63
(24), was downregulated in
RUNX2-silencing spheres exposed to GEM. RT-PCR analyses
demonstrated that GEM-mediated transcriptional activation of mutant
p53, TAp73 and E2F-1 was unaffected by RUNX2 depletion (Fig. 8B). Among the p53 family-target genes
that we examined, RUNX2 knockdown further stimulated GEM-induced
upregulation of p21WAF1 and NOXA. TAp63 and Itch
remained unchanged regardless of GEM treatment or RUNX2 depletion,
raising a possibility that TAp63 was regulated by RUNX2 at the
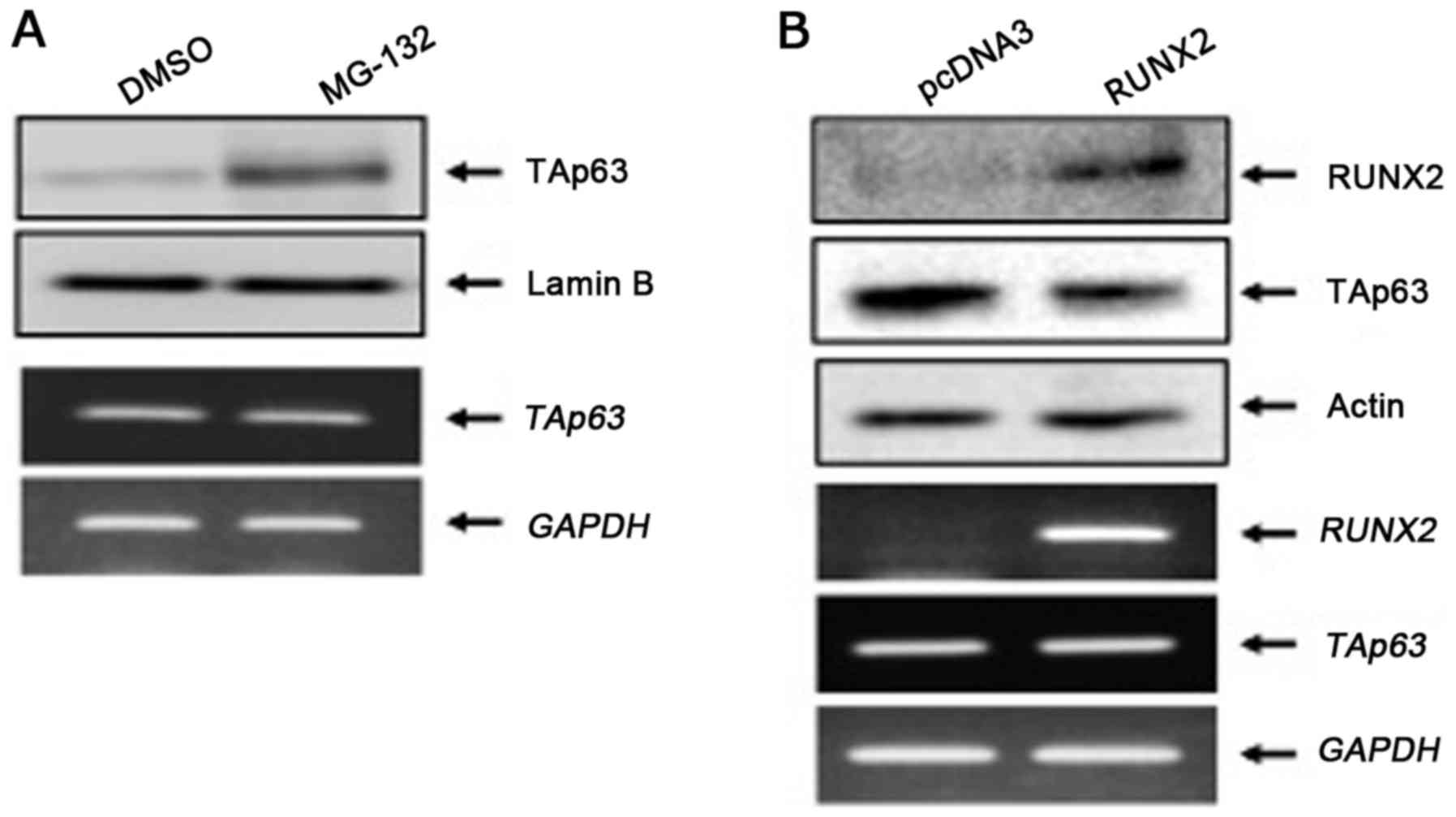
protein level under sphere culture. As displayed in Fig. 9A, TAp63 protein was stabilized in
MiaPaCa-2 spheres in the presence of proteasome inhibitor MG-132.
