Introduction
Breast cancer is the most prevalent cancer in women
worldwide and accounts for approximately 25% of all cancer cases
among women (1,2). In spite of the significant increase in
the survival rates of breast cancer patients during the last
decades, the leading cause of cancer-related mortality among women
remains breast cancer (3). However,
the mechanisms of progression and metastasis of breast cancer are
still poorly understood. Hence, further mechanistic explorations
are crucial to the discovery of new targeted drug treatments for
breast cancer.
The FGF family contains 23 recognized members acting
on 5 FGF receptors that are composed of an intracellular domain, a
transmembrane region and an extracellular portion (4). Members of the FGF family regulate
numerous cellular and physiology processes including cell
differentiation, growth, tissue repair, angiogenesis,
morphogenesis, inflammation, tumor growth and the development of
the embryo and the skeleton (5–8). The
FGF family increases the motility, proliferation, invasiveness, and
migration of many different cells (9–11). The
important role of FGF18 in limb development and skeletal growth,
probably through the modulation of osteoclasts, chondrocytes and
osteoblasts, has been investigated (12). Furthermore, the expression of FGF18
in colon and ovarian tumors was upregulated, and tumor progression
as well as poor overall survival in patients were highly related to
the increased expression of FGF18 mRNA and protein (13–15).
The MAPK (mitogen-activated protein kinase)
signaling pathway is a highly conserved intracellular pathway that
has vital effects in the transmission of signals to the nucleus,
where it transcriptionally mediates genes that participate in
various cellular processes. The MAPK pathway is also correlated
with pancreatic, colon, brain and breast cancer development. It is
commonly abnormally activated, increasing proliferation, migration
and invasion characteristics through a downstream pathway including
extracellular signal-regulated kinase (ERK), PI3K/AKT, p38 and JNK
pathways during neoplastic transformation (16,17).
The ERK signaling pathway, one of the downstream pathways of MAPK,
is involved in cell proliferation, differentiation, and migration
(18). FGFs stimulate specific
signaling pathways after activating FGFRs, such as ERK, AKT,
protein kinase C and phospholipase Cγ. As one of the MYC family of
transcription factors, the c-Myc protein has a fundamental effect
on cellular transformation, apoptosis and cell cycle progression
(19,20).
Epithelial-to-mesenchymal transition (EMT) is a
process in which epithelial cells are converted to migratory and
invasive cells. EMT activation is closely associated with cancer
cell motility and invasiveness (21). A group of EMT-inducing transcription
factors (EMT-TFs) is functionally activated during the EMT cellular
program. Mesenchymal markers, such as N-cadherin, vimentin and
Snail 1, activate EMT cellular programs in epithelial cells.
N-Cadherin is commonly expressed in mesenchymal cells, however high
expression of N-cadherin is associated with increasing motility and
invasion in some cancer cells. N-cadherin is not required for EMT
development, however it promotes EMT by inducing cell migration
abilities (22,23). Vimentin mediates cell migration
ability in numerous cell types. In fibroblasts, vimentin filaments
bind the nucleus to the plasma membrane and play a vital role in
the formation of vimentin-associated matrix adhesions that are
dynamically turned over in migrating cells (24). Snail 1, one of the transcription
suppressors, has been found to participate in regulating the early
development of EMT, and the expression of Snail 1 is highly linked
to metastasis in primary human breast tumors (25,26).
In the present study, we investigated the role of FGF18 in the
growth and metastasis of breast cancer, as well as its possible
mechanisms, and discovered a potential targeted therapeutic agent
for breast cancer.
Materials and methods
Cell culture and treatment
The human breast cancer cell line (MDA-MB-231) was
provided from the American Type Culture Collection (ATCC; Manassas,
VA, USA). Cells were maintained in Dulbecco's modified Eagle's
medium (DMEM; Gibco; Thermo Fisher Scientific, Inc., Grand Island,
NY, USA), supplemented with 10% heat-inactivated fetal bovine serum
(FBS; Gibco) and penicillin-streptomycin solution and incubated at
37°C with 5% CO2.
siRNA transfection
Lipofectamine™ 3000 (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) was used to perform cell transfection, following
the manufacturer's instructions. For the FGF18 functional analysis,
the MDA-MB-231 cells were transfected with negative control FGF18
(FGF18-NC), and FGF18 siRNA, all obtained from Shanghai GenePharma
Co., Ltd. (Shanghai, China). The sequences of the reagent were as
follows: NC, 5′-UUCUCCGAACGUGUCACGUTT-3′; siRNA1,
5′-GCAUUGCCUGUGUUUACATT-3′; siRNA2, 5′-GCAAGGAGUGUGUGUUCAUTT-3′;
and siRNA3, 5′-GCAAGGAGACGGAAUUCUATT-3′. The transfection complexes
of siRNA and Lipofectamine™ 3000 were added into the
cells in serum-free medium for 48 h. RT-qPCR and western blot
analysis were used to screen whether siRNA had been successfully
transfected.
Lentivirus packaging and stable cell
lines
FGF18 overexpression lentivirus (FGF O), FGF18
knockdown (FGF18 KO) lentivirus and control check vectors (FGF18
CK) were designed and packaged by GenePharma. MDA-MB-231 cells were
transfected with lentivirus/medium at a ratio of 1:50. Stable cell
lines were selected by puromycin (Sigma-Aldrich, Merck KGaA,
Darmstadt, Germany) at the concentration of 5 µg/ml for a two-week
period. The lentivirus vectors had enhanced green fluorescent
protein (eGFP) which could provide the means to screen the
efficiency of transfection using a microscope.
CCK-8 proliferation assay
The proliferation of breast cancer cells was
examined by CCK-8 assay (Dojindo Laboratories, Japan). MDA-MB-231
cells were stimulated with 0, 10, 20 and 50 ng/ml FGF18
(MultiSciences Biotech Co., Ltd., Hangzhou, China). ERK inhibitor
(10 µmol/ml, FR180204; Selleck Chemicals, Shanghai, China) was
added to 96-well cell culture plates. Cells (2×103) were
seeded into each well with 100 µl culture media. The same treatment
was used for MDA-MB-231/FGF18-NC, MDA-MB-231/siFGF18 cell lines.
Following 24, 48, 72, 96 and 120 h, 100 µl fresh medium was
replaced with 10% CCK-8 in each well. Subsequently, the cells were
incubated for an additional 2 h at 37°C. The absorbance was
determined at 450 nm wavelength with a microplate reader (Tecan
Austria GmbH, Grödig, Austria). All tests were performed in
triplicate.
Colony formation assay
For the cells to form colonies, a total of 700
MDA-MB-231 cells with 0, 10, 20 and 50 ng/ml FGF18 or 10 µmol/ml
ERK inhibitor were added to 6-well plates and maintained in medium
with 10% FBS. The medium was replaced every 4 days. After 2 weeks,
the colonies were fixed with methanol and stained with 0.1% crystal
violet for 20 min. The visible colonies were manually counted. Each
experiment was repeated at least three times independently.
Wound healing scratch assay
MDA-MB-231 cells with 0, 10, 20 or 50 ng/ml FGF18
were seeded in a 6-well cluster plate (2×106 cells/well)
with 2 ml of complete DMEM. The same treatment was used for
MDA-MB-231/FGF18-NC, and MDA-MB-231/siFGF18 cell lines. A clean and
uniform scratch was made in the single layer of cells with a
sterile pipette at 24 h. The assay was performed three times.
Images were captured from each well every 24 h to determine the
width of the wound.
