Introduction
Hepatocellular carcinoma (HCC) is the fifth most
common malignant tumor worldwide and is the third leading cause of
cancer-related mortality (1). For
the treatment of HCC, a variety of modalities have been used,
including molecular-targeted therapy, transarterial
chemoembolization (TACE), liver transplantation and local tumor
ablation [e.g., irreversible electroporation and radiofrequency
ablation (RFA)] (2). Among these,
RFA is accepted as a potentially curative local treatment for
patients with early-stage HCC (3).
However, tumor tissues experience insufficient radiofrequency
ablation (iRFA) with temperatures (60–42°C) too low to kill all of
the cancer cells, resulting in rapid and aggressive recurrence of
HCC after RFA (4–6). For instance, Yoshida et al
(7) revealed that iRFA promoted HCC
spread and growth by transiently inducing EMT-like changes and an
enhanced malignant potential. Liu et al (8) reported that RFA increased VEGF
expression via CaMKII-induced ERK activation, and then accelerated
residual HCC growth. However, the mechanisms underlying the
RFA-induced tumor promotion remain largely unexplored.
Autophagy is an evolutionarily conserved,
intracellular self-protective mechanism for the degradation of
cytoplasmic material, damaged organelles and aggregate-prone
proteins in lysosomes (9,10). Recently, considerable evidence has
supported that autophagy plays a critical role in many human
diseases, including cancer. In pancreatic cancer, the inhibition of
autophagy suppressed cell growth in vitro and tumor
progression in vivo (11).
In HCC, LC3-II (a key autophagic marker) expression levels were
positively related with the development and a poor prognosis of HCC
(12). Chang et al (13) revealed that inhibition of autophagy
reduced viability of HCC. Moreover, autophagy can act as an
accomplice of survival, malignant progression and distant
metastasis of HCC cells under adverse conditions (11). Peng et al (14) demonstrated that hypoxia-induced
autophagy resulted in resistance of HCC cells to chemotherapeutic
agents. In the present study, our results indicated that LC3B
expression was upregulated in the residual hepatocellular carcinoma
cells after RFA treatment in vivo. However, the role of
autophagy in rapid and aggressive recurrence of HCC after RFA
remains largely unknown. Thus, we sought to determine the potential
role and mechanism of autophagy in rapid and aggressive recurrence
of HCC after RFA. Our results demonstrated that autophagy and the
CD133 feedback loop are indispensable for iRFA-induced tumor cell
progression in HCC.
Materials and methods
Ethics statement
In the present study, the use of human tissue
samples and all experimental procedures and protocols were approved
by the Ethics Committee of the First Affiliated Hospital of Third
Military Medical University with the following reference no.
2013(47).
Patients and samples
Hepatocellular carcinoma (HCC) tissues were obtained
from 31 patients who underwent surgical resection between December
2013 and December 2015 at the Southwest Hospital, Third Military
Medical University. Sixteen specimens were obtained from HCC
patients with radiofrequency ablation (RFA) treatment at our
hospital, and the remaining 15 specimens were obtained from HCC
patients with non-RFA treatment. RFA treatment was performed with
the patient under endotracheal general anesthesia. Then, patients
were submitted to RFA with ultrasonography guidance, utilizing a
generator providing 460 kHz alternating current and a semi-flax
retractable multi-pronged curved electrode-needle (RITA Medical
Systems, Inc., Mountain View, CA, USA). The average target
temperature was set at 100–110°C, and ablation was continued for 25
min depending on the desired ablation size. The process was
monitored by real-time ultrasound to ensure 1-cm margins.
Immunohistochemical analysis
Human HCC tissues were fixed in 4% paraformaldehyde
overnight, and subsequently embedded in paraffin. The
paraffin-embedded tissues were cut into standard 6 µm sections,
deparaffinated in xylene and rehydrated through graded alcohol
solutions. Antigen retrieval was performed 10 min at 92°C in EDTA
(10 mmol/l, pH 8.0) in a water bath. Endogenous peroxidases were
inactivated by immersing the sections in 0.3% hydrogen peroxide
(H2O2) for 12 min. Next, the sections were
blocked with 5% goat serum for 60 min at 37°C. The slides were
incubated with rabbit polyclonal antibody against human LC3B
(1:1,000; cat. no. ab63817; Abcam, Cambridge, MA, USA) and mouse
monoclonal antibody against human CD133 (1:50; cat. no. MAB4399-I;
EMD Millipore, Billerica, MA, USA) overnight at 4°C. Next, the
slides were treated with the appropriate HRP-conjugated goat
antibody against rabbit/mouse (1:1,500; cat. no. KIT-5920; Maixin
Biotechnology, Co., Ltd., Fuzhou, China) for 45 min at 37°C, and
then developed with 3,3′-diaminobenzidine. Finally, the slides were
counterstained with hematoxylin and mounted. The slides were
examined under an Olympus light microscope (Leeds Precision
Instruments, Minneapolis, MN, USA). LC3B and CD133 protein
expression in tissues were determined according to methods
described by Pinheiro et al (15). Sections were semi-quantitatively
scored for the extent of immunoreactions as follows: 0, 0%
immunoreactive cells; 1, <5% immunoreactive cells; 2, 5–50%
immunoreactive cells; and 3, >50% immunoreactive cells.
Additionally, the staining intensity was semi-quantitatively scored
as 0 (negative), 1 (weak), 2 (intermediate), or 3 (strong). The
final immunoreaction score was defined as the sum of both
parameters.
Cell lines and cell culture
Huh-7 and SMMC7721 cells were obtained from the Cell
Bank Type Culture Collection of the Chinese Academy of Sciences
(Shanghai, China). Huh-7 cells were cultured in Dulbecco's modified
Eagle's medium (DMEM; Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) containing 10% fetal bovine serum (FBS; Gibco;
Thermo Fisher Scientific, Scoresby VIC, Australia) and 1%
penicillin-streptomycin (Gibco; Thermo Fisher Scientific) at 37°C
in a humidified atmosphere of 5% CO2. SMMC7721 cells
were cultured in RPMI-1640 medium (Invitrogen; Thermo Fisher
Scientific) containing 10% FBS (Gibco; Thermo Fisher
Scientific).
Heat treatment
iRFA treatment was performed in vitro as
previously described (7). Huh-7 and
SMMC7721 cells were seeded onto 6-well plates (5×104
cells/well) and further incubated for 24 h. Next, the plates were
sealed and submerged in a water bath at a temperature setting of
50°C for 10 min. Thereafter, the cells were maintained at 37°C for
12, 24 and 48 h. The cells that survived the treatment were used in
subsequent experiments.
Autophagy inhibitors and knockdown of
CD133
3-Methyladenine (3-MA) and chloroquine (CQ) were
purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany) and
used to inhibit autophagy in Huh-7 and SMMC7721 cells. Huh-7 and
SMMC7721 cells after heat treatment were incubated at 37°C for 12,
24 or 48 h in the absence or presence of 3-MA (5 mM) or CQ (5 µM)
(16). Then, the cells were used
for western blotting, transmission electron microscopy, confocal
microscopy, CCK-8 and cell invasion assay.
The CD133 siRNA (CD133 KD) and negative control
siRNA (con) were obtained from Shanghai GeneChem Co., Ltd.
(Shanghai, China). The sequences used for the experiments were as
follows: CD133 KD: 5′-CCUUUGUCUUUGGUGCAAA-3′ con:
5′-UUCUCCGAACGUGUCACGU-3′.
Huh-7 and SMMC7721 cells were transfected using
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific) in
96-well or 6-well plates and then were further incubated for 24 or
48 h, according to the manufacturer's instructions.