Notably, forced expression of RUNX2 in MiaPaCa-2 spheres reduced
TAp63 at the protein level but not at the mRNA level (Fig. 9B). Collectively, these results
demonstrated that knockdown of RUNX2 stabilized TAp63, potentiated
TAp63-dependent cell death pathway in MiaPaCa-2 spheres, thereby
increasing their GEM sensitivity.
Discussion
In the present study, we have demonstrated that,
similar to the conventional 2D cell culture model, RUNX2
gene silencing increased the GEM sensitivity of p53-mutated
pancreatic cancer MiaPaCa-2 cells under the non-adherent 3D sphere
system. Therefore, we propose that RUNX2 may serve as a
potential therapeutic target of pancreatic cancer.
According to our results, MiaPaCa-2 cells poorly
responded to GEM compared to SW1990 cells under the adherent 2D
culture conditions. Notably, MiaPaCa-2 cells efficiently formed
spheres, whereas SW1990 cells did not, indicating that p53
status is involved in the formation of pancreatic cancer cell
spheres. In support of this notion, p53-mutated pancreatic
cancer Panc-1 cells also generated the sphere structures under our
experimental conditions (data not shown). Recently, Ren et
al (25) described that
wild-type p53 suppressed epithelial-mesenchymal transition (EMT) of
prostate cancer cells through the downregulation and upregulation
of the expression of mesenchymal and epithelial markers,
respectively. Additionally, they have also revealed that prostate
cancer cell sphere formation was markedly suppressed by wild-type
p53. Alternatively, Cho et al (26) successfully generated spheres from
the adherent pancreatic cancer CFPAC-1 and CAPAN-1 cells bearing
p53 mutation in serum-free medium. Based on their
observations, CFPAC-1 spheres highly expressed pro-oncogenic PAUF
(pancreatic adenocarcinoma upregulated factor) relative to the
adherent CFPAC-1 cells and PAUF gene silencing resulted in a
decrease in the number of spheres and an enhancement of
chemotherapeutic response to GEM in association with downregulation
of MRP5 (multidrug resistant protein 5) and RPM2 (ribonucleotide
reductase M2). Similar results were also obtained in
p53-mutated pancreatic cancer BxPC-3 cells (27). Notably, Di Fiore et al
(28) described that ectopic
expression of mutant p53 (R248W) in osteosarcoma cells promoted
cancer stem cell (CSC)-like features such as sphere formation,
indicating that certain gain of function (GOF) mutants of p53
contributed to efficient sphere generation in the absence of serum.
Therefore, it is likely that certain GOF mutants of p53-induced
sphere-forming ability in malignant cancer cells may be one of
their strategies to survive under the severe environments such as
low nutrient. From our expression analyses, mutant p53 expression
was elevated in MiaPaCa-2 spheres relative to the adherent
MiaPaCa-2 cells accompanied by a significant reduction in
p53-target gene transcription such as p21WAF1,
BAX, PUMA and NOXA. Considering that mutant p53 acts
as a dominant-negative inhibitor against the other pro-apoptotic
p53 family members such as TAp73 and TAp63, it is possible that the
collaboration of mutant p53 with PAUF and/or mutant p53-dependent
inhibition of TAp73/TAp63 activity may participate in the
regulatory mechanism of sphere formation as well as GEM resistance.
Further experiments are required to adequately address this
issue.
