Introduction
Vascular endothelial (VE)-cadherin is the main
transmembrane protein sustaining intercellular adherens junctions
in vascular endothelial cells where it plays a central role in
modulating endothelial integrity and permeability (1). In vivo experimental models have
demonstrated that mice deficient of VE-cadherin die in the embryo
stage by severe vascular defects (2). Although VE-cadherin is considered
specific for endothelial cells, its expression has also been found
in several types of tumor cells such as melanoma, breast cancer and
lung cancer (3–5). VE-cadherin is also expressed in a
subset of acute lymphoblastic leukemia cells, where it contributes
to cell survival (6). Surprisingly,
VE-cadherin can be found in highly aggressive tumor cells but not
in non-aggressive ones, which indicates that VE-cadherin may play
various roles in tumor progression (7). In 1999, Maniotis et al
presented a new concept, vasculogenic mimicry (VM), which describes
the finding that tumor cells are able to self-assemble into
tube-like structure independent of endothelial cells (7). Several endothelial-specific proteins
were found in these VM tumor cells. Among them, VE-cadherin is
considered most critical for VM as it was found that downregulation
of VE-cadherin in melanoma led to the loss of VM formation
(8). Thus, VE-cadherin has been
used as a therapeutic target to control tumor angiogenesis in
vivo (9).
VE-cadherins exhibit Ca2+-dependent
homodimer interaction and cluster across cell membranes though
their extracellular domains. Similar to N-cadherin and E-cadherin
in the classic cadherin family, VE-cadherin also has five
homologous domains in its extracellular region (named EC1 to EC5)
(10). The cytoplasmic part of
VE-cadherin contains domains interacting with β-catenin and
plakoglobin (11,12). There have been several studies that
showed that the N-terminal region corresponding to the EC1 domain
of cadherins is responsible for intercellular hemophilic adhesion
(13–16). Other research has found that there
exists multiple binding sites in the ectodomain for cadherin
adhesion. Antibodies directed to EC1 and EC3 affect VE-cadherin
adhesion and clustering (17).
However, whether other ECs are associated with VE-cadherin adhesion
still needs to be elucidated. In the present study, we generated a
monoclonal antibody binding to a novel epitope in the EC4 domain of
VE-cadherin and demonstrated that the antibody inhibited the
proliferation and vasculogenic mimicry of lung cancer Glc-82 cells
by modulating AKT phosphorylation. Our results suggest that the EC4
domain is involved in VE-cadherin adhesion and the monoclonal
antibody may be promising for development for anti-vasculogenic
mimicry in cancer treatment.
Materials and methods
Recombinant expression of the
extracellular domain of VE-cadherin protein
The coding sequence for the whole extracellular
region of VE-cadherin was amplified by reverse transcriptional PCR
using total RNA extracted from human umbilical vein endothelial
cells (HUVECs). The primer sequences were 5′-CCGGAATTCTGGATTTGGAACCAGATGCAC-3′
and 5′-CCGCTCGAGCAAGATGCTGTACTTGGTCAT-3′
(EcoRI and XhoI sites are underlined). The PCR
product was digested with restriction enzymes and ligated into the
pET41a expression vector (Novagen, San Diego, CA, USA). The
construct was transformed into DH5α and the inserted fragment was
verified by DNA sequencing. The recombinant protein was produced by
transforming the expression construct into E. coli BL21
(DE3) followed by isopropyl β-D-1-thiogalactopyranoside (IPTG, 1
mM) induction. The bacteria were collected by centrifugation at
5,000 rpm for 10 min at 4°C, and then sonicated in lysis buffer (20
mM NaH2PO4, 10 mM imidazole, pH 8.0). The
supernatant which contained the VE-cadherin recombinant protein was
collected by centrifugation at 13,000 rpm for 20 min at 4°C and
incubated with Ni2+-Sepharose beads for 3 h in an
ice-water bath. The beads were washed with lysis buffer and the
bound protein was eluted with elution buffer (20 mM
NaH2PO4, 250 mM imidazole, pH 8.0). The
solution with VE-cadherin fusion protein was dialyzed against PBS
(pH 7.4) overnight at 4°C. The purified protein was analyzed by
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) followed by Coomassie blue staining.
Production and characterization of
monoclonal antibodies to the extracellular domain of
VE-cadherin
BALB/c male mice were purchased from Shanghai SLAC
Laboratory Animal company (Shanghai, China). The mice were housed
in light-controlled room (lights on at 07:00, off at 19:00) at a
room temperature of 23±1°C and a humidity of 50±0% with food and
water available ad libitum. All animal procedures were
conducted in strict accordance with the Animal Ethics Guidelines
and approved by the Ethics Committee of Soochow University. Three
BALB/c male mice (6 weeks old, 18–20 g) were immunized with three
injections of 100 µg of the recombinant extracellular domain of
VE-cadherin protein each time. Freund's complete adjuvant was used
for the initial injection. The Freund's incomplete adjuvant was
used for the second injection 28 days after the initial
immunization. The first two immunizations were both administered
subcutaneously. Four weeks later, 100 µg of the recombinant protein
was injected into the tail vein of the mice without adjuvant for
the final boost. Three days later, mouse spleens were separated and
splenocytes were isolated and fused with SP2/0 myeloma cells using
PEG1500 (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Hybridoma
cells were selected in HAT medium (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Hybridoma cells producing antibodies specific
for recombinant VE-cadherin protein were identified by
enzyme-linked immunosorbent assay (ELISA) and subcloned by two
rounds limiting-dilution in 96-well plates.
Immunofluorescence
Cells (5×103) were inoculated in 96-well
plates the day before and fixed with 4% paraformaldehyde at 4°C for
20 min. Cells were washed with PBS three times and incubated with
2C11, 8A03, or mouse IgG at 37°C for 1 h. After washing, the cells
were labeled with Alexa Fluor 488-conjugated goat anti-mouse IgG
(1:500, Invitrogen; Thermo Fisher Scientific, Inc.). Nuclei were
stained with DAPI (4′,6-diamidino-2-phenylindole). The fluorescent
images were captured using an inverted fluorescence microscope
(DIML; Leica Microsystems, Wetzlar, Germany).
Flow cytometry
EA.hy926 cells (3×106) were trypsinized
to suspension, washed with phosphate-buffered saline (PBS), and
then incubated with 2C11, 8A03, or mouse IgG (1 µg/ml) at room
temperature for 1 h. Cells were washed with PBS and labeled with
Alexa Fluor 488-conjugated goat anti-mouse IgG antibody for 1 h at
room temperature. The reaction was stopped by addition of 500 µl
PBS and fluorescence was measured by flow cytometry (BD
FACSCalibur; BD Biosciences, San Diego, CA, USA).
