Introduction
Histone modification represents one of the main
mechanisms of the epigenetic regulation of gene expression
(1,2). Increasing evidence indicated that
specific members of the histone demethylase family have been
associated with human diseases, especially carcinogenesis (3). Notably, LSD1 was the first discovered
histone demethylase which specifically catalyzed the removal of
mono- and dimethyl groups from methylated histone H3 at lysine 4
(H3K4me1/2) contributing to silencing or activation of target genes
(4). LSD1 is highly expressed in a
variety of carcinomas, including breast (5), lung (6), hepatocarcinoma (7) and ovarian cancer (8). Our previous studies have demonstrated
that the overexpression of LSD1 was associated with an aggressive
phenotype and poor prognosis of ovarian cancer (9). However, the molecular mechanism
underlying the association of the overexpression of LSD1 with the
progression of ovarian cancer remains largely unknown.
Autophagy is an evolutionarily conserved cellular
process for recycling and removal of damaged organelles,
macromolecules and protein aggregates which are encapsulated by
double-membrane structures (autophagosomes) and delivered to
lysosomes for degradation (10–12).
Autophagy is essential for cells to gain energy and maintain
cellular homeostasis, which protects them from nutrient and
environmental stress (13).
Microtubule-associated protein 1 light chain 3 (LC3) is a protein
participating in autophagy through conjugation with
phosphatidylethanolamine (PE) to form LC3-II, which is an
autophagosomal marker (14,15). Activation of autophagy is sensitized
by inactivation of the mammalian target of rapamycin (mTOR), which
is a major cellular nutrition and energy sensor kinase (16). Many studies have revealed that
autophagy plays complex roles in tumorigenesis and tumor
progression (13,17,18).
At present, how autophagy regulates tumorigenesis and tumor
progression is not fully understood.
Numerous studies have revealed that autophagy was
directly or indirectly regulated by epigenetics, either by the
modification of transcriptional factors or chromatin structure
during gene expression (19,20).
Recently, several studies found that LSD1 was involved in the
regulation of autophagy through a variety of mechanisms (21,22).
In the present study, we observed that knockdown of LSD1 caused
accumulation of LC3-II and markedly enhanced the autophagic flux in
ovarian cancer HO8910 cells. Furthermore, depletion of LSD1
significantly promoted the starvation- and rapamycin-induced
activation of autophagy. Notably, we found that serum starvation or
rapamycin treatment downregulated LSD1, indicating that
downregulation of LSD1 may mediate the activation of autophagy by
starvation or rapamycin. Finally, we confirmed that the negative
regulation of autophagy by LSD1 was conducted through the
activation of the mTOR signaling pathway. Our study has provided
direct evidence supporting the negative role of LSD1 in the
regulation of autophagy in ovarian cancer cells.
Materials and methods
Cell lines and cell culture
The human ovarian epithelial cancer cells (HO8910)
were kindly provided by Dr Qixiang Shao of Jiangsu University
(Zhenjiang, China). HO8910 and 293 cells were cultured in
Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal
bovine serum (FBS; both form Gibco, Grand Island, NY, USA) at 37°C
in a humidified atmosphere containing 5% CO2.
Reagents, antibodies and plasmids
pLKO-Tet-On, pHR'-CMV-8.2∆VPR and pHR'-CMV-VSVG
vectors were kindly provided by Dr Changdeng Hu (Purdue University,
West Lafayette, IN, USA). The antibodies were obtained as follows:
the LSD1 (1:1,000; cat. no. 2184S), H3 (1:2,000; cat. no. 4499S),
H3K4me2 (1:1,000; cat. no. 9725S), Beclin1 (1:1,000; cat. no.
3495P), LC3 (1:500; cat. no. 4180S), p70S6K (1:1,000; cat. no.
2708T) and phospho-p70S6K (1:1,000; cat. no. 9234T) antibodies were
purchased from Cell Signaling Technology (Boston, MA, USA);
α-tubulin (1:5,000; cat. no. BS1699) and goat anti-rabbit IgG
(H&L)-HRP (1:5,000; cat. no. BS13278) were obtained from
Bioworld Technology (Shanghai, China); Alexa Fluor® 488
AffiniPure goat anti-rabbit IgG (H+L) (1:100; cat. no. R37116) were
purchased from Invitrogen (Carlsbad, CA, USA).
Electrochemiluminescence (ECL) reagents were purchased from
Millipore (EMD Millipore, Billerica, MA, USA). Bioepitope Nuclear
and Cytoplasmic Extraction kit were purchased from Bioworld
Technology. Polybrene, doxycycline (Dox), chloroquine (CQ),
tranylcypromine (TCP), dimethyl sulfoxide (DMSO), and
dihydrochloride (DAPI) were purchased from Sigma-Aldrich (St.
Louis, MO, USA). Bafilomycin A1 (BafA1) was purchased from GE
Healthcare Life Sciences (Piscataway, NJ, USA). Rapamycin and
MHY1485 were purchased from Selleck Chemicals (Shanghai,
China).
Generation of stable LSD1 knockdown
cell line
pLKO-Tet-On-shLSD1 plasmid and lentiviral particles
were generated as previously described (9). Lipofectamine 2000 reagent (Invitrogen)
was used as the transfection reagent and the transfection was
performed according to the manufacturer's protocol. In brief, 1 ml
lentiviral supernatant and 3 ml complete medium containing 8 µg/ml
polybrene were added to infect the HO8910 cells. After being
infected twice, 2 µg/ml puromycin was added into HO8910 cells for
72 h and then maintained with 1.0 µg/ml puromycin for two weeks to
select stably transfected HO8910-shLSD1 cells.
