Introduction
The use of nanoparticle (NP) technology in cancer
chemotherapy results in a superior performance. Advantageous
properties include enhanced solubility of hydrophobic drugs, the
possibility of surface functionalization for targeted delivery, and
controlled and sustained drug release, which allows a reduction in
drug dosage and thus fewer drug side effects (1,2). In
addition, it has been confirmed that NP therapy could take
advantage of the enhanced permeability and retention (EPR) effect
in solid tumors. The EPR effect results from an abnormal lymphatic
drainage system in tumors, which increases the accumulation of
nanocarrier-delivered drugs, as the tumor fails to remove NPs from
the interstitial space (3).
Liposomes and polymeric NPs are the two most
prevalently advocated nanocarriers for anticancer drug delivery
(4,5). Liposomes show superior
bio-compatibility, enhanced loading efficiency and a favorable
pharmacokinetic profile, including prolonged circulation time. Easy
surface functionalization for targeted delivery is an additional
benefit. Therefore, liposomes represent an increasingly applied
drug delivery vehicle (6). Although
liposomes have the aforementioned advantages, burst release of
drugs, instability and lack of structural integrity represent
limitations of the system (7,8).
Biodegradable polymeric NPs improve the solubility
of hydrophobic drugs, promote stability and enable controlled
release. Thus, this system was developed as a second predominant
platform for drug delivery (9).
However, NPs formed by biodegradable polymers do not possess the
same favorable biocompatibility profile as liposomes, with lower
cellular uptake in comparison. Thus, a delivery system combining
the advantages of liposomes and polymer NPs, while avoiding
disadvantages, would be considered as ideal. Recent studies have
reported that lipid-polymer hybrid NPs (LPNs) with a lipid shell
and a polymer core, improve stability, enhance cellular uptake and
increase encapsulation efficiency (EE) (5,10,11).
LPNs feature a polymer core coated with lipid layers, thereby
combining the superior biocompatible property of lipids with the
structural integrity of polymeric NPs.
Poly(D,L-lactide-co-glycolide) (PLGA), one of the Food and Drug
Administration (FDA)-approved biodegradable polymers, is most
commonly used as the polymer core, where poorly water-soluble drugs
are encapsulated (12). Adding a
lipid layer onto PLGA NPs gives rise to LPNs.
The lipid component always contains natural
phospholipids, including lecithin, soybean phospholipids and
synthetic phospholipids, including
1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-carboxy(polyethylene
glycol)2000 (DSPE-PEG2000). Phospholipids, which contain
hydrophilic headgroups and two hydrophobic chains, form a monolayer