Preparation of recombinant human
VE-cadherin fragments
Five pairs of primers were synthesized for
amplification of the five extracellular domains (EC1-EC5) of
VE-cadherin respectively (primer sequences are listed in Table I). The PCR products were constructed
into the pET41a expression vector. The five EC domains were
recombinantly expressed in BL21 (DE3) cells respectively under the
induction of 1 mM IPTG at 37°C for 3 h. Bacterial cells were
harvested and lysed in 1Χ Laemmli sample buffer (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) by boiling for 5 min and the
soluble proteins were loaded on SDS-PAGE and the binding of 2C11
monoclonal antibody was assessed by western blot analysis.
 | Table I.Primer sequences for EC domain
amplification. |
Table I.
Primer sequences for EC domain
amplification.
| cdh5-d1-f |
CCGGAATTCTGGATTTGGAACCAGATGCAC |
| cdh5-d1-r |
CCGCTCGAGGAACACAGGCCAGTTGTCGTT |
| cdh5-d2-f |
CCGGAATTCACGCATCGGTTGTTCAATGC |
| cdh5-d3-r |
CCGCTCGAGGAAAATGGGGGGCTCGTC |
| cdh5-d4-f |
CCGGAATTCCAGCAGCCTTTCTACCACTTCC |
| cdh5-d4-r |
CCGCTCGAGCGGGGCATTGTCATTCTCAT |
| cdh5-d5-f |
CCGGAATTCGAGTTTGCCAAGCCCTACCAG |
| cdh5-d5-r |
CCGCTCGAGCAAGATGCTGTACTTGGTCAT |
Epitope mapping of the 2C11 monoclonal
antibody
Immunodot blot analysis
Seven consecutive 15-amino acid peptides spanning
the whole EC4 domain were synthesized (Table II). Synthetic peptides (20, 40 and
80 ng) were spotted on a cellulose acetate membrane, the membrane
was blocked with 5% dried milk in PBS for 2 h, and then incubated
with 2C11 (1:500 dilution) for 1 h at room temperature followed by
washing three times with PBS-T, and then incubated with
HRP-conjugated goat anti-mouse IgG (1:10,000 dilution) for 1 h.
Enhanced chemiluminescence (ECL; Biological Industries, Kibbutz
Beit Haemek, Israel) was conducted, and the signal was detected by
Kodak Medical X-ray Processer (Kodak, Rochester, NY, USA) or Tanon
5200 AutoChemi Image system (Tanon Inc., Shanghai, China). The
purified VE-cadherin fusion protein was used as the positive
control.
| Table II.Synthetic peptide sequences used for
epitope mapping. |
Table II.
Synthetic peptide sequences used for
epitope mapping.
| Epitope | Amino acid
sequence |
|---|
| D1 | QQPFYHFQLKENQKK |
| D2 | PLIGTVLAMDPDAAR |
| D3 | HSIGYSIRRTSDKGQ |
| D4 | FFRVTKKGDIYNEKE |
| D5 | LDREVVPWYNLTVEA |
| D6 | KELDSTGTPTGKESI |
| D7 | VQVHIEVLDENDNAP |
Competitive ELISA
The synthetic peptides were preincubated with the
2C11 antibody for 1 h, and then added to the 96-well ELISA plates
which were coated with the purified recombinant VE-cadherin
protein. After reaction for 1 h, the plate was washed three times
with PBST and HRP-conjugated goat anti-mouse IgG (1:10,000
dilution) was added and incubated for 1 h. After three times
washing, TMB substrate was added into each wells to develop color,
and the reaction was stopped by 2N H2SO4. The
absorbance at 450 nm was measured using a spectrophotometer (Epoch;
BioTek Instruments, Winooski, VT, USA).
Western blot analysis and RT-PCR
detection of VE-cadherin expression
To prepare cell lysate, the cells were lysed with
RIPA buffer (50 mM Tris-HCl, 1% Triton X-100, 10% glycerol, 150 mM
NaCl, proteinase inhibitor cocktail) for 30 min on ice and
centrifuged at 13,000 rpm for 10 min at 4°C. The cell lysates
containing a total protein amount of 25 µg were placed onto
SDS-PAGE gels for separation and transferred onto PVDF membranes by
MiniProtean (Bio-Rad Laboratories). The membranes were blocked with
5% non-fat milk in PBS for 2 h, and then incubated with 2C11 (1:500
dilution) or 8A03 (1:200 dilution) for 1 h at room temperature
followed by washing three times with PBS-T, and then incubated with
HRP-conjugated goat anti-mouse IgG (1:10,000 dilution) for 1 h.
Proteins were detected by enhanced chemiluminescence (ECL;
Biological Industries) by Kodak Medical X-ray Processer or Tanon
5200 AutoChemi Image system (Tanon Inc.).
RNA was isolated using TRIzol reagent. cDNA was
obtained using RevertAid™ First Strand cDNA Synthesis kit (Fisher
Scientific France, Illkirch-Graffenstaden, France).
Semi-quantitative RT-PCR (30 cycles) was employed to analyze
VE-cadherin gene expression using the gene-specific forward and
reverse primers: 5′-CCGGAATTCTGGATTTGGAACCAGATGCAC-3′ and
5′-CCGCTCGAGCAAGATGCTGTACTTGGTCAT-3′. The expression level of
β-actin was used as the internal control.
3D cell culture
Tube-like formation in Matrigel was performed as
previously described (18). In
brief, Matrigel (150 µl/well) (BD Biosciences, Bedford, MA, USA)
was added to 48-well plates on ice and allowed to polymerize for 30
min at 37°C. Glc-82 cells and HUVECs were resuspended in DMEM
containing MAb 2C11, 8A03 or mouse IgG (2 µg/ml), which was plated
onto the layer of Matrigel (3×104 cells/well). Matrigel
cultures were incubated at 37°C and capillary tube formation was
photographed at various time-points. Five visual fields (up, down,
left, right and center) were randomly chosen from each well under
an inverted microscope (Carl Zeiss, Oberkochen, Germany) to count
the number of tube-like structures. The area covered by the tube
network was quantified by ImageJ software (National Institutes of
Health, Bethesda, MD, USA).
Cell proliferation assay
Cell proliferation was assessed using Cell Counting
Kit-8 (CCK-8; Beyotime Institute of Biotechnology, Haimen, China)
as previously described (19).