Western blotting
Tissues and cells were lysed in RIPA protein lysis
buffer (Thermo Fisher Scientific, Rockford, IL, USA) containing
protease inhibitors. The protein concentration was determined using
a BCA protein assay (Beyotime Institute of Biotechnology, Jiangsu,
China). Next, the proteins were denatured and separated via
SDS-PAGE gel (15% for separating LC3B and 6% for separating CD133)
and then transferred to nitrocellulose transfer membranes (Whatman,
Piscataway, NJ, USA). The membranes were blocked with 5% non-fat
powdered milk in phosphate-buffered saline (PBS) for 1 h at room
temperature and then incubated with rabbit polyclonal antibody
against human LC3B (1:800; cat. no. L7543; Sigma-Aldrich; Merck
KGaA), mouse monoclonal antibody against human CD133 (1:500; cat.
no. MAB4399-I; EMD Millipore), or rabbit polyclonal antibody
against human GAPDH (1:200; cat. no. sc-25778; Santa Cruz
Biotechnology, Dallas, TX, USA) overnight at 4°C. The membranes
were then washed extensively with TBST and incubated with goat
antibody against mouse (1:10,000; cat. no. ab97040; Abcam) and goat
antibody against rabbit (1:1,000; cat. no. ab7085; Abcam) for 1 h
at room temperature. The signal was detected using an enhanced
chemiluminescence system (Thermo Fisher Scientific) in accordance
with the manufacturer's protocol. The results were normalized to
GAPDH and expressed as relative densities.
Transmission electron microscopy
Huh-7 and SMMC7721 cells were exposed to iRFA
treatment alone or iRFA treatment with 3-MA and then further
incubated for 24 h. Next, the cells were fixed with 2.5%
glutaraldehyde and then rinsed three times (30 min each) with 0.1 M
PBS. Subsequently, the cells were fixed with 1% osmium tetroxide
for 2 h and gradient-dehydrated in acetone, saturated, and embedded
in epoxy 618. Semi-thin slices and then ultra-thin slices were
prepared and stained with uranium and lead. Finally, autophagy was
evaluated using transmission electron microscopy (Tecnai 10;
Philips, Eindhoven, The Netherlands).
Confocal microscopy
Huh-7 and SMMC7721 cells were exposed to iRFA
treatment in the presence of 3-MA or CD133-siRNA and then further
incubated for 24 h. After treatment, the cells were washed with
PBS, incubated for 10 min at 37°C in 4% paraformaldehyde and then
permeabilized with 0.1% Triton X-100. Next, the cells were
incubated for 1 h with a primary antibody at 37°C, washed
extensively with PBS buffer, and then incubated for 1 h with a
secondary antibody. The primary antibodies used were mouse
monoclonal antibody against human CD133 (1:50; cat. no. MAB4399-I;
EMD Millipore) and rabbit polyclonal antibody against human LC3B
(1:200; cat. no. L7543; Sigma-Aldrich; Merck KGaA) and the
secondary antibodies were Alexa Fluor 488-conjugated goat
anti-rabbit (1:500; cat. no. A-11034; Thermo Fisher Scientific) and
Alexa Fluor 568-conjugated goat anti-mouse (1:400; cat. no.
A-21134; Thermo Fisher Scientific). After staining, coverslips were
mounted with Vectashield (cat. no. H1200; Vector Laboratories,
Burlingame, CA, USA) and analyzed by Radiance 2000 laser scanning
confocal microscope (Carl Zeiss Microimaging, Thornwood, NY, USA).
The number of autophagosomes using confocal microscopy were counted
according to the methods described by Kader et al (17). The LC3B puncta were identified as
highly fluorescent green aggregates. The number of LC3B puncta was
quantified using the NIH ImageJ 1.41 software (National Institutes
of Health, Bethesda, MD, USA) and 45 cells/group from 3 independent
experiments were analyzed.
Cell Counting Kit −8 (CCK-8)
assay
Cell viability was detected by CCK-8 assay (Beyotime
Institute of Biotechnology) according to the manufacturer's
instructions. Huh-7 and SMMC7721 cells were exposed to iRFA
treatment in the presence of 3-MA or CD133-siRNA and then further
incubated for 24 h. After treatment, the cells were cultured in
96-well plates (3×103 cells/well) and further incubated
for 12, 24, 36 and 48 h. The CCK-8 solution was added to each well.
Next, the absorbance at 450 nm was measured with a microplate
reader (Thermo Fisher Scientific). All the assays were performed in
triplicate.
Cell invasion assay
Huh-7 and SMMC7721 cells were exposed to iRFA
treatment in the presence of 3-MA or CD133-siRNA and then further
incubated for 24 h. Next, Transwell chambers (Costar; Corning Inc.,
Corning, NY, USA) were coated with 50 µg reconstituted basement
membrane matrix (BD Biosciences, San Jose, CA, USA). Then,
2×104 cells in 100 µl serum-free medium were seeded into
the upper chambers. A total of 600 µl of DMEM containing 10% FBS
was added to the lower chamber. After 48 h of incubation, the cells
on the lower surface of the membrane were stained with crystal
violet for 30 min and washed with PBS. The cells were counted in
six random fields under a light microscope at an ×200
magnification.
Statistical analysis
The data were analyzed using SPSS 18.0 statistical
software (SPSS, Inc., Chicago, IL, USA) and presented as the means
± SD from at least 3 independent experiments. The differences
between groups in the western blot analysis, cell viability and
cell invasion assays were analyzed using either the Student's
t-test or the one-way ANOVA. The association between LC3B and CD133
in HCC tissues was calculated using the Spearman's correlation
coefficient. P<0.05 was considered to indicate a statistically
significant result.
Results
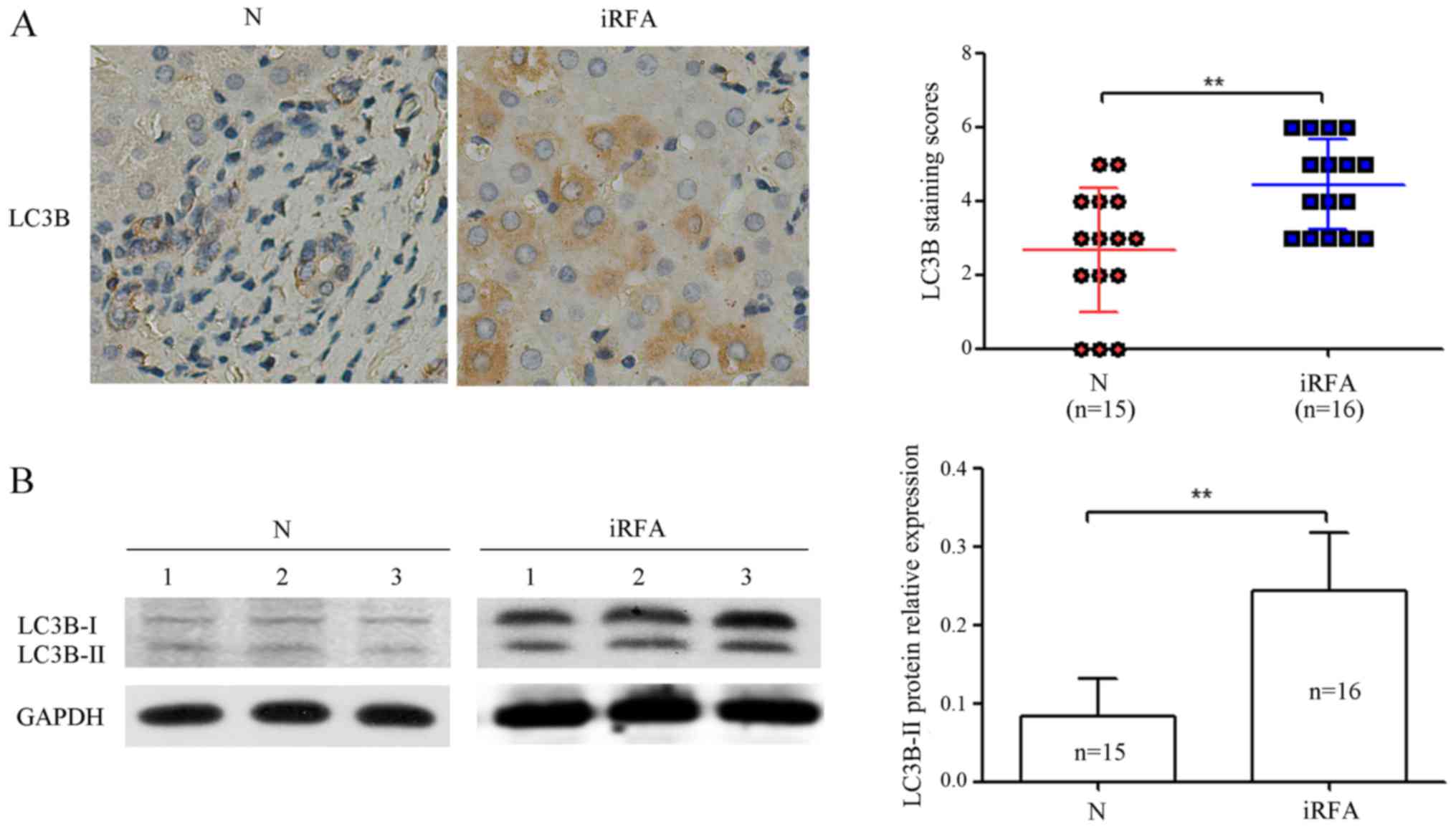
RFA promotes LC3B protein expression
levels in human HCC tissues
To determine the effect of RFA on autophagy in human
HCC tissues, we analyzed the LC3B protein expression levels in 16
HCC specimens with RFA treatment and 15 HCC specimens with non-RFA
treatment by immunohistochemical analysis and western blotting.