RNA extraction and qRT-PCR
RNA was extracted using the RNAiso plus (Takara Bio,
Shiga, Japan). To produce cDNA, 2 µg of total RNA was processed
with the PrimeScript RT Reagent kit (Takara Bio) according to the
manufacturer's instructions. Then the cDNAs were subjected to
qRT-PCR as previously described (23). qRT-PCR was performed using the
comparative cycle threshold (CT) method with SYBR-Green PCR Master
Mix (Takara Bio) according to the manufacturer's instructions on a
Bio-Rad CFX96 system (Bio-Rad Laboratories, Inc., Hercules, CA,
USA). The primer sequences were used as follows: LSD1
(GenBank accession no. NM 015013.3), 5′-CAAGTGTCAATTTGTTCGGG-3′
(forward) and 5′-TTCTTTGGGCTGAGGTACTG-3′ (reverse), GAPDH
(GenBank accession no. NM 001256799.1), 5′-GCAAATTCCATGGCACCGTC-3′
(forward) and 5′-TCGCCCCACTTGATTTTGG-3′ (reverse), LC3
(GenBank accession no. NM 022818.4), 5′-CCAGATCCCTGCACCATG-3′
(forward) and 5′-CTGCTTCTCACCCTTGTATCG-3′ (reverse). The following
PCR conditions were used: initial denaturation at 95°C for 30 sec,
followed by 40 cycles of denaturation at 95°C for 5 sec, annealing
at 58°C for 30 sec and elongation at 72°C for 1 min. The relative
results were analyzed using the comparative cycle threshold method
(2−∆∆CT) with GAPDH as the reference gene
(24).
Western blot analysis
Histones were prepared using the Bioepitope Nuclear
and Cytoplasmic Extraction kit (Bioworld Technology) following the
manufacturer's protocol. Cells were lysed with RIPA buffer
(Kangchen Biotech, Shanghai, China) to extract total protein and
equal amounts of soluble protein samples were subjected to 8–12%
SDS-PAGE gel and transferred to 0.2/0.45-µm pore polyvinylidene
difluoride (PVDF) membranes (EMD Millipore). The membranes were
blocked in 5% non-fat dry milk for 1 h at room temperature (RT).
The membranes were washed in TBST three times and incubated
overnight at 4°C with the corresponding primary antibodies. The
membranes were then washed with TBST and incubated with the
secondary antibody for 1 h at RT. The results were developed by ECL
reagents. To perform densitometry analysis, digital images of the
positive bands were obtained with ChemiDoc XRS and analyzed using
the image analysis program Quantity One (Bio-Rad Laboratories). The
results were expressed as the ratio of target protein/loading
control. Data shown are representative of three independent
experiments.
Immunofluorescence
Cells were plated on glass coverslips in 24-well
tissue culture plates. In brief, cells were washed with PBS, fixed