Glc-82 cells were seeded in 96-well plates at a density of
5×103 cells per well. 2C11 antibody (at concentrations
of 0.5, 2 and 8 µg/ml) was added into the cultured medium and
incubated for 24, 48 and 72 h, respectively. CCK-8 solution (10 µl)
was added into each well and incubated for 3 h at 37°C. The
absorbance at 450 nm was measured with a spectrophotometer (Epoch;
BioTek Instruments). Columns represented the mean percentage of
optical density (OD) values relative to the negative control. All
measurements were performed in triplicate and repeated at least
three separate experiments.
Statistical analysis
Results are expressed as mean ± SD (range) or
percent. The difference between two groups was analyzed using the
Student's paired t-test. A P-value <0.05 was considered
statistically significant. All calculations were performed using
GraphPad Prism software (GraphPad Software, Inc., San Diego, CA,
USA).
Results
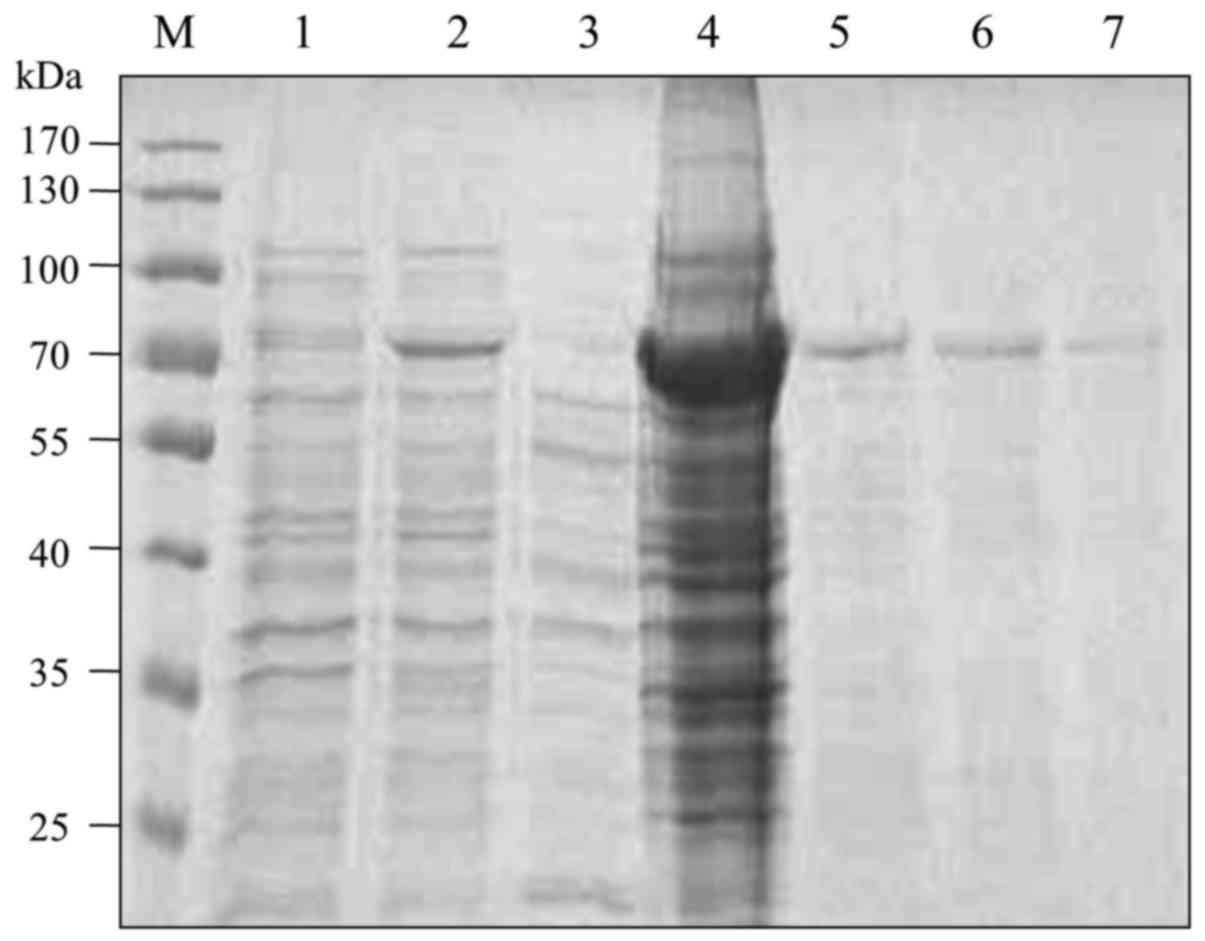
Expression and purification of
recombinant VE-cadherin fusion protein
The cDNA encoding the extracellular domains (EC1 to
EC5) of VE-cadherin was cloned into the pET41a vector. The
histidine-tagged fusion protein was induced in BL21 (DE3) bacteria
with 1 mM IPTG. SDS-PAGE and Coomassie blue staining showed that
the recombinant protein was successfully expressed at a molecular
weight of approximately 70 kDa and was mainly located in the
precipitation fraction. The fusion protein in the precipitation
fraction was purified by Ni2+-Sepharose beads (Fig. 1).