Invasion and migration assays
Cell migration and invasion assays were performed
using a Tanswell chamber (8 mm, 24-well format; Corning;
Sigma-Aldrich) that was coated with or without 150 µl of ice-cold
diluted Matrigel (BD Biosciences, Franklin Lakes, NJ, USA) in DMEM
basal medium and incubated overnight at 37°C. MDA-MB-231 cells with
0, 10, 20 or 50 ng/ml FGF18 at a density of 2×104
(migration)/5×104 (invasion) were seeded on the upper
chamber in serum-free medium. The lower chamber was filled with 600
µl of 10% FBS-supplemented medium; 20 h later, the reduced cells
were removed by a cotton swab in the top chamber, and then were
fixed with methanol. Crystal violet (1%) was used to stain cells
outside the inserts for 20 min. The same treatment was used for
MDA-MB-231/FGF18-NC and MDA-MB-231/siFGF18 cell lines. A light
microscope (Olympus Corp., Tokyo, Japan) was used to count the
number of invading cells on the membrane and the results were
calculated as the means ± SD. The assay was repeated three
times.
Flow cytometry analysis
Cells were collected after stimulating with 0, 10,
20 or 50 ng/ml FGF18 for two days and resuspended in cold PBS, and
then stained with a cell cycle staining kit (MultiSciences Biotech
Co., Ltd.) according to the manufacturer's protocol. The same
treatment was used for the MDA-MB231/FGF18-NC and MDA-MB231/siFGF18
cell lines. Data were analyzed using flow cytometry (BD
Biosciences).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted with TRIzol reagent (Takara
Bio, Inc., Otsu, Japan) and cDNA was synthesized using PrimeScript
RT reagent (Takara Bio) following the manufacturer's protocol. The
PCR program used for amplification was as follows: i) 94°C for 30
sec; ii) 94°C for 30 sec; iii) 55°C for 30 sec; iv) 72°C for 1 min
and; v) 72°C for 10 min. Steps ii) through iv) were repeated 35
times for β-actin and other genes. The following PCR primers were
used: FGF18 forward sequence, 5′-GGACATGTGCAGGCTGGGCTA-3′ and
reverse, 5′-GTAGAATTCCGTCTCCTTGCCCTT-3′. All PCR reactions were
performed using the fluorescent SYBR Green I methodology.
Quantitative RT-PCR (qRT-PCR) was performed on StepOnePlus
Real-Time PCR system (Applied Biosystems; Thermo Fisher Scientific,
Inc.) with FastStart Universal SYBR-Green Master (Roche
Diagnostics, Basel, Switzerland) following the manufacturer's
protocol. The relative quantification was calculated by the
2−ΔΔCt method.
Western blot analysis
All of the proteins were extracted using Total
Protein Extraction kits (KeyGen Biotech, Nanjing, China) following
the manufacturer's protocol. SDS-PAGE (5X; Beyotime Institute of
Biotechnology, Shanghai, China) was used to form a complex with the
extracted protein for storage in −20°C. Equal amounts of protein
were separated by 12% SDS-PAGE and transferred onto a
nitrocellulose membrane (EMD Millipore Corporation, Billerica, MA,
USA). The membranes were first stained to confirm the uniform
transfer of all samples and then incubated in the blocking solution
(5 ml of skimmed milk powder dissolving in 45 ml of TBST) for 2 h
at room temperature. Membranes were then incubated overnight at 4°C
with diluted (1:1,000) primary antibodies against FGF18 (cat. no.
ab169615; Abcam, Cambridge, MA, USA) ERK (cat. no. 4695; Cell
Signaling Technology, Inc., Danvers, MA, USA), p-ERK (cat. no.
4370), c-Myc (cat. no. 13987), N-cadherin (cat. no. 14215),
vimentin (cat. no. 5741), Snail 1 (cat. no. 3879) and GAPDH (cat.
no. 51332; all form Cell Signaling Technology, Inc., Danvers, MA,
USA), followed by incubation with horseradish peroxidase conjugated
anti-rabbit IgG (1:1,000; cat. no. sc-2004; Santa Cruz
Biotechnology, CA, USA) and anti-mouse IgG secondary antibodies
(1:1,000; cat. no. sc-2005; Santa Cruz Biotechnology, CA, USA) for
2 h. Following washing with TBST, the immune-reactive proteins were
detected with a Bio-Rad Western Blotting Detection system (Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Tumor xenograft mouse model
The animal experiments were approved by the Animal
Management Rule of the Chinese Ministry of Health and were in
accordance with the approved guidelines and the experimental
protocols of Nanjing Medical University. All twenty BALB/c nude
female mice used in this experiment were purchased from The Model
Animal Research Center of Nanjing University (Nanjing, China). We
divided the mice into four groups: i) FGF18 control check (FGF18
CK); ii) FGF18 overexpression (FGF18 O); iii) FGF18 knockdown
(FGF18 KO); and iv) ERK inhibitor group. Subsequently, each
5-week-old mouse was subcutaneously injected with MDA-MB-231 cells
(2×106) suspended in 200 µl of PBS. The ERK inhibitor
group was injected with FR180204 at the dose of 20 mg/kg/day,
administered intraperitoneally. The tumor was visible at 2 weeks
after injection. Tumor sizes and body weight were monitored every 3
days. After 29 days, the nude mice were sacrificed by cervical
dislocation and the tumor volume was calculated following the
formula: Ixb2x0.5 (I and b are the largest perpendicular
lengths of the tumor). The longest tumor exhibited by a single
subcutaneous had been indicated, and none of the mice presented
multiple tumors. The tumor tissues were frozen immediately at −80°C
for further study.
Statistical analysis
Each experiment was repeated three times and data
are presented as the mean ± standard error. Data were analyzed
using SPSS 10.0 software (SPSS, Inc., Chicago, IL, USA). One-way
ANOVA was used to determine the difference among at least three
groups, while a t-test was used to analyze differences between two
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
FGF18 improves the proliferation of
MDA-MB-231 cells
To explore the effects of FGF18 on MDA-MB-231 cells,
we used a cell proliferation assay with various FGF18
concentrations and exposure times. Fig.