with 4% paraformaldehyde and permeabilized with ice-cold 100%
methanol (25), and then the cells
were blocked with 3% bovine serum albumin (BSA) for 1 h at RT. The
cells were incubated with primary antibody LC3 (1:200) at 4°C
overnight and Alexa Fluor® 488 AffiniPure goat
anti-rabbit IgG (H+L) (1:100) was used as secondary antibody for 1
h at RT. The number of nuclei was revealed by DAPI staining.
Fluorescence images were captured using the Nikon new Ni Series
microscopes (Nikon, Melville, NY, USA).
Statistical analysis
All the experiments were repeated independently
three times and the data values were presented as the mean ± SEM.
The differences between the groups were analyzed by Student's
t-test, when two groups were compared or by one-way ANOVA, when
more than two groups were compared using SPSS 11.5 software (SPSS,
Inc., Chicago, IL, USA). P-values with a 95% confidence interval
were obtained from at least three independent experiments. A
P-value of <0.05 was considered to indicate a statistically
significant difference.
Results
Knockdown of LSD1 induces accumulation
of LC3-II proteins
To examine whether LSD1 plays an important role in
autophagy, we determined the effect of LSD1 knockdown on the level
of the autophagosomal marker LC3-II. The inducible LSD1 shRNA cell
line HO8910-shLSD1 was generated using lentiviral vector-loaded
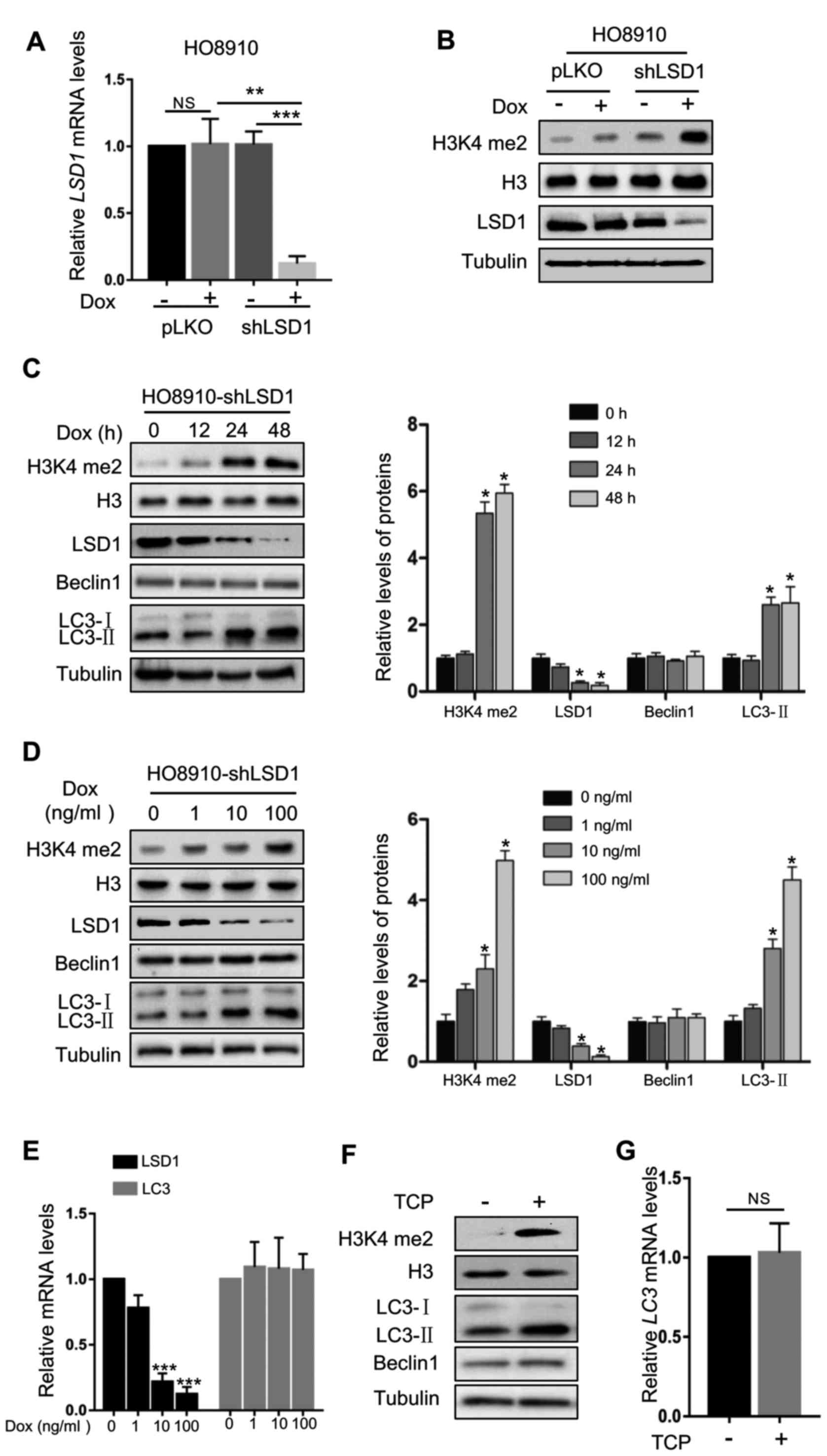
LSD1 shRNA in HO8910 ovarian cancer cells. As displayed in Fig. 1A and B, the protein product and mRNA
levels of LSD1 were effectively silenced in HO8910-shLSD1 cells. In
addition, LSD1 knockdown markedly increased the H3K4me2 levels, a
major substrate of LSD1 (Fig. 1B).
Depletion of LSD1 caused significant accumulation of LC3-II in a
time- and dose-dependent manner, but did not affect the levels of
Beclin1 (Fig. 1C and D), a protein
involved in autophagosomal biogenesis (26). In addition, knockdown of LSD1 did
not significantly alter the LC3 mRNA levels (Fig. 1E), indicating that the effect of
LSD1 downregulation on the LC3-II level was produced by affecting
the protein translation, modification or degradation. To further
confirm this result, we used tranylcypromine (TCP), a specific
inhibitor of LSD1, to suppress the demethylase activity of LSD1
(27). TCP markedly increased the
H3K4me2 levels, indicating that the demethylase activity of LSD1
was effectively inhibited (Fig.
1F). Consistent with the effect of the knockdown of LSD1,
inhibition of LSD1 by TCP markedly increased the LC3-II protein
levels (Fig. 1F), while it did not