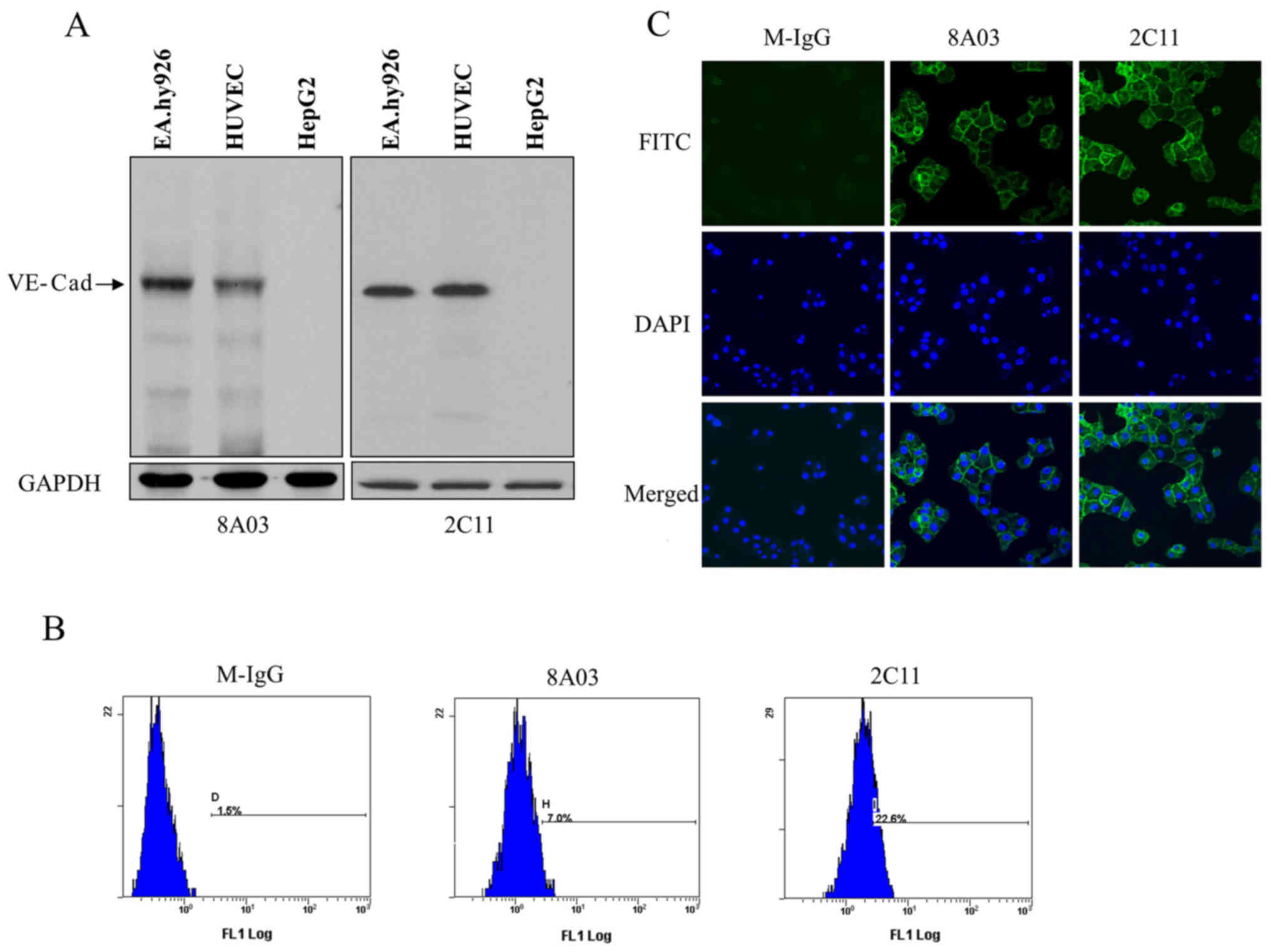
Specificity identification of
monoclonal antibodies to VE-cadherin
The monoclonal antibody in the culture supernatants
of hybridoma cells was detected by indirect ELISA. After two rounds
of subcloning and detection, a stable positive hybridoma clone,
designated as 2C11, was chosen to produce ascites. The monoclonal
antibody in ascites was purified with protein G Sepharose and
stored at a final concentration of 0.35 mg/ml.
To analyze whether 2C11 could bind native
VE-cadherin, EA.hy926 cells, HUVECs and HepG2 cell lysates were
prepared for western blot analysis. The results showed that 2C11
specifically recognized a protein with a molecular weight of
approximately 130 kDa in EA.hy926 and HUVEC cells, but not in HepG2
cells. Commercial antibody 8A03 was used as a positive control
(Fig. 2A).
To further explore the binding specificity of 2C11
antibody to VE-cadherin, immunofluorescence and flow cytometric
analysis were performed. Flow cytometry demonstrated that 2C11
could recognize VE-cadherin located on the endothelial cell surface
(Fig. 2B). Results of the
immunofluorescence showed positive green fluorescence on the
EA.hy926 cell surface labeled with 2C11, while no fluorescence was
detected on cells labeled with mouse IgG (Fig. 2C). These results suggest that the
2C11 antibody recognized the VE-cadherin native protein.
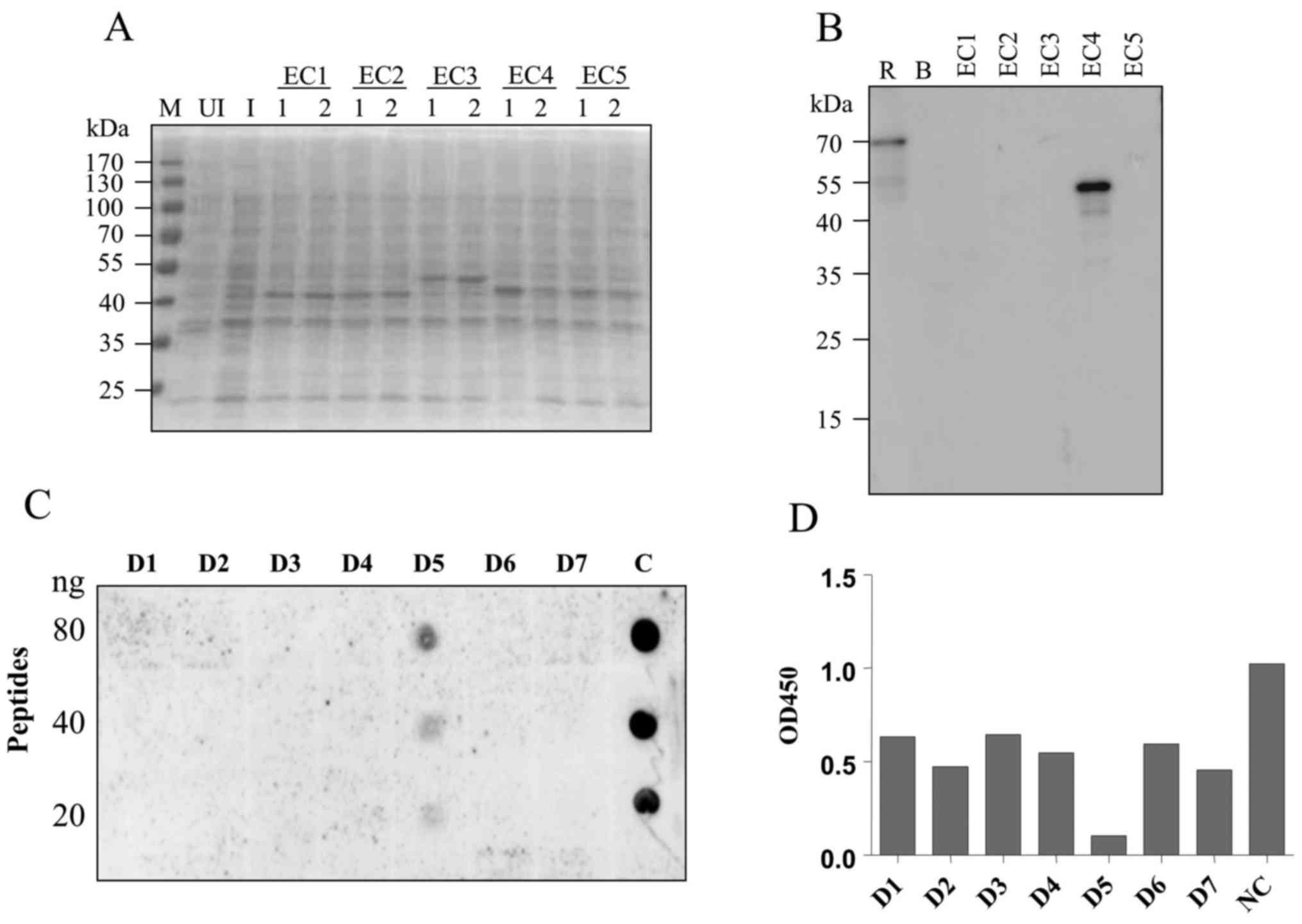
Epitope mapping with synthetic
peptides
To explore the binding epitope of the 2C11 antibody,
five fragments representing a different region of VE-cadherin
extracellular domain (EC1-EC5) were recombinantly expressed in
E. coli. The predicted molecular weights of the resulting
recombinant proteins were 45, 45, 50, 45 and 45 kDa, respectively
(Fig. 3A). Western blot analysis
showed that mAb 2C11 specifically recognized the EC4 fragment
(Fig. 3B) which contains 105 amino
acids. Next, seven consecutive peptides, each comprised of 15 amino
acids, were synthesized (sequences are listed in Table I). Dot blot and competitive ELISA
showed that 2C11 specifically recognized the fifth peptide D5.
