Introduction
Lung cancer is one of the most common cancers
worldwide and is associated with increased morbidity and mortality.
Most patients are diagnosed with lung cancer at the advanced and
non-curable stage since there are no obvious symptoms in the early
stages. Exploring novel genes and epigenetic mechanisms that are
involved in lung tumorigenesis and identification of potential
targets or therapeutic interventions have become increasingly
urgent in cancer research (1).
Interleukin-13 (IL-13) is a proinflammatory cytokine which
regulates immune responses and the microenvironment in cancer as
well as orchestrating normal and abnormal cell behaviors (2,3). The
receptor subunits of IL-13, IL-13Rα1 and IL-13Rα2, are found to be
overexpressed in many cancer types (4–7). Ying
Yang 1 (YY1) is a member of the GLI-Krüppel family of transcription
factors and is widely distributed in eukaryotic cells (8). YY1 activates the expression levels of
other transcription factors such as c-Myc by directly
binding to their promoters and functions as an oncogene (9). Controversially, YY1 has been shown to
inhibit cell proliferation in breast cancer, indicating its
differential roles in different tissues. Previous reports have
demonstrated that IL-13 and YY1 are associated with the PI3K/AKT
signaling pathway (10–13). However, how IL-13 and YY1 regulate
the PI3K/AKT pathway in lung cancer is currently unclear.
Recently, microRNAs (miRNAs) are found to be
involved in every step of tumor progression, including
proliferation, apoptosis, angiogenesis and metastasis (14). miRNAs are endogenous non-coding RNAs
with short hairpin structures found in eukaryotes. They can
complementarily bind with the 3′UTR region of target mRNAs, thus
inhibiting mRNA translation and inducing mRNA degradation. miRNAs
can function as oncogenes known as oncomiRs, and oncomiRs are found
to be overexpressed in malignant tumors and play critical roles in
mediating tumor progression. miRNAs can also function as tumor
suppressors in the reciprocal by suppressing oncogene expression in
cancer cells, but their expression levels are generally
downregulated in tumors (14).
Recently, miRNAs are suggested for their use in new therapeutic
approaches, such as exogenous introduction of tumor suppressive
miRNAs in the clinic. Recently, the miR-29a/b/c family was shown to
have inhibitory roles in lung cancer progression (15–17). A
previous study revealed that miR-29 promoted stem cell
differentiation by targeting YY1 in smooth muscle cells, and showed
the potential regulation of YY1 by miR-29a in cancer stem cells
(16). Other studies have
demonstrated that under regulation of NF-κB, YY1 was inhibited by
miR-29a in smooth muscle cells (15). Since YY1 plays an important role in
mediating IL-13-induced lung cancer progression, how miR-29a is
involved in IL-13-induced lung cancer cell invasion, and how
miR-29a executes its role as tumor suppression remain unclear.
In the present study, we aimed to investigate the
role of miR-29a in cell invasion mediated by IL-13 in lung cancer.
We investigated how miR-29a is involved in the IL-13/PI3K/AKT/YY1
pathway in lung tumorigenesis, and we showed whether miR-29a can
act as the potential therapeutic target in lung cancer.
Materials and methods
Cell culture and drug treatment
Human lung adenocarcinoma cell line A549 was
purchased from Shanghai Cell Bank, Chinese Academy of Sciences
(Shanghai, China). A549 cells were cultured in Dulbecco's modified
Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), which contained 10% fetal bovine serum (FBS),
100 µg/ml penicillin and 50 µg/ml streptomycin at 37°C in an
incubator with 5% CO2. A549 cells were serum-starved for
24 h, and were then treated with IL-13 (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) at different concentrations or for specified
hours to investigate its functions. In addition, pretreatment with
40 µM PI3K/AKT pathway inhibitor LY294002 (Sigma-Aldrich; Merck
KGaA) was also implemented in our studies.
Real-time quantitative PCR
The total RNA was extracted using TRIzol reagent
(Sigma-Aldrich; Merck KGaA). Spectrometer and agarose
electrophoresis were used to measure the RNA concentration and
detect whether or not RNA was degraded. The total RNA was
reverse-transcribed to cDNA and the oligo(dT) was used as a primer
(Reverse Transcription Kit cat. no. AH401-01; Beijing Transgen
Biotech Co., Ltd., Beijing, China). The amplification and detection
were performed using Applied Biosystems 7500 Real-Time PCR Systems
(Thermo Fisher Scientific, Inc.). The thermocycling conditions were
set as follows: 95°C for 10 min followed by 40 cycles of 95°C for
15 sec and 60°C for 1 min. The primers for GAPDH and YY1 gene were
designed by Primer Premier 5.0 software (Premier Biosoft
International, Palo Alto, CA, USA) and were synthesized by Takara
Bio (Shiga, Japan) (Table I). The
relative expression quantities of the target genes were evaluated
using the 2−ΔΔCt method and mRNA and miRNA were
normalized to the expression quantity of GAPDH and U6 snRNA,
respectively.
 | Table I.Primer sequences. |
Table I.
Primer sequences.
| Gene | Primer | Sequence
(5′-3′) |
|---|
| GAPDH | Forward |
CGGAGTCAACGGATTTGGTCGTAT |
|
| Reverse |
AGCCTTCTCCATGGTGGTGAAGAC |
| YY1 | Forward |
GAAGCCCTTTCAGTGCACGTT |
|
| Reverse |
ACATAGGGCCTGTCTCCGGTAT |
| miR-29a | Forward |
CGCGGATCCATGGTTAAAGAGCCCAATGTATGCTG |
|
| Reverse |
CCCAAGCTTTCAGTATAACCATTCATGATATGCTAA |
| U6 | Forward |
CTCGCTTCGGCAGCACA |
|
| Reverse |
AACGCTTCACGAATTTGCGT |
Western blot analysis
The expression levels of YY1, AKT, pAKT and
N-cadherin were examined by western blot assay. Cellular proteins
were extracted using RIPA protein lysate (Applygen Technologies
Inc., Beijing, China). The protein concentration was detected by
the BCA assay (Beyotime Institute of Biotechnology, Shanghai,
China). An equal amount of proteins for each group was loaded,
separated by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE, 15%), and then transferred onto
polyvinylidene fluoride membranes (Beyotime Institute of
Biotechnology), and blocked with 5% skim milk. Membranes were
incubated with primary antibodies: YY1 (cat. no. ab10923; Abcam,
Cambridge, MA, USA), AKT (cat. no. C67E7; Cell Signaling
Technology, Danvers, MA, USA), pAKT Ser473 (cat. no. 9271; Cell
Signaling Technology) and N-cadherin (cat. no. D4R1H; Cell
Signaling Technology) (diluted by 1:2,000) and β-actin antibody
(cat. no. ZB2305; ZEGB-Bio Ltd., Beijing, China), respectively, at
4°C overnight. Membranes were washed with TBST 3 times (10 min
each) and incubated with secondary antibodies (diluted by 1:5,000),
HRP-labeled goat anti-mouse IgG (H+L) and HRP-labeled goat
anti-rabbit IgG (H+L), (cat. no. ZB2301; ZSGB-BIO) at room
temperature for 2 h. After that, the membranes were washed again
with TBST another 3 times (10 min each). The Western blot substrate
kit (Biovision, Inc., Milpitas, CA, USA) was used to detect protein
bands. The band intensities in the western blots were determined by
ImageJ software (version 1.51; NIH, Bethesda, MD, USA).