Immunohistochemical analysis results indicated that the expression
of the LC3B protein was increased by 77.0% in HCC specimens with
RFA treatment compared with that of HCC specimens with non-RFA
treatment (Fig. 1A). As shown in
Fig. 1B, western blotting results
demonstrated that the expression of the LC3B protein was increased
by 1.67-fold in HCC specimens with RFA treatment compared with that
of HCC specimens with non-RFA treatment. These data provided us
with experimental evidence that RFA induced LC3B protein expression
levels in the human HCC tissues.
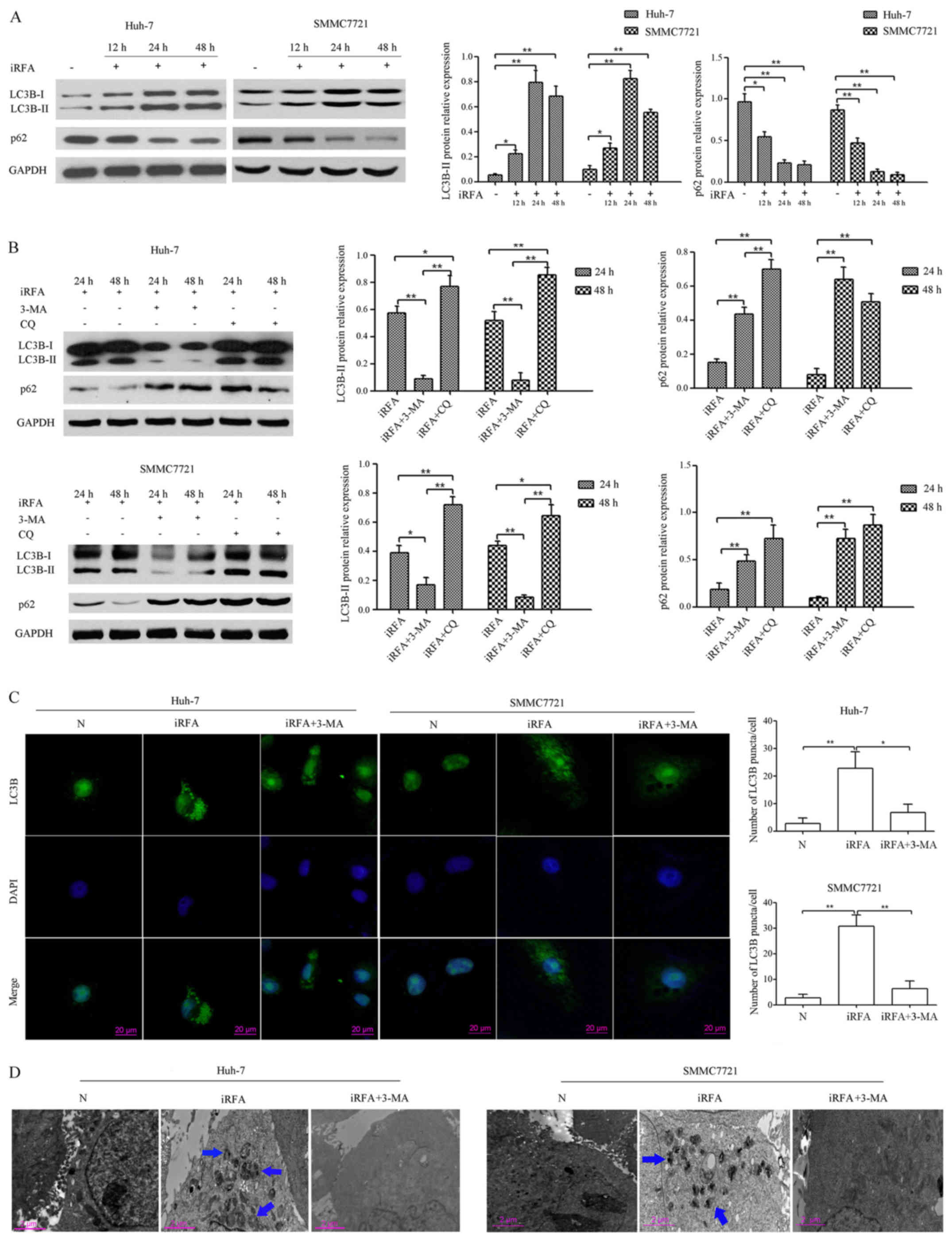
iRFA induces autophagy in HCC cells in
vitro
To further address whether autophagy could be
induced in HCC cells during iRFA, we investigated the LC3B-II
protein expression in vitro, which is considered an accurate
indicator of autophagy (18).
Western blot analysis demonstrated that the ratio of LC3B-II to
GAPDH in Huh-7 cells with iRFA treatment was increased by 2.67-fold
after 12 h, 12.16-fold after 24 h and 10.33-fold after 48 h,
respectively. In the other group, the expression of LC3B-II in
SMMC7721 cells with iRFA treatment was upregulated by 2.8-fold
after 12 h, 8.51-fold after 24 h and 5.72-fold after 48 h,
respectively. In addition, p62 (SQSTM1, an autophagy-specific
substrate) protein expression levels in Huh-7 cells with iRFA
treatment was decreased by ~43.4% after 12 h, 76.2% after 24 h and
78.3% after 48 h, respectively. Moreover, the expression of p62 in
SMMC7721 cells with iRFA treatment was reduced by ~78.3% after 12
h, 87.0% after 24 h and 93.0% after 48 h, respectively (Fig. 2A).
To further evaluate the effect of iRFA on autophagic
flux, the autophagy inhibitors 3-MA and CQ were used. Our results
demonstrated that 3-MA attenuated the levels of LC3B-II protein and
significantly increased the accumulation of p62 protein (Fig. 2B), which is consistent with its
ability to block autophagosome formation (19). Conversely, CQ, which blocks the
maturation of autophagosomes and digestion of autophagic substrates
(20), resulted in significant
accumulation of LC3B-II and p62 protein in Huh-7 and SMMC7721 cells
after iRFA treatment (Fig. 2B).
To further confirm that iRFA induces autophagy in
Huh-7 and SMMC7721 cells, we analyzed autophagy by confocal
microscopy and transmission electron microscopy (TEM). As shown in
Fig. 2C, the formation of LC3B
puncta in Huh-7 and SMMC7721 cells after iRFA treatment was
increased by 7.50-fold and 11.50-fold when compared with the
control group, respectively (Fig.
