Introduction
MicroRNAs (miRNAs) are a type of small (~22-nt
long), non-coding RNA molecules that regulate target gene
expression by translational inhibition or mRNA destabilization
through the combination with the 3′-untranslated region of target
messenger RNAs. The loss of homeostasis in the miRNA/mRNA axis
leads to a number of physiological and pathological events, such as
inflammation, senescence and tumorigenesis (1,2).
Osteosarcoma (OS) is the most common primary bone
malignancy among children and adolescents (3). The exact pathological mechanisms of
this malignancy remain unknown. Acting as the majority of
non-coding RNAs, the effects of miRNAs in OS have been widely
reported (4). For example, miR-224
was reported to be suppressed in OS tissues and was correlated with
shorter survival time of OS patients. Overexpression of miR-224
promoted the sensitivity of OS cells to cisplatin by targeting Rac1
(5). In addition, our previous
study reported that miR-140 was downregulated in OS tissues and
functioned as a tumor suppressor in OS, as overexpression of
miR-140 significantly inhibited cell proliferation in vitro
and tumor growth in vivo by targeting the HDAC4 signaling
pathway (6). Furthermore, miR-186
has been reported to function as a tumor suppressor in a variety of
malignancies and regulate the chemosensitivity of non-small cell
lung cancer cells to paclitaxel by targeting the MAPT-signaling
pathway (7). In idiopathic
pulmonary fibrosis cells, it has been closely related with the
overexpression of collagen V and epithelial-to-mesenchymal
transition (EMT) (8). However, its
exact function in OS has not been identified yet. In the present
study, the expression of miR-186 and its putative biological
functions in OS cells were investigated.
Materials and methods
Clinical samples and cell culture
To explore the expression of miR-186 in OS, a cohort
of 40 OS cases was adopted into the experiment. The detailed
information of these patients are displayed in Table I. All patients had not followed any
therapy before their recruitment to this study with written
consents for their participation in this study. The present study
was approved by The Institutional Ethics Committee of Chongqing
Medical University. Adjacent normal tissues were collected from the
patients as negative controls.
 | Table I.Clinicopathological factors of 40 OS
patients. |
Table I.
Clinicopathological factors of 40 OS
patients.
| Clinicopathological
factors | No. of patients |
|---|
| Sex |
|
| Male | 23 |
|
Female | 17 |
| Age (years) |
|
|
>25 | 18 |
|
<25 | 22 |
| Tumor sites |
|
|
Femur | 16 |
|
Tibiofibules | 9 |
| Other
sites | 15 |
| Pathological
types |
|
|
Osteoblastic | 18 |
|
Chondroblastic | 6 |
|
Fibroblastic | 9 |
| Other
types | 7 |
| Alkaline
phosphatase |
|
|
Normal | 24 |
|
Elevated | 16 |
Human OS cell lines, U2 and HOS, were adopted for
further biological function assays. The cell lines were both
purchased from the Cell Bank of the Chinese Academy of Medical
Sciences (Shanghai, China). The cells were all cultured in
RPMI-1640 medium (Gibco, Grand Island, NY, USA) with 10% fetal
bovine serum (FBS; Gibco) and incubated in a humidified atmosphere
of 5% CO2 at 37°C.
Oligonucleotides and cell
transfection
MicroRNA-186 and relative scramble mimic
oligonucleotides were all purchased from Dharmacon (Lafayette, CO,
USA). A final concentration of 50 nM mimics was transfected into OS
cells using the DhamaFECT 1 transfection reagents according to the
manufacturer's instructions. After 6-h transfection, the medium was
changed and the cells were cultured for 48 h and harvested for
further experiments.
RNA extraction and qRT-PCR
To assess the expression level of miR-186,
quantitative real-time PCR (qRT-PCR) was performed using SYBR Green
(TransGen Biotech, Beijing, China) in the Bio-Rad real-time PCR
system. Total RNA was extracted from the cells and tissues using
TRIzol (Invitrogen Life Technologies, Carlsbad, CA, USA) according
to the manufacturer's instructions. The data of miR-186 and PTTG1
expression were normalized to endogenous U6 snRNA and GAPDH,
respectively. The primer for reverse transcription was used as
follows: 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGAGCCCAA-3′. The
forward and reverse primers for miR-186 RT-PCR were
5′-CGCGGCAAAGAATTCTCCTT-3′ and 5′-GCCGCAGTGGTTTTACCCT-3′,
respectively. The forward and reverse primers for PTTG1 were
5′-TTTGACCTGCCTGAAGAGC-3′ and 5′-CGACAGAATGCTTGAAGGAG-3′
respectively. The forward and reverse primers for U6 and GAPDH were
used as follows: For U6 5′-CTCGCTTCGGCAGCACATATACT-3′ and
5-ACGCTTCACGAATTTGCGTGTC-3′, respectively and for GAPDH,
5′-CTCGCTTCGGCAGCACATATACT-3′ and 5′-ACGCTTCACGAATTTGCGTGTC-3′,
respectively. Subsequently, the PCR data were analyzed using the
2−ΔΔCT method.