Cell transfection and construction of
stable cell line
A549 cells were transfected with the miR-29a
plasmid, miR-29a inhibitor and miRNA NC sequences (Table II) that were all purchased from
Shanghai GenePharma Technology Ltd., Co. Transfection was carried
out using Invitrogen Lipofectamine 2000 (Thermo Fisher Scientific,
Inc.) and cells were cultured at 37°C in an incubator with 5%
CO2. Transfection efficiency was observed under a
fluorescence microscope after 4–8 h and the complete medium was
replaced. Then cell culture was continued for 24–48 h. To establish
a stable transfected RNAi A549 cell line, the cells were
transferred to a 10-cm Petri dish to grow. Blasticidin was added to
select transfected cells. Then the medium was replaced every 3–4
days and the appropriate Blasticidin S was added until the
formation of a resistant monoclonal colony, which took
approximately 14 days. In each group, at least 10 drug-resistant
clones were selected for amplification.
| Table II.Sequences of miR-29a. |
Table II.
Sequences of miR-29a.
| Plasmid | Sequence
(5′-3′) |
|---|
| NC |
AAATGTACTGCGCGTGGAGAC |
| Mimic |
TAGCACCATCTGAAATCGGTTA |
| Inhibitor |
TAACCGATTTCAGATGGTGCTA |
MTT assay
Cell proliferation was evaluated by
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT)
(Sigma-Aldrich; Merck KGaA) assays. The cells need to be measured
were inoculated into 96-well plates with ~5×103
cells/well and each group had 5 replicates. MTT (20 µl, 5 mg/ml)
(Sigma-Aldrich; Merck KGaA) was placed into each well and the
culture process was sustained for 4 h at 37°C in an incubator with
5% CO2. Subsequently, dimethyl sulfoxide (DMSO; 100 µl)
was added into each well and the cells were gently shaken for ~10
min to dissolve the crystals. Samples were detected using a
microplate reader (Thermo Fisher Scientific, Inc.) at a wavelength
of 490 nm.
Wound-healing assay
The scratch wound-healing assay was used to assess
A549 cell migration. Firstly, three uniform transverse lines were
painted at the back of 6-well plates by a marker; and then
5×105 cells were seeded into plates and cultured
overnight. When the cells achieved adherence, they were starved
with medium free of serum for 12 h. Then, the medium was removed,
and sterile tips (200 µl) were used to draw a line which was
vertical to the back-transverse lines, followed by washing the
cells twice with phosphate-buffered saline (PBS). Migration
distances of cells were imaged and monitored using an inverted
microscope (Carl Zeiss AG, Oberkochen, Germany) with 3 randomly
selected fields in the wounded region at 0, 24, 48 and 72 h. The
percentage of wound width was calculated as: Wound width at 0, 24,
48 or 72 h divided by the original wound width measured at 0 h.
Transwell invasion assay
Transwell invasion assay was performed using 24-well
Transwell plates with 8-µm pores and a coating of Matrigel (100
µl/well) (BD Biosciences, San Jose, CA, USA) to measure the
invasion status of A549 cells. In addition, 2×104 cells
were placed in the upper Transwell chamber. Then, 600 µl complete
DMEM was added in the lower chamber as the chemo-attractant. After
cells were incubated for 24 h at 37°C, non-invasive cells in the
top chamber were removed with cotton swabs. Invasive cells at the
bottom of the membrane were fixed with methanol and then were
stained with crystal violet. The number of invasive cells was
counted by hemocytometry to assess the invasion status of the
cells.
Flow cytometry
A549 cells were plated into 6-well tissue culture
plates at ~2×104/well, each group has three duplicates.
After incubation, they were harvested in 1.5 ml EP tubes and washed
with PBS. Binding buffer (500 µl) was used to resuspend the cells.
Then Annexin-FITC and propidium iodide (PI), each 5 µl, were added
in EP tubes, being fully mixed; followed by incubation for 15 min
at room temperature in the dark. Data acquisition was performed
with FACSCalibur flow cytometry (Becton Dickinson, San Jose, CA,
USA).
Statistical analysis
Data presented as bar graphs are the means ± SD/SED
of at least three independent experiments. The statistical
significance of data was analyzed by one-way ANOVA through SPSS
20.0 software (IBM Corp., Armonk, NY, USA). Two-sided P<0.05 was
defined as the cut-off value for evaluating statistical
significance.
Results
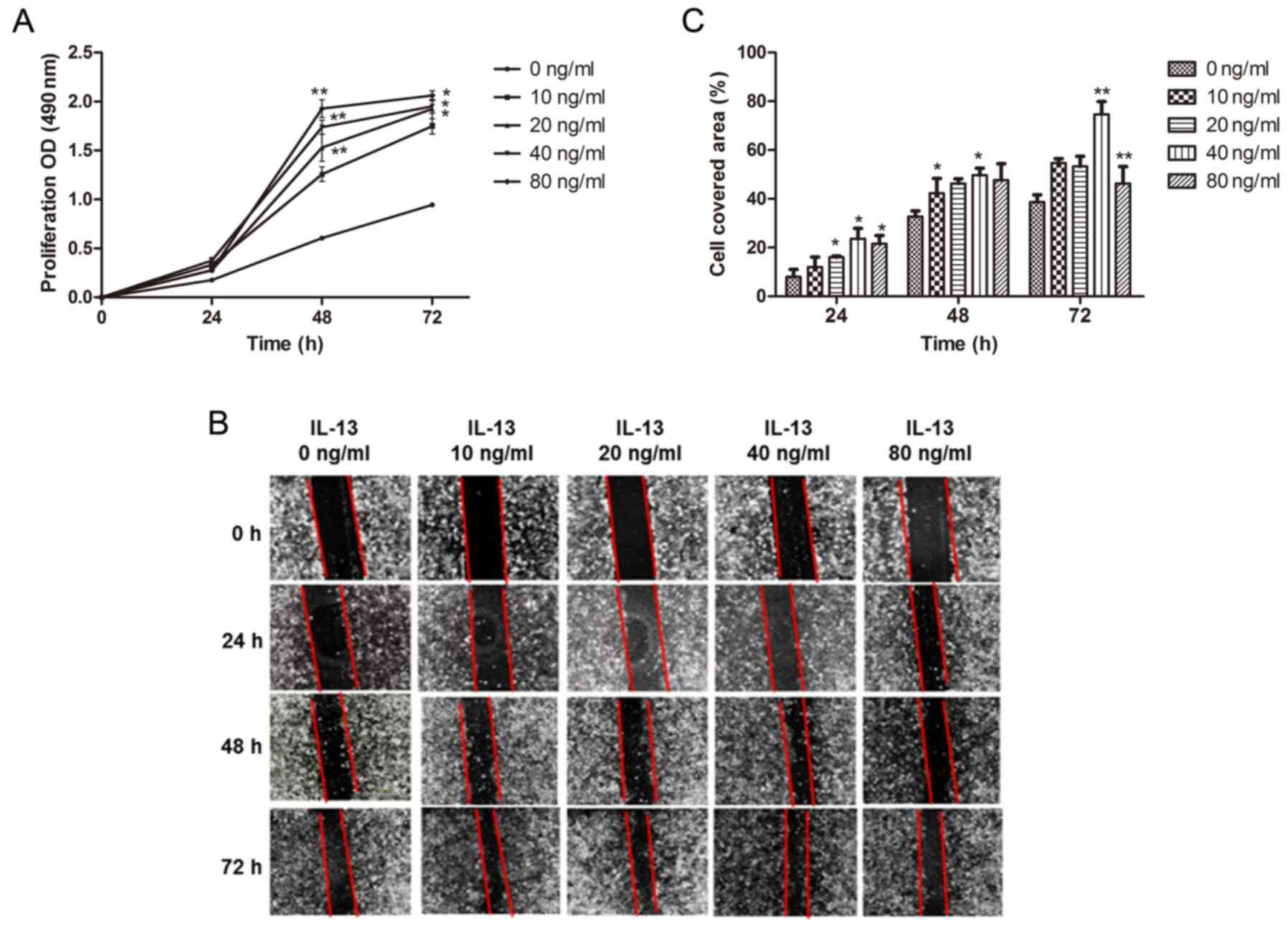
IL-13 promotes the proliferation and
migration of A549 cells
A549 cells were stimulated with different
concentrations of IL-13 (0, 10, 20, 40 and 80 ng/ml) for 24–72 h.
