Introduction
Lung cancer is the most common cancer and the
leading cause of cancer-related deaths in both men and women in the
USA and China, among which the proportion of squamous cell lung
carcinoma was as high as 30–40% (1,2). As a
common type of lung cancer, the histological changes of squamous
cell lung carcinoma have been basically identified, which mainly
included the loss of cilia, squamous metaplasia, atypical
hyperplasia and dysplasia induced by chronic stimulation, and
injury of the bronchial mucosa columnar epithelium (3). Research on etiology and pathogenesis
has confirmed that the occurrence and development of squamous cell
lung cancer is a pathological process involving multiple genes and
pathways. However, genomic alterations in squamous cell lung
cancers have not been comprehensively characterized and no
molecular-targeted agents have been specifically developed for its
treatment (4). Therefore, it is
urgently needed to elucidate the mechanisms underlying the
occurrence and development of squamous cell lung cancer and develop
new therapeutic targets.
Among numerous tumor pathogenesis, methylation of
the functional genes has attracted the interest of many
researchers. Aberrant promoter island methylation of tumor
suppressor genes, including overall low level methylation and
hyper-methylation in some local regions, has been established as a
common epigenetic mechanism underlying the pathogenesis of human
cancers (5–7). The tripartite motif (TRIM) proteins
are a family of proteins with conserved structure, rapid evolution
and E3 ubiquitin ligase activities. TRIM is involved in a broad
range of cellular processes and diseases, including innate
immunity, development process, genetic diseases and cancer
(8). Thus far, more than 60 members
of the TRIM family proteins have been found in humans (9), among which, the tripartite motif
containing 58 (TRIM58) was identified as a candidate tumor
suppressor and a novel methylated gene (7,10).
TRIM58 has been reported to regulate terminal erythroid cell cycles
and enucleation (11), and the
overexpression of TRIM58 has been associated with the prognosis of
early lung cancer (12).
Furthermore, aberrant inactivation of TRIM58 consequent to CpG
island hyper-methylation may stimulate early carcinogenesis of lung
adenocarcinoma (7). However,
further functional research and verification on the association
between TRIM58 and the prognosis of lung cancer are still
needed.
In the present study to elucidate the influence of
TRIM58/cg26157385 methylation on lung cancer prognosis, we used the
large quantities of mRNA-Seq data in lung squamous cell carcinoma
patients published in cBioPortal database (http://www.cbioportal.org/), to screen out candidate
genes related to the methylation of TRIM58/cg26157385, and
construct a prognostic discrimination system based on these genes.
The reliability of the prognostic discrimination system was further
validated in an independent dataset.
Materials and methods
Data analysis
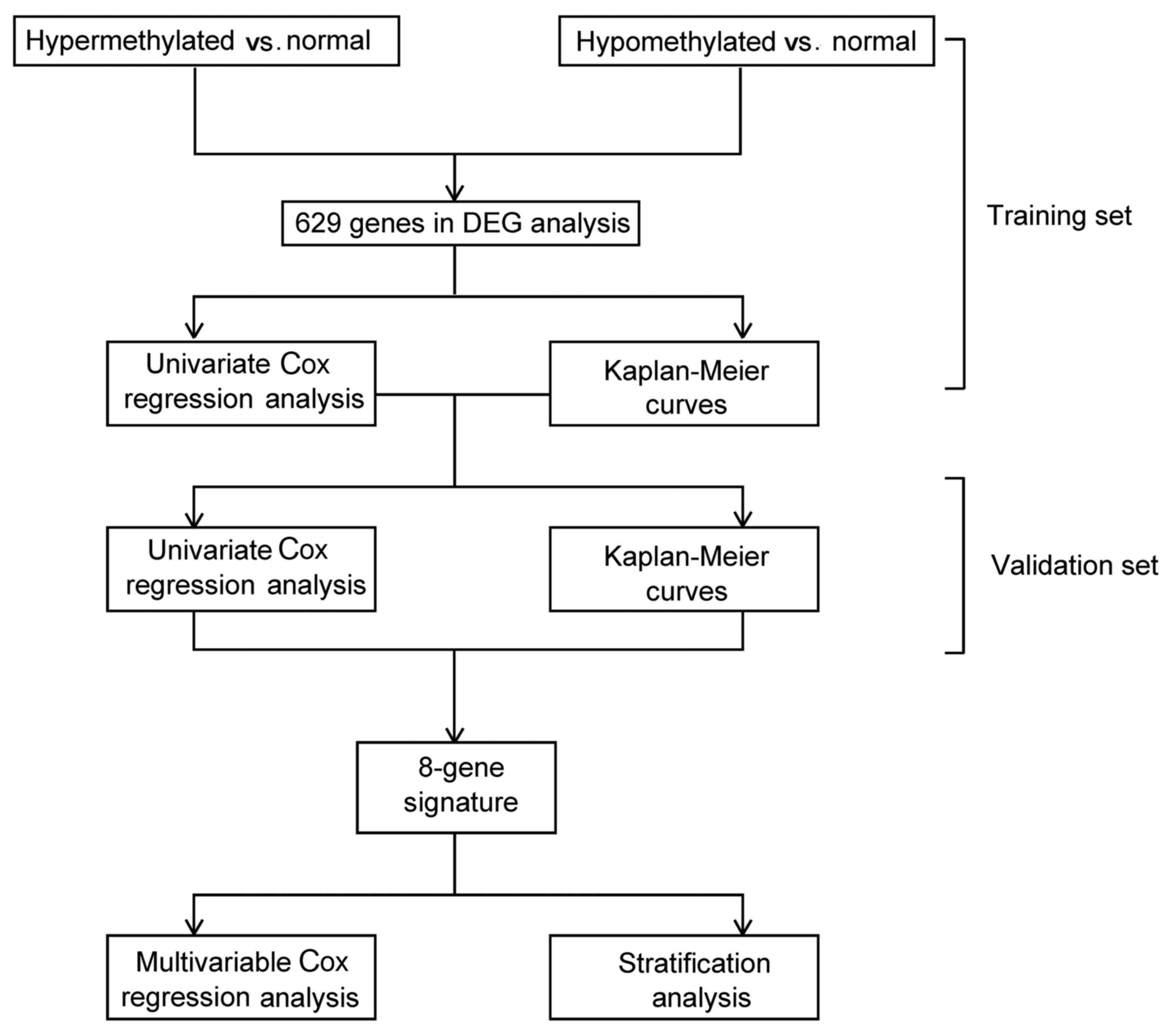
The data analysis flow of the present study is
briefly summarized in Fig. 1.
Collection and pretreatment of
collected data
On February 22, 2017, two sets of squamous cell lung
carcinoma data were downloaded from cBioPortal database (http://www.cbioportal.org/). One dataset was selected
as the training dataset, in which there were 370 squamous cell lung
carcinoma samples with both methylation sequencing information (the
platform was Illumina Methylation450) and mRNA expression profiling
data (the platform was IlluminaHiSeq2000). The other dataset was
selected as the validation dataset, including 178 squamous cell
lung carcinoma samples with mRNA expression profiling data (the
platform was IlluminaHiSeq2000). Furthermore, 51 normal control
samples were obtained from the cBioPortal database.