Luciferase reporter assay and vector
construction
To identify the direct target role of PTTG1 on
miR-186 by connecting with its 3′-UTR, the Dual-Luciferase Reporter
assays were adopted. The full-length 3′-UTR of the PTTG1 mRNA was
amplified from genomic DNA and then cloned into the pGL-3 vector
(Promega, Madison, WI, USA). Mutations of PTTG1 3′-UTR sequence
were created through the QuickChange Site-Directed Mutagenesis kit
according to the manufacturer's instructions (Stratagene; Agilent
Technologies, Inc., Santa Clara, CA, USA). Before transfection,
about 1×105 cells/well were seeded into 24-well plates
for 24 h. The cells were transfected with 10 ng pRL-TK
Renilla Luciferase reporter, 50 ng pGL-3 firefly Luciferase
reporter and 50 nM miRNA-186/scramble mimic. The pRL-TK vector
served as internal control and the luciferase reporter construct
containing the miRNA-186 consensus target sequence served as
positive control. All transfections were carried out with
Lipofectamine 2000 (Invitrogen Life Technologies). At 48 h after
transfection, the cells were collected and lysed using the Passive
Lysis Buffer (Promega). Finally, the Luciferase activity was
assessed using the Dual-Luciferase Reporter assay (Promega). The
results were normalized to the Renilla Luciferase.
Cell proliferation assay
The effects of miR-186 on the cell proliferation
rate of OS cells were assessed using Cell Counting Kit-8 (CCK-8)
assays. At 24 h after transfection 0.5×104 HOS or U2
cells were seeded into the 96-well plates. The cells were incubated
in 10% CCK-8 (Dojindo Molecular Technologies, Inc., Kumamoto,
Japan) and diluted in normal culture medium at 37°C until visual
color conversion occurred. The proliferation rate was determined at
0, 24, 48 and 72 h after the cells were seeded into 96-well plates
and quantification was performed on a microtiter plate reader
according to the manufacturer's instructions. All experiments were
performed in triplicate.
Cell cycle and apoptosis analysis
For the analysis of the cell cycle, HOS or U2 cells
were harvested, diluted to a concentration of 5×105
cells/ml and washed twice with ice-cold PBS 48 h after
transfection. Then the cells were fixed in ice-cold 70% ethanol and
incubated with propidium iodide (PI) and RNase A. For the analysis
of apoptosis, the cells were incubated with PE Annexin V and 7-AAD
according to the PE Annexin V Apoptosis Detection kit I (BD
Biosciences, San Jose, CA, USA) protocol. All cells in both assays
were then analyzed by fluorescence-activated cell sorting (FACS).
Cells that bound to PE Annexin V underwent early apoptosis, while
cells that bound to both PE Annexin V and 7-AAD were either in the
late stages of apoptosis or already dead. All experiments were run
in triplicate.
Cell Matrigel assay
For analyzing the effects of miR-186 on cell
migration and invasion, the Matrigel chamber assays were adopted.
After a 24-h transfection, 2×105 HOS or U2 cells
suspended in serum-free medium were added to the upper chamber
(8-µm pore filter). Medium containing 20% FBS was added to the
lower chamber as a chemoattractant. For the invasion assays, the
Transwell chambers were coated with Matrigel (BD Biosciences) and
then incubated at 37°C for 4 h and inserted in 2×4-well plates.
After incubation for 24 h, the non-invasive cells on the upper
surface of the chamber were gently removed with a cotton swab. The
cells passing through the chambers and located on the lower surface
were stained with 0.05% crystal violet, air dried and photographed.
All experiments were performed in triplicate.
Immunoblot assays
After 48 h transfection, HOS and U2 cells were
harvested in PBS at 4°C and lysed on ice in cold-modified
radioimmunoprecipitation buffer supplemented with protease
inhibitors. Protein concentration was assessed using the BCA
Protein Assay kit (Beyotime Institute of Biotechnology, Haimen,
China) and equal amounts of protein were analyzed by SDS-PAGE. The
gels were electroblotted onto nitrocellulose membranes (EMD
Millipore, Billerica, MA, USA). The membranes were blocked for 1 h
with 5% fat-free dry milk in Tris-buffered saline containing 0.1%
Tween-20 and incubated at 4°C overnight with primary antibody.
Detection was assessed using peroxidase-conjugated secondary
antibodies (1:1,000; anti-rabbit IgG; cat. no. 7054; Cell Signaling
Technology, Inc., Danvers, MA, USA) and the enhanced
chemiluminescence system (ECL; EMD Millipore). The primary
antibodies used were PTTG1 (1:1,000; rabbit, monoclonal; cat. no.
13445S), HIF-1 (1:1,000; rabbit, polyclonal; cat. no. 3716S) and
GAPDH (1:1,000; rabbit, monoclonal; cat. no. 5174S; Cell Signaling
Technology). The experiments were performed three times.
Cell glucose uptake and lactate
production assays
For the analysis of the glucose uptake, the Glucose
Test kit (BioVision, Milpitas, CA, USA) was used. After
transfection, HOS and U2 cells were seeded into 6-well plates at a
density of 106 cells/well at 37°C for 48 h. The medium
at 0 h was collected as background glucose concentration. The
glucose concentration reduction of the medium was considered as
cellular glucose uptake. Thus, glucose uptake was calculated as
follows: glucose uptake=(background concentration-reading
concentration)/protein concentration. For assessing the
extracellular lactic acid production, the culture medium of cells
was explored using the Lactate Assay kit (BioVision) according to
the manufacturer's instructions. The values were normalized to the
protein concentration.