Compared to the control group (0 ng/ml), proliferation of A549
cells was accelerated after being treated with IL-13, and the
concentration of IL-13 at 40 ng/ml showed the most significant
induction (P<0.01, 48 h; P<0.05, 72 h) (Fig. 1A). We found that IL-13 promoted the
migration of A549 cells, and the optimal concentration of IL-13 at
40 ng/ml was used and it exhibited the strongest ability to induce
cell migration, while the concentration of IL-13 at 80 ng/m only
showed a slight increase in cell migration (Fig. 1B and C). These results indicated
that IL-13 promoted proliferation and migration of A549 cells in a
concentration- and time-dependent manner.
IL-13 activates the PI3K/AKT/YY1
pathway in A549 cells
To investigate the mechanism underlying the
induction of cell proliferation and migration by IL-13 in A549
cells, we examined the regulation of the PI3K/AKT pathway and the
transcription factor YY1 by IL-13. The PI3K inhibitor LY294002 was
used to block the PI3K/AKT pathway and to examine the YY1
expression level in this pathway. Our data determined that IL-13
stimulation of all concentrations (with 40 ng/ml the strongest)
promoted YY1 expression at the mRNA level compared with the control
group (P<0.01) (Fig. 2A). We
further examined the protein expression level of YY1 by western
blot analysis, and it showed that YY1 and pAKT protein levels were
increased after IL-13 stimulation compared with the control group.
Consistently, the protein expression levels of YY1 and pAKT
indicated highest induction following treatment with IL-13 at 40
ng/ml in the A549 cells (P<0.05) (Fig. 2B and C). The mRNA level of YY1
showed highest induction after being treated with 40 ng/ml IL-13
stimulation for 12 h (Fig. 2D and
E). This showed that IL-13 can upregulate YY1 expression level
in A549 cells and this induction was correlated with the
concentration of IL-13 and time period. The increased level of AKT
phosphorylation in A549 cells treated with IL-13 demonstrated
activation of the PI3K/AKT pathway triggered by IL-13 (Fig. 2E and F). To determine whether YY1
expression level was regulated by the PI3K/AKT pathway, we used 40
µM LY294002 in addition to IL-13 (40 ng/ml) to stimulate the A549
cells in the experimental group. Following inhibition of the
PI3K/AKT pathway, we did not observe the induction of YY1 at both
the mRNA and protein level after IL-13 treatment in the lung cancer
cells. This indicated that upregulation of YY1 by IL-13 was
mediated by activation of the PI3K/AKT pathway (Fig. 2G-I).
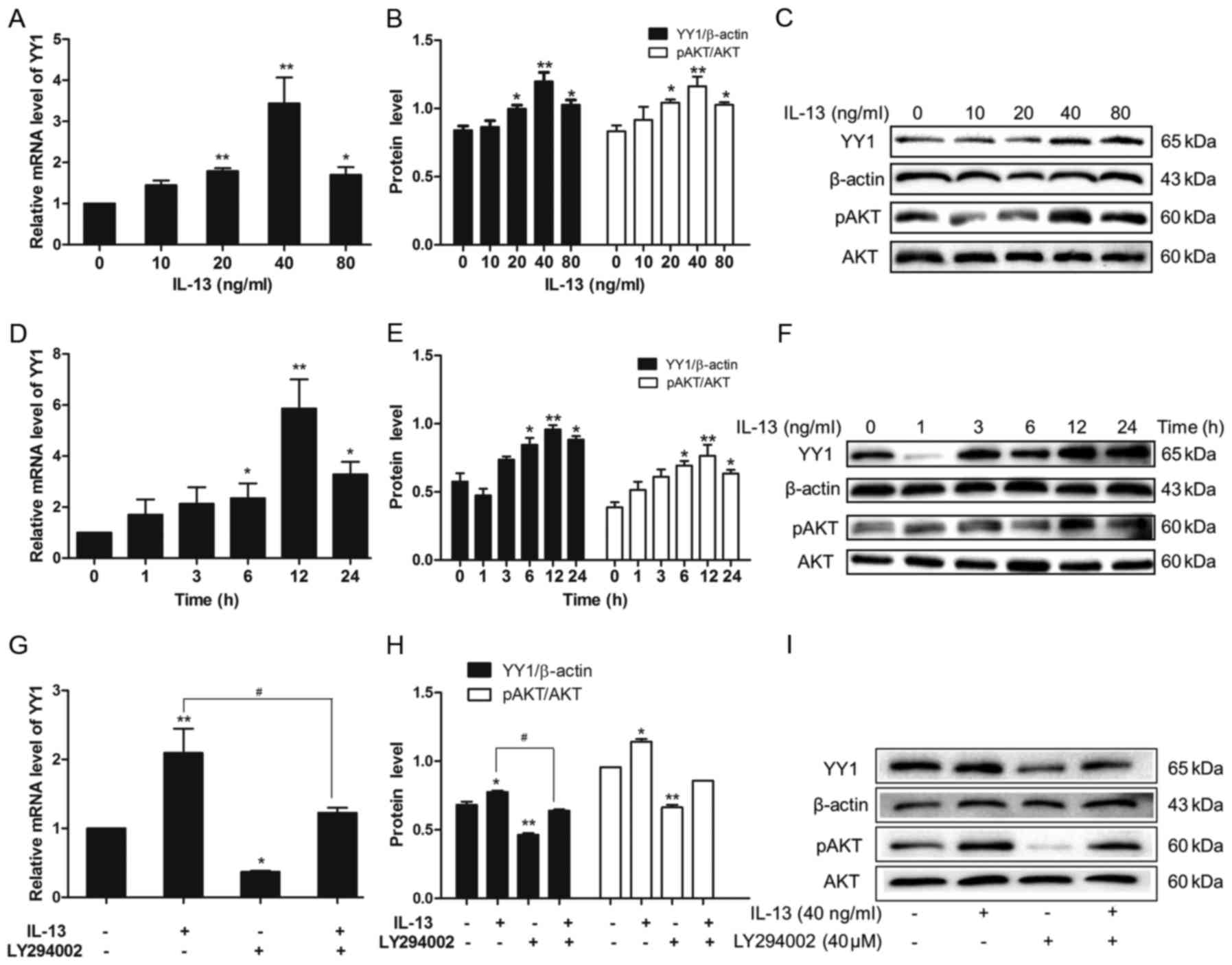 | Figure 2.Effect of IL-13 on the PI3K/AKT/YY1