Clinical data
Information of samples in the training
dataset
The 370 patients with squamous cell lung carcinoma
consisted of 267 males (74.17%) and 93 females (25.83%) with mean
age of 67.71±8.65 years. There were 114 smokers (32.11%), 13
non-smokers (3.66%) and 228 cases (64.23%) who had quit smoking.
The number of patients who received radiotherapy and those who had
not received radiotherapy was 36 (11.92%) and 266 (88.08%),
respectively. The number of patients who received targeted therapy
and those who had not received targeted therapy was 93 (30.69%) and
210 (69.31%), respectively, with a mean survival time of
26.99±28.92 months. A total of 135 patients died at the end of the
follow-up period, accounting for 33.24% of the total
population.
Information of samples in the
validation dataset
The 178 patients with squamous cell lung carcinoma
consisted of 133 males (74.72%) and 45 females (25.28%) with mean
age of 67.86±8.25 years. There were 27 smokers (16.56%), 4
non-smokers (2.45%) and 132 cases (80.98%) who had quit smoking.
The number of patients who received radiotherapy and those who had
not received radiotherapy was 14 (12.5%) and 98 (87.5%),
respectively. The number of patients who received targeted therapy
and those who had not received targeted therapy was 35 (30.43%) and
80 (69.57%), respectively, with mean survival time of 29.96±31.85
months. A total of 78 patients died at the end of the follow-up
period, accounting for 43.82% of the total population.
Information of normal control
samples
The 51 patients in the normal control samples
consisted of 35 males (68.63%) and 16 females (31.37%) with mean
age of 68.34±8.49 years. There were 13 smokers (25.49%), 34 cases
(66.67%) who had quit smoking and 4 cases (7.84%) without any
information. The number of patients who received radiotherapy,
those who had not received radiotherapy and the patients without
any information was 3 (5.88%), 35 (68.63%) and 13 (25.49%),
respectively. The number of patients who received targeted therapy,
who had not received targeted therapy, and those without any
information was 13 (25.49%), 25 (49.02%) and 13 (25.49%),
respectively, with mean survival time of 23.93±27.97 months. A
total of 27 subjects died at the end of the follow-up period,
accounting for 52.94% of the total population. The information of
samples in the training, validation and normal control dataset is
listed in Table I.
 | Table I.Clinical information of the patients
in the training and the validation datasets. |
Table I.
Clinical information of the patients
in the training and the validation datasets.
|
| Training
dataset | Validation
dataset | Normal control
dataset |
|---|
| Sample count | 370 | 178 | 51 |
| Sex
(Male/female) | 267/93 | 133/45 | 35/16 |
| Age (mean ±
SD) | 67.71±8.65 | 67.86±8.25 | 68.34±8.49 |
| Smoking status
(Yes/reform/no/NA) | 114/228/13/15 | 27/132/4/15 | 13/34/0/4 |
| Radiotherapy
(Yes/no/NA) | 36/266/68 | 14/98/66 | 3/35/13 |
| Targeted-therapy
(Yes/no/NA) | 93/210/67 | 35/80/63 | 13/25/13 |
| Death (Yes/no) | 135/235 | 78/100 | 27/24 |
| Overall survival
months (mean ± SD) | 26.99±28.92 | 29.96±31.85 | 23.93±27.97 |
Grouping of methylation samples
DNA methylation β-values are continuous variables
between 0 (completely unmethylated) and 1 (completely methylated),
representing the ratio of the intensity of the methylated bead type
to the combined locus intensity (13). The methylation data of
TRIM58/cg26157385 in each sample were included in the Illumina
Methylation450 platform, which consisted of two peaks, 0.1 and 0.9,
corresponding to the low and high degree of methylation,
respectively. As higher β-values represent higher level of DNA
methylation, we divided the samples into low-methylation samples
with β-values of 0.1–0.5 and high-methylation samples with β-values
of 0.5–0.9. Finally, 133 high-methylation samples and 237
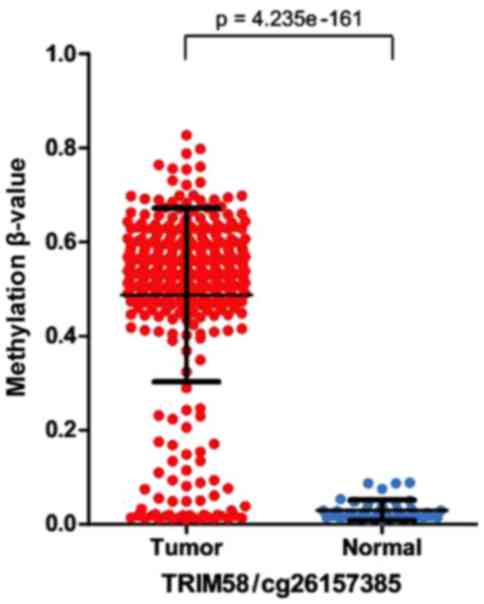
low-methylation samples were obtained. Furthermore, there were
significant differences of the β-values between all the tumor
samples and the normal control samples (Fig. 2).
Screening of differentially expressed
genes
The mRNA expression profiling data corresponding to
the high and low degrees of methylation samples in the training
dataset were analyzed for the differentially expressed genes using
the limma and multtest package (http://bioconductor.org/packages/release/bioc/html/multtest.html)
in R language version 3.0.1 (14).
FDR value <0.05 and fold change value >1.5 were selected as
threshold.
Screening of genes significantly
related to prognosis and calculation of risk score
There were corresponding survival information for
347 out of 370 samples with squamous cell lung carcinoma. Screening
of genes significantly related to prognosis was performed using Cox
regression analysis and the significant P-value and the prognostic
correlation coefficient β-value were calculated using the log-rank
test. According to the prognostic correlation coefficient β-value
and the expression value of the gene, the risk score computing
model was defined as follows:
Risk
score=βgene1×exprgene1+βgene2×exprgene2+⋯+βgenen×exprgenen
The prognostic correlation coefficient β-value was
indicated by β and the expression value of the gene was indicated
by expr. The corresponding risk score value of each sample was
calculated and the samples were divided into the high-risk and the
low-risk group according to the risk score median which was set as
the boundary (15–17).
Correlation analysis of the risk score
with the clinical features
Correlation analysis of the risk score with the
clinical features, including age (≥60 and <60), sex (male and
female), radiotherapy (yes or no), targeted therapy (yes or no)
were performed using the univariate and multivariable Cox
regression analyses for survival package. The screened clinical
features significantly associated with prognosis were analyzed
using the Kaplan-Meier survival curve.
Verification of the risk scoring
system
The expression values of signature genes were first
extracted from the validation dataset. The corresponding risk score
values of each sample were also calculated according to the
expression value of each gene and the samples were divided into the
high-risk group and the low-risk group using the risk score median
as boundary. The relationship between the high-risk and the
low-risk samples and the clinical features were verified using the
risk scoring system.