2C). TEM results of Huh-7 and SMMC7721 cells after iRFA
treatment also revealed an increase in the number of autophagosomes
(Fig. 2D). In addition, 3-MA
treatment reversed the effect of iRFA on the number of
autophagosomes in Huh-7 and SMMC7721 cells (Fig. 2C and D).
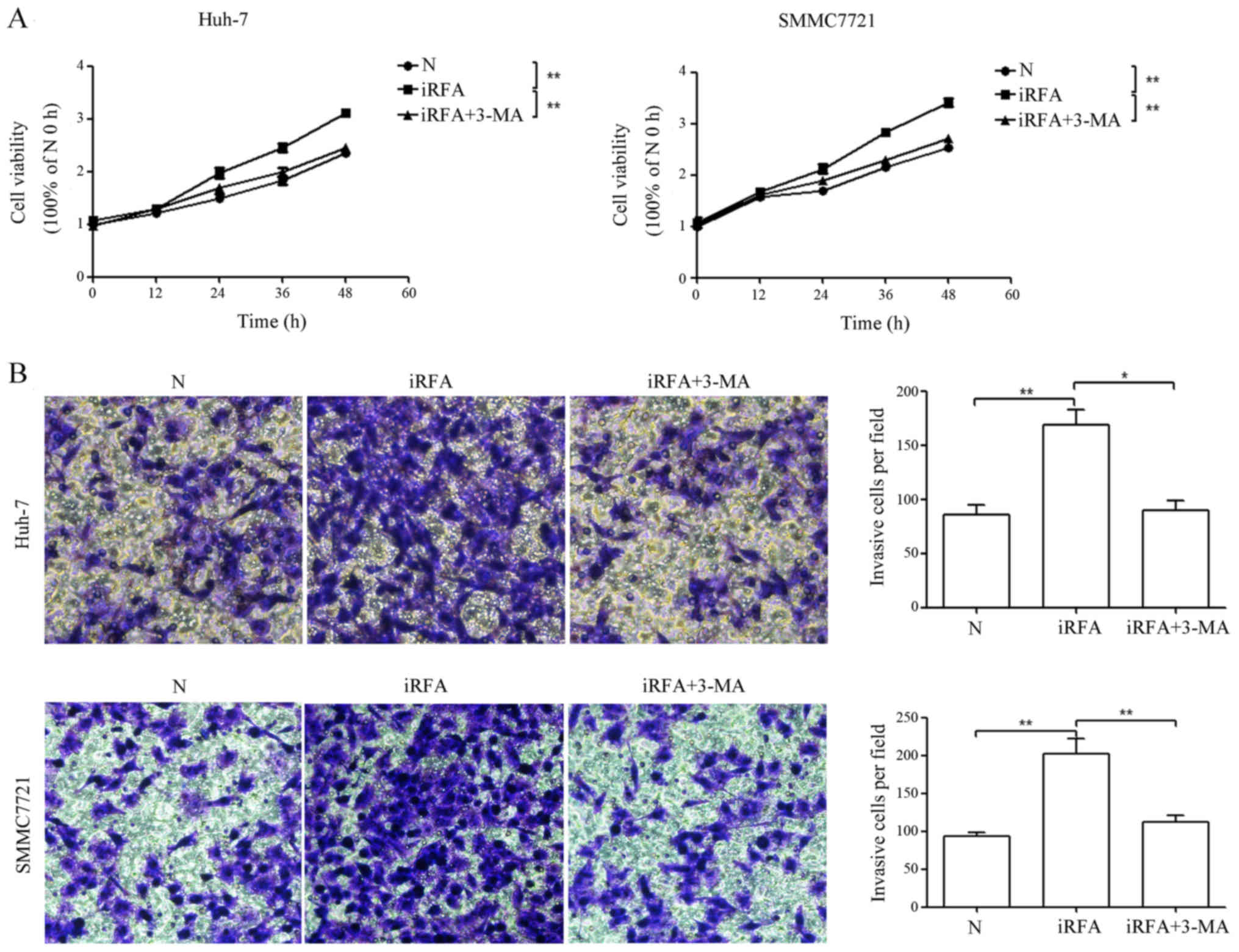
Inhibition of autophagy suppresses the
enhanced cell viability and invasion of HCC cells after iRFA
treatment
To determine the effect of iRFA on HCC progression,
the viabilities and invasion abilities of Huh-7 and SMMC7721 cells
were assessed by CCK-8 and Transwell assays, respectively. The
results of CCK-8 assay indicated that the cell viability of
iRFA-treated Huh-7 and SMMC7721 cells was increased by 32.1 and
33.8%, respectively, when compared with the control group (Fig. 3A, P<0.01). Similarly, the
invasive potential of iRFA-treated Huh-7 and SMMC7721 cells was
increased by 0.97- and 1.15-fold, respectively, when compared with
the control group (Fig. 3B,
P<0.01). Next, to determine whether iRFA-induced autophagy
played an important role in enhanced cell viability and invasion of
HCC cells, autophagy was inhibited by 3-MA treatment. The results
revealed that the cell viability of Huh-7 and SMMC7721 cells
exposed to iRFA treatment with 3-MA was decreased by 21.0 and 19.6%
when compared with the viability of cells exposed to iRFA treatment
alone, respectively (Fig. 3A).
Moreover, the invasive potential of Huh-7 and SMMC7721 cells
exposed to iRFA treatment with 3-MA was suppressed by 47.1 and
40.1% when compared with the invasive potential of cells exposed to
iRFA treatment alone, respectively (Fig. 3B).
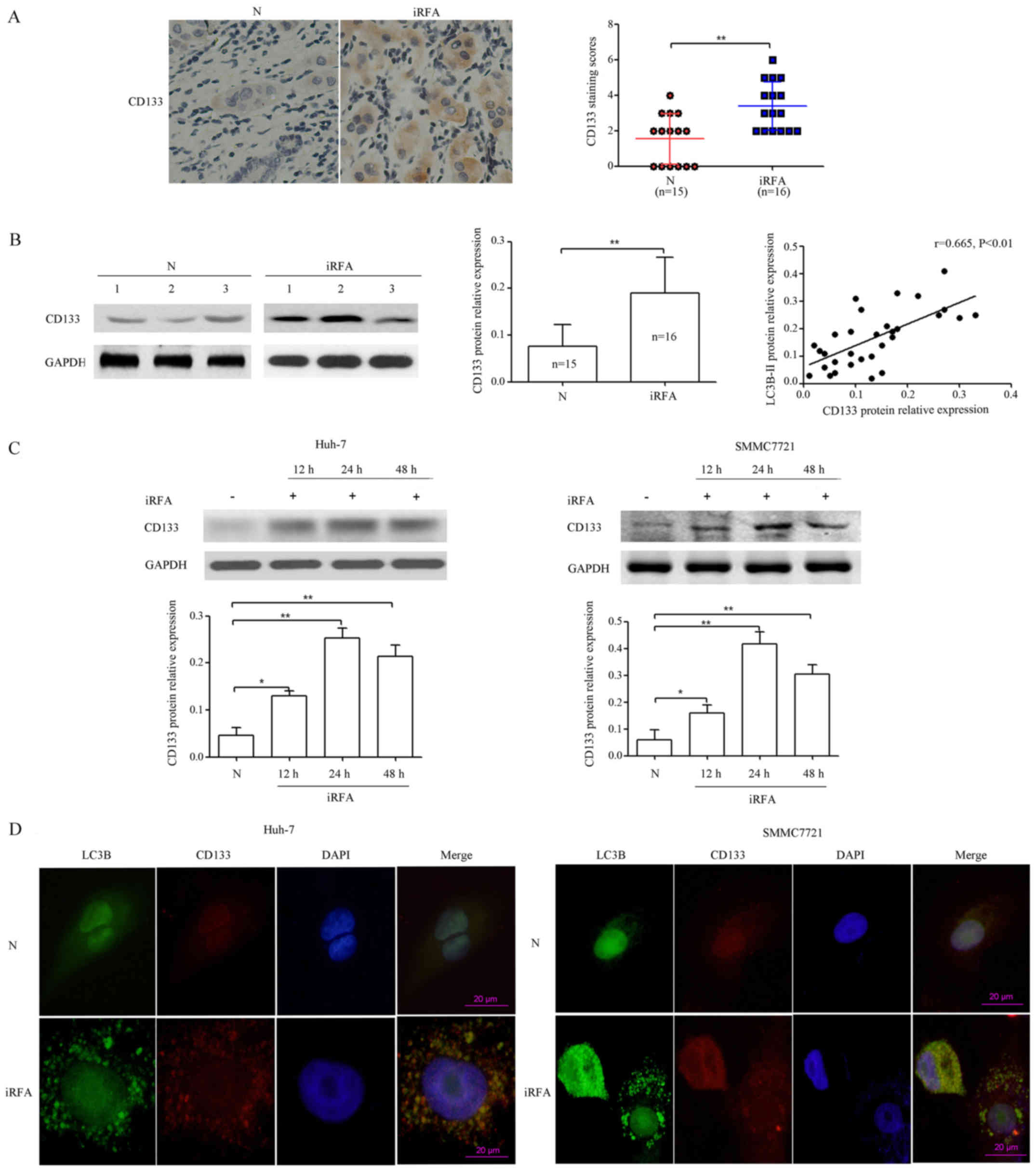
CD133 is upregulated after iRFA treatment. Previous
studies have demonstrated that tumor initiation and maintenance and
high metastatic potential in several cancers have been linked to
cancer stem cells (CSCs) (21). In
pancreatic cancer, blockade of autophagy by pharmacological or
genetic inhibitors reduced CSC populations, sphere-forming ability,
drug resistance and tumor formation (22). However, the relationship between
autophagy and CSCs in rapid and aggressive recurrence of HCC after
RFA remains to be explored. To determine whether CD133 could be
induced in HCC cells during iRFA, we investigated CD133 protein
expression in vivo, which has recently been identified as a
critical CSC marker (23).