pathway in A549 cells. (A) qPCR was applied to detect YY1 at the
mRNA level. The mRNA level of YY1 was promoted after IL-13
stimulation; and the effect was highest at 40 ng/ml IL-13. (B and
C) Western blot analysis was applied to detect YY1 and pAKT protein
levels. As analyzed by ImageJ software, the protein levels of YY1
and pAKT were promoted after IL-13 stimulation; these effects were
highest at 40 ng/ml IL-13 (*P<0.05, **P<0.01, compared with
the 0 ng/ml group). (D) The mRNA level of YY1 was promoted after
the stimulation of 40 ng/ml IL-13 for 24 h while at 12 h this
effect was the highest. (E and F) As analyzed by ImageJ software,
the protein levels of YY1 and pAKT were promoted after IL-13
stimulation while the effect at 12 h was the highest (*P<0.05,
**P<0.01, compared with the 0 h group). (G) Addition of LY294002
inhibited YY1 expression at the mRNA level. (H and I) As analyzed
by ImageJ software, the protein levels of YY1 and pAKT were
inhibited by LY294002 [*P<0.05, **P<0.01, blank group;
#P<0.05, IL-13 group by IL-13(+) LY294002(−)/IL-13(−)
LY294002(−) and IL-13(+) LY294002(+)/IL-13(−) LY294002(+)]. In G
and H and Fig. 3C, background
signals in the YY1 expression and cell migration are at an unequal
level when compared treatment with LY294002 (−) and (+). |
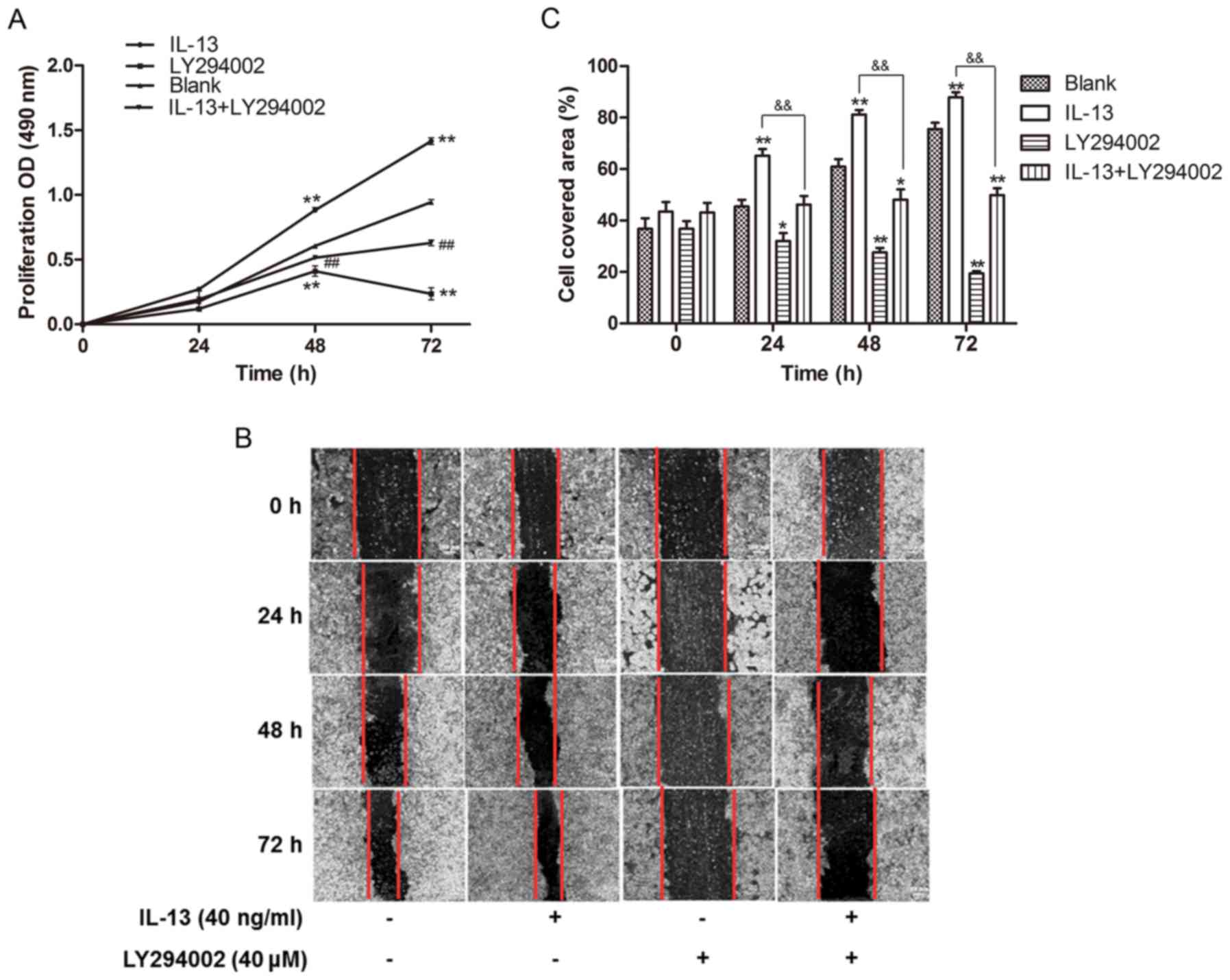
IL-13 promotes the proliferation and
migration of A549 cells through the PI3K/AKT/YY1 pathway
To determine whether IL-13-induces cell
proliferation and migration in lung cancer depends on activation of
the PI3K/AKT signaling pathway, we co-treated A549 cells with
LY294002 (40 µM) and IL-13 (40 ng/ml) for 24–72 h. Our results
showed that treatment with IL-13 induced cell proliferation, but
blocking the PI3K/AKT pathway by LY294002 inhibited cell growth in
the A549 cells (P<0.01) (Fig.
3A). IL-13 stimulation activated PI3K/AKT signaling in the A549
cells and significantly promoted cell migration as determined by a
wound-healing assay (Fig. 3B and
C). We further assessed the cell migration effects induced by
IL-13 by blocking the PI3K/AKT pathway using LY294002. We observed
a reduced cell covered area in the LY294002 group, which suggested
that inhibition of the PI3K/AKT pathway weakened the migratory
ability in original A549 cells comparing to the control group.
Deactivating the PI3K/AKT pathway impaired cell migration induced
by IL-13 in the A549 cells (P<0.01) (Fig. 3B and C), which indicated that IL-13
induced cell proliferation and migration mainly by activating the
PI3K/AKT signaling pathway.