alter the mRNA levels (Fig. 1G).
These data indicated that LSD1 was a negative regulator for
production of LC3-II, indicating that LSD1 may play an important
role in autophagy.
LSD1 depletion results in the
induction of autophagic flux
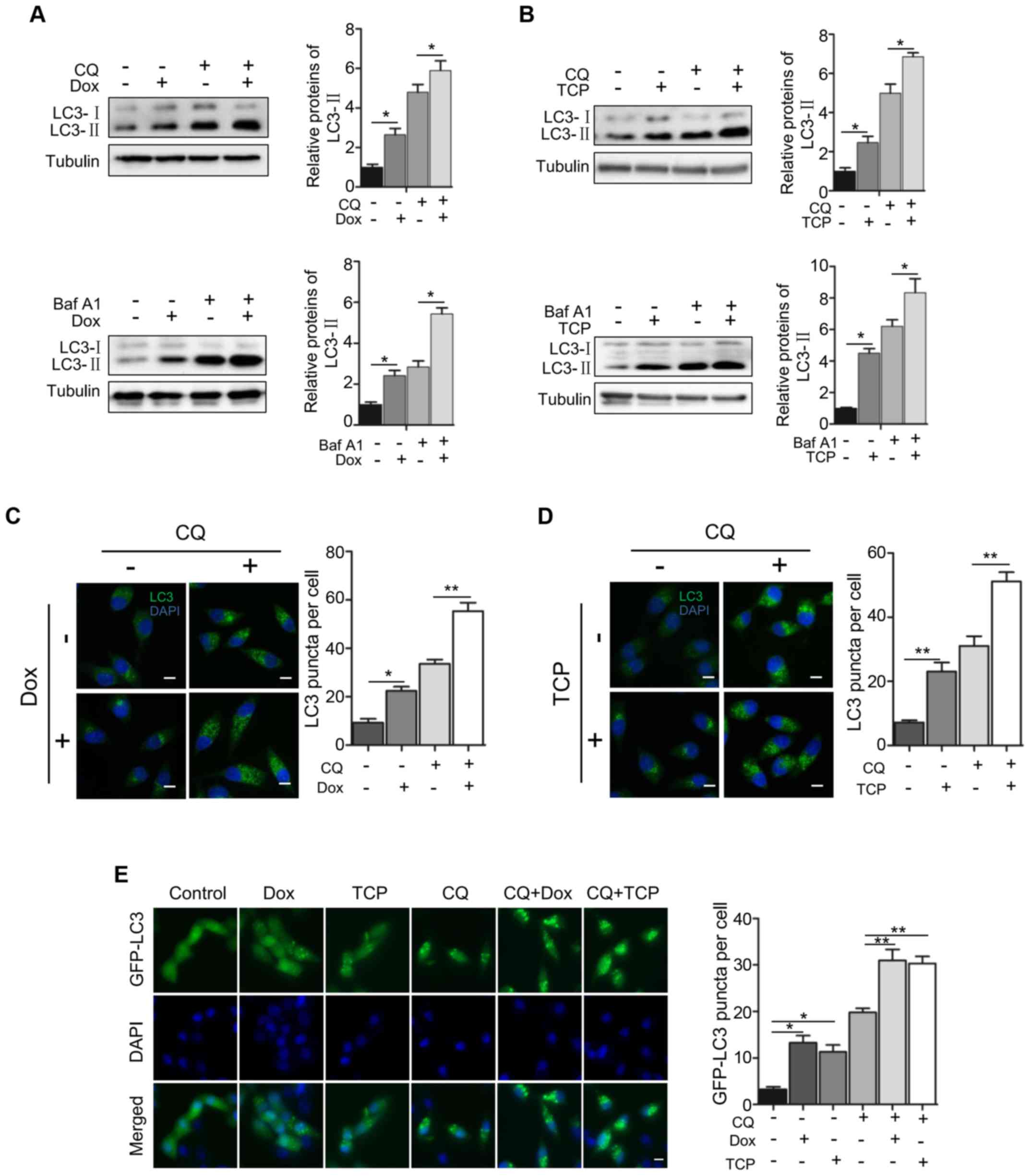
To confirm the role of LSD1 in autophagy, we
examined the effect of the downregulation or inhibition of LSD1 on
autophagic flux. We used the V-ATPase inhibitor bafilomycin A1
(BafA1) and the lysosomal inhibitor chloroquine (CQ) to block the
autophagic flux. As displayed in Fig.
2, BafA1 or CQ alone with the expression of LSD1 shRNA produced
a significant increase of LC3-II (Fig.
2A). Similarly, BafA1 or CQ with the LSD1 inhibitor TCP
significantly induced higher levels of LC3-II (Fig. 2B), indicating that the
downregulation of the LSD1 activity or protein level stimulated
autophagy. Subsequently, we determined the effect on autophagy flux
by immunofluorescence staining of LC3. As displayed in Fig. 2C, knockdown of LSD1 increased the
amount of LC3 puncta, which was consistent with the immunoblotting
data displayed in Fig. 2A,
indicating an inhibitory role of LSD1 in autophagy. Treatment with
CQ alone resulted in the accumulation of LC3 puncta, indicating
that CQ blocks the autophagic flux. Knockdown of LSD1 plus
treatment with CQ caused a greater increase in the amount of LC3
puncta than that of LSD1 knckdown or CQ treatment alone (Fig. 2C), indicating that depletion of LSD1
activated the autophagic flux. Treatment of the cells with the LSD1
inhibitor TCP revealed the same effect as that of LSD1 knockdown
(Fig. 2D), indicating that the
demethylase activity of LSD1 was required for the inhibitory effect
on autophagy flux. We further confirmed the inhibitory role of LSD1
in autophagy flux by visualizing the effect of LSD1 knockdown or
treatment with TCP on the amount of GFP-LC3 puncta in HO8910-shLSD1
cell line, as displayed in Fig. 2E.
Collectively, these results clearly indicated that LSD1 was a
negative regulator in autophagy.
Inhibition of LSD1 enhances the
activation of autophagy
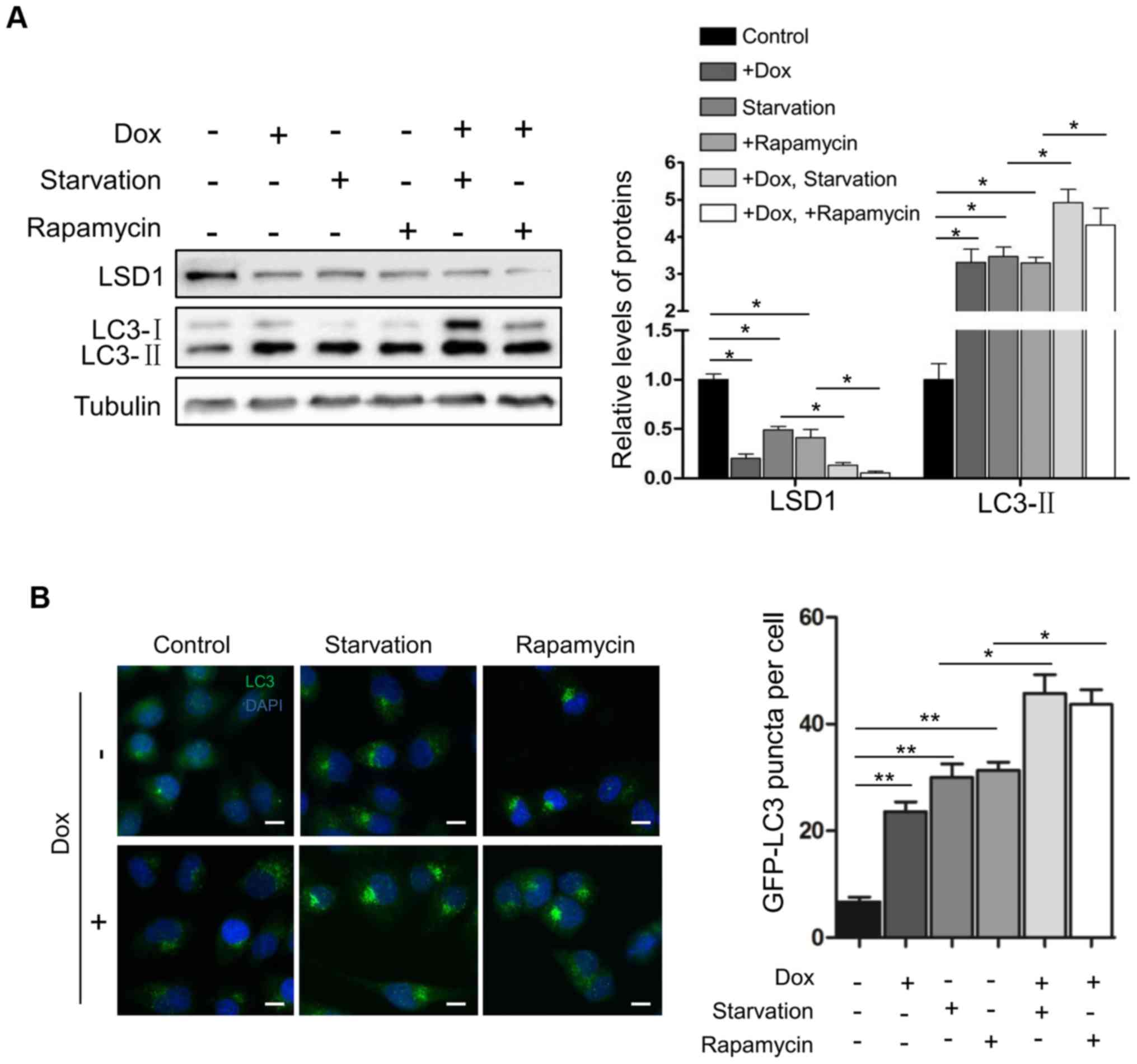
Subsequently, we determined the effect of LSD1 on
the activation of autophagy. Serum starvation and rapamycin are two
known physiologic stimuli for the activation of autophagy (12,28).
As expected, serum withdrawal and rapamycin treatment induced the
elevation of LC3-II (Fig. 3A) and
increased the number of LC3 puncta (Fig. 3B) in HO8910 cells, indicating that
serum starvation and rapamycin activated autophagy. Knockdown of