GO analysis and pathway
enrichment
The biological process and KEGG pathway enrichment
analysis of the selected prognostic genes were performed using the
Fisher algorithm using the clusterProfiler package in R language
(18). The specific algorithm is as
follows:
p=1-∑i=0x-1(Mi)(N-MK-i)(NK)
N represents the total number of genes in the
genome, M represents the number of genes in the pathway and K
indicates the number of differentially expressed genes. The
Fisher's score indicates the ratio of genes (number X) belonging to
the functional pathway out of the total differentially expressed
genes (number K).
Results
Detection and validation of eight
prognostic genes associated with TRIM58/cg26157385 methylation
Except the death numbers and the overall survival
months, there were no significant differences among the clinical
features between the samples in the training set and the samples in
the normal controls (Table II).
Compared with the 51 samples in the normal group, there were 527
and 449 significantly different expression genes in the 133
high-methylation samples and 237 low-methylation samples,
respectively. A total of 629 significantly differentially expressed
genes related to TRIM58 methylation were screened.
| Table II.Comparison of the clinical features
between samples in the training dataset and samples in the normal
control. |
Table II.
Comparison of the clinical features
between samples in the training dataset and samples in the normal
control.
|
| P-value |
|---|
| Sex | 0.9843a |
| Age (years) | 0.6335b |
| Smoking status | 0.301b |
| Radiotherapy | 0.5961b |
|
Targeted-therapy | 0.7108b |
| Death | 0.0163b |
| Overall survival
months | 0.0489a |
Based on the expression level of these 629 genes and
the corresponding survival information of the tumor samples, 183
genes significantly associated with prognosis (P<0.05) were
screened using the Cox regression analysis. Eight genes with
P-value >10−5 were selected as candidate signature
genes, including alpha-2-macroglobulin-like 1 (A2ML1), cyclin-E1
(CCNE1), COBL, establishment of sister chromatid cohesion
N-acetyltransferase 2 (ESCO2), G protein-coupled receptor 115
(GPR115), matrix metalloproteinases 10 (MMP10), OVO homologue-like
1 (OVOL1) and secretoglobin family 1A member 1 (SCGB1A1).
According to the prognostic correlation coefficient
β-value (Table III) and the
expression value of genes in each sample, the corresponding risk
score of each sample was calculated as follows:
| Table III.The P-value and the β-value of
signature genes. |
Table III.
The P-value and the β-value of
signature genes.
| Gene | P-value | β-value |
|---|
| A2ML1 | 9.56E-08 | 0.659 |
| COBL | 2.30E-07 | 0.823 |
| OVOL1 | 2.49E-07 | 1.355 |
| CCNE1 | 1.49E-06 | 1.502 |
| MMP10 | 2.02E-06 | 0.442 |
| SCGB1A1 | 7.20E-06 | 0.411 |
| ESCO2 | 7.44E-06 | −2.815 |
| GPR115 | 7.93E-06 | −0.941 |
Risk
score=0.659×exprA2ML1+0.823×exprCOBL+1.355×exprOVOL1+1.502×exprCCNE1+0.442×exprMMP10+0.411×exprSCGB1A1-2.815×exprESCO2-0.941×exprGPR115
The samples in the training dataset were divided
into the high-risk group and the low-risk group according to the
risk score, and the screened clinical features significantly
associated with prognosis were analyzed using the Kaplan-Meier
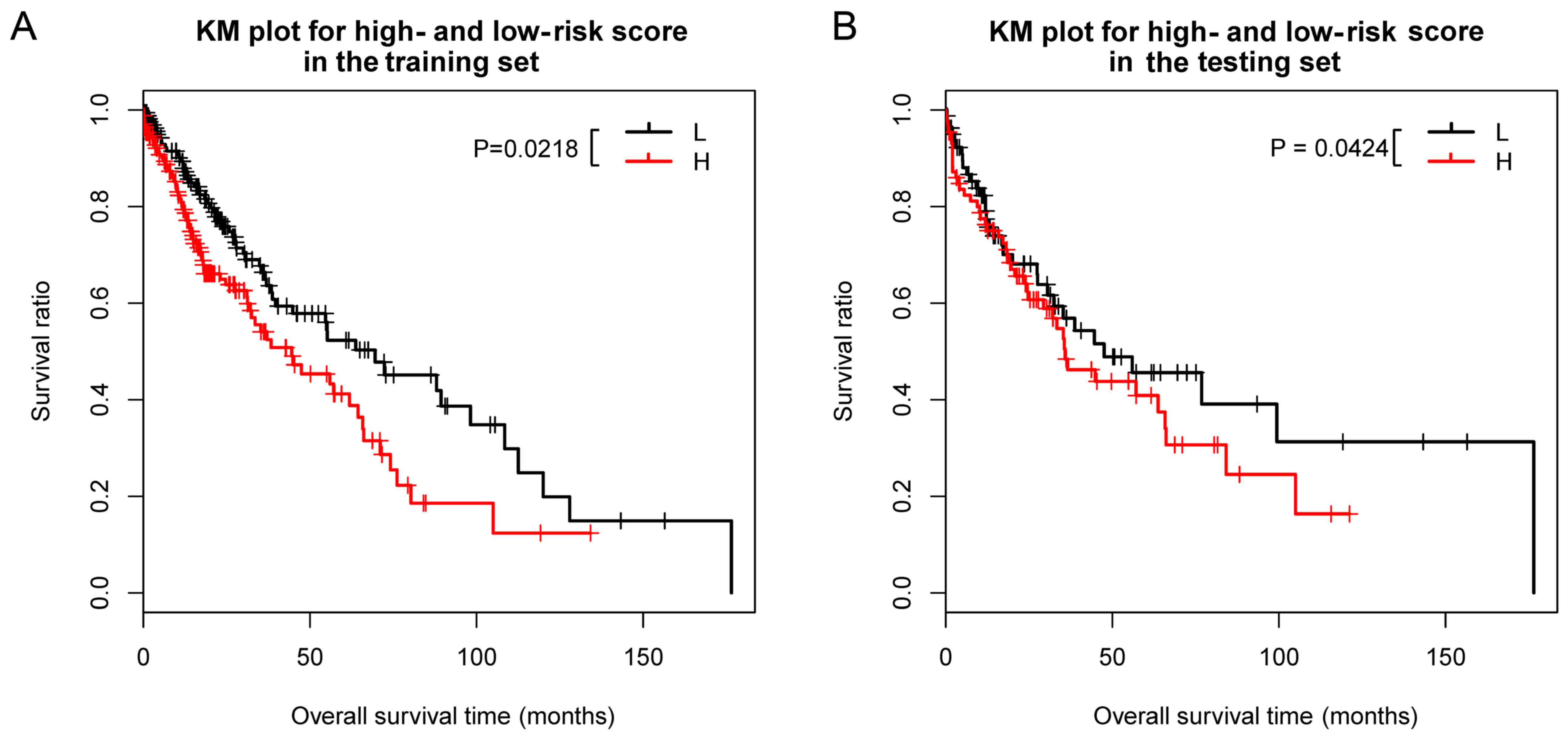
survival curve (Fig. 3A). The
results indicated that the risk scoring system successfully
classified different samples with different prognosis
(P=0.0218).
Among the 178 samples in the validation dataset, 168
had survival information. The overall survival analysis result was
similar to the result of the training dataset (P=0.0424) (Fig. 3B).