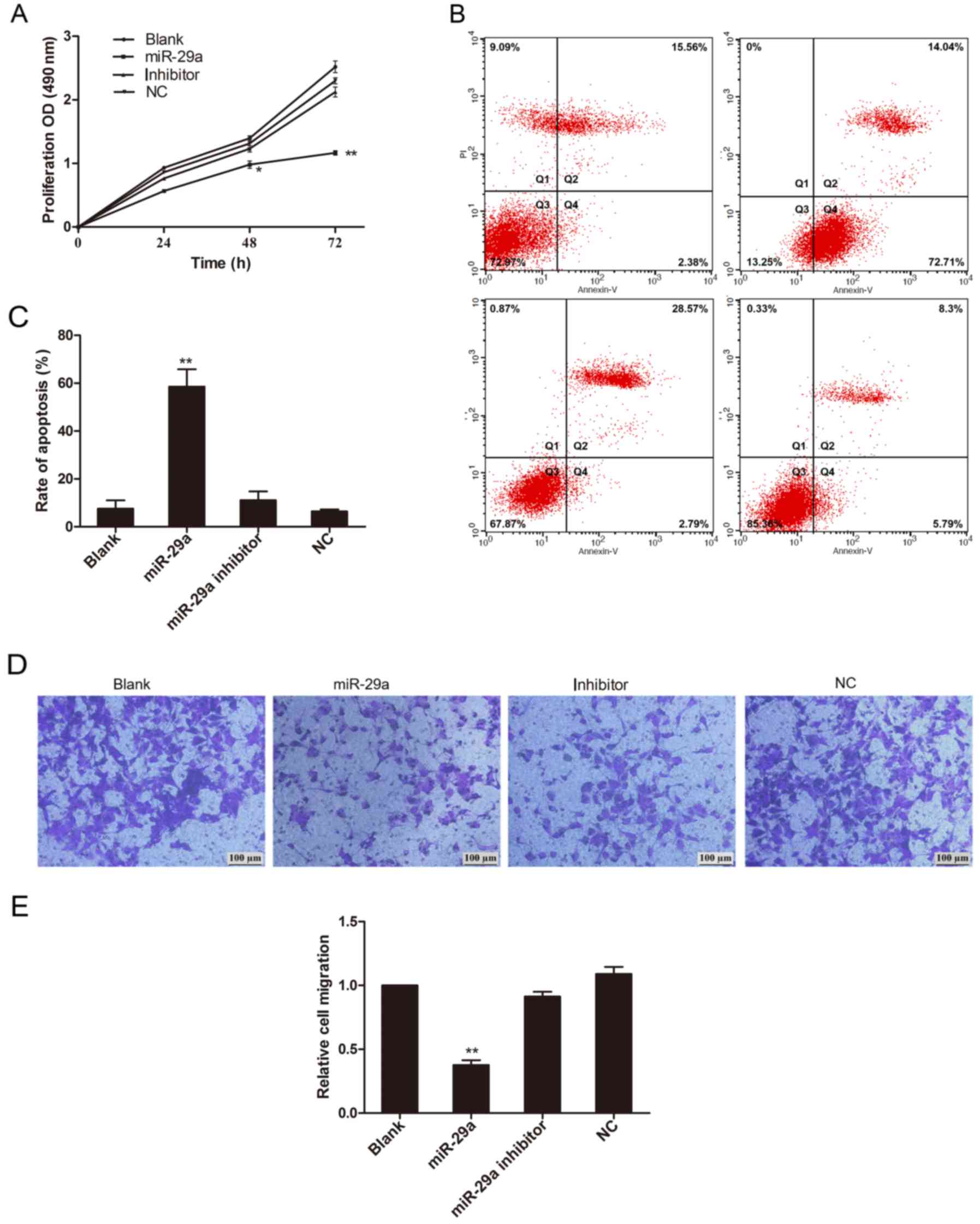
miR-29a inhibits proliferation and
migration of A549 cells
Previous reports have shown that miR-29a is involved
in cell growth in other types of cancer. To test whether miR-29a
exhibits an inhibitory function in lung cancer induced by IL-13, we
overexpressed miR-29a expression in vitro by transfecting
A549 cells with the miR-29a sequence. By MTT assay, we showed that
cell proliferation in the A549-miR-29a group was inhibited with the
most extensive inhibitory effects at 48 and 72 h (P<0.05 and
P<0.01 respectively) (Fig. 4A).
Overexpression of miR-29a promoted cell apoptosis and inhibited
cell invasion in the A549 cells (Fig.
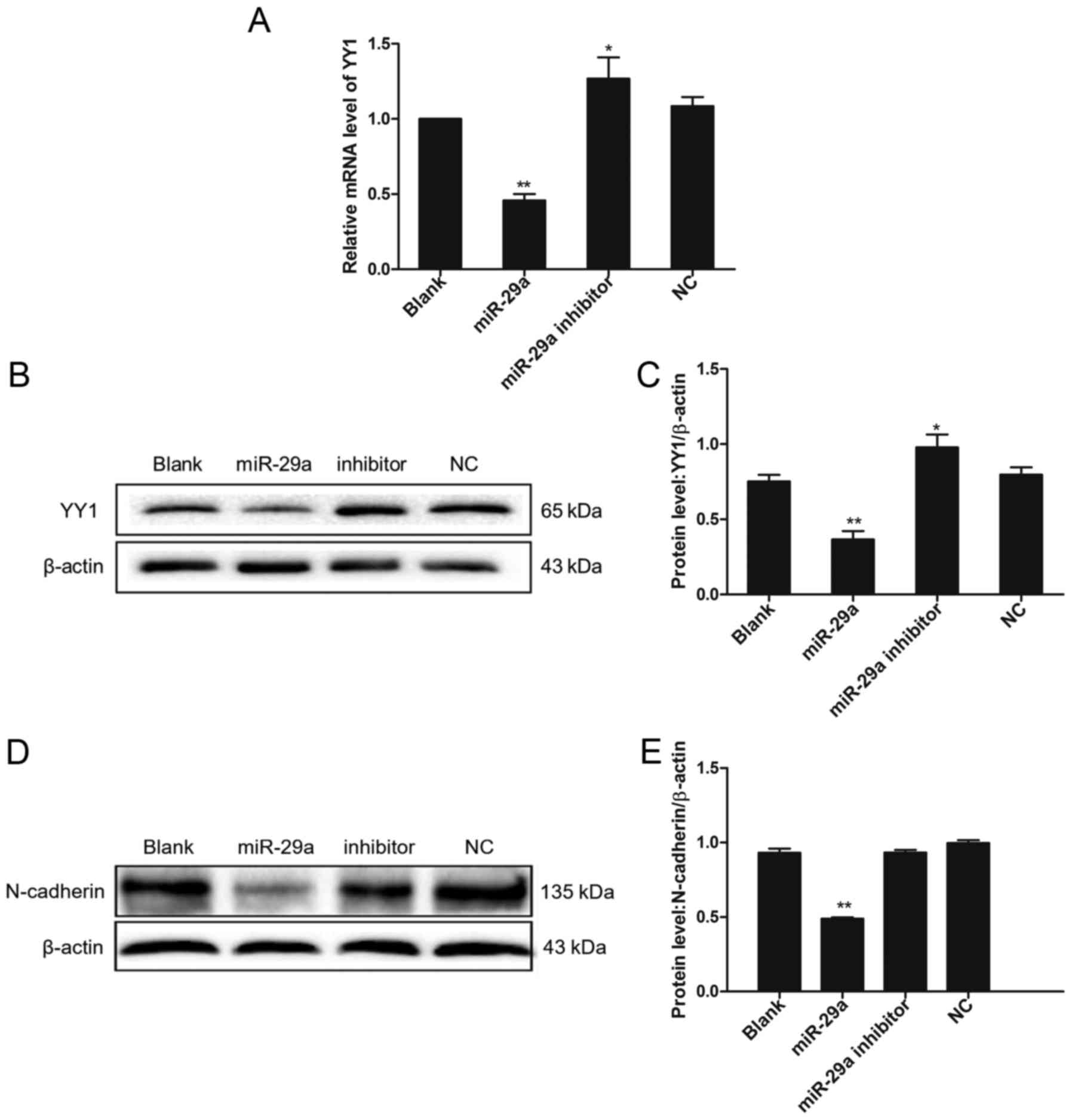
4B-E). To examine the molecular mechanism of miR-29a in lung
cancer induced by IL-13, we measured the expression level of YY1 in
the miR-29a-overexpressing A549 cells. Our results showed that a
decline in YY1 expression was observed at both mRNA and protein
level under miR-29a treatment (Fig.
5A-C). In contrast, transfection with the miR-29a inhibitor
upregulated the expression level of YY1 (Fig. 5A-C). We found that YY1 expression
was negatively correlated with miR-29a. Furthermore, we measured
the expression levels of N-cadherin (a biomarker for invasive
cells), and our results showed that N-cadherin was significantly
downregulated in the miR-29a-overexpression group, compared to the
control (Fig. 5D and E), which
validated the inhibitory role of miR-29a in cell invasion in lung
cancer.