Statistical analysis
All data were derived from no less than three
independent experiments, and are presented as the mean ± SD.
Statistical analysis was carried out using SPSS 18.0 software (IBM,
New York, NY, USA). The Student's t-test was used for comparisons
between two groups. P≤0.05 was considered to indicate a
statistically significant difference.
Results
miR-186 is suppressed in OS
tissues
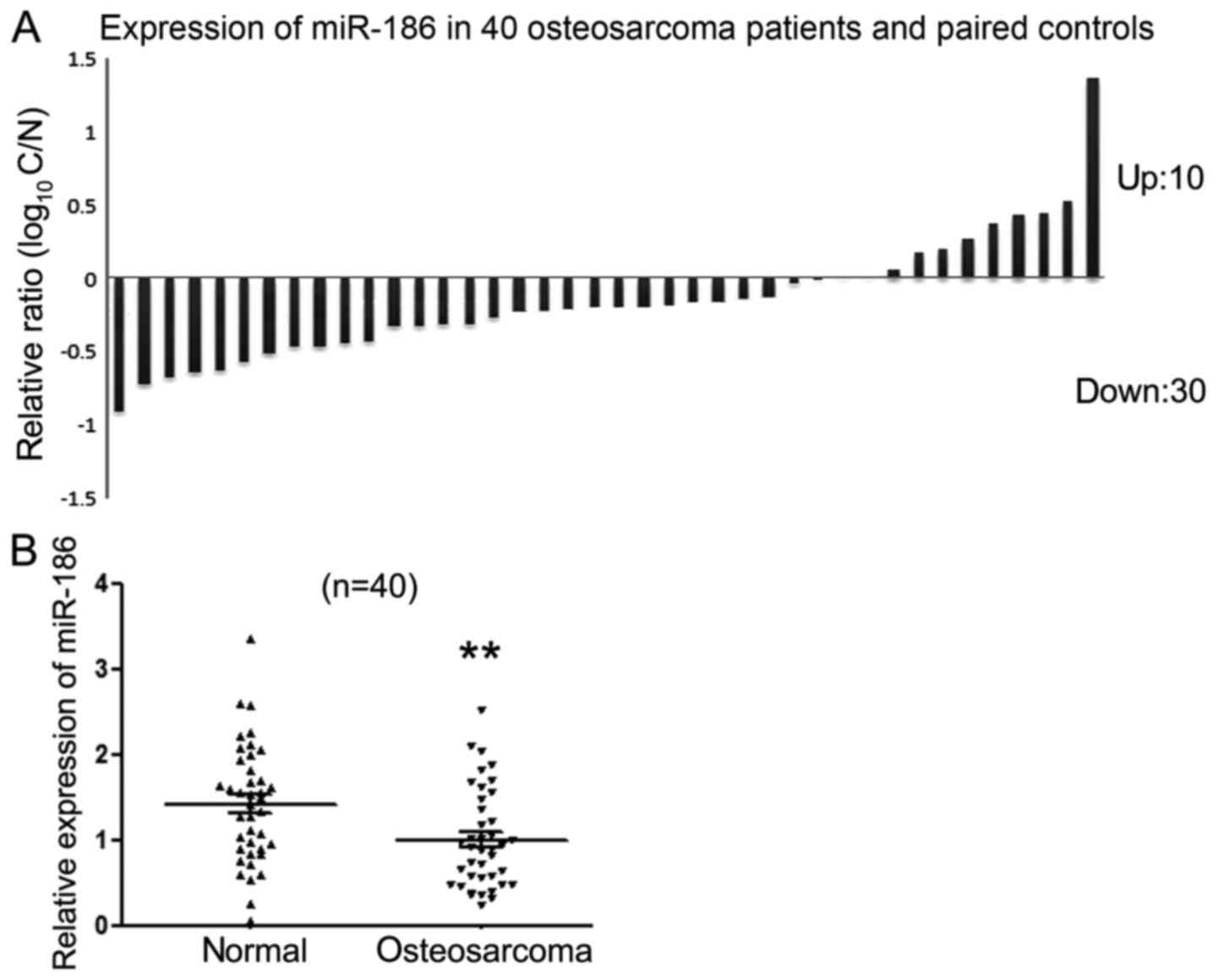
To explore the effects of miR-186 in OS, the
expression level of miR-186 was detected in a cohort of 40 OS
tissues and adjacent normal tissues through quantitative RT-PCR
assays. Compared to the adjacent normal tissues, 30 out of 40 (75%)
OS tissues exhibited lower level of miR-186 expression (Fig. 1A). To further identify the
expression tendency of miR-186 in OS, we analyzed its average level
between OS tissues and normal tissues. As expected, the average
level of miR-186 was significantly attenuated in OS tissues
(Fig. 1B). The suppression of
miR-186 in OS indicated its tumor-suppressive role in OS.