In the training dataset, the median overall survival
time of the high-risk group and the low-risk group was 450 and 608
days, respectively, and in the validation dataset, the median
overall survival time of the high-risk group and the low-risk group
were 460 and 665 days, respectively.
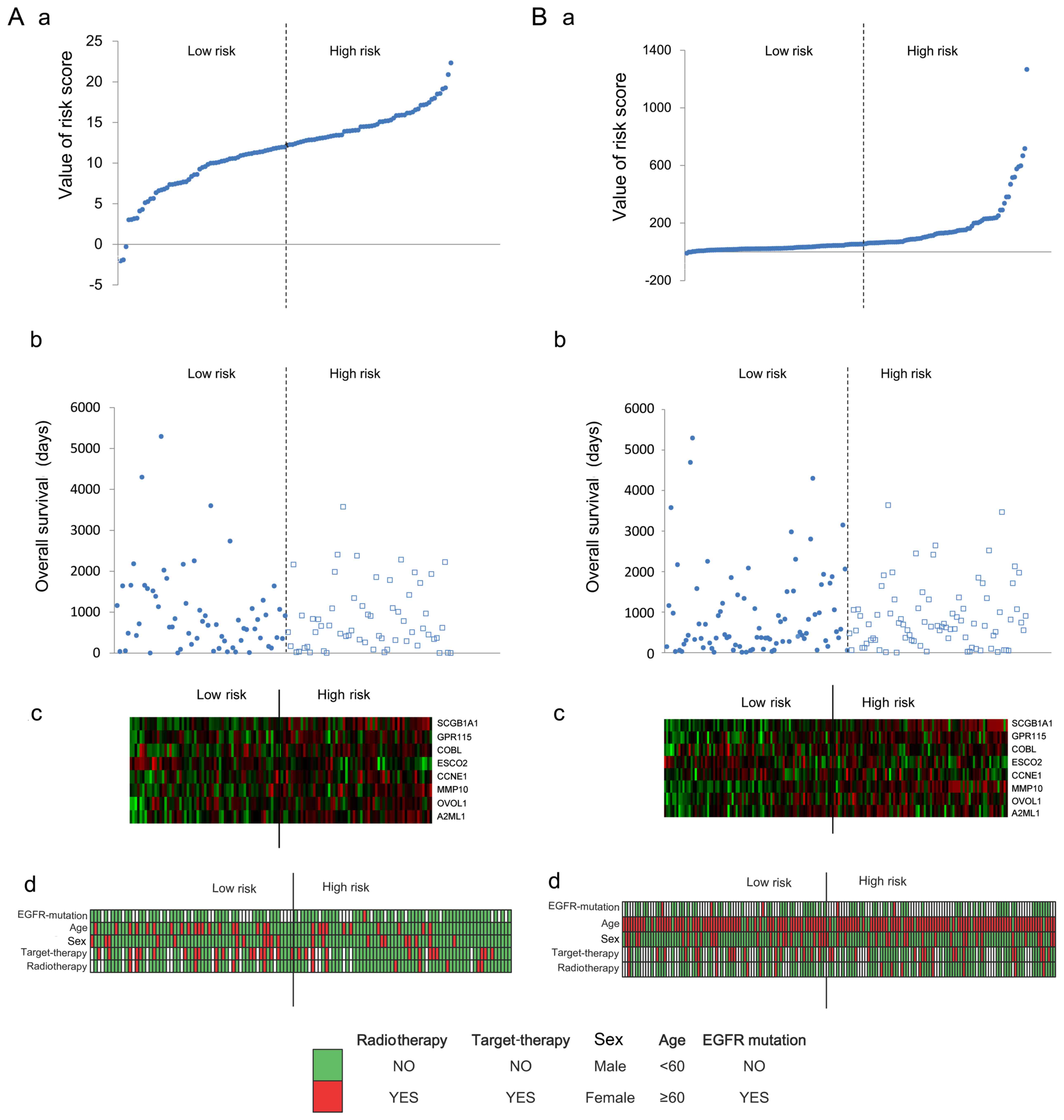
Clinical and molecular characteristics
of patients with high and low risk of lung squamous cell carcinoma
driven by TRIM58 methylation
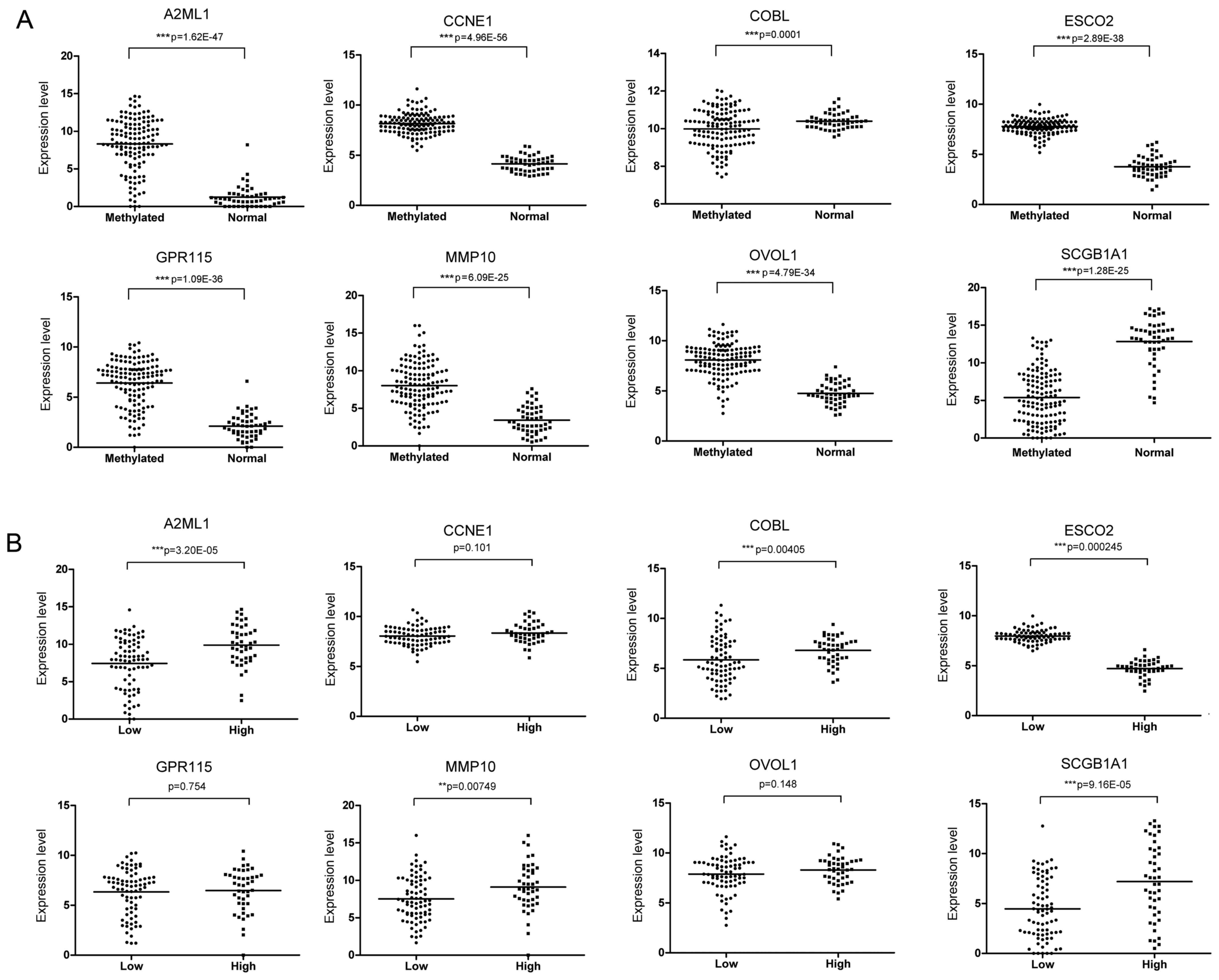
The expression values of eight signature genes in
different sets of the training dataset are displayed in Fig. 4. Risk score, overall survival days,
expression value of eight signature genes and clinical information
of the corresponding samples in the training dataset and validation
dataset are displayed in Fig.