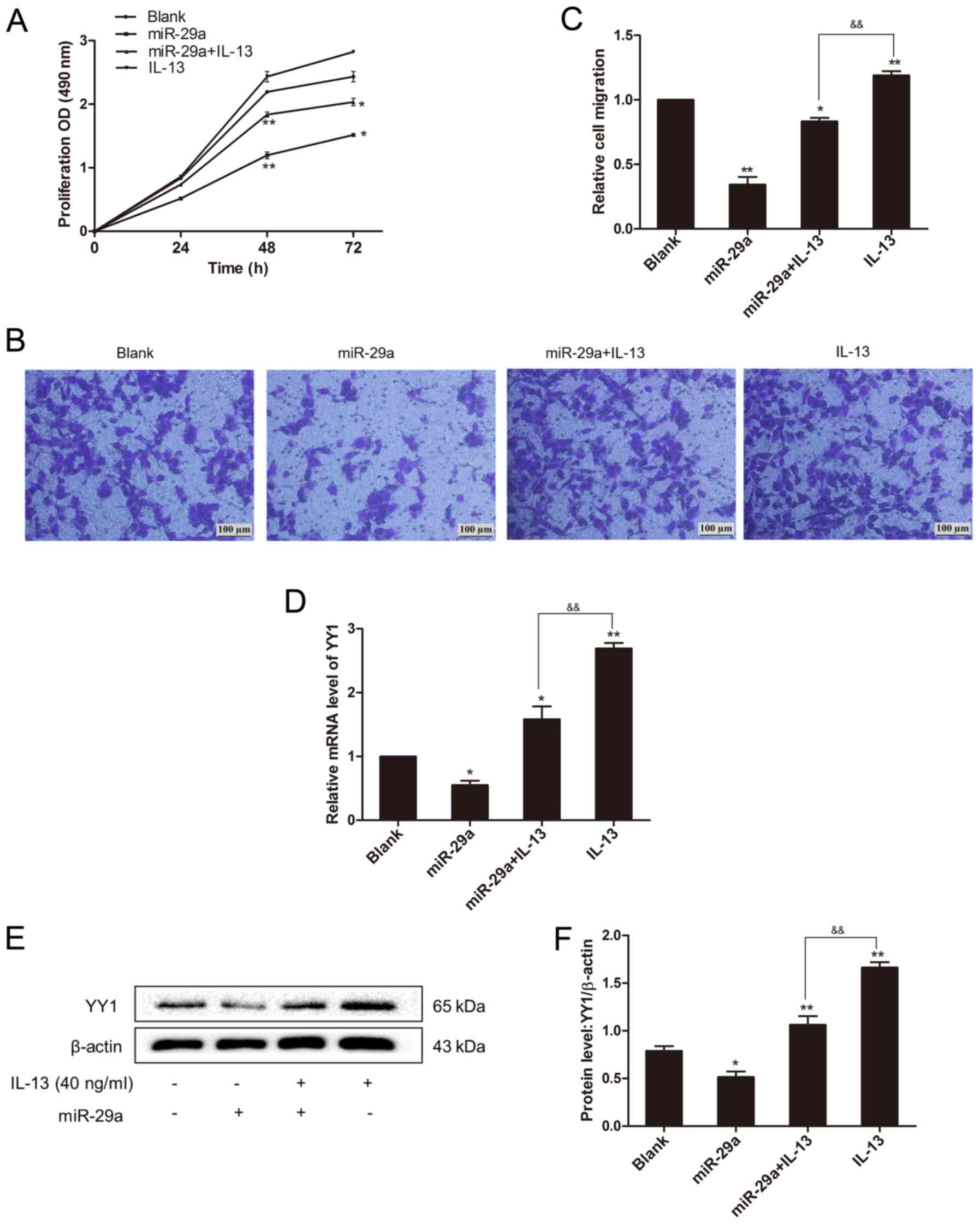
miR-29a inhibits IL-13-induced cell
invasion by targeting YY1
Since YY1 expression level was correlated with IL-13
and miR-29a, we determine whether miR-29a affects the role of IL-13
in lung cancer A549 cells. Overexpression of miR-29a in the
IL-13-treated group significantly downregulated cell proliferation
and invasion in A549 cells, as compared to the control group
treated with IL-13 (Fig. 6A-C). We
measured the related YY1 expression levels in the groups, and we
found that overexpression of miR-29a significantly reduced YY1
expression (P<0.01) (Fig. 6D-F).
This showed that under stimulation of IL-13, miR-29a overexpression
repressed YY1 expression and inhibited the malignant behaviors of
A549 cells mediated by IL-13.
Discussion
The phosphoinositide 3-kinase/serine-threonine
kinase (PI3K/AKT) signaling pathway plays a pivotal role in
tumorigenesis due to its regulation of cellular functions,
including proliferation, differentiation and metastasis of cancer
cells (18,19). Excessive activation of the PI3K/AKT
pathway in normal cells could trigger their transformation into
cancer cells, and the expression levels of PI3K and AKT are
increased in NSCLC (20). Previous
studies have shown that IL-13 activates the PI3K/AKT signaling
pathway in porcine endothelial cells and human dermal fibroblasts
(10,11), but how IL-13 regulates lung cancer
cells remains largely unknown. Our results showed that within a
certain range, rising concentrations of IL-13 (from 0 to 40 ng/ml)
and increasing periods of timeC (from 0 to 12 h) were associated
with the gradually increased amount of pAKT in A549 cells,
indicating that IL-13 is one of the simulative factors of the
PI3K/AKT pathway in human lung cancer. Furthermore, we demonstrated
that IL-13 increased cell proliferation and migration abilities in
lung cancer cells by activating the PI3K/AKT pathway. Our findings
are consistent with a previous study which showed that IL-13Rα2 is
able to activate the Scr/PI3K/AKT/mTOR pathway (21).
YY1 is one of the direct downstream effectors of AKT
(22–24). Silencing of AKT can decrease the YY1
protein level to approximately 50% (12). Consistently, we showed that
treatment of A549 cells with LY294002 decreased the pAKT level
subsequently decreasing the YY1 level at both the mRNA and protein
levels. The identification of the IL-13/PI3K/AKT/YY1 signaling
transduction pattern was reported previously in lung fibroblasts
(13), but it has not been
confirmed in lung cancer cells. By using lung cancer cell line
A549, we showed that IL-13 stimulation was positively correlated
with the levels of pAKT and YY1 in a time- and
concentration-dependent manner, indicating the importance of the
IL-13/PI3K/AKT/YY1 pathway in lung tumorigenesis. Compared with the
blank group, proliferation of A549 cells decreased obviously when
treated with LY294002. AKT controls cell proliferation by directly
phosphorylating the glycogen synthase kinase-3β (GSK3β) (25) and negatively influences expression
of CKIs to accumulate CDK complexes (26). We further showed that the increased
migration of A549 cells treated with IL-13 and promotion of the
processes of epithelial-mesenchymal transition (EMT) and inhibition
the PI3K/AKT pathway by LY294002 could attenuate the EMT process.
Recently, EMT has been reported to play a key role in the migration
of cancer cells. Others have reported that AKT forms a positive
loop with TWIST in EMT-induced events, and AKT inhibition causes
downregulation of Snail and upregulation of E-cadherin (27).
Recently, miRNAs have been shown to play important
roles in every step of cancer progression. In our results,
transfection with miR-29a inhibited the proliferation of A549 cells
and enhanced their apoptosis by mediating expression of YY1 in cell
cycle regulation. Activation of cyclin D1 was found to coincide
with the release of YY1 transcriptional repressor complex in
estrogen-responsive human breast cancer (28). Previous groups have shown that YY1
is inhibited by p53 (29).
YY1 activates both endogenous and exogenous c-Myc promoter
when overexpressed, thus inhibiting the function of p21
(30,31). YY1 was found to activate
transcription of the NF-κB subunits, Rel-A and Rel-B, and also
compete with NF-κB for binding the cytokine response unit within
the serum amyloid A gene promoter (32–34).