Overexpression of miR-186 inhibits
cell proliferation and promotes cell apoptosis in OS cells
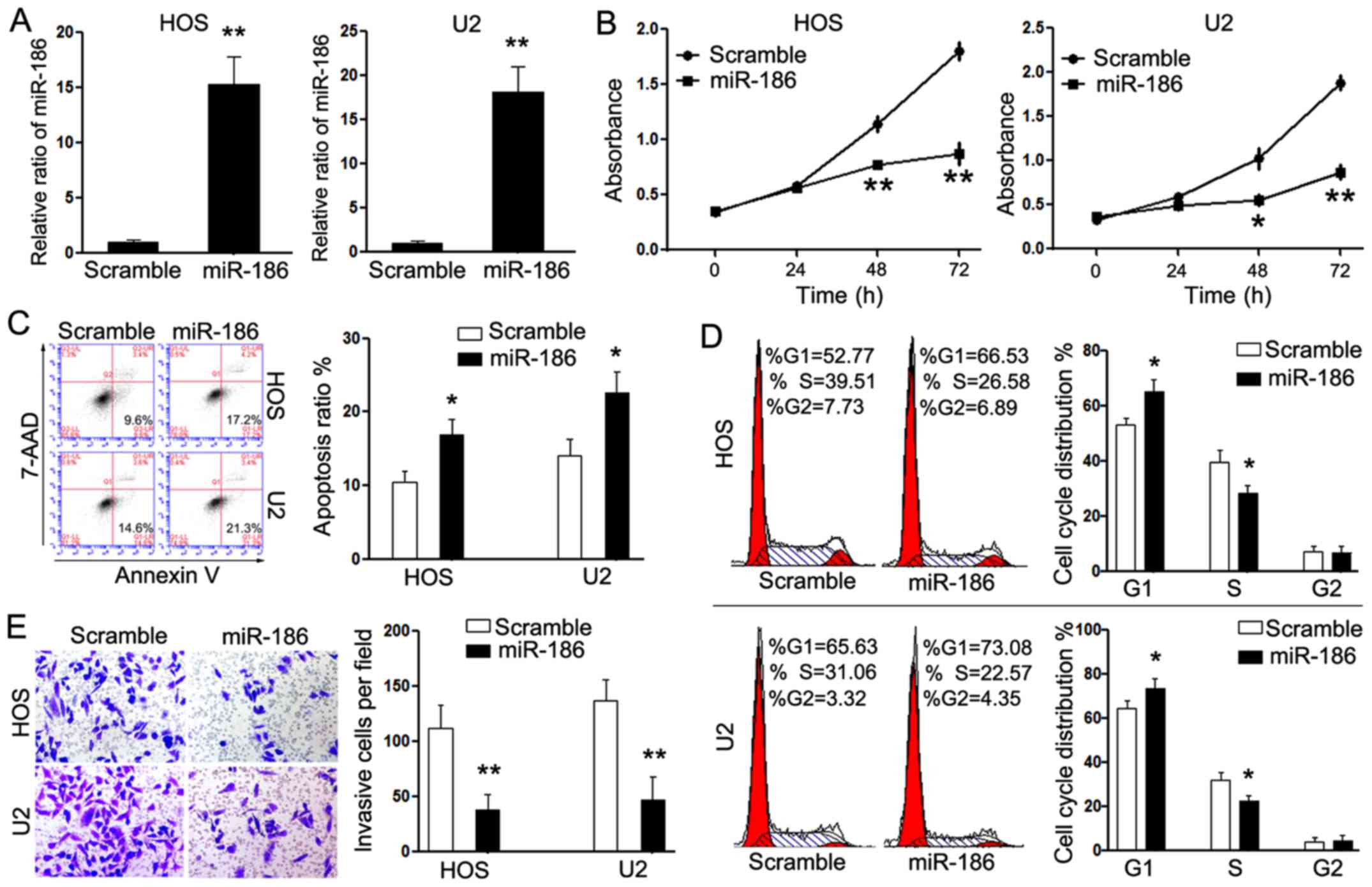
To identify the biological function of miR-186 in
OS, the overexpression model of OS cells was established through
transiently transfecting miR-186 mimic into the OS cell lines, HOS
and U2. The cells transfected with scramble mimic were used as
negative control. Upon transfection, the expression of miR-186 was
~15-fold higher in HOS cells (designed as HOS/186) and 18-fold
higher in U2 cells (designed as U2/186) compared with control
groups (Fig. 2A).
Subsequently, we explored the effects of miR-186 on
the proliferation, cell cycle progression and apoptosis of OS
cells. The rate of cell proliferation was assessed in both cell
lines in vitro using the CCK-8 assay. The doubling time of
HOS/186 and U2/186 cells was significant lower than that of HOS/Scr
and U2/Scr cells, indicating that overexpression of miR-186
inhibited the growth rate of OS cells (Fig. 2B). Further FACS assays revealed that
overexpression of miR-186 resulted in a significant upregulation of
early apoptotic cells (Fig. 2C),
associated with a simultaneous increased amount of cells in the
G1-phase and a reduced number of cells in the S-phase of the cell
cycle (Fig. 2D), which indicated
that overexpression of miR-186 inhibited cell proliferation and
induced cell apoptosis of OS cells.