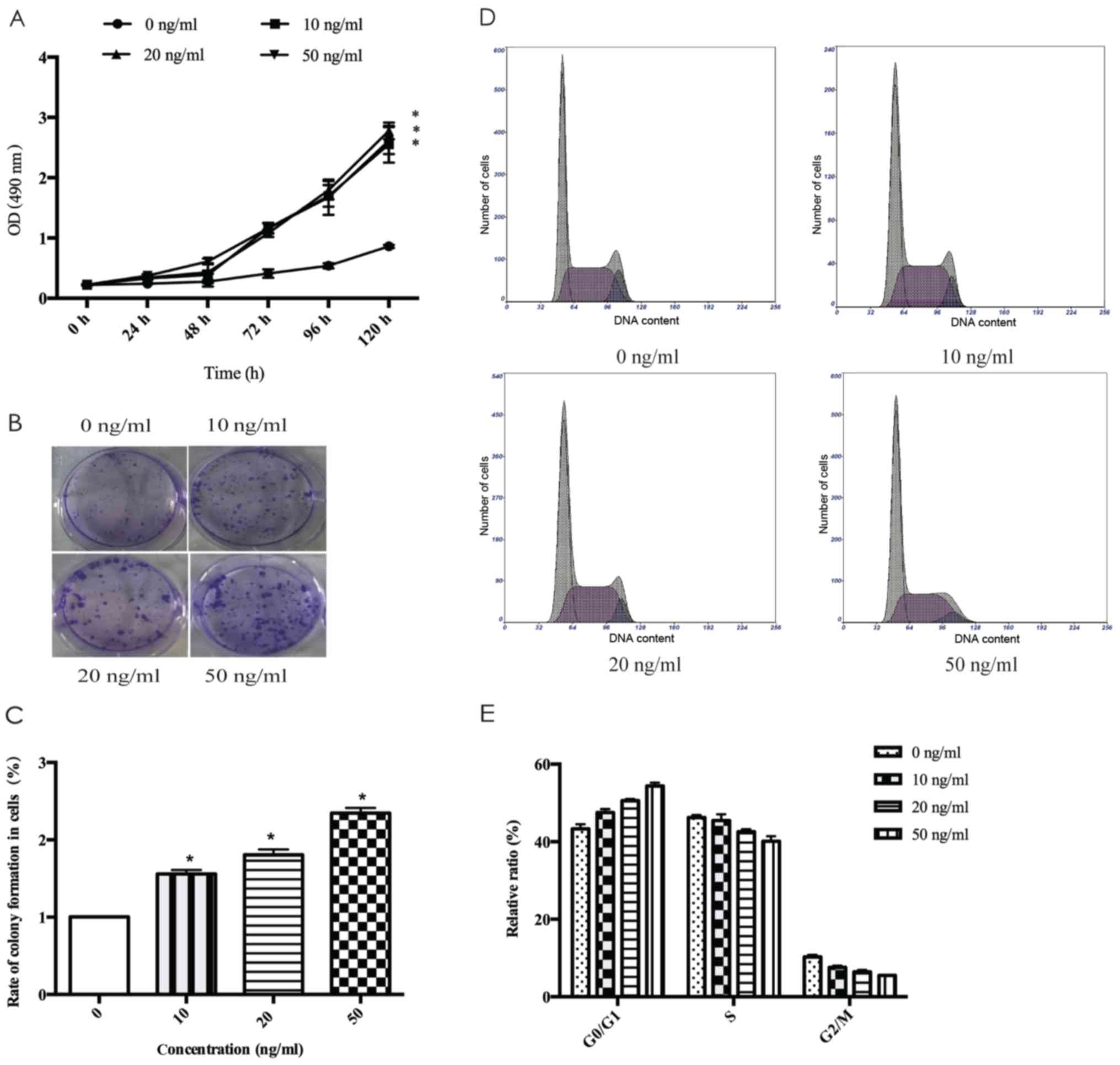
1A displays that MDA-MB-231 cell proliferation was stimulated
by FGF18. However, the proliferation did not differ with different
concentrations of FGF18. Colony formation assays (Fig. 1B and C) revealed the proliferative
promotion ability of FGF18, and the number of colonies formed in
MDA-MB-231 cells with the number of colonies being markedly higher
with 50 ng/ml FGF18 than with lower doses. These results revealed
that FGF18 has an important effect in increasing the proliferation
of breast cancer cells.
FGF18 regulates the cell cycle
progression of MDA-MB-231 cells
To further investigate how FGF18 stimulated
MDA-MB-231 cell proliferation, the role of FGF18 on cell cycle
kinetics was analyzed to explore how FGF18 mediated the cell cycle.
We found that the percentage of G0/G1 phase
increased and the percentage of S phase decreased from FGF18
stimulation relative to the untreated MDA-MB231 cells (Fig. 1D and E). These results suggest that
FGF18 promotes the proliferation of breast cancer cells by
increasing the percentage of cells in G0/G1
phase.
FGF18 mediates the promotion of cancer
cell proliferation via the ERK/c-Myc signaling pathway in MDA-MB231
cells
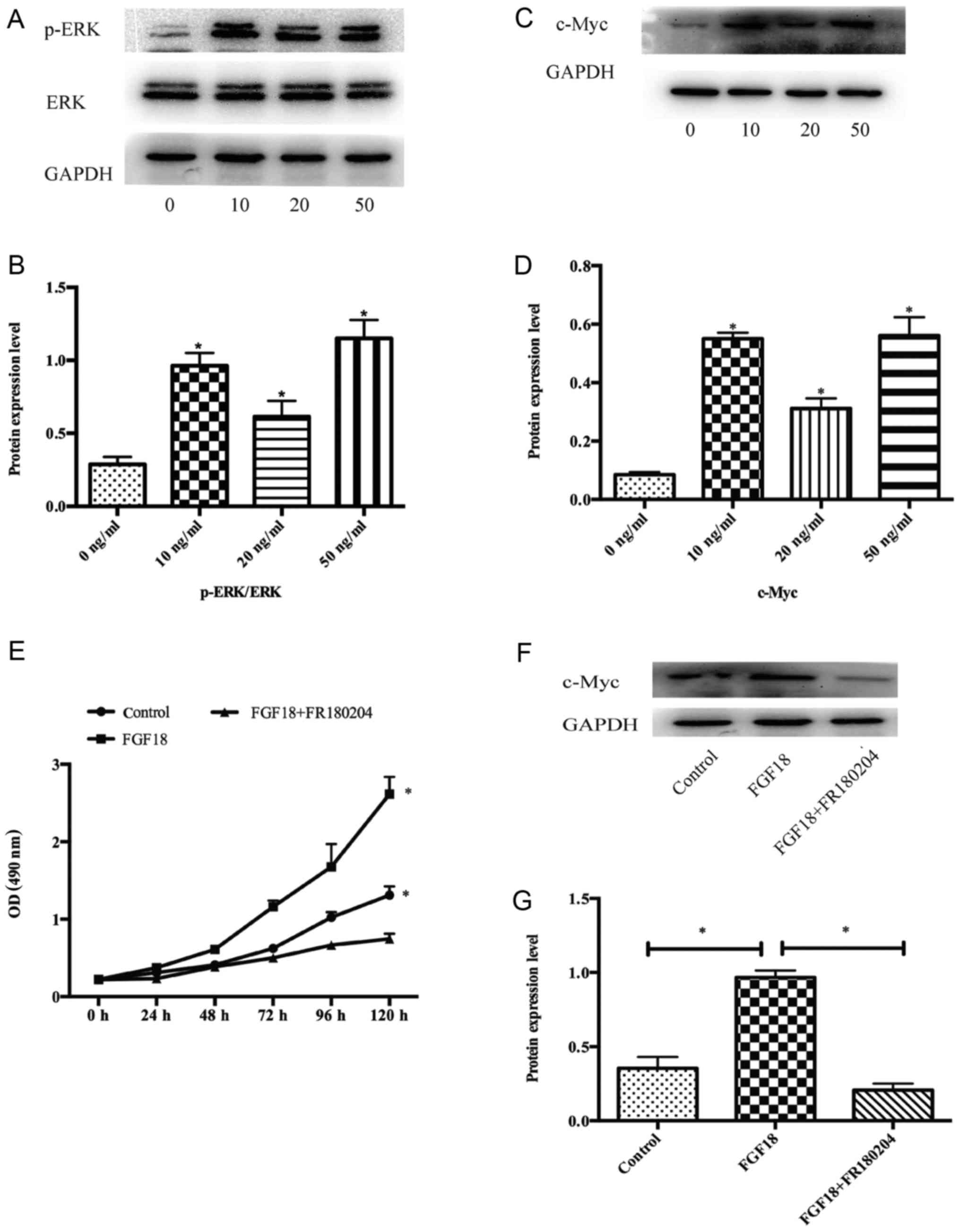
We demonstrated that FGF18 significantly stimulated
the proliferation of MDA-MB231 cells, however the underlying
mechanisms were unclear. Therefore, a western blot analysis was
used to investigate the effects of FGF18 on signal transduction in
the MAPK pathway. We incubated the MDA-MB231 cells with 0, 10, 20
and 50 ng/ml FGF18 for 48 h, and collected the total protein
lysates. These protein lysates were subjected to western blotting
with p-ERK1/2 and ERK1/2 antibodies. The level of p-ERK1/2
increased with FGF18 stimulation (Fig.