As suggested by Kuo et al (22), 3D sphere cultures may reflect the
in vivo-like microenvironments more effectively than the
conventional 2D adherent ones. This sphere culture system takes
into account the critical interaction of cells with their neighbors
and their environments. According to our results, at most 20% of
MiaPaCa-2 spheres underwent cell death in response to GEM. The
adherent MiaPaCa-2 cells displayed a lower sensitivity to GEM than
the monolayer SW1990 cells. Under the sphere culture conditions,
GEM exposure resulted in an upregulation of mutant p53, TAp73,
TAp63 proteins and p53 family-target genes such as
p21WAF1 and NOXA, while, RUNX2 protein
level remained unchanged regardless of the GEM treatment. As
previously described (13,14), RUNX2 was capable of attenuating the
pro-apoptotic activity of TAp73/TAp63, therefore it is likely that,
in addition to mutant p53, RUNX2 prohibited TAp73/TAp63 activity
under non-adherent 3D cultures. In a good agreement with this
notion, RUNX2 gene silencing improved GEM sensitivity of
MiaPaCa-2 spheres in association with a further augmentation of
GEM-induced accumulation of TAp63 protein but not of TAp73 protein.
In contrast to MiaPaCa-2 spheres, RUNX2 depletion in
MiaPaCa-2 monolayer cultures increased their GEM sensitivity
through the potentiation of TAp73-dependent cell death pathway
(14). At present, we do not know
the precise molecular mechanisms behind RUNX2-mediated differential
regulation of TAp73/TAp63 under 2D and 3D sphere culture
conditions.
Since RUNX2 knockdown in MiaPaCa-2 spheres
did not affect the TAp63 transcription level following GEM
exposure, TAp63 may be regulated at the protein level. Indeed,
TAp63 protein was stabilized in MiaPaCa-2 spheres treated with
proteasome inhibitor, and the overexpression of RUNX2 in MiaPaCa-2
spheres reduced TAp63 at the protein level. Notably, Itch which
acts as an E3 ubiquitin protein ligase for TAp63 (24), was downregulated in
RUNX2-depleted MiaPaCa-2 spheres exposed to GEM, indicating
that the reduced expression of Itch facilitated the stabilization
of TAp63 protein. Levy et al (29) indicated that Itch promoter
contained the consensus RUNX-binding site, and RUNX1 stimulateds
the transcription of Itch. Based on our results, the
knockdown of RUNX2 had an undetectable effect on Itch
mRNA level, indicative that, unlike RUNX1, RUNX2 may regulate Itch
at the protein level but not at the mRNA level. As proposed
(30), Itch was also a potential
molecular target of chemotherapy.
Another finding of the present study was that
RUNX2 gene silencing further induced the accumulation of
γH2AX in response to GEM. Since ATM-mediated phosphorylation of
histone variant H2AX is one of the early molecular events of DNA
damage response, RUNX2 may participate in the initial regulatory
mechanisms of DNA damage response. Recently, we have also observed
a similar phenomenon under 2D cultures demonstrating that depletion
of RUNX2 in MiaPaCa-2 cells increased the accumulation of
γH2AX following GEM or SAHA exposure (14,31),
indicating that RUNX2 attenuated the expansion of DNA lesions
irrespective of 2D or 3D culture systems. Notably, Yang et
al (32) described that RUNX2
binds to γH2AX and siRNA-mediated knockdown of RUNX2
increases γH2AX in response to UV. Further studies are required to
elucidate the functional significance of RUNX2/γH2AX interaction
during DNA damage response.
In conclusion, the results of the present study
demonstrated that, similar to 2D cultures, RUNX2 depletion
improved GEM sensitivity of MiaPaCa-2 spheres, and thus strongly
indicated that RUNX2 gene silencing may provide a promising
strategy for the treatment of GEM-resistant pancreatic cancer with
p53 mutation.
Acknowledgements
We thank Dr Toshiko Yoshida, Dr Motonori Okabe and
Dr Chika Soko for their valuable discussions.
Funding
The present study was supported in part by a grant
from the Ministry of Education, Culture, Sports, Science and
Technology of Japan (no. 23501278).