around the hydrophobic polymer core, while the DSPE-PEG2000 embeds
in the lipid monolayer to form a PEGylation stealth layer outside
the lipid shell, thus promoting electrostatic and steric stability
and prolonged circulation times (Fig.
1A) (13).
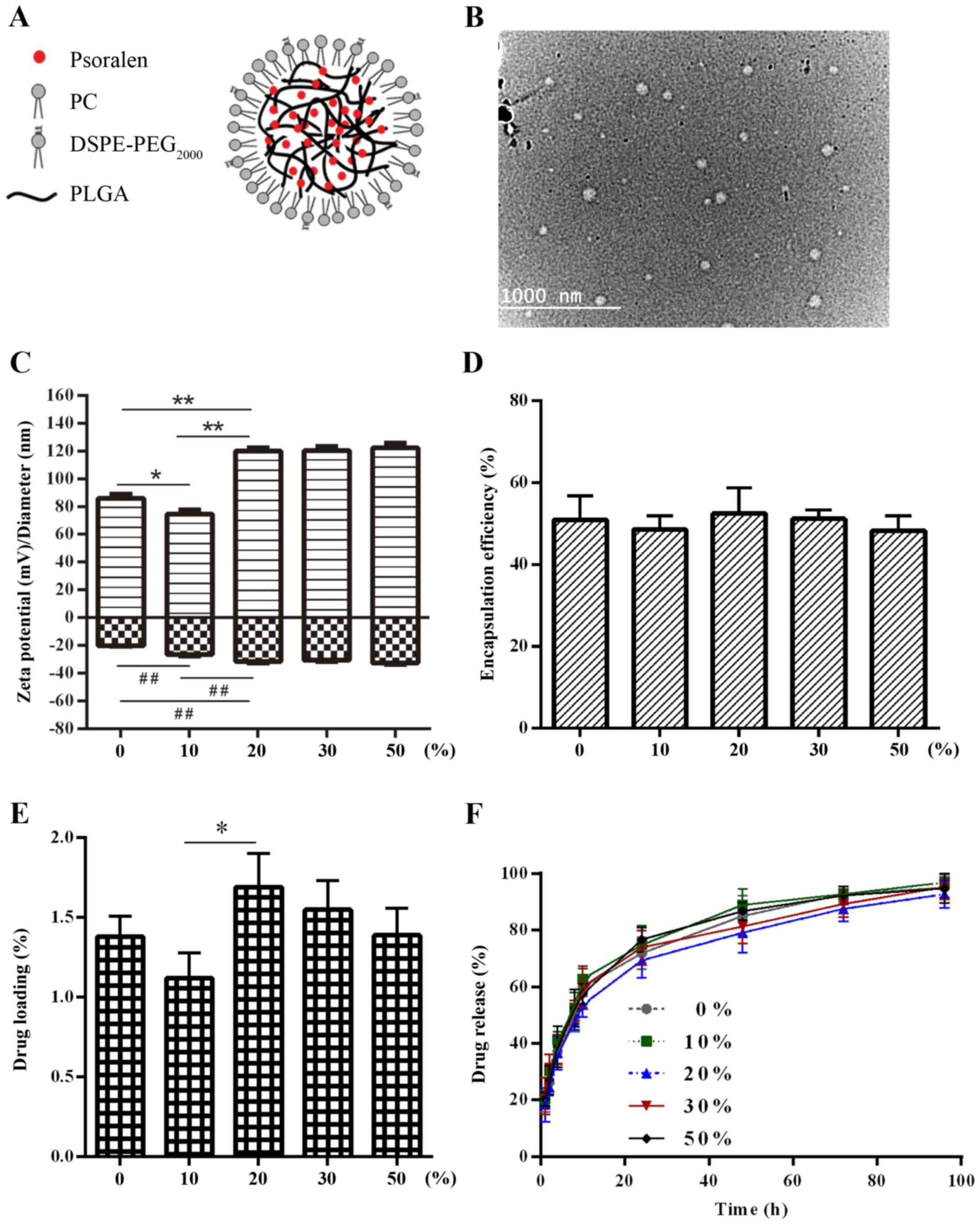 | Figure 1.(A) Schematic illustration of the
P-LPN structure featuring a lipid shell and a polymer core. (B)
Transmission electron micrograph imaging of LPNs. Characteristics
of the P-LPNs. (C) Changes in NP size and ζ potential, (D) effect
on encapsulation efficiency, (E) influence on drug loading and (F)
in vitro drug release profiles at various
DSPE-PEG2000/lipid weight ratios. *P<0.05,
**P<0.01 and ##P<0.01. NP, nanoparticle; PC,
phosphatidylcholine; DSPE-PEG2000,
1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-carboxy(polyethylene
glycol)2000; PLGA, poly(D,L-lactide-co-glycolide); P-LPN,
psoralen-loaded lipid-polymer hybrid nanoparticles. |
Psoralen (PSO), a natural coumarin compound, which
is popular in traditional Chinese medicine, has antitumor,
estrogen-like and multi-drug resistance modulating properties
(14–16). However, its use has been limited due
to poor solubility. Recently, various drug delivery systems have
been developed to improve solubility and controlled release.
Jambhrunkar et al (17)
developed mesoporous silica nanoparticles to encapsulate
griseofulvin, with low water solubility. Results revealed that a
charged surface improved the solubility and drug release of
griseofulvin, while hydrophobic modification on the nanoparticle
led to sustained drug release. Resveratrol cross-linked chitosan
nanoparticles modified with phospholipids were synthesized by Jeong
et al (18). Data revealed
that the chitosan nanoparticles increased the water solubility of
resveratrol and displayed slow and sustained release. A
methotrexate loaded lipid-polymer hybrid nano-carrier consisting of
PLGA and Lipoid S100 was prepared, which showed superior controlled
action compared with the free drug solution (19). In the present study, an
emulsification-solvent-evaporation (ESE) method was employed to
fabricate PSO-loaded LPNs (P-LPNs) (20). Emphasis was focused on optimizing
the parameters for production of LPNs and finding an optimal
formulation with respect to the in vitro drug release,
storage properties and cytotoxicity. The size, poly-dispersity and
ζ potential of the LPNs were characterized by dynamic light
scattering (DLS). Transmission electron microscopy (TEM) was used
to monitor surface morphology and structure. EE, drug loading (DL)
and drug-release kinetics were determined by high-performance
liquid chromatography (HPLC). The resistance reversal effect of
P-LPNs proved superior to free PSO, which was shown by in
vitro cytotoxicity assays in K562 and HepG2 cells.