2A and B), and the expression of c-Myc rose significantly
relative to the untreated control MDA-MB231 cells (Fig. 2C and D). To further confirm the
effects on the ERK pathway, we used the specific inhibitor FR180204
(ERK inhibitor) and found that at a concentration of 10 µmol/ml,
FR180204 could significantly inhibit the proliferation of MDA-MB231
cells with the stimulation of FGF18 (Fig. 2E). Furthermore, c-Myc protein was
also changed by the inhibitors (Fig.
2F-G). These results indicated that FGF18 promoted the
proliferation of MDA-MB231 cells via modulations of the ERK/c-Myc
signaling pathway.
FGF18 promotes the migration and
invasion of MDA-MB-231 cells and regulates EMT-inducing
transcription factors
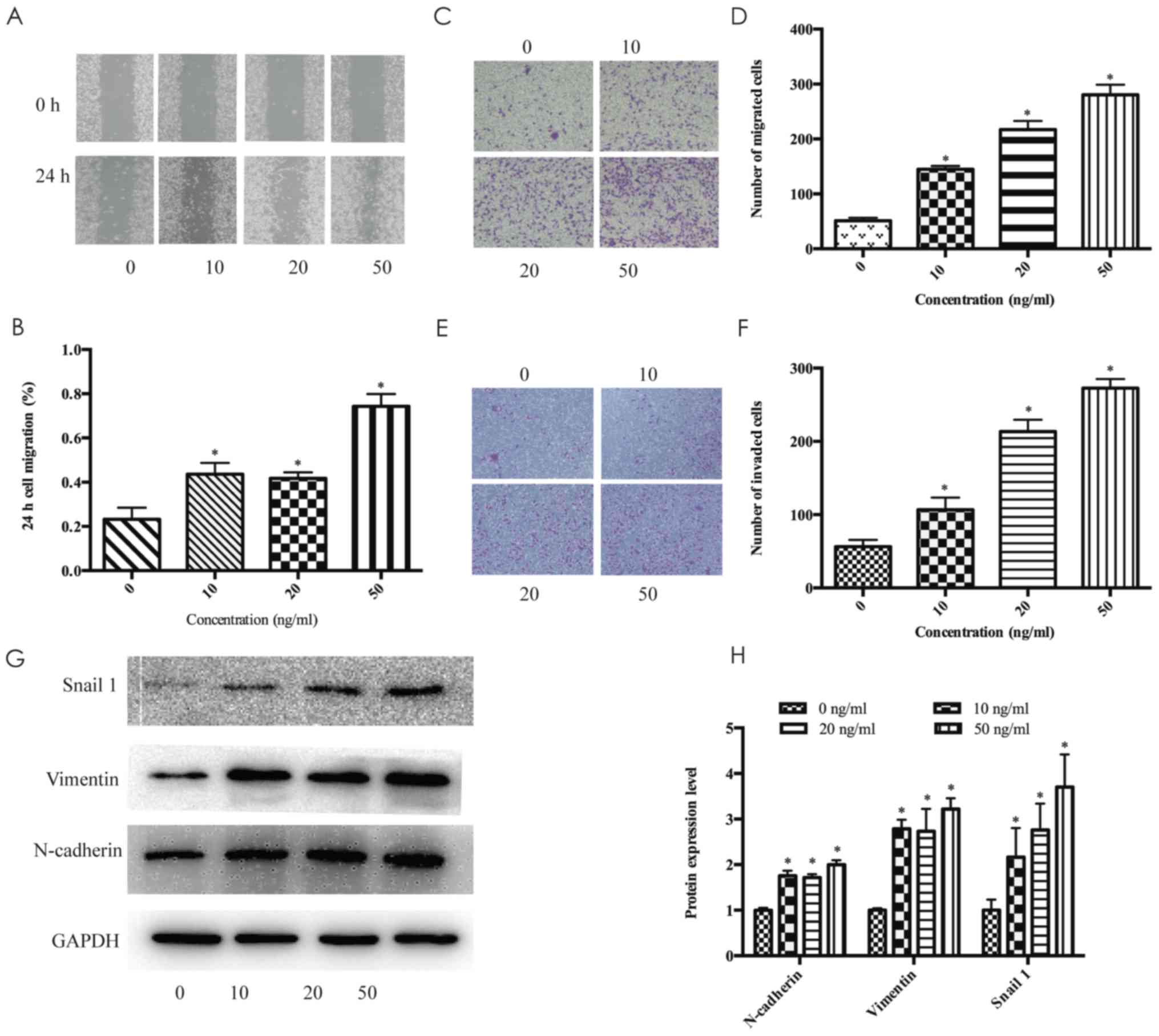
The wound healing scratch assays revealed that after
treatment with 10, 20 and 50 ng/ml FGF18 for 24 h, the migration
distance of MDA-MB-231 cells significantly increased (Fig. 3A and B). Furthermore, Transwell
assays also confirmed our findings on the effect of FGF18 on the
migration enhancement of MDA-MB-231 cells (Fig. 3C and D). High concentration of FGF18
significantly enhanced the invasiveness of the MDA-MB-231 cells
(Fig. 3E and F). Furthermore, the
protein expression levels of N-cadherin, vimentin and Snail 1
increased after stimulation with FGF18 in a dose-dependent manner,
particularly at the concentration of 50 ng/ml (Fig. 3G and H). These results indicated
that FGF18 promoted the invasion and migration of MDA-MB-231 cells
by increasing EMT-inducing transcription factors.