Overexpression of miR-186 suppresses
cell invasion in OS cells
Tumor metastasis is a pivotal reason for the poor
clinical outcome of OS. Using neoadjuvant chemotherapy with
surgery, the five-year survival rate of patients with
non-metastatic OS has increased >50%, while the prognosis of
patients with tumor metastasis is still poor (9). Thus, we further explored the effects
miR-186 on cell invasion of OS cells using the Matrigel chamber
assays. The OS cells transfected with miR-186 (HOS/186 and U2/186
cells) exhibited less cells passing through the chambers coated
with Matrigel (Fig. 2E), which
means that transfection with miR-186 significantly suppressed the
invasive capacity of OS cells.
miR-186 modulates the expression of
PTTG1 in OS cells
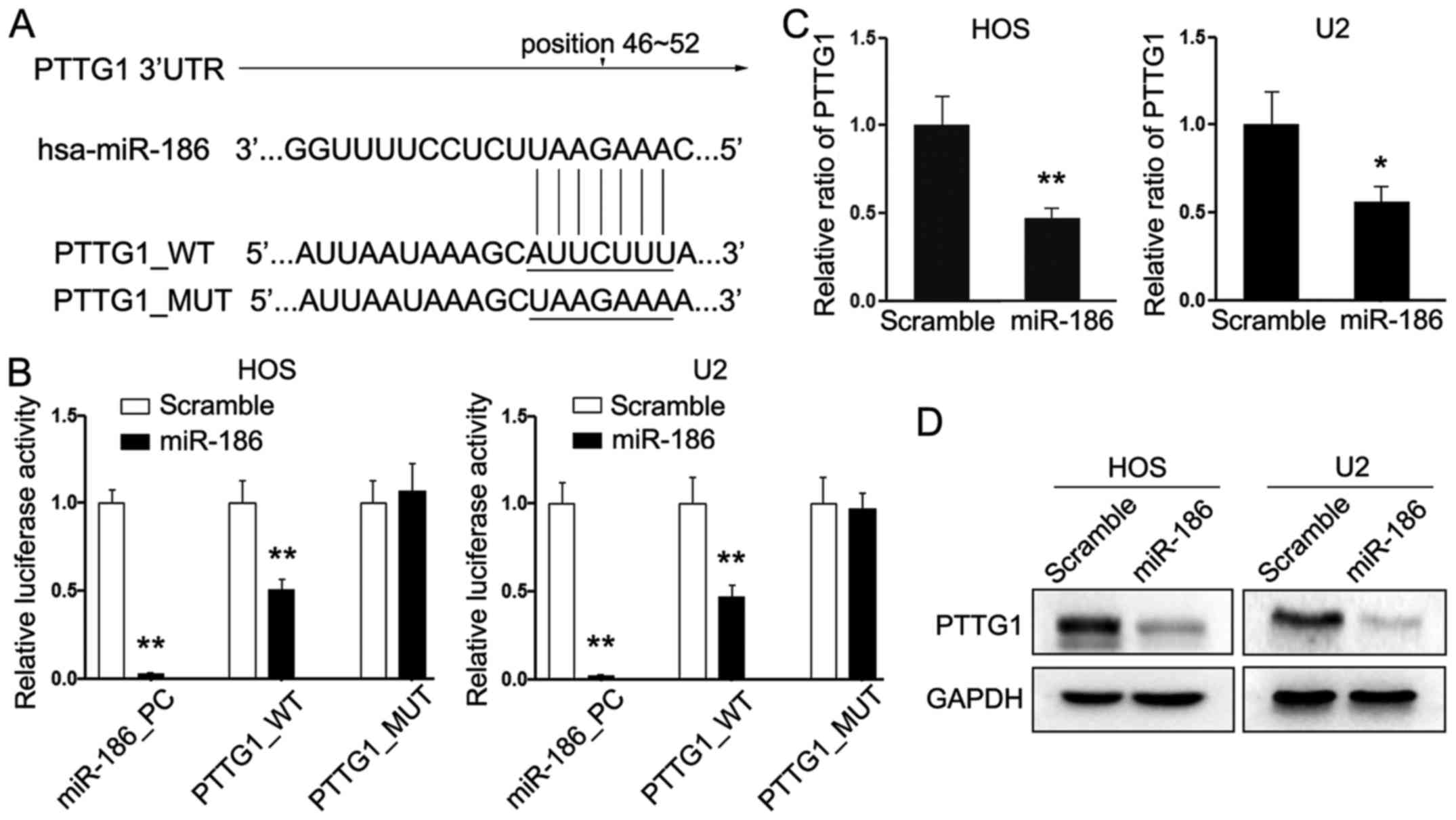
The aforementioned data strongly indicated that
miR-186 functioned as a tumor suppressor in OS cells. To elucidate
the putative mechanisms involved in miR-186-mediated tumor
suppressive effects in OS, we investigated its target genes using
the target prediction software, miRanda (http://www.targetscan.org/vert_71/) and TargetScan
(http://34.236.212.39/microrna/microrna/home.do). Both
these tools indicated that PTTG1 was a target gene of miR-186
(Fig. 3A). PTTG1 has been reported
to function as an oncogene in a cohort of cancers, including OS
(10). Inhibition of PTTG1 in MG-63
cells caused cell cycle arrest and subsequent apoptosis (11). To identify the target role of PTTG1
in OS cells, the dual-luciferase reporter assays were adopted.