Materials and methods
Materials
PSO (>98% purity) was purchased from Chengdu
Pufeide Biotechnology Co., Ltd. (Sichuan, Chengdu, China). DOX
hydrochloride was obtained from Selleck Chemicals (Houston, TX,
USA). Soybean phospholipids [injection grade; phosphatidylcholine
(PC)] were produced by Shanghai Tywei Pharmaceutical Co., Ltd.
(Shanghai, China).
1,2-Distearoyl-sn-glycero-3-phosphocholine-N-[methoxy(polyethylene
glycol)-2000 (DSPE-PEG2000) was provided by Shanghai AVT
Pharmaceutical Technology Co., Ltd. (Shanghai, China).
Poly(d,l-lactide-co-glycolide) (PLGA; 50:50; molecular weight,
8,000-12,000) was obtained from Jinan Daigang Biomaterial Co., Ltd.
(Shandong, Jinan, China). Glucose, lactose, mannitol, sucrose and
D-trehalose were provided by Tianjin Damao Chemical Company
(Tianjin, China). Cell Counting Kit-8 (CCK-8) was obtained from
Dojindo Molecular Technologies, Inc. (Kumamoto, Japan). RPMI-1640
cell culture medium, penicillin-streptomycin and trypsin-EDTA were
purchased from Thermo Fisher Scientific, Inc. (Waltham, MA, USA).
Fetal bovine serum (FBS) was provided by AusGenex Pty, Ltd.
(Molendinar, Queensland, Australia). Dialysis membranes with a
molecular weight cutoff of 8,000–14000 KDa were purchased from
Sangon Biotech, Co., Ltd. (Shanghai, China). The chronic myeloid
leukemia K562 and hepatoblastoma HepG2 cell lines were provided by
Nanjing KeyGen Biotech Co., Ltd. (Nanjing, Jiangsu, China).
Ethanol, acetonitrile and all other chemicals were HPLC grade.
Preparation of the NPs
The ESE technique was used to prepare the P-LPNs.
Briefly, PLGA (1.9 to 75 mg/ml) and PSO (0.75–2.25 mg/ml) were
dissolved in acetonitrile at room temperature, which served as the
organic phase. Phospholipids and DSPE-PEG2000 (4:1 mass ratio) were
first dissolved in ethanol at 100% of the PLGA polymer weight, then
water was added to obtain a 4% ethanol solution, which acted as the
aqueous phase. The mixture was heated to 70°C and the organic phase
was added dropwise into the preheated aqueous phase, with an
oil-water ratio of 1:10 (v/v). The solution was stirred for 90 min
to evaporate the organic solvent and allow self-assembly. The NP
emulsion was filtered through a 0.22-µm membrane. P-LPNs were used
immediately, stored at 4°C or lyophilized for later use.
Characterization of NPs
Particle size, polydispersity and ζ potential were
measured by DLS using a Zetasizer Nano ZS90 (Malvern Panalytical,
Malvern, England). The dispersion of NPs was diluted with ultrapure
water and subsequently analyzed.
EE and DL capacity
The EE and DL capacity were measured using an
ultrafiltration method. The NP emulsion was centrifuged at 11,000 ×
g for 30 min at 4°C to collect free PSO, which was then determined
by HPLC (Agilent 1260; Agilent Technologies, Inc., Santa Clara, CA,
USA) at an absorbance wavelength of 245 nm. Another aliquot of the
P-LPN solution was extracted by organic solvent to obtain total PSO
in the NP emulsion. The HPLC was equipped with a reverse-phase
column (Agilent Technologies, Inc.; 150×4.6 mm; 5 µm). The mobile
phase was a mixture of acetonitrile and water (55:45 v/v) and the
flow rate was 1.0 ml/min. The DL capacity and EE were calculated as
follows (5,8): EE% = (total drug in NP - free
drug)/total drug in NP × 100. DL% = encapsulated drug / total drug
in NP × 100.
TEM
The morphology of the NPs was determined by TEM
(TECNAI 10; Philips Healthcare, Amsterdam, The Netherlands) at an
accelerating voltage of 200 kV. One drop of the NP dispersion was
deposited onto the carbon-coated copper grid, followed by
air-drying at room temperature overnight prior to imaging. Samples
on the copper grid were scanned at ×37,000 magnification.