LSD1 in cells further enhanced serum starvation- or
rapamycin-induced elevation of LC3-II (Fig. 3A) and increased the number of LC3
puncta in shLSD1 cells (Fig. 3B),
indicating that depletion of LSD1 promoted the starvation- and
rapamycin-induced activation of autophagy. Collectively, these
results demonstrated that LSD1 was a suppressor for autophagy
activation.
Notably, serum starvation or rapamycin treatment
caused a significant downregulation of LSD1 to the same level as
that of shLSD1 (lanes 3 and 4; top panel; Fig. 3A), and additional downregulation of
LSD1 was observed when the treatment was combined with the
induction of the shLSD1 knockdown (lanes 5 and 6; the top panel;
Fig. 3A), raising a possibility
that serum starvation or rapamycin activated autophagy through the
downregulation of LSD1, or LSD1 mediated the inactivation of
autophagy by mTOR signaling.
LSD1 regulates autophagy through the
mTOR signaling pathway
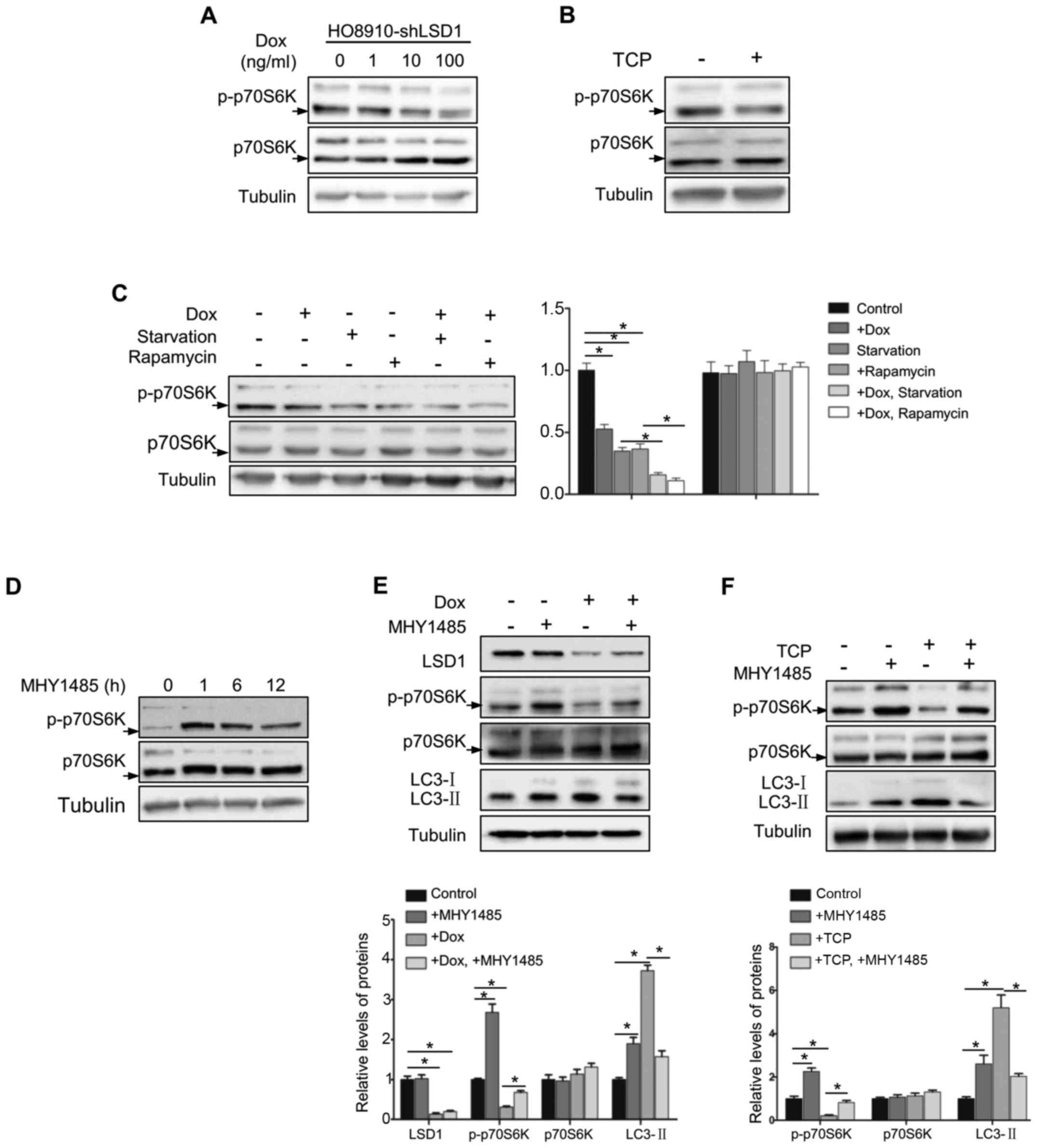
To explore the mechanism underlying the inhibitory
effect of LSD1 on autophagy, we determined whether LSD1 was
involved in the mTOR signaling pathway, a known pathway
inactivating autophagy (16). As
p70S6K is a known downstream substrate of mTOR (29), we detected phosphorylation of Thr389
in p70S6K to evaluate the mTOR activation. As displayed in Fig. 4A, phosphorylation of p70S6K
(Thr389), was decreased in LSD1-depleted cells in a dose-dependent
manner. Similarly, inhibition of mTOR signaling was also detected
with treatment of the LSD1 inhibitor TCP (Fig. 4B). To further confirm these results,
we determined the phosphorylation of p70S6K (Thr389) following
starvation or rapamycin treatment in HO8920-shLSD1 cells and
observed that knockdown of LSD1 resulted in further suppression of
the mTOR pathway during starvation or rapamycin treatment (Fig. 4C), indicating that LSD1 regulated
autophagy was indispensable through the mTOR signaling. To
determine whether LSD1 knockdown induced autophagy directly via the
mTOR pathway, we used an mTOR activator MHY1485 (30), which directly bound to mTOR and
induced the phosphorylation of p70S6K (Thr389) to activate mTOR. As
displayed in Fig. 4D, treatment
with MHY1485 reached the maximum to activate mTOR at 1 h and was
sustained for 12 h. Notably, MHY1485 treatment abolished the effect
of LSD1 knockdown on p70S6K phosphorylation and LC3-II accumulation
(Fig. 4E), indicating that LSD1
knockdown-induced autophagy was blocked by mTOR activation. Similar
results were also observed with TCP treatment (Fig. 4F). These results indicated that