These results demonstrated that the D5 peptide LDREVVPWYNLTVEA was
the binding epitope of the 2C11 antibody (Fig. 3C and D).
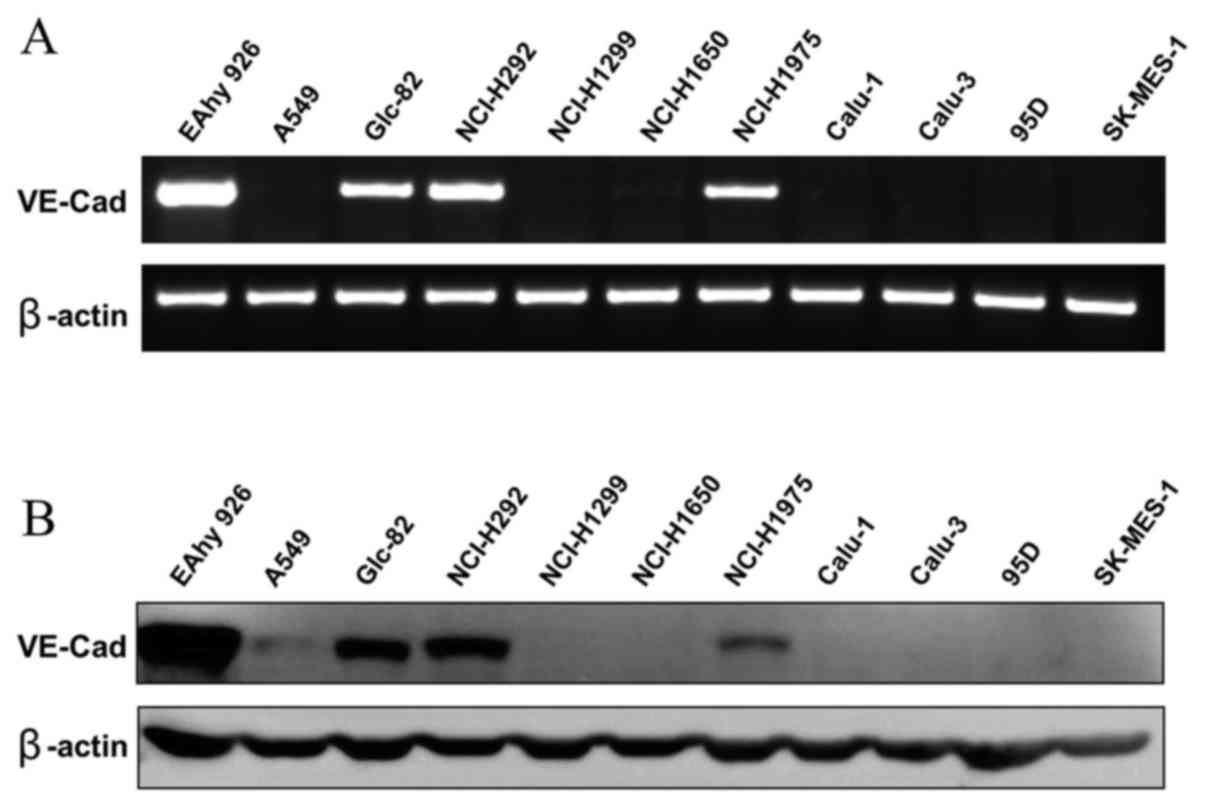
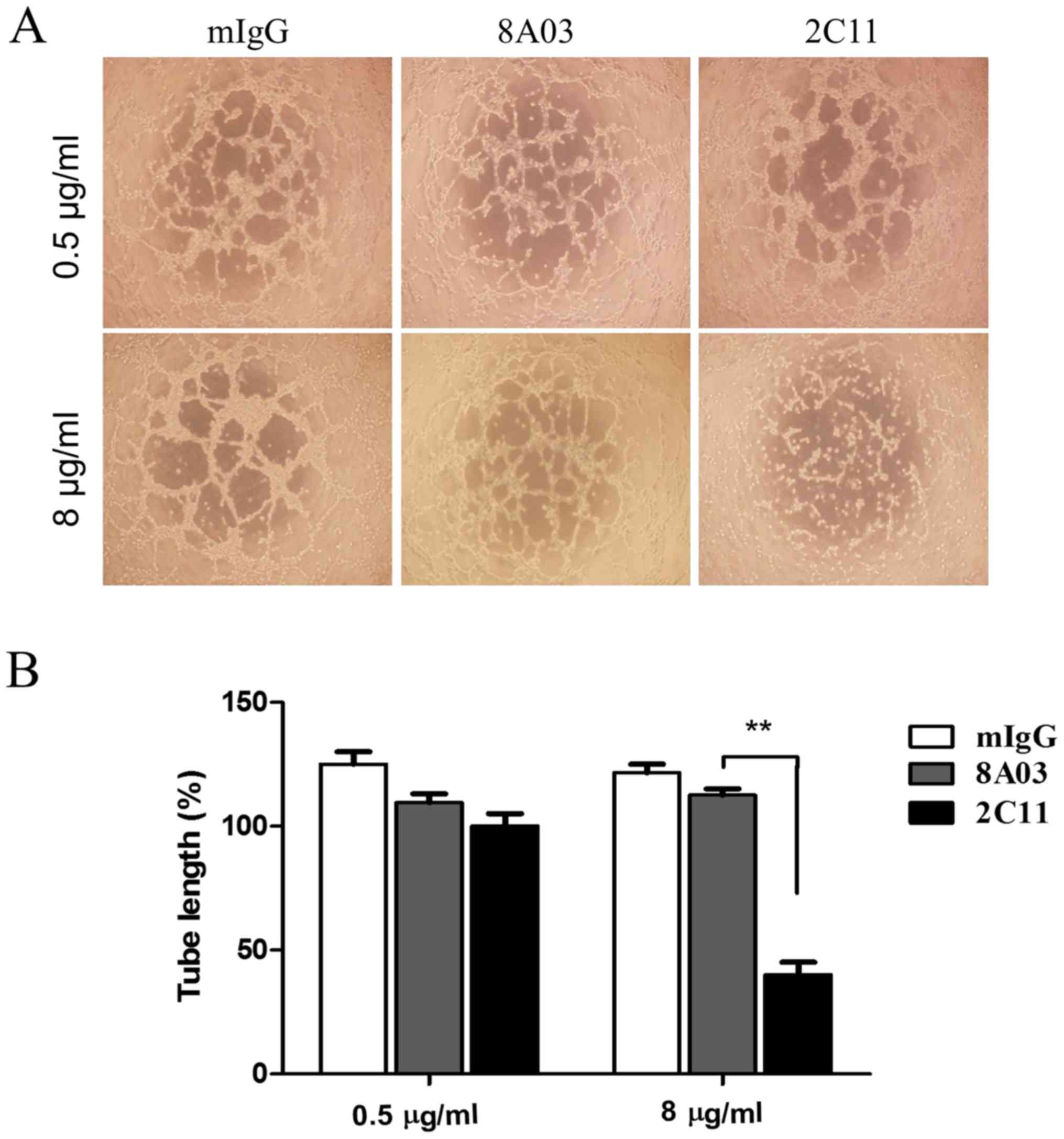
2C11 inhibits vasculogenic mimicry
formation of Glc-82 cells in vitro
Vasculogenic mimicry was observed in some aggressive
tumor cells. Surface expression of VE-cadherin on tumor cells is
considered most important for VM formation. Whether the 2C11
monoclonal antibody has biological activity on VM of tumor cells is
unknown. Here we examined several poorly differentiated lung
adenocarcinoma cell lines; among them, Glc-82 cells were found to
express VE-cadherin by RT-PCR and western blot analysis (Fig. 4A and B). Next, we tested the effect
of 2C11 on tube-like structure formation of Glc-82 lung cancer
cells. The results showed that 2C11, but not mouse IgG and 8A03
antibody, significantly prevented tubular network formation and