In vitro PSO release
The dialysis-bag method was used to investigate PSO
release. Phosphate-buffered saline (PBS; 0.1 M, pH 7.4) containing
0.1% (w/v) Tween-80, was used as the release medium to improve the
solubility of PSO. Dialysis bags containing 2 ml NP solution were
placed into a beaker filled with 200 ml PBS. Subsequently the
beaker was incubated at 37°C in a water bath under continuous
stirring at 100 rpm. At the indicated times, a 1 ml aliquot of the
release medium was removed for determining PSO content by HPLC
analysis. The release medium was exchanged periodically during the
dialysis process.
Stability studies
In order to investigate the in vitro
stability of NPs in solution, an aliquot of NP solution was
filtered through a 0.22-µm membrane and incubated in five volumes
of PBS, RPMI-1640 medium, RPMI-1640 medium with 10% FBS, 10% (v/v)
human plasma in PBS and 100% human plasma at 37°C for 120 h.
Particle size and polydispersity were measured by dynamic light
scattering at 24-h intervals.
Optimizing the formulation of
lyoprotectant
Freeze-dried drugs can be stored for a prolonged
period of time and easily be re-dissolved to restore activity.
Thus, freeze-drying technology is widely used in the preparation of
NPs. In order to develop appropriate storage conditions, five
different lyoprotectants, including glucose, lactose, mannitol,
sucrose and D-trehalose, were tested. NPs were frozen and kept at
−80°C overnight, then lyophilized at −80°C for 24 h. The
lyophilized NP powder with different lyoprotectants or
concentrations (2–10%) was resuspended in water to detect the
ability to re-dissolve, and particle size and ζ potential were
measured by DLS.
Cell lines and culture conditions
Sensitive K562 and HepG2 cells were cultured in
RPMI-1640 medium supplemented with 10% (v/v) FBS and 1% (v/v)
penicillin-streptomycin in a humidified atmosphere with 5% CO2 at
37°C. For the DOX-resistant K562 and HepG2 cell cultures, 1 µM DOX
was added in the RPMI-1640 medium every three passages to maintain
drug resistance. The cells were used for experiments once they
reached 80% confluency.
In vitro cytotoxicity assay
Cells were incubated at 37°C overnight in 96-well
plates (Corning Inc., Corning, NY, USA) at a cell density of
5×103 cells/well. Medium was then removed and replaced
with fresh medium containing blank NPs, free DOX, free PSO, DOX +
PSO or DOX + P-LPNs, and further incubated for 48 h. PSO or the
PSO-loaded NPs were at the equivalent PSO dose of 50, 25, 12.5,
6.25 and 3.12 µM, respectively. The amount of blank NPs was
equivalent to the blank carrier in P-LPNs, which ranged from 6.25
to 50 µM. DOX used for sensitive K562 and HepG2 cells ranged from
0.006 to 12.5 µM, and for resistant K562 and HepG2 cells from 0.78
to 200 µM. A non-toxic concentration of 20 µM PSO only was used in
co-administration protocols with DOX in either the free form or in
the NP formulations. Medium was removed at the end of the
incubation period, and 20 µl CCK-8 solution was added to each well.
Plates were further incubated for 3 h. Then absorbance was measured
at a wavelength of 420 nm using a 96-well plate reader (BioTek
Instruments, Inc., Winooski, VT, USA). Cell toxicity was calculated
according to the following equation: Cell viability (%) =
Abssample/Abscontrol × 100, where
Abssample is the absorbance of cells in the presence of
different formulations and Abscontrol is the absorbance
of cells in the absence of drug. The half maximal inhibitory
concentration (IC50) values were determined by fitting
hyperbolic concentration response curves to data points.
Statistical analysis
Quantitative data are expressed as the mean ±
standard deviation. The statistical significance was determined by
Student's t-test (between 2 groups) or by one-way analysis of
variance (≥3 groups) followed by Tukey's multiple comparison test,
using GraphPad Prism 6.0 software (GraphPad Software, La Jolla, CA,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results and Discussion
Preparation and characterization of
LPNs
PC and pegylated phospholipids
(DSPE-PEG2000) were dissolved in ethanol to form the
aqueous phase, which was heated at 37°C for dispersion. PLGA or
PLGA/PSO was dissolved in acetonitrile to form the oil phase, which
was then added to the preheated aqueous phase. The solution was
subsequently stirred for 90 min to allow for the lipids to
self-assemble and the organic solvent to evaporate. This allows a
lipid monolayer to be formed around the PLGA/PSO core. The PLGA and
PEG, as well as soybean PC, have been proven to be non-toxic and
biocompatible by the FDA.