5.
Eight significant expression factors
associated with treatment
As displayed in Table
IV, the results of single factor Cox regression demonstrated
that radiotherapy, targeted therapy and risk score were
significantly related to prognosis and the results of further
multivariate Cox regression analysis indicated that targeted
therapy and risk score were significantly correlated with the
prognosis of lung squamous cell carcinoma.
| Table IV.Univariate and multivariate cox
regression analysis of prognosis between samples and clinical
information in the training dataset. |
Table IV.
Univariate and multivariate cox
regression analysis of prognosis between samples and clinical
information in the training dataset.
|
| Univariate cox | Multivariable
cox |
|---|
|
|
|
|
|---|
| Variables | P-value | HR (95% CI) | P-value | HR (95% CI) |
|---|
| Age (years) | 0.519 | 1.41
(0.49–2.06) |
|
|
| Sex | 0.703 | 1.19
(0.48–2.91) |
|
|
| EGFR mutation | 0.934 | 0.921
(0.722–1.006) |
|
|
| Radiotherapy | 0.0049 | 0.67
(0.22–2.05) | 0.0739 | 0.605
(0.412–0.72) |
|
Targeted-therapy | 0.0188 | 1.06
(0.47–2.42) | 0.0341 | 0.871
(0.388–0.957) |
| Risk score | 0.0393 | 1.105
(1.005–1.22) | 0.0121 | 1.122
(1.026–1.228) |
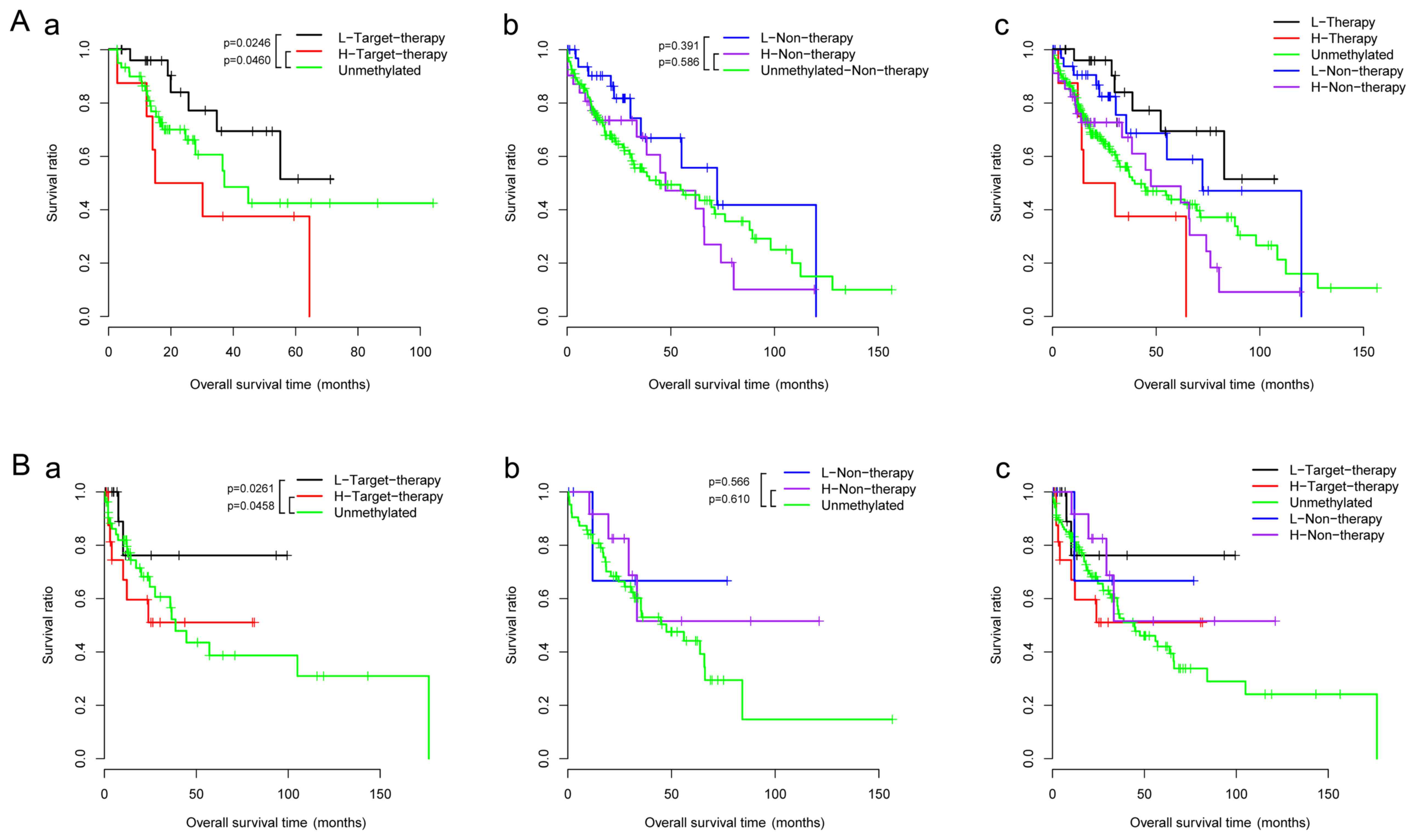
Further survival analysis of two clinical features,
targeted therapy and risk score, were performed in the training
dataset. As displayed in Fig. 6A-a,
the survival rate of high risk-targeted therapy group was
significantly lower than the unmethylated samples, while that of
low-risk-targeted therapy group was significantly higher than the
unmethylated samples. No similar results were observed in the
non-therapy group (Fig. 6A-b),
which further confirmed the relationship between the targeted
therapy and prognosis. Based on the results in the training
dataset, the data of the targeted-therapy group corresponding to
the validation dataset were collected and the correlation between
the clinical information in the high-risk and the low-risk group
was verified by the results of the survival rate. As displayed in
Fig. 6B-a, the results were
consistent with the training set, and the survival rate of the
high-risk group was significantly lower than that of the low-risk
group.