Immunohistochemical analysis results indicated that CD133 protein
expression was increased by 1.06-fold in HCC specimens with RFA
treatment compared with that of HCC specimens with non-RFA
treatment (P<0.01; Fig. 4A). In
line with the previous data, western blot analysis demonstrated
that CD133 protein expression was increased by 1.10-fold and was
positively correlated with LC3B protein expression in HCC specimens
(r=0.636, P<0.01; Fig. 4B).
Next, CD133 protein levels were determined in Huh-7 and SMMC7721
cells after iRFA treatment for 12, 24 and 48 h at 50°C. Our results
demonstrated that CD133 protein expression in Huh-7 cells with iRFA
treatment was increased by 1.79-fold after 12 h, 4.43-fold after 24
h and 3.57-fold after 48 h, respectively. In the other group, CD133
protein expression in SMMC7721 cells exposed to iRFA treatment was
upregulated by 1.67-fold after 12 h, 5.94-fold after 24 h and
4.06-fold after 48 h, respectively (Fig. 4C). Furthermore, confocal microscopy
results indicated that the CD133 protein colocalized with the
autophagy protein LC3B in Huh-7 and SMMC7721 cells after iRFA
treatment (Fig. 4D). These results
demonstrated that CD133 played an important role in rapid and
aggressive recurrence of HCC after RFA.
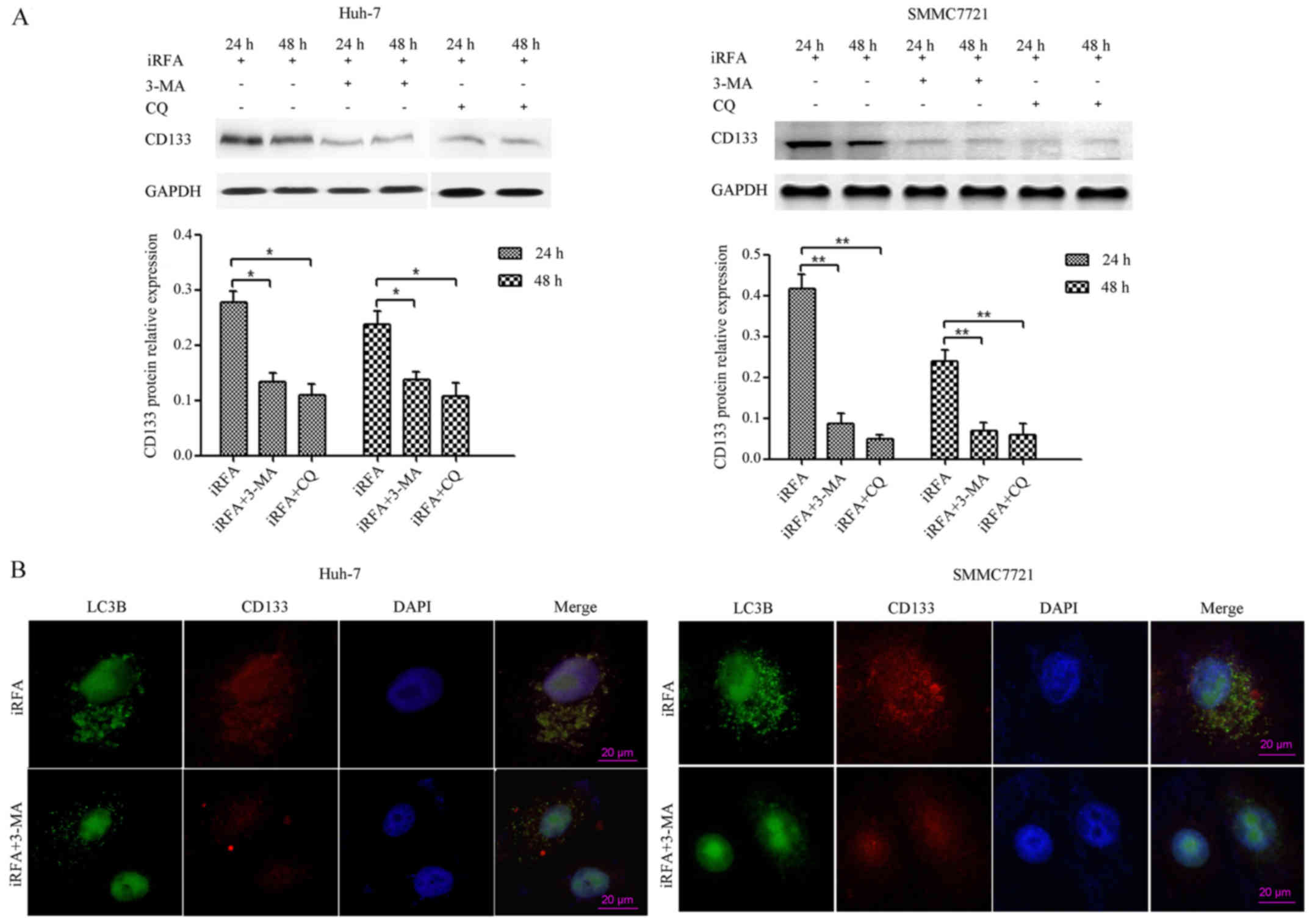
Inhibition of autophagy suppresses
CD133 expression of HCC cells after iRFA treatment
To determine whether iRFA-induced autophagy played
an important role in regulating the expression of the CD133
protein, autophagy was inhibited by 3-MA or CQ treatment. Western
blotting results demonstrated that the expression of the CD133
protein in Huh-7 cells exposed to iRFA treatment with 3-MA or CQ)
was suppressed by 61.4% after 24 h and 60.2% after 48 h,
respectively, when compared with that of cells exposed to iRFA
treatment alone (Fig. 5A,
P<0.05). In addition, the decrease in CD133 protein expression
of Huh-7 cells exposed to iRFA treatment with CQ exceeded 63% when
compared with that of cells exposed to iRFA treatment alone
(Fig. 5A, P<0.05). A similar
effect occurred in SMMC7721 cells, wherein there was a significant
decrease in the CD133 protein expression (Fig. 5A). Furthermore, confocal microscopy
results indicated that both the CD133 protein and the autophagy
protein LC3B in iRFA-treated Huh-7 and SMMC7721 cells after
inhibition of autophagy (3-MA) was significantly decreased
(Fig. 5B).