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
TO and TN organized the present study. MS, MN, TO
and DS performed the experiments. OS contributed to the
interpretation of the results. MS and TO were the majorcontributors
in writing the manuscript. All authors read and approved the
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
DTT
|
dithiothreitol
|
|
EGF
|
epidermal growth factor
|
|
EMT
|
epithelial-mesenchymal transition
|
|
bFGF
|
fibroblast growth factor-basic
|
|
GEM
|
gemcitabine
|
|
MRP5
|
multidrug resistant protein 5
|
|
PARP
|
poly (ADP-ribose) polymerase
|
|
PI
|
propidium iodide
|
|
RPM2
|
ribonucleotide reductase M2
|
|
RUNX2
|
runt-related transcription factor
2
|
|
siRNA
|
small interfering RNA
|
|
TBS
|
Tris-buffered saline
|
References
|
1
|
Vincent A, Herman J, Schulick R, Hruban RH
and Goggins M: Pancreatic cancer. Lancet. 378:607–620. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Neesse A, Algül H, Tuveson DA and Gress
TM: Stromal biology and therapy in pancreatic cancer: A changing
paradigm. Gut. 64:1476–1484. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Burris HA III, Moore MJ, Andersen J, Green
MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo
AM, Tarassoff P, et al: Improvements in survival and clinical
benefit with gemcitabine as first-line therapy for patients with
advanced pancreas cancer: A randomized trial. J Clin Oncol.
15:2403–2413. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ryan DP, Hong TS and Bardeesy N:
Pancreatic adenocarcinoma. N Engl J Med. 371:1039–1049. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
von Hoff DD, Ervin T, Arena FP, Chiorean
EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et
al: Increased survival in pancreatic cancer with nab-paclitaxel
plus gemcitabine. N Engl J Med. 369:1691–1703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cicenas J, Kvederaviciute K, Meskinyte I,
Meskinyte-Kausiliene E, Skeberdyte A and Cicenas J: KRAS, TP53,
CDKN2A, SMAD4, BRCA1, and BRCA2 mutations in pancreatic cancer.
Cancers. 9:E422017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Komori T, Yagi H, Nomura S, Yamaguchi A,
Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, et al:
Targeted disruption of Cbfa1 results in a complete lack of
bone formation owing to maturational arrest of osteoblasts. Cell.
89:755–764. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Otto F, Thornell AP, Crompton T, Denzel A,
Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen
BR, et al: Cbfa1, a candidate gene for cleidocranial
dysplasia syndrome, is essential for osteoblast differentiation and
bone development. Cell. 89:765–771. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Franceschi RT and Xiao G: Regulation of
the osteoblast-specific transcription factor, Runx2: Responsiveness
to multiple signal transduction pathways. J Cell Biochem.
88:446–454. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chimge NO and Frenkel B: The RUNX family
in breast cancer: Relationships with estrogen signaling. Oncogene.
32:2121–2130. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cohen-Solal KA, Boregowda RK and Lasfar A:
RUNX2 and the PI3K/AKT axis reciprocal activation as a driving
force for tumor progression. Mol Cancer. 14:1372015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ozaki T, Wu D, Sugimoto H, Nagase H and
Nakagawara A: Runt-related transcription factor 2 (RUNX2) inhibits
p53-dependent apoptosis through the collaboration with HDAC6 in
response to DNA damage. Cell Death Dis. 4:e6102013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sugimoto H, Nakamura M, Yoda H, Hiraoka K,
Shinohara K, Sang M, Fujiwara K, Shimozato O, Nagase H and Ozaki T:
Silencing of RUNX2 enhances gemcitabine sensitivity of
p53-deficient human pancreatic cancer AsPC-1 cells through
the stimulation of TAp63-mediated cell death. Cell Death Discov.
6:e19142015. View Article : Google Scholar
|
|
14
|
Nakamura M, Sugimoto H, Ogata T, Hiraoka
K, Yoda H, Sang M, Sang M, Zhu Y, Yu M, Shimozato O and Ozaki T:
Improvement of gemcitabine sensitivity of p53-mutated
pancreatic cancer MiaPaCa-2 cells by RUNX2
depletion-mediated augmentation of TAp73-dependent cell death.
Oncogenesis. 5:e2332016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ozaki T, Nakamura M, Ogata T, Sang M, Yoda
H, Hiraoka K, Sang M and Shimozato O: Depletion of pro-oncogenic
RUNX2 enhances gemcitabine (GEM) sensitivity of p53-mutated
pancreatic cancer Panc-1 cells through the induction of
pro-apoptotic TAp63. Oncotarget. 7:71937–71950. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vousden KH and Lu X: Live or let die: The
cell's response to p53. Nat Rev Cancer. 2:594–604. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Melino G, De Laurenzi V and Vousden KH:
p73: Friend or foe in tumorigenesis. Nat Rev Cancer. 2:605–615.