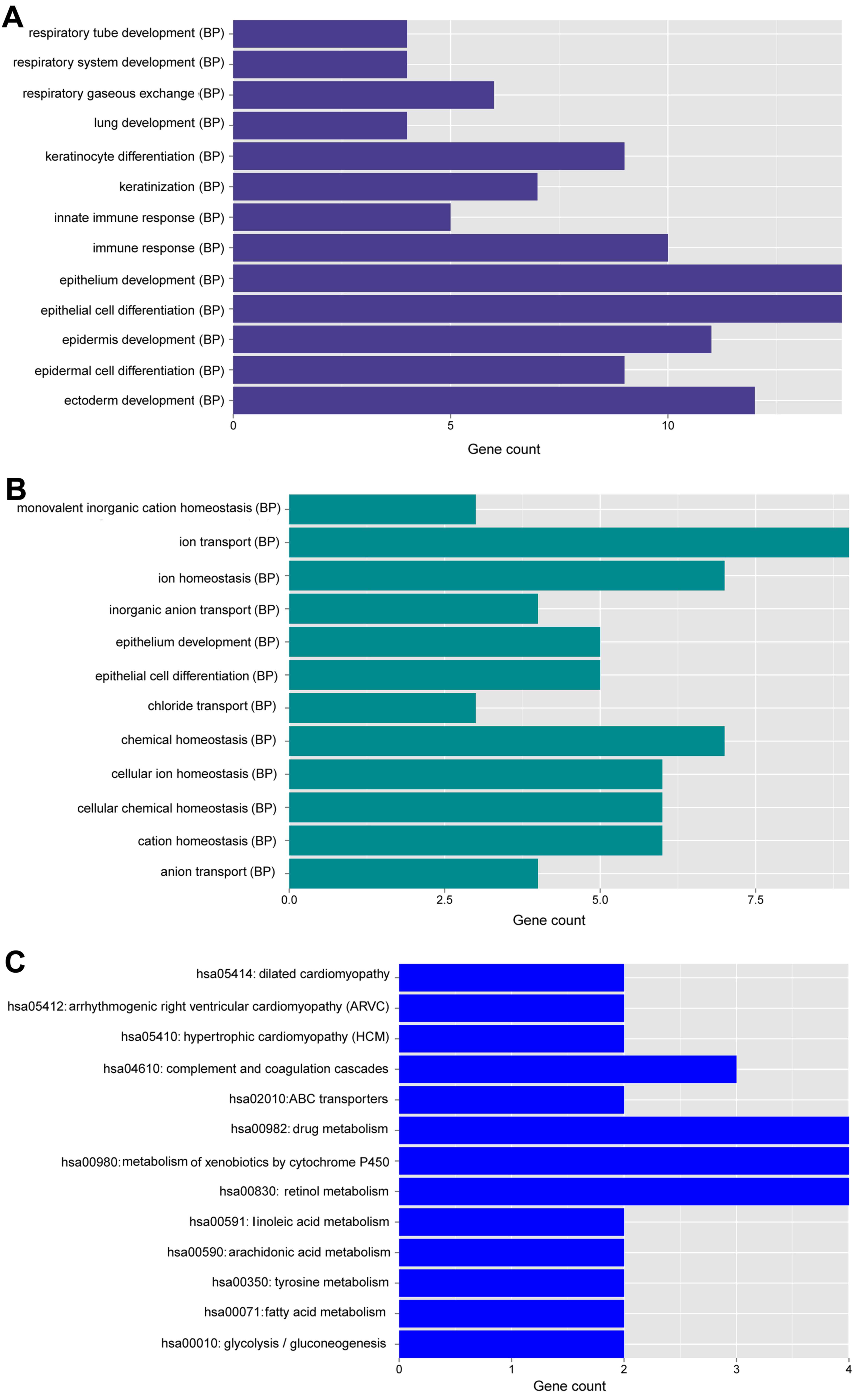
Functional enrichment analysis of
genes associated with different prognosis
Totally, 181 differentially expressed genes between
the high-risk group and the low-risk group in the training dataset
were obtained with an FDR value of <0.05. The hierarchical
clustering chart analysis was performed based on gene expression
level (Fig. 7). GO function and
KEGG pathway enrichment analysis of these genes were performed,
(Fig. 8). Upregulated genes were
mainly related to the functions of epithelial cell development,
differentiation and keratinization, respiratory system development
and immune response, while downregulated genes were mainly related
to inorganic anion transport, (cellular) ion homeostasis and
(cellular) chemical homeostasis. A total of 13 pathways were
enriched, such as drug metabolism, metabolism of xenobiotics by
cytochrome P450, retinol metabolism and complement and coagulation
cascades.
Discussion
Aberrant promoter island methylation of tumor
suppressor genes has been established as a common epigenetic
mechanism underlying the pathogenesis of human cancers, and may be
used as diagnostic marker for tumorigenesis (5–7). In
the present study, we tried to use the large quantities of mRNA-Seq
data in lung squamous cell carcinoma patients published in
cBioPortal database (http://cbioportal.org) to screen out candidate genes
related to the methylation of TRIM58/cg26157385. Compared with the
other large-scale cancer genomic projects, such as The Cancer
Genome Atlas (TCGA) and the International Cancer Genome Consortium
(ICGC) (19,20), the cBioPortal for Cancer Genomics
was specifically designed to lower the barriers of access to the
complex datasets and thereby accelerate the translation of genomic
data into new biological insights, therapies and clinical trials
(20,21). Based on the genomic data types
integrated by cBioPortal, which include somatic mutations, DNA
copy-number alterations, mRNA and microRNA expression, DNA
methylation, protein abundance and phosphoprotein abundance, users
achieved the exploration of multidimensional cancer genomic data
and biological pathway, survival analysis, analysis of mutual
exclusivity between genomic alterations, selective data download,
programmatic access and publication-quality summary visualization
(20). The present study was
performed using 370 squamous cell lung carcinoma samples in the
training dataset with methylation sequencing information and mRNA
expression profiling data, and 178 squamous cell lung carcinoma
samples in the validation dataset with mRNA expression profiling
data. According to the DNA methylation β-values of
TRIM58/cg26157385, the 370 samples in the training dataset were
divided into 133 high-methylation samples and 237 low-methylation
samples.
Significantly different expression genes, especially
markedly upregulated or downregulated ones, often help reveal the
mechanisms of various biological progresses and are usually
preferred potential candidate genes for researchers to study. In
the present study, 527 and 449 significantly different expression
genes were gained in the high-methylation samples and
low-methylation samples, respectively, and 629 significantly
differentially expressed genes related to TRIM58 methylation were
gained after the extraction of intersection between the two sets of
differentially expressed genes. As a candidate tumor suppressor and
a novel methylated gene (7,10,12),
aberrant inactivation of TRIM58 consequent to CpG island
hyper-methylation may stimulate the early carcinogenesis of lung
adenocarcinoma, and furthermore, TRIM58 methylation may be a
possible early diagnostic and epigenetic therapeutic target in lung
adenocarcinoma (7). Therefore, the
association between the significantly differentially expressed
genes related to TRIM58 methylation and lung cancer should be
documented by combining with clinical information. After
integration with the corresponding survival information for 347 out
of 370 samples with squamous cell lung carcinoma, a total of 183
genes significantly associated with prognosis were gained using the
Cox regression analysis of the survival package, and eight genes
including A2ML1, GPR115, MMP10, CCNE1, ESCO2, COBL, OVOL1 and
SCGB1A1 were selected as candidate signature genes.