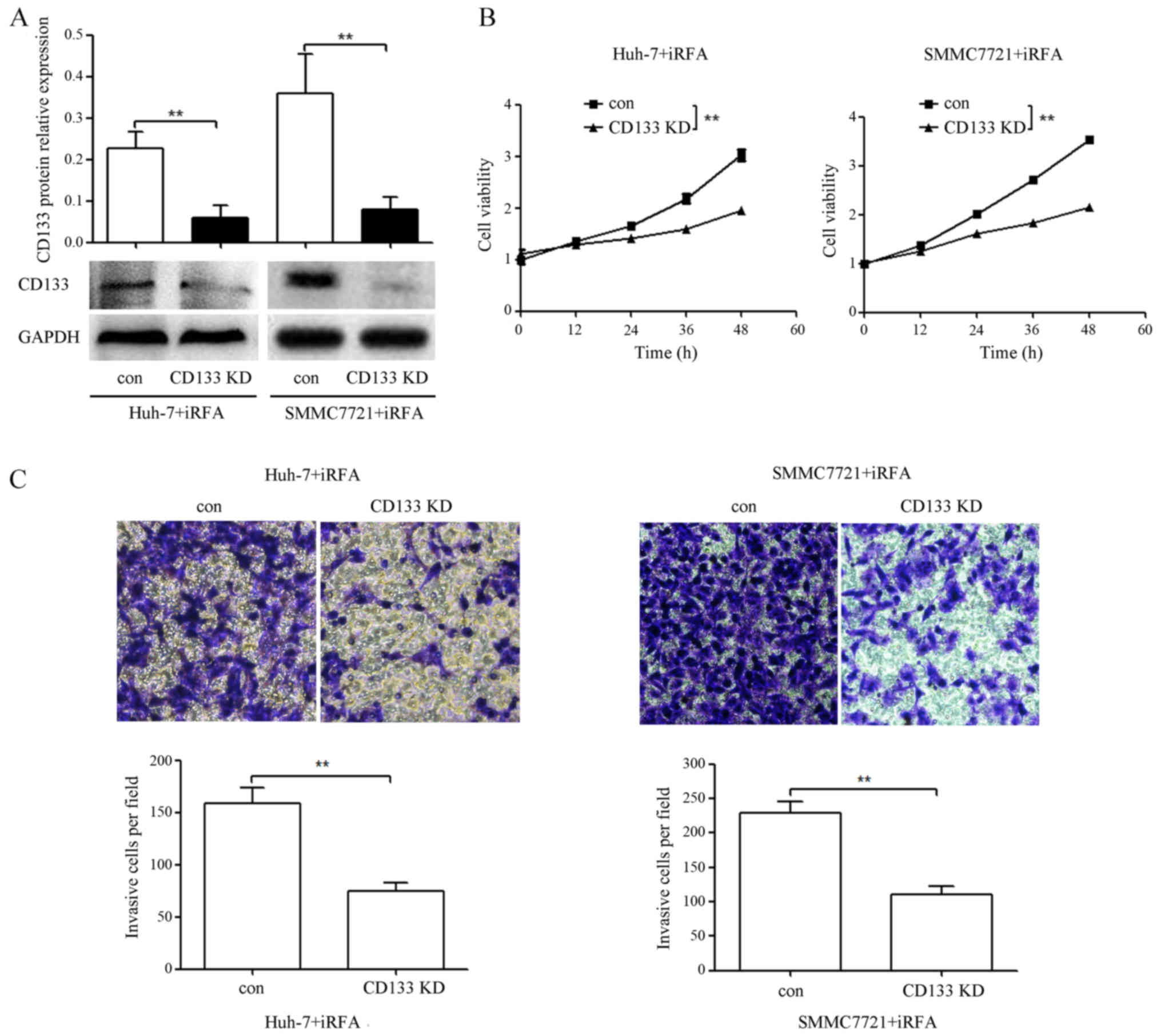
CD133 knockdown suppresses the
enhanced cell viability and invasion of HCC cells after iRFA
treatment
To assess whether iRFA-induced CD133 played an
important role in enhanced cell viability and invasion of HCC
cells, we transfected the iRFA-treated Huh-7 and SMMC7721 cells
with CD133-siRNA or negative control siRNA. As shown in Fig. 6A, CD133 protein expression in
iRFA-treated Huh-7 and SMMC7721 cells transfected with CD133-siRNA
was downregulated by 73.5 and 88.9% when compared with that in
cells transfected with the negative control siRNA, respectively
(Fig. 6A). Moreover, cell viability
in iRFA-treated Huh-7 and SMMC7721 cells after CD133 knockdown was
suppressed by 35.6 and 38.0% when compared with that in cells
transfected with the negative control siRNA, respectively (Fig. 6B). Similarly, the invasion abilities
determined using Transwell assay in iRFA-treated Huh-7 and SMMC7721
cells after CD133 knockdown were suppressed by 53.2 and 51.9%,
respectively (Fig. 6C).
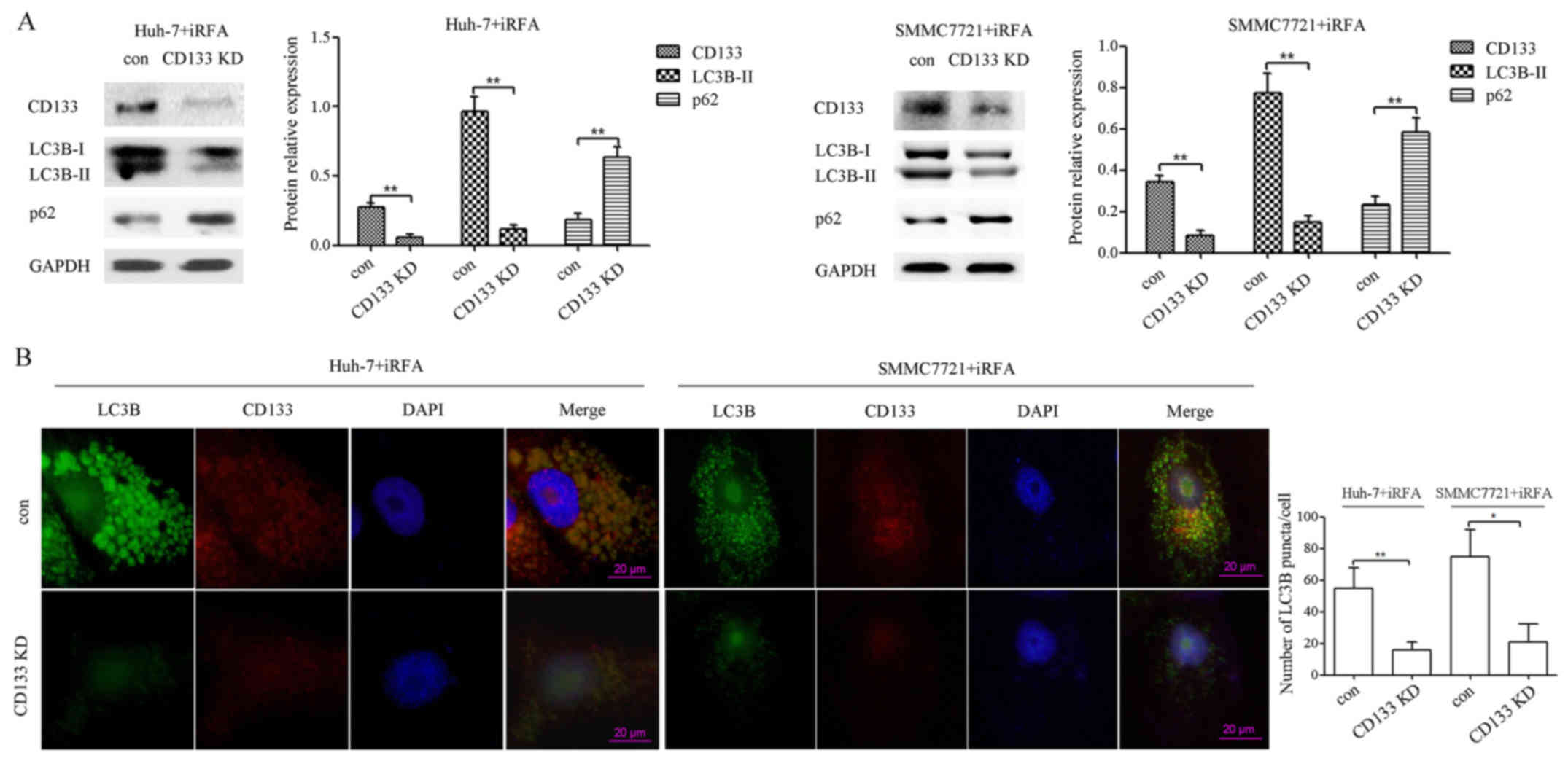
CD133 knockdown suppresses autophagy
of HCC cells after iRFA treatment
In HCC, CD133 is involved in cell survival through
regulation of autophagy and glucose uptake (24). To provide evidence that CD133 plays
a role in the regulation of autophagy after iRFA treatment, we
assessed the effect of CD133 knockdown on the expression of LC3B-II
and p62 protein in iRFA-treated Huh-7 and SMMC7721 cells. As shown
in Fig. 7A, LC3B-II protein
expression determined using western blotting in iRFA-treated Huh-7
and SMMC7721 cells transfected with CD133-siRNA was decreased by
87.5 and 79.5% when compared with that in cells transfected with
the negative control siRNA, respectively (Fig. 7A). However, the p62 protein in
iRFA-treated Huh-7 and SMMC7721 cells after CD133 knockdown was
increased by 2.47- and 1.50-fold when compared with that in cells
transfected with the negative control siRNA, respectively (Fig. 7A). Furthermore, confocal microscopy
results revealed that the formation of LC3B puncta in iRFA-treated
Huh-7 and SMMC7721 cells after inhibition of the CD133 protein was
decreased by 71.5 and 72.0% when compared with the control group,
respectively (Fig. 7B).
Discussion
Radiofrequency ablation (RFA) is accepted as a safe
and effective therapy for the early stages of primary HCC (25). However, iRFA treatment has been
reported as a risk factor of local recurrence (4). Thus, it is urgently required to
understand the mechanisms by which local recurrence is induced to
improve prognosis of HCC patients.
Recently, autophagy was revealed to be important in
maintaining cancer cell survival through conferring stress
tolerance and limiting damages (26). For instance, sorafenib-induced
autophagy acts as a chemoresistance mechanism in HCC (27). In the present study, we revealed
that LC3B (a key autophagic marker) expression was significantly
induced in the residual HCC cells after RFA treatment in
vivo (Fig. 1). In addition,
iRFA treatment leaded to autophagy, autophagosome formation and
autophagic flux in Huh-7 and SMMC7721 cells in vitro
(Fig. 2). Therefore, we concluded
that autophagy may play an important role in promoting rapid and
aggressive recurrence of HCC after iRFA treatment. To determine the
effect of autophagy on iRFA-induced rapid and aggressive recurrence
of HCC, we blocked autophagy using 3-MA in Huh-7 and SMMC7721 cells
exposed to iRFA treatment. Our results revealed that the inhibition
of autophagy by 3-MA suppressed iRFA treatment-induced cell
viability and invasion of HCC cells (Fig. 3). Thus, these findings demonstrated
that autophagy acted as an accomplice of survival, malignant
progression of HCC cells under iRFA treatment. In contrast,
numerous studies have revealed that autophagy also functioned as a