downregulation of LSD1 induced autophagy via the inhibition of mTOR
signaling.
Discussion
In the present study, we revealed that LSD1, a
lysine-specific demethylase, was a negative regulator of autophagy
protein in HO8910 ovarian cancer cells. LSD1 reduction led to the
activation of autophagy and the promotion of starvation- and
rapamycin-induced autophagy via the mTOR signaling pathway, which
could be a therapeutic target for treatment of ovarian cancer due
to the negative role of LSD1 in autophagy.
We have previously identified that LSD1 was
overexpressed in ovarian cancer cells and its overexpression
correlated with high migration and invasion (9,31).
Furthermore, it has been reported that the autophagy level was
lower in ovarian cancers than benign tumors and the induction of
autophagy in this type of cancer was associated with prolonged
overall survival of patients (32,33).
Thus, determining the regulatory mechanism of LSD1-mediated
autophagy in ovarian cancer is urgently required for developing new
therapeutic approaches. In fact, recent research has pointed the
vital role of LSD1 in autophagy. For example, the FGF19-SHP-LSD1
regulatory axis mediated a postprandial epigenetic suppression of
hepatic autophagy (34).
Furthermore, a recent study reported that LSD1 overexpression
promoted tumorigenesis and inhibited autophagy via decreasing the
p62 protein stability in a demethylation-independent manner in
gynecologic malignancies. In addition, the higher p62 and
autophagic flux levels elicited by LSD1 inhibition could reduce
tumor growth in vivo (21),
indicating that LSD1 was an important regulator of autophagy in
tumors, and further potential mechanisms of these effects need to
be elucidated. In the present study, we demonstrated that LSD1
knockdown induced the accumulation of LC3-II proteins in HO8910
ovarian cancer cells. To confirm whether the LSD1 knockdown
stimulated the production of autophagosomes or caused a blockage of
autophagic degradation, we used two lysosomal inhibitors BafA1,
that prevented maturation of autophagic vacuoles by inhibiting
fusion between autophagosomes and lysosomes (35) and CQ, that raised the lysosomal pH,
which also inhibited fusion of autophagosomes with lysosomes
(36). Upon treatment with these
lysosomal inhibitors, an increased accumulation of LC3-II and
autophagic flux were observed during LSD1 depletion, indicating
that the LC3-II accumulation mediated by LSD1 knockdown was
involved in the autophagy induction process.