Compared to the cells transfected with scramble mimic, cells
treated with miR-186 mimic demonstrated significantly attenuated
luciferase activity when co-transfected with the reporter gene
containing wild-type 3′-UTR of PTTG1. However, transfection with
miR-186 mimic did not affect the luciferase activity when
co-transfected with gene containing the mutant 3′-UTR in HOS and U2
cells (Fig. 3B). Furthermore,
western blot analysis and qRT-PCR analysis confirmed that the PTTG1
protein as well as mRNA levels were indeed reduced drastically in
HOS/186 and U2/186 cells consistently (Fig. 3C and D). Collectively, these data
strongly indicated that PTTG1 is a target gene of miR-186 in OS
cells.
PTTG1 is involved in miR-186-mediated
suppressive effects
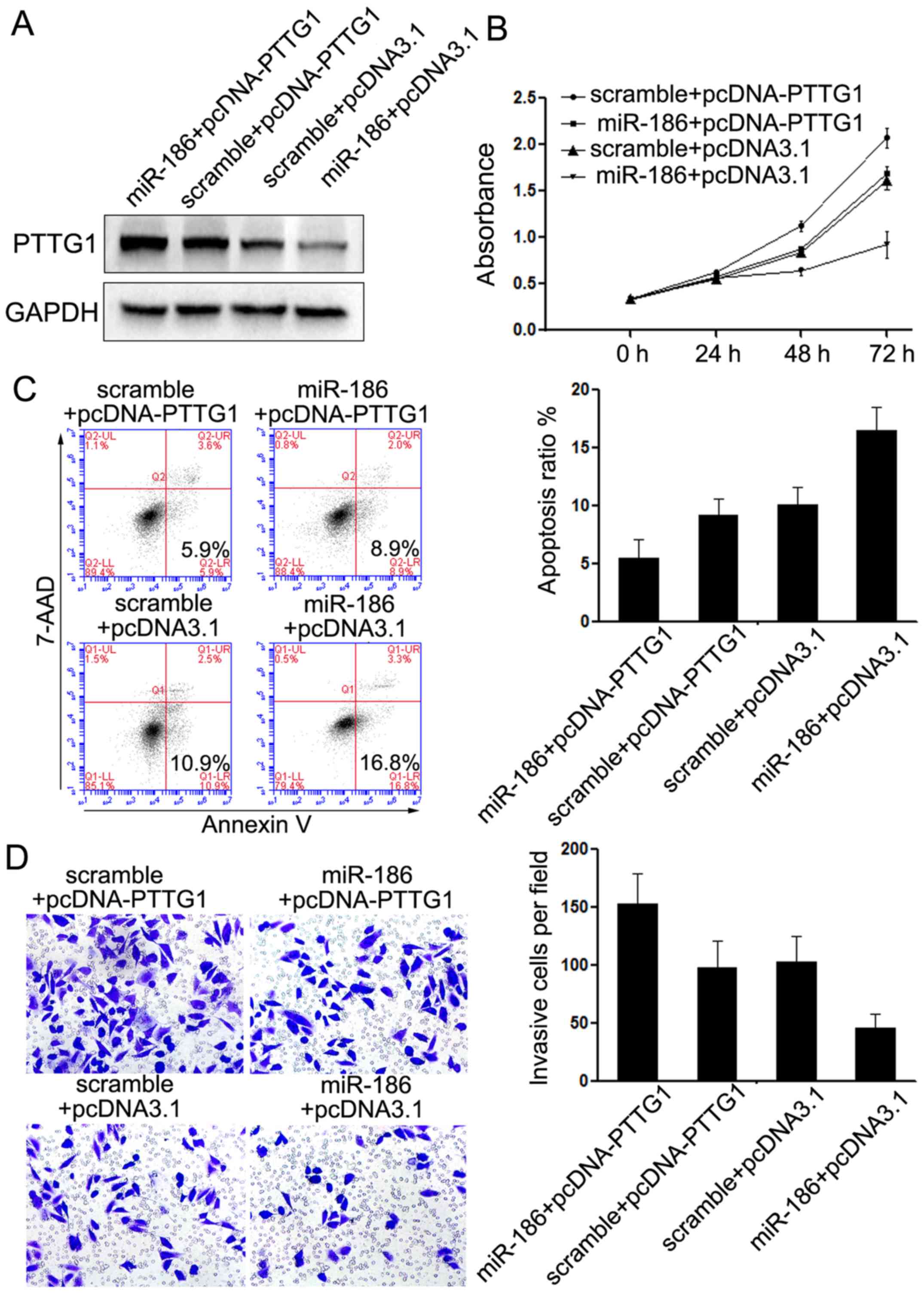
As aforementioned, PTTG1 functions as an oncogene in
a cohort of cancer cells. We hypothesized that PTTG1 may be
involved in the miR-186-mediated suppressive effects in OS cells.