reduced the size and length of the cords of Glc-82 cells (Fig. 5A and B). These results demonstrated
that the anti-VE-cadherin mAb 2C11 inhibited VM formation of lung
cancer cells and may be promising to be developed as a therapeutic
antibody for aggressive lung cancer.
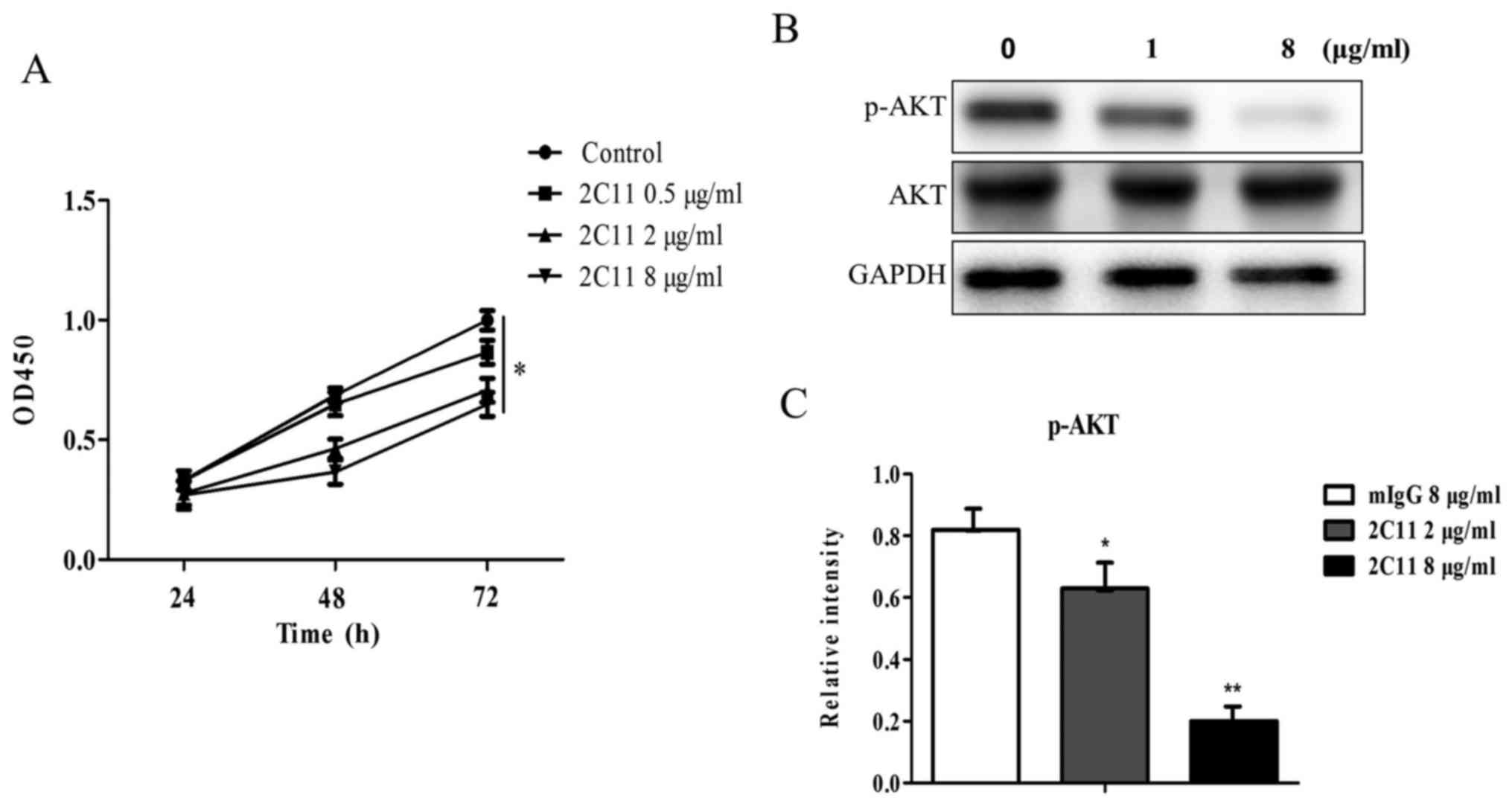
Effect of 2C11 on Glc-82 cell
proliferation
To examine the effect of 2C11 on cell proliferation,
a proliferation assay was performed using CCK-8 reagent. The 2C11
antibody was added into the Glc-82 cell culture medium and
incubated for different time periods. Mouse IgG (10 µg/ml) was used
as the negative control. Data showed that the proliferation rates
of Glc-82 cells were slower after treatment of the 2C11 antibody
compared to the negative control (100% proliferation, Fig. 6A). The result indicated that the
2C11 antibody inhibited the proliferation of Glc-82 cells.