YY1 has been shown to repress expression of Fas and DR5, increasing
resistance to apoptosis (35,36).
In the present study, we demonstrated that invasion of lung cancer
was positively related with YY1 expression. Previous reports have
demonstrated that YY1 binds to the matrix metalloproteinases-2
(MMP-2) promoter to promote its expression, causing the remodeling
of the extracellular matrix (ECM) (37,38).
We showed that the reduction in N-cadherin after transfecting
miR-29a implied that EMT may participate in the YY1-regulated tumor
cell biological behavior. miR-29a is predicted to have a
complementary site in YY1 3′UTR (position 774–780 of YY1 3′UTR:
5′-UGGUGCUC; hsa-miR-29a-3p: 3′-ACCACGAG). We indicated that
miR-29a repressed YY1 expression at both the mRNA and protein
levels, leading to inhibition of cell proliferation and invasion of
A549 cells. In Fig. 4, we did not
observe any significant differences between miR-29a
inhibitor-transfected and NC group based on our data. It may be due
to the fact that the endogenous level of miR-29a in A549 cells was
very low, and it did not show any difference when we further
knocked down its level. But instead, it showed significant effects
when we overexpressed it in cells. miR-29a plays an inhibitory role
in EMT, in particular it inhibits transition from epithelial to
mesenchymal form of cells. N-cadherin is the chosen marker for
mesenchymal cells and suppression of N-cadherin confirms our
hypothesis that miR-29a acts as a tumor suppressor miRNA to inhibit
cell invasion. While E-cadherin is a marker for epithelial cells, a
further study of EMT may utilize both N-cadherin and E-cadherin
expression levels to monitor the process of EMT.
Previously, miR-29a has been shown to inhibit
tumorigenicity in NSCLC by downregulating DNA methyltransferase
(DNMT)3A and 3B, which silence tumor suppressor genes such as
FHIT and WWOX (39).
Overexpression of miR-29a was found to reduce the proliferation,
migration, and invasion of NSCLC cells, which could be partially
attributed to LASP-1 (a cAMP and cGMP dependent signaling protein)
inhibition (40). In the present
study, we further examined the role of miR-29a in IL-13-induced
lung cancer progression, and we showed that miR-29a inhibited lung
cancer progression by targeting YY1 in the PI3K/AKT pathway. We
hypothesized that miR-29a plays an important role in regulating
lung cancer tumorigenicity through repression of signaling protein
LASP-1, transcription factor YY1, and epigenetic factors DNMT3A and
3B. In our data, we significantly showed that miR-29a transfection
greatly weakened IL-13-induced lung cancer progression and led to
YY1 repression, causing lower proliferation and weaker invasive
ability in A549 cells. These results indicated that miR-29a could
act as a promising therapeutic target for NSCLC. We may use miR-29a
to antagonize the effects induced by IL-13 on tumor cell behavior
and inhibit expression of YY1 in lung cancer. A previous study
showed that miR-29a regulated expression of YY1, but this was shown
in lung fibroblasts (16). In the
present study, we further demonstrated the role of miR-29a in lung
cancer cells instead of lung fibroblasts, which provides new
evidence that miR-29a is involved in IL-13-induced cell invasion in
lung cancer. In this study, we elucidated the role of miR-29a in
the IL-13/PI3K/AKT/YY1 signaling pathway in lung cancer that
represents a novel finding. We further examined the activation of
the PI3K/AKT pathway in lung cancer A549 cells, and measured the
phosphorylation of AKT following IL-13 stimulation. In addition, we
investigated whether blocking the PI3K/AKT pathway would affect the
effects of YY1 mediated by miR-29a and IL-13. However, our studies
were limited to in vitro experiments, which cannot entirely
represent these mechanisms at the individual level. Further
research on miR-29a in a corresponding animal model may provide
more evidence for clinical molecular-targeted treatment of lung
cancer.
In summary, we identified the activation of the
PI3K/AKT/YY1 signaling pathway in IL-13-induced lung cancer
progression. By overexpression miR-29a, we blocked IL-13-induced
YY1 to inhibit its tumor-promoting functions in A549 cells. Further
research on miR-29a including in vitro and animal model
studies may provide more evidence for clinical molecular-targeted
treatment of lung cancer.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (nos. 81200069 and 31660287);
the National Natural Science Foundation of Jiangxi Province (no.
20161BAB205204); the Science and Technology Plan of Education
Department of Jiangxi Province (no. 150217); and the Postgraduates
Innovation Special Fund Project of Nanchang University (no.
cx2016356).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YZ was in charge for thesis writing and
modification. SH and RM were responsible for conducting the
experiments and in charge of constructing the graphs. All authors
contributed equally to this study. All authors read and approved
the manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
The study used cell lines and thus ethical approval
was waived.
Consent for publication
Not applicable.
Authors' information
ORCID ID: 0000-0003-3769-2496 for LX.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lemjabbar-Alaoui H, Hassan OU, Yang YW and
Buchanan P: Lung cancer: Biology and treatment options. Biochim
Biophys Acta. 1856:189–210. 2015.PubMed/NCBI
|
|
2
|
Joshi BH, Hogaboam C, Dover P, Husain SR
and Puri RK: Role of interleukin-13 in cancer, pulmonary fibrosis,
and other TH2-type diseases. Vitam Horm. 74:479–504.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP,
Wang J, Zhang Y and Elias JA: Pulmonary expression of
interleukin-13 causes inflammation, mucus hypersecretion,
subepithelial fibrosis, physiologic abnormalities, and eotaxin
production. J Clin Invest. 103:779–788. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fujisawa T, Joshi B, Nakajima A and Puri
RK: A novel role of interleukin-13 receptor alpha2 in pancreatic
cancer invasion and metastasis. Cancer Res. 69:8678–8685. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fujisawa T, Joshi BH and Puri RK: IL-13
regulates cancer invasion and metastasis through IL-13Rα2 via
ERK/AP-1 pathway in mouse model of human ovarian cancer. Int J
Cancer. 131:344–356. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jakubzick C, Kunkel SL, Joshi BH, Puri RK
and Hogaboam CM: Interleukin-13 fusion cytotoxin arrests
Schistosoma mansoni egg-induced pulmonary granuloma formation in
mice. Am J Pathol. 161:1283–1297. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shimamura T, Fujisawa T, Husain SR, Joshi
B and Puri RK: Interleukin 13 mediates signal transduction through
interleukin 13 receptor alpha2 in pancreatic ductal adenocarcinoma:
Role of IL-13 Pseudomonas exotoxin in pancreatic cancer
therapy. Clin Cancer Res. 16:577–586. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Erkeland SJ, Valkhof M,
Heijmans-Antonissen C, Delwel R, Valk PJ, Hermans MH and Touw IP:
The gene encoding the transcriptional regulator Yin Yang 1 (YY1) is
a myeloid transforming gene interfering with neutrophilic
differentiation. Blood. 101:1111–1117. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hsu KW, Hsieh RH, Lee YH, Chao CH, Wu KJ,
Tseng MJ and Yeh TS: The activated Notch1 receptor cooperates with
alpha-enolase and MBP-1 in modulating c-myc activity. Mol
Cell Biol. 28:4829–4842. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Grehan JF, Levay-Young BK, Fogelson JL,
François-Bongarçon V, Benson BA and Dalmasso AP: IL-4 and IL-13
induce protection of porcine endothelial cells from killing by
human complement and from apoptosis through activation of a
phosphatidylinositide 3-kinase/Akt pathway. J Immunol.