The experiments presented in our study added a new
mechanism underlying the LSD1-mediated suppression of autophagy. We
revealed for the first time, that knockdown or inhibition of LSD1
induced autophagic flux in HO8910 ovarian cancer cells. While
autophagy can be potently activated by serum starvation and
rapamycin, we demonstrated that depletion of LSD1 obviously
promoted the starvation- and rapamycin-induced activation of
autophagy, which clearly demonstrated that LSD1 was a suppressor
for autophagy activation. Notably, we observed that serum
starvation or rapamycin treatment downregulated LSD1, indicating
that downregulation of LSD1 may mediate the activation of autophagy
or may have a feedback effect on LSD1 expression in starvation- and
rapamycin-induced autophagy.
The mTOR signaling pathway plays a crucial role in
regulating autophagy, and mTOR is inhibited in response to various
cellular conditions such as serum starvation, stress stimulation
and growth factor deprivation (16). Several studies have indicated that
the mTOR signaling pathway may be involved in epigenetic changes
during autophagy. For example, methyltransferase EZH2
epigenetically suppressed several negative regulators of mTOR
pathway via MTA2 to inhibit autophagy (37). Furthermore, LSD1 has been found to
be a regulator of the mTOR pathway in neuroblastoma cells. Ambrosio
et al (39) found that
SESN2, which serves as a key positive regulator of the autophagic
pathway by directly inhibiting the mTORC1 activity (38), played an important role in
LSD1-mediated autophagy. SESN2 is an LSD1-suppressed target gene
and LSD1 inhibition triggered the transcriptional activation of
SESN2, which resulted in the decrease of mTORC1 activity (39). Collectively, these findings provided
a rationale for our hypothesis and indicated that the molecular
mechanism of LSD1-mediated autophagy in ovarian cancer should be
urgently explored. In the present study, we demonstrated that LSD1
depletion-induced autophagy in ovarian cancer cells was dependent
on mTOR inactivation which was characterized by decreasing the
phosphorylation of p70S6K (Thr389), a downstream effector and
marker of mTOR. Furthermore, we noticed that the mTOR activator
MHY1485 rescued the phosphorylation levels of p70S6K in response to
LSD1 deficiency, which further supported our hypothesis. In
addition, LSD1 depletion-induced LC3-II accumulation was attenuated
upon mTOR activation. Thus, LSD1 appeared to regulate autophagy in
an mTOR-dependent way.
In conclusion, our data identified LSD1 as an
autophagy suppressor in HO8910 cells, which involved both basal and
starvation- or rapamycin- induced autophagic progress through the
mTOR pathway. Further studies are warranted to elucidate the
potential mechanisms of how starvation and rapamycin alter the
expression of LSD1, as well as uncover the LSD1-dependent
epigenetic changes which may mediate autophagic process in ovarian
cancer. Our results established a novel link among epigenetic
regulation, mTOR pathway and autophagy in HO8910 ovarian cancer
cells, providing new insights for future cancer treatment.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from the
National Natural Science Foundation of China (no. 81170573).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
GS and QL conceived and designed the study. YW
performed the experiments. YW and TH wrote the paper. TH, RW, JW
and KP were involved in the conception of the study. All authors
read and approved the manuscript and agree to be accountable for
all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
References
|
1
|
Lawrence M, Daujat S and Schneider R:
Lateral Thinking: How histone modifications regulate gene
expression. Trends Genet. 32:42–56. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Baek SH: When signaling kinases meet
histones and histone modifiers in the nucleus. Mol Cell.
42:274–284. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shi Y and Whetstine JR: Dynamic regulation
of histone lysine methylation by demethylases. Mol Cell. 25:1–14.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lan F, Nottke AC and Shi Y: Mechanisms
involved in the regulation of histone lysine demethylases. Curr
Opin Cell Biol. 20:316–325. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Serce N, Gnatzy A, Steiner S, Lorenzen H,
Kirfel J and Buettner R: Elevated expression of LSD1
(Lysine-specific demethylase 1) during tumour progression from
pre-invasive to invasive ductal carcinoma of the breast. BMC Clin
Pathol. 12:132012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hayami S, Kelly JD, Cho HS, Yoshimatsu M,
Unoki M, Tsunoda T, Field HI, Neal DE, Yamaue H, Ponder BA, et al:
Overexpression of LSD1 contributes to human carcinogenesis through
chromatin regulation in various cancers. Int J Cancer. 128:574–586.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Guo WH, Yuan LH, Xiao ZH, Liu D and Zhang
JX: Overexpression of SUMO-1 in hepatocellular carcinoma: A latent
target for diagnosis and therapy of hepatoma. J Cancer Res Clin
Oncol. 137:533–541. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shao G, Wang J, Li Y, Liu X, Xie X, Wan X,
Yan M, Jin J, Lin Q, Zhu H, et al: Lysine-specific demethylase 1
mediates epidermal growth factor signaling to promote cell
migration in ovarian cancer cells. Sci Rep. 5:153442015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li Y, Wan X, Wei Y, Liu X, Lai W, Zhang L,
Jin J, Wu C, Shao Q, Shao G, et al: LSD1-mediated epigenetic
modification contributes to ovarian cancer cell migration and
invasion. Oncol Rep. 35:3586–3592. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Klionsky DJ: Autophagy: From phenomenology
to molecular understanding in less than a decade. Nat Rev Mol Cell
Biol. 8:931–937. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mizushima N: Autophagy in protein and
organelle turnover. Cold Spring Harb Symp Quant Biol. 76:397–402.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Johansen T and Lamark T: Selective
autophagy mediated by autophagic adapter proteins. Autophagy.
7:279–296. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tanida I, Ueno T and Kominami E: LC3
conjugation system in mammalian autophagy. Int J Biochem Cell Biol.