The eight candidate signature genes were divided
into four groups. The first group included three genes encoding
enzyme and protease inhibitors, including MMP10, GPR115 and A2ML1.
MMPs, a group of enzymes that are collectively capable of cleaving
all components of the extracellular matrix, are involved in the
epithelial migration, neoangiogenesis, matrix degradation and
formation, among which, MMP10 was potential oral cancer marker
(22–24). GPR115 is a member of the adhesion G
protein-coupled receptors (GPCR) family, which are membrane-bound
receptors with long N-terminus, and several adhesion GPCRs are
known to have important physiological functions in CNS development
and immune system response mediated by large cell surface ligands,
however, there has not been discovered a function for GPR115
(25). A2ML1 is a kind of protease
inhibitor belonging to the alpha-macroglobulin superfamily and
displays a unique trap mechanism of inhibition, by which the A2M
inhibitor undergoes a major conformational change upon its cleavage
by a protease, thereby trapping the protease and blocking it from
subsequent substrate binding (26,27).
The second group included two genes, CNE1 and ESCO2, related to
gene amplification. Gene amplification represents one of the
molecular genetic hallmarks of human cancer and elucidating the
molecular mechanisms of how amplified genes initiate and maintain
malignant phenotypes and drive tumor progression was fundamental
for understanding the molecular etiology of human cancer and its
therapeutic implications (28).
CCNE1 is the most frequent amplified gene in ovarian serous
carcinomas and its gene amplification is related to poor survival
and potential therapeutic target in ovarian cancer, and therefore,
CCNE1-targeted therapy may benefit ovarian cancer patients with
CCNE1 amplification (28,29). ESCO2 gene encoded a protein which
may have acetyltransferase activity and be required for the
establishment of sister chromatid cohesion during the S phase of
the cell cycle, and furthermore, it may function in transcription
repression through modulation of the chromatin structure (30). Transcriptional control plays a key
role in regulating epidermal proliferation and differentiation, and
several transcription factors are known to regulate the balance
between the mesenchymal to epithelial transition and the opposite
program, such as COBL and OVOL1 in the third group (31–34).
COBL is a WH2 domain-based actin nucleator, which has been
demonstrated to play a critical role in dendrite formation and
dendritic arborisation, and its spatial control is indispensable
for proper establishment and plasticity of cell morphology
(31,34). OVOL1, encoding a zinc finger protein
homologous to Drosophila melanogaster Ovo, is expressed in
embryonic epidermal progenitor cells that are transiting from
proliferation to terminal differentiation and regulates the growth
arrest of embryonic epidermal progenitor cells and suppresses c-myc
transcription (32,33). There was one gene in the last group,
SCGB1A1, an anti-inflammatory protein predominantly expressed by
Clara cells in the lung, and a certain number of results indicated
that low SCGB1A1 level may play a key role in the pathophysiology
of asthma (35,36). As MMP10, CNE1, ESCO2, COBL and OVOL1
had been reported to be related to cancer and other human lung
diseases, we had reason to believe that the 8 candidate genes
identified in the present study may be potential factors related to
squamous cell lung carcinoma. Further validation of these 8
prognostic genes associated with TRIM58 methylation were performed
using risk score, clinical and molecular characteristics of
patients in the training dataset. The reliability of the prognostic
discrimination system was further validated in an independent
dataset. The present study provided potential diagnosis markers for
the clinical diagnosis of lung squamous cell carcinoma and was
helpful to explore the possible pathogenesis of lung squamous cell
carcinoma.
Functional annotations of the significantly
different expression genes according to GO and KEGG databases would
provide ample numbers of candidate genes and more information about
the pathogenesis of lung squamous cell carcinoma. In the present
study, upregulated genes were mainly related to epithelial cell
development, differentiation and keratinization, respiratory system
development and immune response, while downregulated genes were
mainly related to inorganic anion transport, (cellular) ion
homeostasis and (cellular) chemical homeostasis. This indicated
that the proliferation and differentiation of epidermal cells in
lung squamous cell carcinoma patients were abnormal and the
homeostasis was disturbed, and these findings were consistent with
the histological changes of squamous cell lung carcinoma (32,33).
Further KEGG pathway analysis demonstrated that these genes were
mainly involved in 13 KEGG pathways. The expression alternation of
genes involved in 8 metabolism related pathways in the present
study again indicated the abnormal metabolism in lung squamous cell
carcinoma patients, and this may give a clue for further clarifying
the pathogenesis of lung squamous cell carcinoma.
It should be noted that the present study is an
extensive bioinformatic study based on published data. The results
of these studies should also be further validated in in
vitro or in vivo models. We hope that these useful
results will help other researchers perform relevant studies.
In conclusion, our data provided a comprehensive
bioinformatic analysis of A2ML1, CCNE1, COBL, ESCO2, GPR115, MMP10,
OVOL1 and SCGB1A1 as well as their corresponding pathways which may
be involved in lung squamous cell carcinoma using two independent
datasets. The results indicated that all of them were significantly
related to the methylation of TRIM58/cg26157385 and treatment of
lung squamous cell carcinoma. Therefore, these genes may be used as
potential diagnostic markers and the present study would be helpful
to elucidate the influence of TRIM58/cg26157385 methylation on lung
cancer prognosis.
Acknowledgements
Not applicable.
Funding
The present study was supported by the general
program of Wuxi municipal health and family planning commision
(MS201715).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
WZ conceived and designed the study. QC collected
and pretreated the data, WQ, DJ and HL performed the data analysis.
QC and WQ wrote the manuscript. WZ, QC, and HL reviewed and edited
the manuscript. All authors read and approved the manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wistuba II, Behrens C, Milchgrub S, Bryant
D, Hung J, Minna JD and Gazdar AF: Sequential molecular
abnormalities are involved in the multistage development of
squamous cell lung carcinoma. Oncogene. 18:643–650. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cancer Genome Atlas Research Network:
Comprehensive genomic characterization of squamous cell lung
cancers. Nature. 489:519–525. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Imoto I, Izumi H, Yokoi S, Hosoda H,
Shibata T, Hosoda F, Ohki M, Hirohashi S and Inazawa J: Frequent
silencing of the candidate tumor suppressor PCDH20 by epigenetic
mechanism in non-small-cell lung cancers. Cancer Res. 66:4617–4626.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Izumi H, Inoue J, Yokoi S, Hosoda H,
Shibata T, Sunamori M, Hirohashi S, Inazawa J and Imoto I: Frequent
silencing of DBC1 is by genetic or epigenetic mechanisms in
non-small cell lung cancers. Hum Mol Genet. 14:997–1007. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kajiura K, Masuda K, Naruto T, Kohmoto T,
Watabnabe M, Tsuboi M, Takizawa H, Kondo K, Tangoku A and Imoto I:
Frequent silencing of the candidate tumor suppressor TRIM58 by
promoter methylation in early-stage lung adenocarcinoma.