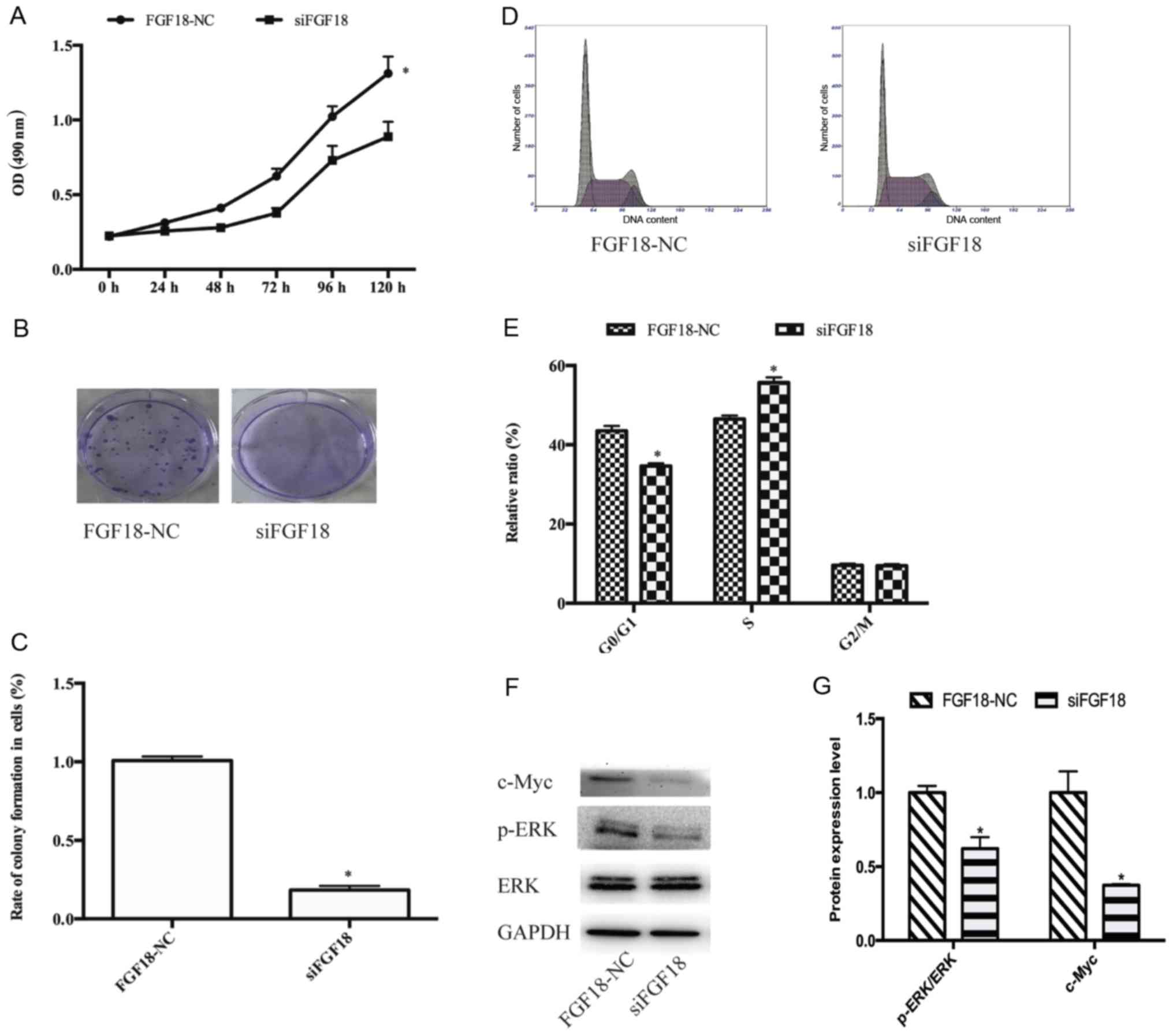
FGF18 siRNA inhibits cell
proliferation, migration and invasion of MDA-MB-231 cells
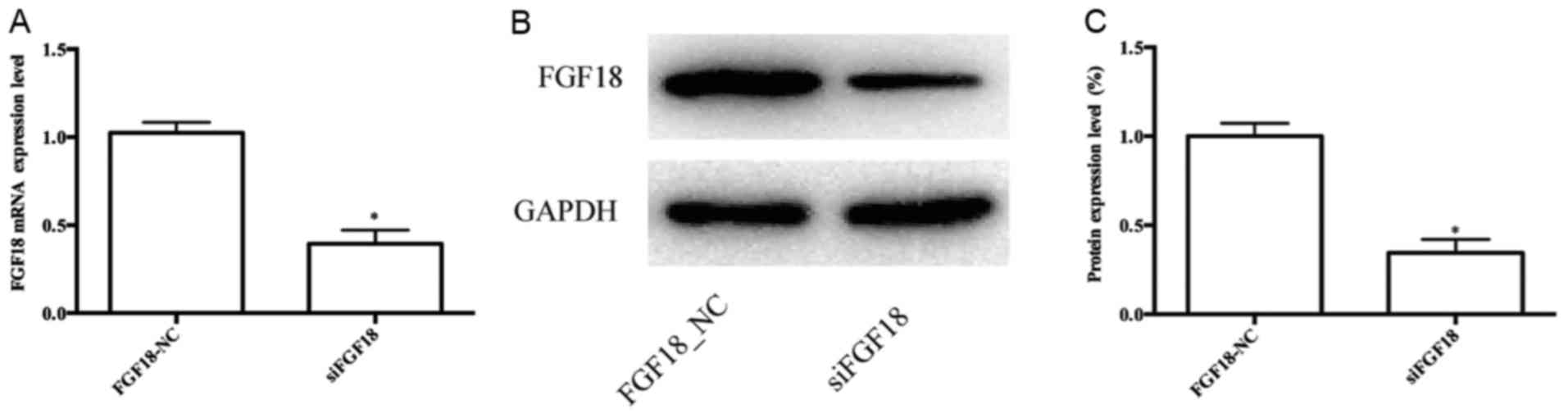
RT-qPCR indicated that siFGF18 significantly reduced
the expression of FGF18 in MDA-MB-231 cells compared with the
control group (Fig. 4A). In
addition, the results of the western blot analyses were consistent
with the mRNA data, demonstrating that the FGF18 was successfully
knocked down using siRNA (Fig. 4B and
C). Following cell transfection, we evaluated the cell
proliferation of MDA-MB-231 cells. The OD values were significantly
reduced in the siFGF18 group in comparison with the control group
(Fig. 5A), and colony formation
assays revealed that the number of colonies of MDA-MB-231 cells
with siFGF18 decreased (Fig. 5B and
C), indicating that siFGF18 inhibited cell proliferation. The
proportion of MDA-MB-231 cells in the G0/G1
phase also decreased (Fig. 5D and
E). The expression of c-Myc protein decreased in the siFGF18
group, with a 58% protein inhibition rate. The level of p-ERK
protein was obviously reduced in the siFGF18 group in comparison to
the control group (Fig. 5F and G).
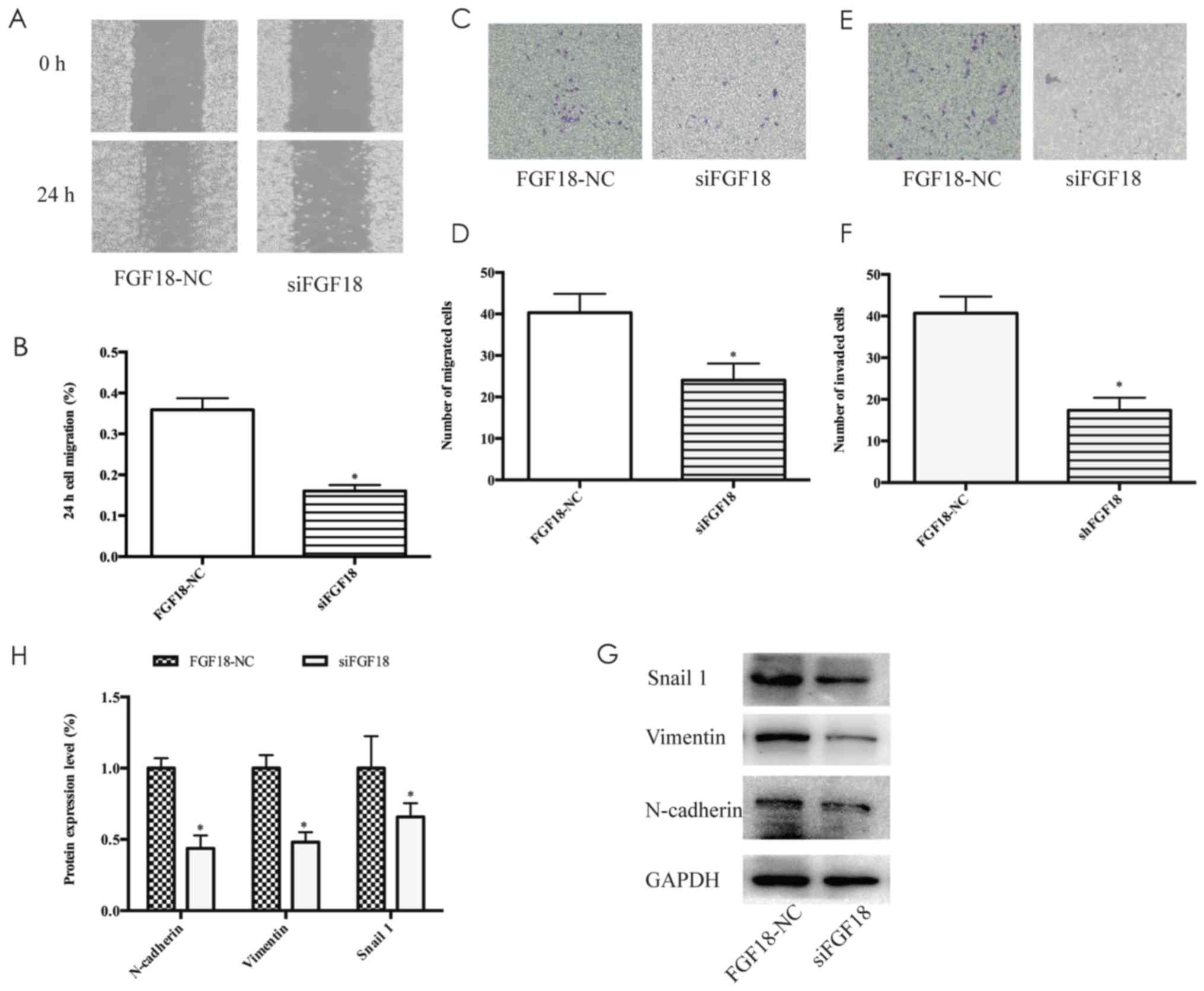
The wound healing and Transwell assays also revealed that the
migration ability in the siFGF18 group decreased in comparison with
the control group (Fig. 6A-D).