175:1903–1910. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Moriya C, Jinnin M, Yamane K, Maruo K,
Muchemwa FC, Igata T, Makino T, Fukushima S and Ihn H: Expression
of matrix metalloproteinase-13 is controlled by IL-13 via PI3K/Akt3
and PKC-delta in normal human dermal fibroblasts. J Invest
Dermatol. 131:655–661. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Petrella BL and Brinckerhoff CE: PTEN
suppression of YY1 induces HIF-2 activity in von-Hippel-Lindau-null
renal-cell carcinoma. Cancer Biol Ther. 8:1389–1401. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guo J, Yao H, Lin X, Xu H, Dean D, Zhu Z,
Liu G and Sime P: IL-13 induces YY1 through the AKT pathway in lung
fibroblasts. PLoS One. 10:e01190392015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lages E, Ipas H, Guttin A, Nesr H, Berger
F and Issartel JP: MicroRNAs: Molecular features and role in
cancer. Front Biosci. 17:2508–2540. 2012. View Article : Google Scholar
|
|
15
|
Larsen JE and Minnar JD: Molecular biology
of lung cancer: Clinical implications. Clin Chest Med. 32:703–740.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jin M, Wu Y, Wang Y, Yu D, Yang M, Yang F,
Feng C and Chen T: MicroRNA-29a promotes smooth muscle cell
differentiation from stem cells by targeting YY1. Stem Cell Res.
17:277–284. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang H, Garzon R, Sun H, Ladner KJ, Singh
R, Dahlman J, Cheng A, Hall BM, Qualman SJ, Chandler DS, et al:
NF-kappaB-YY1-miR-29 regulatory circuitry in skeletal myogenesis
and rhabdomyosarcoma. Cancer Cell. 14:369–381. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ebrahimi S, Hosseini M, Shahidsales S,
Maftouh M, Ferns GA, Ghayour-Mobarhan M, Hassanian SM and Avan A:
Targeting the Akt/PI3K signaling pathway as a potential therapeutic
strategy for the treatment of pancreatic cancer. Curr Med Chem.
24:1321–1331. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bahrami A, Khazaei M, Hasanzadeh M,
ShahidSales S, Mashhad Joudi M, Farazestanian M, Sadeghnia HR,
Rezayi M, Maftouh M, Hassanian SM and Avan A: Therapeutic potential
of targeting PI3K/AKT pathway in treatment of colorectal cancer:
Rational and progress. J Cell Biochem. 119:2460–2469. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Heavey S, O'Byrne KJ and Gately K:
Strategies for co-targeting the PI3K/AKT/mTOR pathway in NSCLC.
Cancer Treat Rev. 40:445–456. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tu M, Wange W, Cai L, Zhu P, Gao Z and
Zheng W: IL-13 receptor alpha2 stimulates human glioma cell growth
and metastasis through the Src/PI3K/Akt/mTOR signaling pathway.
Tumour Biol. 37:14701–14709. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Berns KI and Bohenzky RA: Adeno-associated
viruses: An update. Adv Virus Res. 32:243–306. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shi Y, Seto E, Chang LS and Shenk T:
Transcriptional repression by YY1, a human GLI-Kruppel-related
protein, and relief of repression by adenovirus E1A protein. Cell.
67:377–388. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Park K and Atchison ML: Isolation of a
candidate repressor/activator, NF-E1 (YY-1, delta), that binds to
the immunoglobulin kappa 3′ enhancer and the immunoglobulin
heavy-chain mu E1 site. Proc Natl Acad Sci USA. 88:9804–9808. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Diehl JA, Cheng M, Roussel MF and Sherr
CJ: Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis
and subcellular localization. Genes Dev. 12:3499–3511. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Graff JR, Konicek BW, McNulty AM, Wang Z,
Houck K, Allen S, Paul JD, Hbaiu A, Goode RG, Sandusky GE, et al:
Increased AKT activity contributes to prostate cancer progression
by dramatically accelerating prostate tumor growth and diminishing
p27Kip1 expression. J Biol Chem. 275:24500–24505. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xu W, Yang Z and Lu N: A new role for the
PI3K/Akt signaling pathway in the epithelial-mesenchymal
transition. Cell Adh Migr. 9:317–324. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cicatiello L, Scafoglio C, Altucci L,
Cancemi M, Natoli G, Facchiano A, Iazzetti G, Calogero R, Biglia N,
De Bortoli M, et al: A genomic view of estrogen actions in human
breast cancer cells by expression profiling of the
hormone-responsive transcriptome. J Mol Endocrinol. 32:719–775.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sui G, Affar el B, Shi Y, Brignone C, Wall
NR, Yin P, Donohoe M, Luke MP, Calvo D, Grossman SR, et al: Yin
Yang 1 is a negative regulator of p53. Cell. 117:859–872. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Riggs KJ, Saleque S, Wong KK, Merrell KT,
Lee JS, Shi Y and Calame K: Yin-yang 1 activates the I promoter.
Mol Cell Biol. 13:7487–7495. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vella P, Barozzi I, Cuomo A, Bonaldi T and
Pasini D: Yin Yang 1 extends the Myc-related transcription factors
network in embryonic stem cells. Nucleic Acids Res. 40:3403–3418.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lu SY, Rodriguez M and Liao WS: YY1
represses rat serum amyloid A1 gene transcription and is
antagonized by NF-kappa B during acute-phase response. Mol Cell
Biol. 14:6253–6263. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sepulveda MA, Emelyanov AV and Birshtein
BK: NF-kappa B and Oct-2 synergize to activate the human 3′ Igh hs4
enhancer in B cells. J Immunol. 172:1054–1064. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang H, Hertlein E, Bakkar N, Sun H,
Acharyya S, Wang J, Carathers M, Davuluri R and Guttridge DC:
NF-kappaB regulation of YY1 inhibits skeletal myogenesis through
transcriptional silencing of myofibrillar genes. Mol Cell Biol.
27:4374–4387. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Martínez-Paniagua MA, Baritaki S,
Huerta-Yepez S, Ortiz-Navarrete VF, González-Bonilla C, Bonavida B
and Vega MI: Mcl-1 and YY1 inhibition and induction of DR5 by the
BH3-mimetic Obatoclax (GX15-070) contribute in the sensitization of
B-NHL cells to TRAIL apoptosis. Cell Cycle. 10:2792–2805. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Reséndiz-Martínez J, Asbun-Bojalil J,
Huerta-Yepez S and Vega M: Correlation of the expression of YY1 and
Fas cell surface death receptor with apoptosis of peripheral blood
mononuclear cells, and the development of multiple organ
dysfunction in children with sepsis. Mol Med Rep. 15:2433–2442.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Naruse K, Lash GE, Innes BA, Otun HA,
Searle RF, Robson SC and Bulmer JN: Localization of matrix
metalloproteinase (MMP)-2, MMP-9 and tissue inhibitors for MMPs
(TIMPs) in uterine natural killer cells in early human pregnancy.
Hum Reprod. 24:553–561. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tian FJ, Cheng YX, Li XC, Wang F, Qin CM,
Ma XL, Yang J and Lin Y: The YY1/MMP2 axis promotes trophoblast
invasion at the maternal-fetal interface. J Pathol. 239:36–47.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fabbri M, Garzon R, Cimmino A, Liu Z,
Zanesi N, Callegari E, Liu S, Alder H, Costinean S,
Fernandez-Cymering C, et al: MicroRNA-29 family reverts aberrant
methylation in lung cancer by targeting DNA methyltransferases 3A
and 3B. Proc Natl Acad Sci USA. 104:15805–15810. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hu Z, Cui Y, Zhou Y, Zhou K, Qiao X, Li C
and Wang S: MicroRNA-29a plays a suppressive role in non-small cell
lung cancer cells via targeting LASP1. Onco Targets Ther.
9:6999–7009. 2016. View Article : Google Scholar : PubMed/NCBI
|