Oncotarget. 8:2890–2905. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Napolitano LM and Meroni G: TRIM family:
Pleiotropy and diversification through homomultimer and
heteromultimer formation. IUBMB Life. 64:64–71. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nisole S, Saieb SA and Saïb A: TRIM family
proteins: Retroviral restriction and antiviral defence. Nat Rev
Microbiol. 3:799–808. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Qiu X, Huang Y, Zhou Y and Zheng F:
Aberrant methylation of TRIM58 in hepatocellular carcinoma and its
potential clinical implication. Oncol Rep. 36:811–818. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Thom CS, Traxler EA, Khandros E, Nickas
JM, Zhou OY, Lazarus JE, Silva AP, Prabhu D, Yao Y, Aribeana C, et
al: Trim58 degrades dynein and regulates terminal erythropoiesis.
Dev Cell. 30:688–700. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
White DE, Rafalskametcalf IU, Ivanov AV,
Corsinotti A, Peng H, Lee SC, Trono D, Janicki SM and Rauscher FJ
III: The ATM substrate KAP1 controls DNA repair in heterochromatin:
Regulation by HP1 proteins and Serine 473/824 phosphorylation. Mol
Cancer Res. 10:401–414. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang X, Zhu H, Snieder H, Su S, Munn D,
Harshfield G, Maria BL, Dong Y, Treiber F, Gutin B and Shi H:
Obesity related methylation changes in DNA of peripheral blood
leukocytes. BMC Med. 8:872010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bao ZS, Li MY, Wang JY, Zhang CB, Wang HJ,
Yan W, Liu YW, Zhang W, Chen L and Jiang T: Prognostic value of a
nine-gene signature in glioma patients based on mRNA expression
profiling. CNS Neurosci Ther. 20:112–118. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cheng W, Ren X, Cai J, Zhang C, Li M, Wang
K, Liu Y, Han S and Wu A: A five-miRNA signature with prognostic
and predictive value for MGMT promoter-methylated glioblastoma
patients. Oncotarget. 6:29285–29295. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang CB, Zhu P, Yang P, Cai JQ, Wang ZL,
Li QB, Bao ZS, Zhang W and Jiang T: Identification of high risk
anaplastic gliomas by a diagnostic and prognostic signature derived
from mRNA expression profiling. Oncotarget. 6:36643–36651.
2015.PubMed/NCBI
|
|
18
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
International Cancer Genome Consortium, .
Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, Bernabé RR, Bhan
MK, Calvo F, Eerola I, et al: International network of cancer
genome projects. Nature. 464:993–998. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gao J, Aksoy BA, Dogrusoz U, Dresdner G,
Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al:
Integrative analysis of complex cancer genomics and clinical
profiles using the cBioPortal. Sci Signal. 6:pl12013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cerami E, Gao J, Dogrusoz U, Gross BE,
Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et
al: The cBio cancer genomics portal: An open platform for exploring
multidimensional cancer genomics data. Cancer Discov. 2:401–404.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Okada A, Tomasetto C, Lutz Y, Bellocq JP,
Rio MC and Basset P: Expression of matrix metalloproteinases during
rat skin wound healing: Evidence that membrane type-1 matrix
metalloproteinase is a stromal activator of pro-gelatinase A. J
Cell Biol. 137:67–77. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vaalamo M, Karjalainen-Lindsberg ML,
Puolakkainen P, Kere J and Saarialhokere U: Distinct expression
profiles of stromelysin-2 (MMP-10), collagenase-3 (MMP-13),
macrophage metalloelastase (MMP-12), and tissue inhibitor of
metalloproteinases-3 (TIMP-3) in intestinal ulcerations. Am J
Pathol. 152:1005–1014. 1998.PubMed/NCBI
|
|
24
|
Yen CY, Chen CH, Chang CH, Tseng HF, Liu
SY, Chuang LY, Wen CH and Chang HW: Matrix metalloproteinases (MMP)
1 and MMP10 but not MMP12 are potential oral cancer markers.
Biomarkers. 14:244–249. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Haitina T, Olsson F, Stephansson O, Alsiö
J, Roman E, Ebendal T, Schiöth HB and Fredriksson R: Expression
profile of the entire family of Adhesion G protein-coupled
receptors in mouse and rat. BMC Neurosci. 9:1–14. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Galliano MF, Toulza E, Gallinaro H, Jonca
N, Ishida-Yamamoto A, Serre G and Guerrin M: A novel protease
inhibitor of the alpha2-macroglobulin family expressed in the human
epidermis. J Biol Chemist. 281:5780–5789. 2006. View Article : Google Scholar
|
|
27
|
Vissers LE, Bonetti M, Overman
Paardekooper J, Nillesen WM, Frints SG, de Ligt J, Zampino G,
Justino A, Machado JC, Schepens M, et al: Heterozygous germline
mutations in A2ML1 are associated with a disorder clinically
related to Noonan syndrome. Eur J Hum Genet. 23:317–324. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nakayama K, Nakayama N, Jinawath N, Salani
R, Kurman RJ, Shih IeM and Wang TL: Amplicon profiles in ovarian
serous carcinomas. Int J Cancer. 120:2613–2617. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nakayama N, Nakayama K, Shamima Y,
Ishikawa M, Katagiri A, Iida K and Miyazaki K: Gene amplification
CCNE1 is related to poor survival and potential therapeutic target
in ovarian cancer. Cancer. 116:2621–2634. 2010.PubMed/NCBI
|
|
30
|
Kim BJ, Kang KM, Jung SY, Choi HK, Seo JH,
Chae JH, Cho EJ, Youn HD, Qin J and Kim ST: Esco2 is a novel
corepressor that associates with various chromatin modifying
enzymes. Biochem Biophys Res Commun. 372:298–304. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ahuja R, Pinyol R, Reichenbach N, Custer
L, Klingensmith J, Kessels MM and Qualmann B: Cordon-bleu is an
actin nucleation factor and controls neuronal morphology. Cell.
131:337–350. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nair M, Teng A, Bilanchone V, Agrawal A,
Li B and Dai X: Ovol1 regulates the growth arrest of embryonic
epidermal progenitor cells and represses c-myc transcription. J
Cell Biol. 173:253–264. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Roca H, Hernandez J, Weidner S, McEachin
RC, Fuller D, Sud S, Schumann T, Wilkinson JE, Zaslavsky A, Li H,
et al: Transcription factors OVOL1 and OVOL2 induce the mesenchymal
to epithelial transition in human cancer. PloS One. 8:e767732013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schwintzer L, Koch N, Ahuja R, Grimm J,
Kessels MM and Qualmann B: The functions of the actin nucleator
Cobl in cellular morphogenesis critically depend on syndapin I.
Embo J. 30:3147–3159. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nie W, Xue C, Chen J and Xiu Q:
Secretoglobin 1A member 1 (SCGB1A1)+38A/G polymorphism is
associated with asthma risk: A meta-analysis. Gene. 528:304–308.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Peri A, Cordella-Miele E, Miele L and
Mukherjee AB: Tissue-specific expression of the gene coding for
human Clara cell 10-kD protein, a phospholipase A2-inhibitory
protein. J Clin Invest. 92:2099–2109. 1993. View Article : Google Scholar : PubMed/NCBI
|