Additionally, the invasion ability of shFGF18-transfected
MDA-MB-231 cells was reduced significantly (Fig. 6E and F). The expression of
N-cadherin, vimentin and Snail 1 proteins decreased significantly
following siFGF18, with 55, 46 and 39% protein inhibition rate
respectively (Fig. 6G and H). These
results indicated that FGF18 siRNA suppressed cell proliferation
through the ERK/c-Myc signaling pathway, and reduced the migration
and invasion abilities of MDA-MB-231 cells by mediating
EMT-inducing transcription factors.
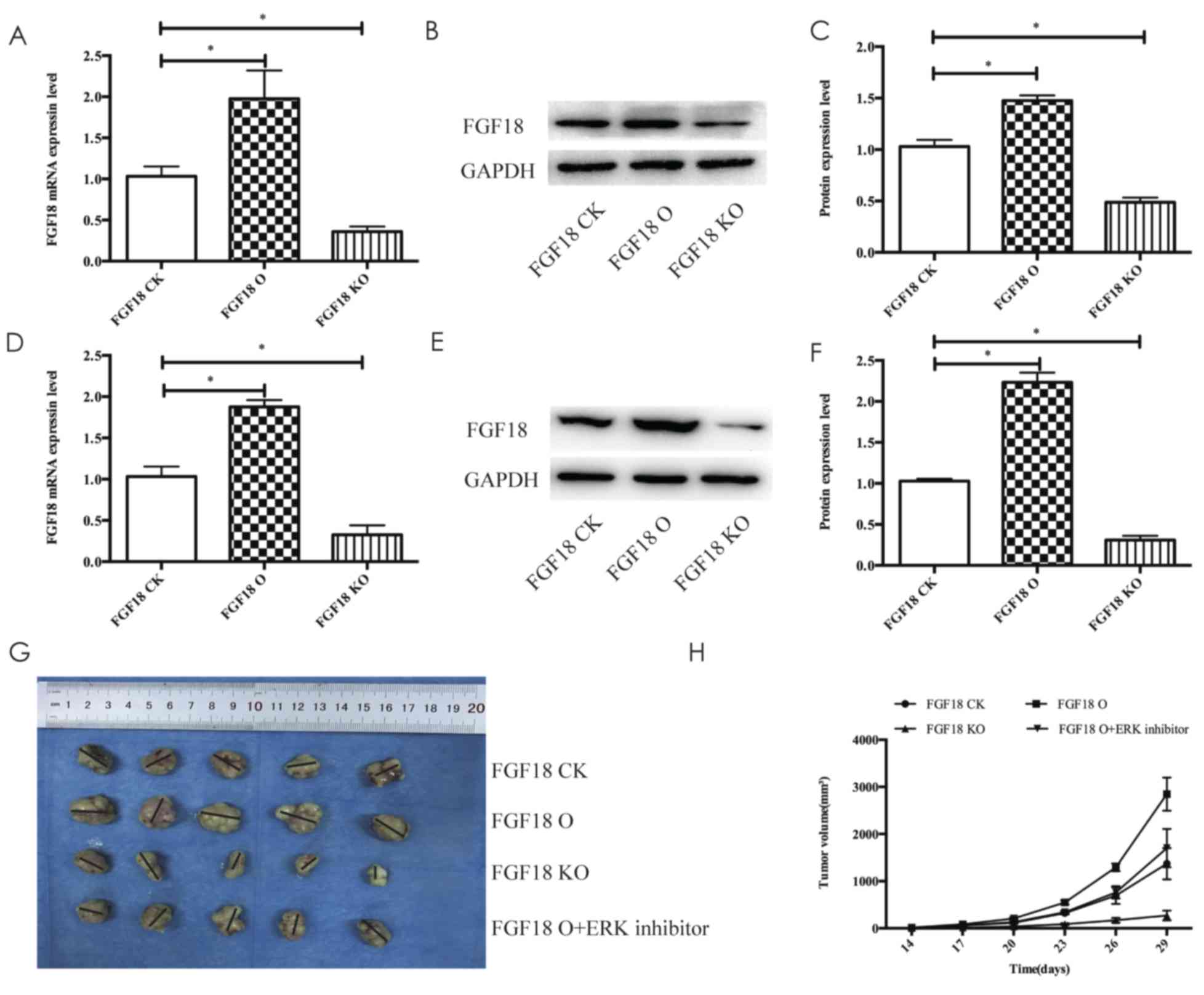
Efficacy of FGF18 xenograft models of
MDA-MB-231 cells
Xenograft models to investigate tumor growth
promotion via FGF18 in vivo, using RT-qPCR revealed that the
expression of FGF18 mRNA in the FGF18 O group transfected with
lentivirus (the overexpression group) increased in comparison with
the FGF18 CK group (control check) and the expression of FGF18 in
the FGF18 KO (knockdown) group decreased compared with the FGF18 CK
group (Fig. 7A). The results of the
expression of FGF18 protein examined by western blot analysis were
consistent with the mRNA data. Furthermore, the expression of FGF18
in tumor tissues was also examined by RT-qPCR and western blot
analysis, and the findings were still consistent with the in
vitro results (Fig. 7D-F). As
above-mentioned, the model of overexpression and knockdown of FGF18
could be successfully built following lentivirus transfection. As
displayed in Fig. 7G and H, the
xenograft tumor sizes of the FGF18 O group were significantly
larger than in the other groups, and the tumor sizes of the FGF18
KO group were markedly smaller than the other groups. In addition,
the FGF18 O group was regarded as the control of ERK inhibitor and
the FGF18 O + ERK inhibitor group had markedly smaller tumor sizes
compared with the FGF18 O group (Fig.