mAb 2C11 inhibits the phosphorylation
of AKT
The 2C11 antibody inhibits Glc-82 cell
proliferation, while the molecular mechanism is unknown. PI3K/AKT
signaling is the classical pathway involved in cell proliferation.
In the present study, we examined the effect of 2C11 on the AKT
signaling pathway. We found that following addition of different
concentrations of the 2C11 antibody, the phosphorylation level of
AKT was decreased (Fig. 6B and C).
This result demonstrated that the 2C11 antibody inhibited Glc-82
cell proliferation via reducing AKT phosphorylation.
Discussion
Cadherins are a family of calcium-dependent,
cell-cell adhesion molecules, which are located at adherens
junctions that mediate adhesive intercellular interactions and
provide a dynamic cell-cell contact and positional clues to cells
during development. VE-cadherin, a unique member of the cadherin
family, is expressed specifically on endothelial cells during the
development of embryo vasculature which has been shown to play a
central role in controlling endothelial monolayer integrity,
junction permeability, cell growth and angiogenesis. However, the
molecular mechanism of VE-cadherin homophilic binding has not yet
been fully elucidated. Previous research has shown that two
monoclonal antibodies BV6 and Cad 5 to VE-cadherin could increase
paracellular permeability and inhibit VE-cadherin reorganization.
Epitope mapping studies defined the sequences TIDLRY located on EC3
and KVFRVDAETGDVFAI on EC1 as the binding domain of BV6 and Cad 5,
respectively (17), indicating that
EC1 and EC3 extracellular regions are important for VE-cadherin
intercellular homophilic binding. However, whether other
extracellular domains are involved in the interaction is not yet
clear. In the present study, we prepared a monoclonal antibody
(2C11) to VE-cadherin and mapped its binding epitope to
LDREVVPWYNLTVEA in the EC4 domain of VE-cadherin, and functional
assays revealed that this antibody was able to block
VE-cadherin-mediated cell adhesion, suggesting that besides EC1 and
EC3, EC4 domain is also critical for the molecular function of
VE-cadherin.
Vasculogenic mimicry is found in certain aggressive
tumors, in which VE-cadherin plays a key role. The effect of 2C11
antibody on VM is not known. In this study, we examined the
VE-cadherin expression in several lung cancer cell lines both at
the mRNA and protein levels and found that VE-cadherin was
expressed in Glc-82 cells. Moreover, our data demonstrated that the
2C11 antibody inhibited VM-like structure of Glc-82 cells on 3D
Matrigel. Mechanistically, 2C11 may inhibit cell-cell adhesion
necessary for VM by blockage of intercellular VE-cadherin
interaction. Furthermore, 2C11 may also affect Glc-82 cell
biological functions. It has been reported that knockdown of
VE-cadherin inhibited the proliferation and promoted the apoptosis
of esophageal squamous cell carcinoma cells (ESCCs) and
osteosarcoma cells (20,21). Here we examined the effect of the
2C11 antibody on cell proliferation using CCK-8 assay. Our result
showed that the 2C11 antibody inhibited Glc-82 cell
proliferation.
PI3K/AKT signaling is the most classic pathway
involved in cell proliferation. In vascular endothelial cells,
VE-cadherin interacts with VEGFR2 and inhibits VEGFR2 activation
and downstream AKT phosphorylation (22). In contrast, Taddei et al
reported that VE-cadherin clustering induced claudin-5 expression
in endothelial cells by activation of PI3K/AKT signaling, and AKT
phosphorylation was reduced when VE-cadherin was knocked down
(23). Our result showed that the
2C11 antibody inhibited phosphorylation of AKT in Glc-82 cells,
which may account for the mechanism of the growth inhibitory effect
of the 2C11 antibody on Glc-82 lung cancer.
In conclusion, we generated a novel monoclonal
antibody to VE-cadherin and mapped its binding epitope to the
fourth extracellular domain (EC4). We found that this antibody
inhibited vasculogenic mimicry of aggressive lung cancer Glc-82
cells, suggesting that the antibody blocked VE-cadherin
intercellular clustering. This is the first report to show that the
EC4 domain is involved in VE-cadherin clustering. Furthermore, we
found that Glc-82 cell growth and AKT phosphorylation were
inhibited by the 2C11 antibody. These results may provide a
candidate approach to control VE-cadherin-positive aggressive
tumors.
Acknowledgements
Not applicable.
Funding
The present study was supported in part by grants
from the Priority Academic Program Development of Jiangsu Higher
Education Institutions (PAPD), the National Natural Science
Foundation of China (nos. 81773356, 81673096, 81600565, 81700129
and 81700235), the Natural Science Foundation (nos. BK20171204 and
BK20150296), the Science and Technology Department (no.
BY2015039-C03) of Jiangsu and the Science and Technology Department
of Suzhou (no. SYS201704).
Availability of data and materials
Available from the corresponding author upon
reasonable request.
Authors' contributions
Acquisition, analysis and interpretation of data and
the drafting of the article were carried out by JD and XJ.