2002. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hollstein M, Sidransky D, Vogelstein B and
Harris CC: p53 mutations in human cancers. Science. 253:49–53.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Reynolds BA and Weiss S: Generation of
neurons and astrocytes from isolated cells of the adult mammalian
central nervous system. Science. 255:1707–1710. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hickman JA, Graeser R, de Hoogt R, Vidic
S, Brito C, Gutekunst M and van der Kuip H: IMI PREDECT Consortium:
Three-dimensional models of cancer for pharmacology and cancer cell
biology: Capturing tumor complexity in vitro/ex vivo.
Biotechnol J. 9:1115–1128. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Deng L, Li D, Gu W, Liu A and Cheng X:
Formation of spherical cancer stem-like cell colonies with
resistance to chemotherapy drugs in the human malignant fibrous
histiocytoma NMFH-1 cell line. Oncol Lett. 10:3323–3331. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kuo CT, Wang JY, Lin YF, Wo AM, Chen BPC
and Lee H: Three-dimensional spheroid culture targeting versatile
tissue bioassays using a PDMS-based hanging drop array. Sci Rep.
7:43632017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lissy NA, Davis PK, Irwin M, Kaelin WG and
Dowdy SF: A common E2F-1 and p73 pathway mediates cell death
induced by TCR activation. Nature. 407:642–645. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rossi M, Aqeilan RI, Neale M, Candi E,
Salomoni P, Knight RA, Croce CM and Melino G: The E3 ubiquitin
ligase Itch controls the protein stability of p63. Proc Natl Acad
Sci USA. 103:12753–12758. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ren D, Wang M, Guo W, Zhao X, Tu S, Huang
S, Zou X and Peng X: Wild-type p53 suppresses the
epithelial-mesenchymal transition and stemness in PC-3 prostate
cancer cells by modulating miR-145. Int J Oncol. 42:1473–1481.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cho JH, Kim SA, Park SB, Kim HM and Song
SY: Suppression of pancreatic adenocarcinoma upregulated factor
(PAUF) increases the sensitivity of pancreatic cancer to
gemcitabine and 5FU, and inhibits the formation of pancreatic
cancer stem like cells. Oncotarget. 8:76398–76407. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gao CC, Xu XL, Li F, Gong BG, Liu S, Cui
YQ, Sun HC, Xu PY, Zheng YM and Jiang H: Silencing pancreatic
adenocarcinoma upregulated factor (PAUF) increases the sensitivity
of pancreatic cancer cells to gemcitabine. Tumour Biol. 37:555–564.
2016. View Article : Google Scholar
|
|
28
|
Di Fiore R, Marcatti M, Drago-Ferrante R,
D'Anneo A, Giuliano M, Carlisi D, De Blasio A, Querques F, Pastore
L, Tesoriere G and Vento R: Mutant p53 gain of function can be at
the root of dedifferentiation of human osteosarcoma MG63 cells into
3AB-OS cancer stem cells. Bone. 60:198–212. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Levy D, Reuven N and Shaul Y: A regulatory
circuit controlling Itch-mediated p73 degradation by Runx. J Biol
Chem. 283:27462–27468. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hansen TM, Rossi M, Roperch JP, Ansell K,
Simpson K, Taylor D, Mathon N, Knight RA and Melino G: Itch
inhibition regulates chemosensitivity in vitro. Biochem
Biophys Res Commun. 361:33–36. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ogata T, Nakamura M, Sang M, Yoda H,
Hiraoka K, Yin D, Sang M, Shimozato O and Ozaki T: Depletion of
runt-related transcription factor 2 (RUNX2) enhances SAHA
sensitivity of p53-mutated pancreatic cancer cells through
the regulation of mutant p53 and TAp63. PLoS One. 12:e01798842017.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang S, Quaresma AJ, Nickerson JA, Green
KM, Shaffer SA, Imbalzano AN, Martin-Buley LA, Lian JB, Stein JL,
van Wijnen AJ and Stein GS: Subnuclear domain proteins in cancer
cells support the functions of RUNX2 in the DNA damage response. J
Cell Sci. 128:728–740. 2015. View Article : Google Scholar : PubMed/NCBI
|