7G and H). These results indicated that FGF18 promoted
MDA-MB-231 cell growth in vivo, and ERK inhibitor could
significantly prevent the growth of MDA-MB-231 cells in response to
the FGF18 stimulation.
Discussion
Research has demonstrated that FGF and FGFR family
have an important relationship with the progression of breast
cancer. FGF1 could have an important role in human breast cancer
growth and patients with high levels of bFGFR may have a more
favorable prognosis (27).
FGF2/FGFR interaction led to complex signal transduction pathways
and to the activation of a ‘proangiogenic phenotype’ in the
endothelium, which regulated the proliferation, migration and
survival of breast cancer cells (28). The growth of lung and colon cancer
cells were induced by the high expression of FGF18 (29,30)
and FGF18 increased ovarian tumor growth and metastasis (14). In the present study we observed that
FGF18 promoted the growth and metastasis of MDA-MB-231 cells. To
the best of our knowledge, this study is the first to report the
effects of FGF18 in breast cancer cells.
MAPKs (JNK1/2, ERK1/2 and p38) participate in the
growth and metastasis of many tumors, such as breast, lung,
ovarian, colorectal and prostate cancer (31–33).
Similarly, the ERK signaling pathway may mediate factors related to
the and poor prognosis and progression in breast cancer (34). The c-Myc proto-oncogene is a
senior administrator of the cell, helping to allocate resources and
direct proliferation, apoptosis, differentiation and growth
(35). A recent study also revealed
that the c-Myc gene was involved in most aspects of the
cellular function, such as the growth, replication, apoptosis,
differentiation and metabolism in breast cancer (36). FR180204 (specific inhibitor for ERK)
was used to identify whether ERK was involved in the progression of
MDA-MB-231 cells. We found that the proliferation in response to
FGF18 was reduced with the inhibition of ERK, and the expression of
the target gene c-Myc decreased. These investigations indicated
that the activation of ERK induced the proliferation of MDA-MB-231
cells by increasing the expression of the target gene c-Myc. In
addition, in vivo, the tumor sizes of mice in the FGF18 O +
ERK inhibition group were similar to the tumor sizes of the
FGF18-NC group. These findings indicated that the ERK/c-Myc
signaling pathway was activated by FGF18 in the progression of
breast cancer. For this reason, we infered that the ERK/c-Myc
signaling pathway may induce proliferative signals in breast cancer
cells.
EMT plays an important role in the acquisition of
migration and invasion capabilities by improving mesenchymal
phenotypes and motility (37).
FGF18 mediates Wnt-dependent stimulation of CD44-positive human
colorectal adenoma cells (30) and
the Wnt signaling pathway is involved in the progression of EMT
(38,39). We observed that FGF18 increased the
expression of EMT-inducing transcription factors N-cadherin,
vimentin and Snail 1, indicating that FGF18 may induce the
progression of EMT in breast cancer cells and then promote the
migration and invasion capabilities of MDA-MB-231 cells. However,
EMT progression can be induced through several other signaling
pathways including TGF-β and Notch (40,41).
The underlying mechanism of EMT-inducing factors mediated by FGF18
has not been investigated. Therefore, further studies exploring the
mechanisms of migration and invasion in MDA-MB-231 cells should be
undertaken.
Furthermore, it was confirmed that the transfection
of siFGF18 could suppress the expression of FGF18 gene and reduce
the effects of growth and metastasis of MDA-MB-231 cells. The
expression of ERK, c-Myc, N-cadherin, vimentin and Snail 1 in human
MDA-MB-231 cells was detected by western blot analysis following
siRNA-FGF18 transfection. These results indicated that the use of
siFGF18 can be a potential treatment for breast cancer.
However, in the preliminary experiment of this
study, we observed that the effect of FGF18 only functioned in the
MDA-MB-231 cells compared with several other cell lines
(SUM1315MO2, SKBR3 and MCF 7). All of these results is not
mentioned in the present study. The ERK signaling pathway may be
involved in these differences. Our future study would be to explore
the underlying molecular mechanisms of the above-mentioned
phenomenon. Using only one cell line was a limitation of the
present study, and a greater number of cell lines would further
support our conclusions.
In conclusion, the present study revealed that
through the ERK/c-Myc signaling pathway and EMT transition, FGF18
had a significant effect on the growth and metastasis of breast
cancer cells, demonstrating that FGF18 provided a potential target
for the effective treatment of breast cancer. Further studies of
breast cancer, exploring the link between FGF18 and the survival,
relapse and metastasis of patients are required.
Acknowledgements
Not applicable.
Funding
The present study was supported in part by a grant
from Talents Planning of Six Summit Fields of Jiangsu Province
(WSW-026), the Maternal and Child Health Research Projects of
Jiangsu Province (F201678) and the Priority Academic Program
Development of Jiangsu Higher Education Institutions (PAPD,
JX10231801).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
ZYY and LQL conceived and designed the study. ZYY
and LQL performed the experiments. ZYY wrote the paper. ZYY and LQL
reviewed and edited the manuscript. All authors read and approved
the manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
All experimental protocols were approved by the
Nanjing Medical University (Shanghai, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
FGF18
|
fibroblast growth factor 18
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
EMT
|
epithelial-to-mesenchymal
transition
|
|
FGF
|
fibroblastic growth factors
|
|
FGFR
|
fibroblastic growth factor
receptor
|
|
MAPK
|
mitogen activated protein kinase
|
|
siRNA
|
short interfering RNA
|
References
|
1
|
Greenlee H, DuPont-Reyes MJ, Balneaves LG,
Carlson LE, Cohen MR, Deng G, Johnson JA, Mumber M, Seely D, Zick
SM, et al: Clinical practice guidelines on the evidence-based use
of integrative therapies during and after breast cancer treatment.
CA Cancer J Clin. 67:194–232. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Polychemotherapy for early breast cancer:
An overview of the randomised trials. Early Breast Cancer
Trialists' Collaborative Group. Lancet. 352:930–942. 1998.
|
|
4
|
Antoine M, Wirz W, Tag CG, Mavituna M,
Emans N, Korff T, Stoldt V, Gressner AM and Kiefer P: Expression
pattern of fibroblast growth factors (FGFs), their receptors and
antagonists in primary endothelial cells and vascular smooth muscle
cells. Growth Factors. 23:87–95. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Diez Del Corral R and Morales AV: The
Multiple roles of FGF signaling in the developing spinal cord.
Front Cell Dev Biol. 5:582017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Majidinia M, Sadeghpour A and Yousefi B:
The roles of signaling pathways in bone repair and regeneration. J
Cell Physiol. 233:2937–2948. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Presta M, Chiodelli P, Giacomini A,
Rusnati M and Ronca R: Fibroblast growth factors (FGFs) in cancer:
FGF traps as a new therapeutic approach. Pharmacol Ther.
179:171–187. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Itoh N and Ornitz DM: Evolution of the Fgf
and Fgfr gene families. Trends Genet. 20:563–569. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Elo T, Lindfors PH, Lan Q, Voutilainen M,
Trela E, Ohlsson C, Huh SH, Ornitz DM, Poutanen M, Howard BA, et
al: Ectodysplasin target gene Fgf20 regulates mammary bud growth
and ductal invasion and branching during puberty. Sci Rep.
7:50492017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Feng S, Wang J, Zhang Y, Creighton CJ and
Ittmann M: FGF23 promotes prostate cancer progression. Oncotarget.