36:2503–2518. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ravikumar B, Vacher C, Berger Z, Davies
JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, et
al: Inhibition of mTOR induces autophagy and reduces toxicity of
polyglutamine expansions in fly and mouse models of Huntington
disease. Nat Genet. 36:585–595. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pimkina J and Murphy ME: ARF, autophagy
and tumor suppression. Autophagy. 5:397–399. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Janji B, Viry E, Baginska J, Moer KV and
Berchem G: Role of autophagy in cancer and tumor progression.
autophagy - A double-edged sword - Cell Survival or Death? 189–190.
2013.
|
|
19
|
Polager S, Ofir M and Ginsberg D: E2F1
regulates autophagy and the transcription of autophagy genes.
Oncogene. 27:4860–4864. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Settembre C, Di Malta C, Polito VA,
Arencibia Garcia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina
D, Colella P, et al: TFEB links autophagy to lysosomal biogenesis.
Science. 332:1429–1433. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chao A, Lin CY, Chao AN, Tsai CL, Chen MY,
Lee LY, Chang TC, Wang TH, Lai CH and Wang HS: Lysine-specific
demethylase 1 (LSD1) destabilizes p62 and inhibits autophagy in
gynecologic malignancies. Oncotarget. 8:74434–74450. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Byun S, Kim YC, Zhang Y, Kong B, Guo G,
Sadoshima J, Ma J, Kemper B and Kemper JK: A postprandial
FGF19-SHP-LSD1 regulatory axis mediates epigenetic repression of
hepatic autophagy. EMBO J. 36:1755–1769. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shao GB, Wang J, Zhang LP, Wu CY, Jin J,
Sang JR, Lu HY, Gong AH, Du FY and Peng WX: Aging alters histone H3
lysine 4 methylation in mouse germinal vesicle stage oocytes.
Reprod Fertil Dev. 27:419–426. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−∆ ∆ C(T)) Method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ren S, Ouyang DY, Saltis M, Xu LH, Zha QB,
Cai JY and He XH: Anti-proliferative effect of
23,24-dihydrocucurbitacin F on human prostate cancer cells through
induction of actin aggregation and cofilin-actin rod formation.
Cancer Chemother Pharmacol. 70:415–424. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liang XH, Jackson S, Seaman M, Brown K,
Kempkes B, Hibshoosh H and Levine B: Induction of autophagy and
inhibition of tumorigenesis by beclin 1. Nature. 402:672–676. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Schenk T, Chen WC, Göllner S, Howell L,
Jin L, Hebestreit K, Klein HU, Popescu AC, Burnett A, Mills K, et
al: Inhibition of the LSD1 (KDM1A) demethylase reactivates the
all-trans-retinoic acid differentiation pathway in acute myeloid
leukemia. Nat Med. 18:605–611. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kobayashi S, Kishimoto T, Kamata S, Otsuka
M, Miyazaki M and Ishikura H: Rapamycin, a specific inhibitor of
the mammalian target of rapamycin, suppresses lymphangiogenesis and
lymphatic metastasis. Cancer Sci. 98:726–733. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ding J, Zhang ZM, Xia Y, Liao GQ, Pan Y,
Liu S, Zhang Y and Yan ZS: LSD1-mediated epigenetic modification
contributes to proliferation and metastasis of colon cancer. Br J
Cancer. 109:994–1003. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Choi YJ, Park YJ, Park JY, Jeong HO, Kim
DH, Ha YM, Kim JM, Song YM, Heo HS, Yu BP, et al: Inhibitory effect
of mTOR activator MHY1485 on autophagy: Suppression of lysosomal
fusion. PLoS One. 7:e434182012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lv T, Yuan D, Miao X, Lv Y, Zhan P, Shen X
and Song Y: Over-expression of LSD1 promotes proliferation,
migration and invasion in non-small cell lung cancer. PLoS One.
7:e350652012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shen Y, Li DD, Wang LL, Deng R and Zhu XF:
Decreased expression of autophagy-related proteins in malignant
epithelial ovarian cancer. Autophagy. 4:1067–1068. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bartholomeusz C, Rosen D, Wei C, Kazansky
A, Yamasaki F, Takahashi T, Itamochi H, Kondo S, Liu J and Ueno NT:
PEA-15 induces autophagy in human ovarian cancer cells and is
associated with prolonged overall survival. Cancer Res.
68:9302–9310. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Byun S, Kim YC, Zhang Y, Kong B, Guo G,
Sadoshima J, Ma J, Kemper B and Kemper JK: A postprandial FGF19 -
SHP - LSD1 regulatory axis mediates epigenetic repression of
hepatic autophagy. EMBO J. 36:1755–1769. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yamamoto A, Tagawa Y, Yoshimori T,
Moriyama Y, Masaki R and Tashiro Y: Bafilomycin A1 prevents
maturation of autophagic vacuoles by inhibiting fusion between
autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E
cells. Cell Struct Funct. 23:33–42. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mizushima N and Yoshimori T: How to
interpret LC3 immunoblotting. Autophagy. 3:542–545. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Füllgrabe J, Klionsky DJ and Joseph B: The
return of the nucleus: Transcriptional and epigenetic control of
autophagy. Nat Rev Mol Cell Biol. 15:65–74. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wolfson RL, Chantranupong L, Saxton RA,
Shen K, Scaria SM, Cantor JR and Sabatini DM: Sestrin2 is a leucine
sensor for the mTORC1 pathway. Science. 351:43–48. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ambrosio S, Saccà CD, Amente S, Paladino
S, Lania L and Majello B: Lysine-specific demethylase LSD1
regulates autophagy in neuroblastoma through SESN2-dependent
pathway. Oncogene. 36:6701–6711. 2017. View Article : Google Scholar : PubMed/NCBI
|