To validate this hypothesis, rescue assays were performed. The
PTTG1/control constructs were transfected to the OS cells which had
been previously transfected with miR-186/scramble mimic. As
displayed in Fig. 4A, the
expression of PTTG1 was partially restored upon transfection with
PTTG1 constructs. Notably, accompanied with the restored expression
of PTTG1, an increased cell proliferation (Fig. 4B) as well as suppressed cell
apoptosis (Fig. 4C) were observed
in OS cells transfected with PTTG1 constructs and miR-186 mimic.
Furthermore, co-transfected with PTTG1 constructs also partially
blocked miR-186-mediated suppression of invasion (Fig. 4D). All these results indicated that
PTTG1 was involved in miR-186-mediated tumor suppressive effects in
OS.
miR-186 suppresses the expression of
HIF-1α and glycolysis of OS cells
Aerobic glycolysis is the major method of energy
supply and thus, one of the characteristic phenotypes of tumor
cells. It was reported that miR-186 significantly inhibited aerobic
glycolysis in gastric cancer by suppressing hypoxia inducible
factor 1α (HIF-1α) (12), which has
been demonstrated to function as an oncogene in OS (6). Thus, we investigated whether
HIF-1α-mediated dysregulation of aerobic glycolysis was also
involved in the suppressive effects of miR-186. As expected,
overexpression of miR-186 significantly suppressed the expression
of HIF-1α in HOS and U2 cells (Fig.
5A). Consistently, HOS and U2 cells that were treated with
miR-186 had less intracellular glucose and lactate production
(Fig. 5B and C). These results
indicated that HIF-1α-regulated glycolysis may be involved in the
miR-186-mediated suppressive effects on OS cells.
Discussion
Research on the current mechanisms underlying the
progression of OS is still limited. Acting as one of the most
important non-coding RNAs, miRNAs have been widely explored in the
tumorigenesis and tumor progression of malignancies, including OS.
miR-186 is a newly reported miRNA and its effects in OS remain
unknown. In the present study, we assessed the expression of
miR-186 in OS tissues and detected its biological functions on the
OS cells and the regulatory mechanisms involved.
miR-186 is a conservative gene, which resides
between exon 8 and 9 of the zinc finger RAN-binding domain
containing 2 (ZRANB2) gene (13).
Despite being found to perform a pivotal role in muscle
differentiation (14), miR-186 has
also been found to participate in many biological and pathological
processes, including cell proliferation, apoptosis and invasion
(7,15). Recently, the tumor suppressive role
of miR-186 has been widely reported in many cancers, such as in
prostate cancer (PC), where miR-186 inhibited the expression of
VEGF and reduced tumor growth by influencing the PGE-2-induced
VEGF-signaling pathway (16). In
addition, in gastric cancer cells, miR-186 inhibited cell
proliferation, migration and invasion by targeting Twist1 (15). In the present study, we investigated
the expression of miR-186 in 40 pairs of OS tissues and relative
normal tissues. As expected, the expression of miR-186 was
consistently suppressed in OS tissues and the restored expression
of miR-186 inhibited cell proliferation, arrested cell cycle
progression, induced cell apoptosis and inhibited cell invasion of
OS cells. These results revealed the tumor-suppressive role of
miR-186 in OS.
To investigate the mechanisms involved in
miR-186-mediated suppressive effects, we searched for its target
genes. Among the predicted genes, PTTG1 attracted most attention.
PTTG1 is a newly identified oncogene and it is highly expressed in
pituitary tumors and other neoplasms. The expression of PTTG1 is
cell cycle-dependent; it peaks at the G2/M phase of the cell cycle
and is degraded at the initiation of anaphase (13). Furthermore, overexpression of PTTG1
causes p53-dependent and p53-independent apoptosis and inhibits
cell mitosis (11). In OS cells,
overexpression of PTTG caused cell cycle arrest and subsequent
apoptosis, while inhibition of PTTG promoted cell survival.
Interestingly, OS cells expressing PTTG demonstrated signs of
aneuploidy including the presence of micronuclei and multiple
nuclei (11). The oncogenic role of
PTTG1 in OS combined with the fact that PTTG1 was reported to be
involved in miR-186-mediated tumor suppressive effects on non-small
cell lung cancer cells led to the hypothesis that it may
participate in miR-186-mediated effects on OS cells. Further
experiments strongly supported our hypothesis as the rescued
expression of PTTG1 in the OS cells which had been previously
transfected with miR-186 partially abolished its suppressive
effects on cell proliferation and invasion. In combination with
previous research results, it suggested that PTTG1 partially
participated in miR-186-mediated tumor suppressive effects.
A hypoxic microenvironment is common in many types
of solid tumors, including OS (17,18).
Hypoxia may induce aerobic glycolysis, which is one of the
characteristic phenotypes of malignant cells and is the major way
of energy supply (12). HIF-1α may
be involved in this mechanism (19). We found that overexpression of
miR-186 significantly suppressed the expression of HIF-1 in OS
cells and inhibited glucose uptake and lactate production. These
results indicated that HIF-1α-mediated dysregulation of aerobic
glycolysis may be involved in the miR-186-mediated suppressive
effects on OS cells and are partially consistent with our previous
study that HIF-1α acts as an oncogene in OS cells (12). However more studies are warranted in
order to identify the underlying mechanisms that participate in the
suppressive effects on aerobic glycolysis.