Acquisition of data was undertaken by BZ and JH. Design of the
experiments, acquisition, analysis and interpretation of data,
drafting and revising the article were conducted by JY and YH. All
authors read and approved the manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Approval has been obtained by the Ethics Committees
of Soochow University, Suzhou.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of
interests.
References
|
1
|
Dejana E: Endothelial adherens junctions:
Implications in the control of vascular permeability and
angiogenesis. J Clin Invest. 98:1949–1953. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Carmeliet P, Lampugnani MG, Moons L,
Breviario F, Compernolle V, Bono F, Balconi G, Spagnuolo R,
Oosthuyse B, Dewerchin M, et al: Targeted deficiency or cytosolic
truncation of the VE-cadherin gene in mice impairs VEGF-mediated
endothelial survival and angiogenesis. Cell. 98:147–157. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hendrix MJ, Seftor EA, Hess AR and Seftor
RE: Vasculogenic mimicry and tumour-cell plasticity: Lessons from
melanoma. Nat Rev Cancer. 3:411–421. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rong X, Huang B, Qiu S, Li X, He L and
Peng Y: Tumor-associated macrophages induce vasculogenic mimicry of
glioblastoma multiforme through cyclooxygenase-2 activation.
Oncotarget. 7:83976–83986. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Williamson SC, Metcalf RL, Trapani F,
Mohan S, Antonello J, Abbott B, Leong HS, Chester CP, Simms N,
Polanski R, et al: Vasculogenic mimicry in small cell lung cancer.
Nat Commun. 7:133222016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang L, O'Leary H, Fortney J and Gibson
LF: Ph+/VE-cadherin+ identifies a stem cell
like population of acute lymphoblastic leukemia sustained by bone
marrow niche cells. Blood. 110:3334–3344. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Maniotis AJ, Folberg R, Hess A, Seftor EA,
Gardner LM, Pe'er J, Trent JM, Meltzer PS and Hendrix MJ: Vascular
channel formation by human melanoma cells in vivo and in vitro:
Vasculogenic mimicry. Am J Pathol. 155:739–752. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Delgado-Bellido D, Serrano-Saenz S,
Fernández-Cortés M and Oliver FJ: Vasculogenic mimicry signaling
revisited: Focus on non-vascular VE-cadherin. Mol Cancer.
16:652017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Corada M, Zanetta L, Orsenigo F, Breviario
F, Lampugnani MG, Bernasconi S, Liao F, Hicklin DJ, Bohlen P and
Dejana E: A monoclonal antibody to vascular endothelial-cadherin
inhibits tumor angiogenesis without side effects on endothelial
permeability. Blood. 100:905–911. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yagi T and Takeichi M: Cadherin
superfamily genes: Functions, genomic organization, and neurologic
diversity. Genes Dev. 14:1169–1180. 2000.PubMed/NCBI
|
|
11
|
Garrett JP, Lowery AM, Adam AP, Kowalczyk
AP and Vincent PA: Regulation of endothelial barrier function by
p120-catenin·VE-cadherin interaction. Mol Biol Cell. 28:85–97.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Venkiteswaran K, Xiao K, Summers S,
Calkins CC, Vincent PA, Pumiglia K and Kowalczyk AP: Regulation of
endothelial barrier function and growth by VE-cadherin,
plakoglobin, and beta-catenin. Am J Physiol Cell Physiol.
283:C811–C821. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Renaud-Young M and Gallin WJ: In the first
extracellular domain of E-cadherin, heterophilic interactions, but
not the conserved His-Ala-Val motif, are required for adhesion. J
Biol Chem. 277:39609–39616. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Patel SD, Ciatto C, Chen CP, Bahna F,
Rajebhosale M, Arkus N, Schieren I, Jessell TM, Honig B, Price SR,
et al: Type II cadherin ectodomain structures: Implications for
classical cadherin specificity. Cell. 124:1255–1268. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Harrison OJ, Bahna F, Katsamba PS, Jin X,
Brasch J, Vendome J, Ahlsen G, Carroll KJ, Price SR, Honig B, et
al: Two-step adhesive binding by classical cadherins. Nat Struct
Mol Biol. 17:348–357. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dalle Vedove A, Lucarelli AP, Nardone V,
Matino A and Parisini E: The X-ray structure of human P-cadherin
EC1-EC2 in a closed conformation provides insight into the type I
cadherin dimerization pathway. Acta Crystallogr F Struct Biol
Commun. 71:371–380. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Corada M, Liao F, Lindgren M, Lampugnani
MG, Breviario F, Frank R, Muller WA, Hicklin DJ, Bohlen P and
Dejana E: Monoclonal antibodies directed to different regions of
vascular endothelial cadherin extracellular domain affect adhesion
and clustering of the protein and modulate endothelial
permeability. Blood. 97:1679–1684. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Collet G, Szade K, Nowak W, Klimkiewicz K,
El Hafny-Rahbi B, Szczepanek K, Sugiyama D, Weglarczyk K,
Foucault-Collet A, Guichard A, et al: Endothelial precursor
cell-based therapy to target the pathologic angiogenesis and
compensate tumor hypoxia. Cancer Lett. 370:345–357. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang J, Wang J, Jiang JY, Liu SD, Fu K
and Liu HY: Tanshinone IIA induces cytochrome c-mediated
caspase cascade apoptosis in A549 human lung cancer cells via the
JNK pathway. Int J Oncol. 45:683–690. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tang NN, Zhu H, Zhang HJ, Zhang WF, Jin
HL, Wang L, Wang P, He GJ, Hao B and Shi RH: HIF-1α induces
VE-cadherin expression and modulates vasculogenic mimicry in
esophageal carcinoma cells. World J Gastroenterol. 20:17894–17904.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang LZ, Mei J, Qian ZK, Cai XS, Jiang Y
and Huang WD: The role of VE-cadherin in osteosarcoma cells. Pathol
Oncol Res. 16:111–117. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tian Y, Gawlak G, O'Donnell JJ III,
Birukova AA and Birukov KG: Activation of vascular endothelial
growth factor (VEGF) receptor 2 mediates endothelial permeability
caused by cyclic stretch. J Biol Chem. 291:10032–10045. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Taddei A, Giampietro C, Conti A, Orsenigo
F, Breviario F, Pirazzoli V, Potente M, Daly C, Dimmeler S and
Dejana E: Endothelial adherens junctions control tight junctions by
VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol.
10:923–934. 2008. View
Article : Google Scholar : PubMed/NCBI
|