tumour suppressor in HCC. Li et al (28) revealed that blocking of autophagy
reversed the proliferation inhibition and cell death effects of
IFN-γ on HCC cells. In addition, Tay et al (29) revealed that lncRNA PTENP1-induced
autophagy may act as an inhibitory factor for HCC cell survival.
These findings suggest that the role of autophagy in cancer is
complex and is likely dependent on the microenvironment and genetic
context.
Recent studies have demonstrated that high
expression levels of putative hepatic stem/progenitor cell
biomarkers such as CD133 confer enhanced malignant potential in HCC
(30,31). In addition, iRFA treated-HCC cells
displayed a higher CD133 expression and malignant potential
(7). Moreover, Yang et al
(22) found that blockade of
autophagy by pharmacological or genetic inhibitors reduced CSC
populations, sphere-forming ability, drug resistance and tumor
formation in pancreatic cancer. In HCC, autophagy is essential for
the survival and maintenance of CD133+ liver cancer stem
cells (32). Thus, we proposed that
induction of autophagy mediated by iRFA treatment contributes to
aggressive recurrence of HCC via the promotion of progenitor
characteristics. Further investigation supported this conclusion.
First, we found that CD133 protein expression was significantly
upregulated in vivo and in vitro and was positively
correlated with LC3B protein expression in HCC specimens (Fig. 4B; r=0.636, P<0.01). Second, CD133
protein colocalized with the autophagy protein LC3B in Huh-7 and
SMMC7721 cells after iRFA treatment (Fig. 4D). Third, inhibition of autophagy by
3-MA or CQ significantly suppressed the expression of the CD133
protein in iRFA-treated Huh-7 and SMMC7721 cells (Fig. 5A and B). Fourth, CD133 knockdown by
CD133-siRNA significantly suppressed cell viability and invasion
ability in iRFA-treated Huh-7 and SMMC7721 cells (Fig. 6).
Notably, CD133 has been indicated to improve the
resistance of glioma cells to a nutrient-deprived microenvironment
by participating in autophagosome biogenesis (33). Meanwhile, Chen et al
(24) found that CD133 is involved
in cell survival through regulation of autophagy and glucose uptake
in HCC. To further explore whether CD133 could regulate
autophagosome biogenesis in iRFA-treated Huh-7 and SMMC7721 cells,
we downregulated CD133 protein expression by CD133-siRNA. Our
results indicated that CD133 downregulation significantly
suppressed LC3B-II protein expression and autophagosome biogenesis,
and induced p62 protein expression in iRFA-treated Huh-7 and
SMMC7721 cells (Fig. 7). Thus, our
results strongly indicated that iRFA treatment could also promote
autophagy by inducing CD133 protein expression.
In conclusion, we identified a new mechanism by
which iRFA promotes rapid growth and invasion of HCC cells by
regulating autophagy and the CD133 feedback loop. These findings
provide novel effective targets for the prevention and treatment of
this undesirable effect during RFA therapy.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Surface
Project of the National Natural Science Foundation of the People's
Republic of China (no. 81272688) and the Key Project of Application
Development of Chongqing (no. cstc2014yykfB10002).
Availability of data and materials
All data generated in this study are included in
this published article.
Authors' contributions
KM and PB designed this research. XW and QD
performed the research. KF, SC, JJ and FX analysed the data. XW and
FX edited the manuscript. All authors read and approved the
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
In the present study, the use of human tissue
samples and all experimental procedures and protocols were approved
by the Ethics Committee of the First Affiliated Hospital of Third
Military Medical University with the following reference no.
2013(47).