The findings of the present study strongly
demonstrated that miR-186 functions as a tumor suppressor in OS
cells through, at least partially, the suppression of the
PTTG1-mediated oncogenic effects on the malignant phenotypes of OS
cells and HIF-1-mediated suppression on aerobic glycolysis. To the
best of our knowledge, this is the first comprehensive study to
explore the role of miR-186 in OS. A combined miRNA-based and
epigenetic treatment may be a novel potential therapeutic target
for OS.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81502329), the
Program of Science and Technology of Chongqing Commission (grant
no. KJ1600228), the Programs of Yongchuan Hospital of Chongqing
Medical University (grant nos. YJZQN 201514 and YCZQN 201511).
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Olivieri F, Capri M, Bonafè M, Morsiani C,
Jung HJ, Spazzafumo L, Viña J and Suh Y: Circulating miRNAs and
miRNA shuttles as biomarkers: Perspective trajectories of healthy
and unhealthy aging. Mech Ageing Dev. 165:162–170. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cao Q, Li YY, He WF, Zhang ZZ, Zhou Q, Liu
X, Shen Y and Huang TT: Interplay between microRNAs and the STAT3
signaling pathway in human cancers. Physiol Genomics. 45:1206–1214.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim HJ, Chalmers PN and Morris CD:
Pediatric osteogenic sarcoma. Curr Opin Pediatr. 22:61–66. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kumar Ram RM, Boro A and Fuchs B:
Involvement and clinical aspects of MicroRNA in osteosarcoma. Int J
Mol Sci. 17:E8772016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Geng S, Gu L, Ju F, Zhang H, Wang Y, Tang
H, Bi Z and Yang C: MicroRNA-224 promotes the sensitivity of
osteosarcoma cells to cisplatin by targeting Rac1. J Cell Mol Med.
20:1611–1619. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xiao Q, Huang L, Zhang Z, Chen X, Luo J,
Zhang Z, Chen S, Shu Y, Han Z and Cao K: Overexpression of miR-140
inhibits proliferation of osteosarcoma cells via suppression of
histone deacetylase 4. Oncol Res. 25:267–275. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ye J, Zhang Z, Sun L, Fang Y, Xu X and
Zhou G: miR-186 regulates chemo-sensitivity to paclitaxel via
targeting MAPT in non-small cell lung cancer (NSCLC). Mol Biosyst.
12:3417–3424. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lei GS, Kline HL, Lee CH, Wilkes DS and
Zhang C: Regulation of collagen V expression and
epithelial-mesenchymal transition by miR-185 and miR-186 during
idiopathic pulmonary fibrosis. Am J Pathol. 186:2310–2316. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ando K, Heymann MF, Stresing V, Mori K,
Rédini F and Heymann D: Current therapeutic strategies and novel
approaches in osteosarcoma. Cancers (Basel). 5:591–616. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cai SQ, Dou TT, Li W, Li SQ, Chen JQ, Zhou
J, Zheng M and Man XY: Involvement of pituitary tumor transforming
gene 1 in psoriasis, seborrheic keratosis, and skin tumors. Discov
Med. 18:289–299. 2014.PubMed/NCBI
|
|
11
|
Yu R, Heaney AP, Lu W, Chen J and Melmed
S: Pituitary tumor transforming gene causes aneuploidy and
p53-dependent and p53-independent apoptosis. J Biol Chem.
275:36502–36505. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu L, Wang Y, Bai R, Yang K and Tian Z:
miR-186 inhibited aerobic glycolysis in gastric cancer via
HIF-1alpha regulation. Oncogenesis. 6:e3182017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu R, Ren SG, Horwitz GA, Wang Z and
Melmed S: Pituitary tumor transforming gene (PTTG) regulates
placental JEG-3 cell division and survival: Evidence from live cell
imaging. Mol Endocrinol. 14:1137–1146. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Antoniou A, Mastroyiannopoulos NP, Uney JB
and Phylactou LA: miR-186 inhibits muscle cell differentiation
through myogenin regulation. J Biol Chem. 289:3923–3935. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cao C, Sun D, Zhang L and Song L: miR-186
affects the proliferation, invasion and migration of human gastric
cancer by inhibition of Twist1. Oncotarget. 7:79956–7996. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Terzuoli E, Donnini S, Finetti F, Nesi G,
Villari D, Hanaka H, Radmark O, Giachetti A and Ziche M: Linking
microsomal prostaglandin E Synthase-1/PGE-2 pathway with miR-15a
and −186 expression: Novel mechanism of VEGF modulation in prostate
cancer. Oncotarget. 7:44350–44364. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Koh MY, Spivak-Kroizman TR and Powis G:
HIF-1alpha and cancer therapy. Recent Results Cancer Res.
180:15–34. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Masoud GN and Li W: HIF-1alpha pathway:
Role, regulation and intervention for cancer therapy. Acta Pharm
Sin B. 5:378–389. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cheng SC, Quintin J, Cramer RA, Shepardson
KM, Saeed S, Kumar V, Giamarellos-Bourboulis EJ, Martens JH, Rao
NA, Aghajanirefah A, et al: mTOR- and HIF-1α-mediated aerobic
glycolysis as metabolic basis for trained immunity. Science.
345:12506842014. View Article : Google Scholar : PubMed/NCBI
|