6:17291–17301. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu XL, Gu MM, Huang L, Liu XS, Zhang HX,
Ding XY, Xu JQ, Cui B, Wang L, Lu SY, et al: Multiple synostoses
syndrome is due to a missense mutation in exon 2 of FGF9
gene. Am J Hum Genet. 85:53–63. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ohbayashi N, Shibayama M, Kurotaki Y,
Imanishi M, Fujimori T, Itoh N and Takada S: FGF18 is required for
normal cell proliferation and differentiation during osteogenesis
and chondrogenesis. Genes Dev. 16:870–879. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sonvilla G, Allerstorfer S, Stättner S,
Karner J, Klimpfinger M, Fischer H, Grasl-Kraupp B, Holzmann K,
Berger W, Wrba F, et al: FGF18 in colorectal tumour cells:
Autocrine and paracrine effects. Carcinogenesis. 29:15–24. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wei W, Mok SC, Oliva E, Kim SH, Mohapatra
G and Birrer MJ: FGF18 as a prognostic and therapeutic biomarker in
ovarian cancer. J Clin Invest. 123:4435–4448. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shimokawa T, Furukawa Y, Sakai M, Li M,
Miwa N, Lin YM and Nakamura Y: Involvement of the FGF18 gene
in colorectal carcinogenesis, as a novel downstream target of the
beta-catenin/T-cell factor complex. Cancer Res. 63:6116–6120.
2003.PubMed/NCBI
|
|
16
|
MacCorkle RA and Tan TH: Mitogen-activated
protein kinases in cell-cycle control. Cell Biochem Biophys.
43:451–461. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Martin GS: Cell signaling and cancer.
Cancer Cell. 4:167–174. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kohno M and Pouyssegur J: Targeting the
ERK signaling pathway in cancer therapy. Ann Med. 38:200–211. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Baudino TA, McKay C, Pendeville-Samain H,
Nilsson JA, Maclean KH, White EL, Davis AC, Ihle JN and Cleveland
JL: c-Myc is essential for vasculogenesis and angiogenesis during
development and tumor progression. Genes Dev. 16:2530–2543. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liao DJ and Dickson RB: c-Myc in breast
cancer. Endocr Relat Cancer. 7:143–164. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Neelakantan D, Zhou H, Oliphant MU, Zhang
X, Simon LM, Henke DM, Shaw CA, Wu MF, Hilsenbeck SG, White LD, et
al: EMT cells increase breast cancer metastasis via paracrine GLI
activation in neighbouring tumour cells. Nat Commun. 8:157732017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hazan RB, Phillips GR, Qiao RF, Norton L
and Aaronson SA: Exogenous expression of N-cadherin in breast
cancer cells induces cell migration, invasion, and metastasis. J
Cell Biol. 148:779–790. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Maeda M, Johnson KR and Wheelock MJ:
Cadherin switching: Essential for behavioral but not morphological
changes during an epithelium-to-mesenchyme transition. J Cell Sci.
118:873–887. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vuoriluoto K, Haugen H, Kiviluoto S,
Mpindi JP, Nevo J, Gjerdrum C, Tiron C, Lorens JB and Ivaska J:
Vimentin regulates EMT induction by Slug and oncogenic H-Ras and
migration by governing Axl expression in breast cancer. Oncogene.
30:1436–1448. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Canesin G, Cuevas EP, Santos V,
López-Menéndez C, Moreno-Bueno G, Huang Y, Csiszar K, Portillo F,
Peinado H, Lyden D and Cano A: Lysyl oxidase-like 2 (LOXL2) and E47
EMT factor: Novel partners in E-cadherin repression and early
metastasis colonization. Oncogene. 34:951–964. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Neal CL, Henderson V, Smith BN, McKeithen
D, Graham T, Vo BT and Odero-Marah VA: Snail transcription factor
negatively regulates maspin tumor suppressor in human prostate
cancer cells. BMC Cancer. 12:3362012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Blanckaert VD, Hebbar M, Louchez MM,
Vilain MO, Schelling ME and Peyrat JP: Basic fibroblast growth
factor receptors and their prognostic value in human breast cancer.
Clin Cancer Res. 4:2939–2947. 1998.PubMed/NCBI
|
|
28
|
Cai Y, Zhang J, Lao X, Jiang H, Yu Y, Deng
Y, Zhong J, Liang Y, Xiong L and Deng N: Construction of a
disulfide-stabilized diabody against fibroblast growth factor-2 and
the inhibition activity in targeting breast cancer. Cancer Sci.
107:1141–1150. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen T, Gong W, Tian H, Wang H, Chu S, Ma
J, Yang H, Cheng J, Liu M, Li X and Jiang C: Fibroblast growth
factor 18 promotes proliferation and migration of H460 cells via
the ERK and p38 signaling pathways. Oncol Rep. 37:1235–1242. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Koneczny I, Schulenburg A, Hudec X,
Knöfler M, Holzmann K, Piazza G, Reynolds R, Valent P and Marian B:
Autocrine fibroblast growth factor 18 signaling mediates
Wnt-dependent stimulation of CD44-positive human colorectal adenoma
cells. Mol Carcinog. 54:789–799. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang XL, Liu KY, Lin FJ, Shi HM and Ou ZL:
CCL28 promotes breast cancer growth and metastasis through
MAPK-mediated cellular anti-apoptosis and pro-metastasis. Oncol
Rep. 38:1393–1401. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hrustanovic G, Olivas V, Pazarentzos E,
Tulpule A, Asthana S, Blakely CM, Okimoto RA, Lin L, Neel DS,
Sabnis A, et al: RAS-MAPK dependence underlies a rational
polytherapy strategy in EML4-ALK-positive lung cancer. Nat Med.
21:1038–1047. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Urosevic J, Garcia-Albéniz X, Planet E,
Real S, Céspedes MV, Guiu M, Fernandez E, Bellmunt A, Gawrzak S,
Pavlovic M, et al: Colon cancer cells colonize the lung from
established liver metastases through p38 MAPK signalling and PTHLH.
Nat Cell Biol. 16:685–694. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Whyte J, Bergin O, Bianchi A, McNally S
and Martin F: Key signalling nodes in mammary gland development and
cancer. Mitogen-activated protein kinase signalling in experimental
models of breast cancer progression and in mammary gland
development. Breast Cancer Res. 11:2092009.
|
|
35
|
Wilde BR and Ayer DE: Interactions between
Myc and MondoA transcription factors in metabolism and
tumourigenesis. Br J Cancer. 113:1529–1533. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fallah Y, Brundage J, Allegakoen P and
Shajahan-Haq AN: MYC-driven pathways in breast cancer subtypes.
Biomolecules. 7:E532017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nieto MA, Huang RY, Jackson RA and Thiery
JP: EMT: 2016. Cell. 166:21–45. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nelson WJ and Nusse R: Convergence of Wnt,
beta-catenin, and cadherin pathways. Science. 303:1483–1487. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang JJ, Li ZF, Li XJ, Han Z, Zhang L and
Liu ZJ: Effects of microRNA-136 on melanoma cell proliferation,
apoptosis and epithelial mesenchymal transition by targeting PMEL
through the Wnt signaling pathway. Biosci Rep. 37:BSR20170743.
2017. View Article : Google Scholar
|
|
40
|
Derynck R, Muthusamy BP and Saeteurn KY:
Signaling pathway cooperation in TGF-β-induced
epithelial-mesenchymal transition. Curr Opin Cell Biol. 31:56–66.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Huber MA, Kraut N and Beug H: Molecular
requirements for epithelial-mesenchymal transition during tumor
progression. Curr Opin Cell Biol. 17:548–558. 2005. View Article : Google Scholar : PubMed/NCBI
|