Consent for publication
Written informed consent was obtained from all of
the patients prior to treatment.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Forner A, Llovet JM and Bruix J:
Hepatocellular carcinoma. Lancet. 379:1245–1255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kang TW, Lim HK and Cha DI: Aggressive
tumor recurrence after radiofrequency ablation for hepatocellular
carcinoma. Clin Mol Hepatol. 23:95–101. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Germani G, Pleguezuelo M, Gurusamy K,
Meyer T, Isgrò G and Burroughs AK: Clinical outcomes of
radiofrequency ablation, percutaneous alcohol and acetic acid
injection for hepatocelullar carcinoma: A meta-analysis. J Hepatol.
52:380–388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee HY, Rhim H, Lee MW, Kim YS, Choi D,
Park MJ, Kim YK, Kim SH and Lim HK: Early diffuse recurrence of
hepatocellular carcinoma after percutaneous radiofrequency
ablation: Analysis of risk factors. Eur Radiol. 23:190–197. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Obara K, Matsumoto N, Okamoto M, Kobayashi
M, Ikeda H, Takahashi H, Katakura Y, Matsunaga K, Ishii T, Okuse C,
et al: Insufficient radiofrequency ablation therapy may induce
further malignant transformation of hepatocellular carcinoma.
Hepatol Int. 2:116–123. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ke S, Ding XM, Kong J, Gao J, Wang SH,
Cheng Y and Sun WB: Low temperature of radiofrequency ablation at
the target sites can facilitate rapid progression of residual
hepatic VX2 carcinoma. J Transl Med. 8:732010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yoshida S, Kornek M, Ikenaga N, Schmelzle
M, Masuzaki R, Csizmadia E, Wu Y, Robson SC and Schuppan D:
Sublethal heat treatment promotes epithelial-mesenchymal transition
and enhances the malignant potential of hepatocellular carcinoma.
Hepatology. 58:1667–1680. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu Z, Dai H, Jia G, Li Y, Liu X and Ren
W: Insufficient radiofrequency ablation promotes human hepatoma
SMMC7721 cell proliferation by stimulating vascular endothelial
growth factor overexpression. Oncol Lett. 9:1893–1896. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ma S: Biology and clinical implications of
CD133(+) liver cancer stem cells. Exp Cell Res. 319:126–132. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tong CM, Ma S and Guan XY: Biology of
hepatic cancer stem cells. J Gastroenterol Hepatol. 26:1229–1237.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang S, Wang X, Contino G, Liesa M, Sahin
E, Ying H, Bause A, Li Y, Stommel JM, Dell'antonio G, et al:
Pancreatic cancers require autophagy for tumor growth. Genes Dev.
25:717–729. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu DH, Jia CC, Chen J, Lin ZX, Ruan DY, Li
X, Lin Q, Min-Dong, Ma XK, Wan XB, et al: Autophagic LC3B
overexpression correlates with malignant progression and predicts a
poor prognosis in hepatocellular carcinoma. Tumour Biol.
35:12225–12233. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chang Y, Yan W, He X, Zhang L, Li C, Huang
H, Nace G, Geller DA, Lin J and Tsung A: miR-375 inhibits autophagy
and reduces viability of hepatocellular carcinoma cells under
hypoxic conditions. Gastroenterology. 143:177–87.e8. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Peng WX, Xiong EM, Ge L, Wan YY, Zhang CL,
Du FY, Xu M, Bhat RA, Jin J and Gong AH: Egr-1 promotes
hypoxia-induced autophagy to enhance chemo-resistance of
hepatocellular carcinoma cells. Exp Cell Res. 340:62–70. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pinheiro C, Longatto-Filho A, Scapulatempo
C, Ferreira L, Martins S, Pellerin L, Rodrigues M, Alves VA,
Schmitt F and Baltazar F: Increased expression of monocarboxylate
transporters 1, 2, and 4 in colorectal carcinomas. Virchows Arch.
452:139–146. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen YJ, Chi CW, Su WC and Huang HL:
Lapatinib induces autophagic cell death and inhibits growth of
human hepatocellular carcinoma. Oncotarget. 5:4845–4854. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kader M, Alaoui-El-Azher M, Vorhauer J,
Kode BB, Wells JZ, Stolz D, Michalopoulos G, Wells A, Scott M and
Ismail N: MyD88-dependent inflammasome activation and autophagy
inhibition contributes to Ehrlichia-induced liver injury and toxic
shock. PLoS Pathog. 13:e10066442017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Klionsky DJ, Cuervo AM and Seglen PO:
Methods for monitoring autophagy from yeast to human. Autophagy.
3:181–206. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fan T, Chen L, Huang Z, Wang W, Zhang B,
Xu Y, Mao Z, Hu H and Geng Q: Autophagy activation by rapamycin
before hypoxia-reoxygenation reduces endoplasmic reticulum stress
in alveolar epithelial cells. Cell Physiol Biochem. 41:79–90. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mizushima N: Methods for monitoring
autophagy. Int J Biochem Cell Biol. 36:2491–2502. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Clevers H: The cancer stem cell: Premises,
promises and challenges. Nat Med. 17:313–319. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang MC, Wang HC, Hou YC, Tung HL, Chiu TJ
and Shan YS: Blockade of autophagy reduces pancreatic cancer stem
cell activity and potentiates the tumoricidal effect of
gemcitabine. Mol Cancer. 14:1792015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mizrak D, Brittan M and Alison M: CD133:
Molecule of the moment. J Pathol. 214:3–9. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen H, Luo Z, Dong L, Tan Y, Yang J, Feng
G, Wu M, Li Z and Wang H: CD133/prominin-1-mediated autophagy and
glucose uptake beneficial for hepatoma cell survival. PLoS One.
8:e568782013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bruix J and Sherman M: American
Association for the Study of Liver Diseases: Management of
hepatocellular carcinoma: An update. Hepatology. 53:1020–1022.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Van den Broeck A, Gremeaux L, Topal B and
Vankelecom H: Human pancreatic adenocarcinoma contains a side
population resistant to gemcitabine. BMC Cancer. 12:3542012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu L, Liao JZ, He XX and Li PY: The role
of autophagy in hepatocellular carcinoma: Friend or foe.
Oncotarget. 8:57707–57722. 2017.PubMed/NCBI
|
|
28
|
Li P, Du Q, Cao Z, Guo Z, Evankovich J,
Yan W, Chang Y, Shao L, Stolz DB, Tsung A, et al: Interferon-γ
induces autophagy with growth inhibition and cell death in human
hepatocellular carcinoma (HCC) cells through interferon-regulatory
factor-1 (IRF-1). Cancer Lett. 314:213–222. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tay Y, Kats L, Salmena L, Weiss D, Tan SM,
Ala U, Karreth F, Poliseno L, Provero P, Di Cunto F, et al:
Coding-independent regulation of the tumor suppressor PTEN by
competing endogenous mRNAs. Cell. 147:344–357. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kohga K, Tatsumi T, Takehara T, Tsunematsu
H, Shimizu S, Yamamoto M, Sasakawa A, Miyagi T and Hayashi N:
Expression of CD133 confers malignant potential by regulating
metalloproteinases in human hepatocellular carcinoma. J Hepatol.
52:872–879. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang XR, Xu Y, Yu B, Zhou J, Qiu SJ, Shi
GM, Zhang BH, Wu WZ, Shi YH, Wu B, et al: High expression levels of
putative hepatic stem/progenitor cell biomarkers related to tumour
angiogenesis and poor prognosis of hepatocellular carcinoma. Gut.
59:953–962. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Song YJ, Zhang SS, Guo XL, Sun K, Han ZP,
Li R, Zhao QD, Deng WJ, Xie XQ, Zhang JW, et al: Autophagy
contributes to the survival of CD133+ liver cancer stem
cells in the hypoxic and nutrient-deprived tumor microenvironment.
Cancer Lett. 339:70–81. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sun H, Zhang M, Cheng K, Li P, Han S, Li
R, Su M, Zeng W, Liu J, Guo J, et al: Resistance of glioma cells to
nutrient-deprived microenvironment can be enhanced by
CD133-mediated autophagy. Oncotarget. 7:76238–76249. 2016.
View Article : Google Scholar : PubMed/NCBI
|