DLS was applied to characterize NP size,
poly-dispersity and ζ potential in each preparation. The mean
diameter of the NPs ranged from 60 to 80 nm, while the ζ potential
ranged between −20 and −30 mV. PSO loading had an effect on the
particle size and ζ potential. The schematic composition of a
hybrid lipid-polymer NP and the TEM image of a preparation are
shown in Fig. 1A and B,
respectively. TEM images indicated that the NPs exhibited a
well-defined spherical shape with mono-dispersion and no adhesion.
Incorporation of PSO caused no significant morphological
changes.
Determining the optimal lipid-polymer
ratio in the preparation
The mean size, poly-dispersity and ζ potential of
the LPNs at different lipid/PLGA ratios between 10 and 400% (w/w)
are shown in Table I. Lipids,
functioning as emulsifiers, stayed at the oil-water interface to
lower the surface tension, and thus aid in producing the NP. At the
weight (W)PC/WPLGA ratio of 10–200%, the particle size ranged from
70 to 100 nm, with polydispersity of 0.1–0.25. However, NPs formed
at 10% WPC/WPLGA were observed to sediment to
the bottom of the bottle overnight (data not shown), likely due to
coalescence and precipitation of the polymer core resulting from
incomplete lipid coverage (8,13).
Increasing the WPC/WPLGA ratio above 2
significantly increased the size, while the ζ potential decreased.
An increasing lipid-PLGA ratio thus influenced size and ζ potential
in an inverse manner.
 | Table I.Characteristics of the lipid-polymer
hybrid nanoparticle at varying lipid/PLGA mass ratios. |
Table I.
Characteristics of the lipid-polymer
hybrid nanoparticle at varying lipid/PLGA mass ratios.
|
WPC/WPLGA, % | Diameter, nm | ZP, mV | PDI |
|---|
| 10 | 77.62±1.24 | −28.8±1.95 | 0.248±0.024 |
| 50 | 80.73±2.48 | −33.8±3.48 | 0.221±0.019 |
| 100 | 77.02±2.96 | −35.4±5.52 | 0.235±0.037 |
| 150 | 75.96±3.60 | −29.7±2.57 | 0.184±0.017 |
| 200 | 98.37±2.56 | −20.4±4.23 | 0.117±0.023 |
| 300 | 115.7±6.89 | −16.2±1.38 | 0.132±0.041 |
| 400 | 147.7±4.88 | −17.3±1.96 | 0.109±0.027 |
An appropriate amount of lipids shielded the
hydrophobic PLGA polymer core to increase stability. However, an
excess of lipids thickened the lipid layer, thereby increasing the
particle size and reducing the ζ potential (8). Thus, in terms of NP size and ζ
potential, 100% WPC/WPLGA was determined as
optimal and chosen for subsequent experiments.
Influence of the lipid-PEG/lipid ratio
on NP properties
Next, the effect of the lipid-PEG/lipid
(DSPE-PEG2000/PC) mass ratio on NP characteristics was
investigated, with the PLGA/lipid weight ratio constant kept at
100%. Data in Fig. 1C-E show the NP
size, EE and DL at different lipid-PEG/lipid mass ratios. The
amount of total lipids was kept constant, while the ratio between
the two components was changed from only PC to a 1+1 ratio of PC
and DSPE-PEG2000. NP size decreased at 10%
DSPE-PEG2000. This decrease was statistically
significant with a P-value of <0.05. At 20, 30 and 50% the NP
size increased (P<0.01). Values at 20, 30 and 50% did not
exhibit a significant difference. By contrast, a decrease in ζ
potential was observed in the samples containing 10 and 20%
DSPE-PEG2000, but no further decrease was observed at 30
and 50%. Drug load varied with NP size, whereby statistical
significance was only reached for the 20% data point. While DL and
NP size changed, the EE was close in all samples.
The in vitro drug release profiles of the
P-LPNs with different lipid-PEG/lipid mass ratios are presented in
Fig. 1F. When the lipid-PEG/lipid
mass ratio was varied from 0 to 50% (w/w), only small differences
in the in vitro drug release rates were observed, which
demonstrated that different amounts of PEGylated lipid in the
lipid-polymer NPs did not significantly affect PSO release. The
half-lives for PSO release were 11.2, 9.5, 11.9, 10.4 and 10.0 h,
respectively, when the DSPE-PEG2000/PC ratio was
increased from 0 to 50%. Thus, the ratio of
DSPE-PEG2000/PC had a minor effect on the drug release
properties of the NPs. At 120 h, >90% of the PSO had been
released in a sustained manner. Considering the EE and DL, an
intermediate lipid-PEG/lipid ratio of 1+4 was chosen for further
characterization of the formulations.
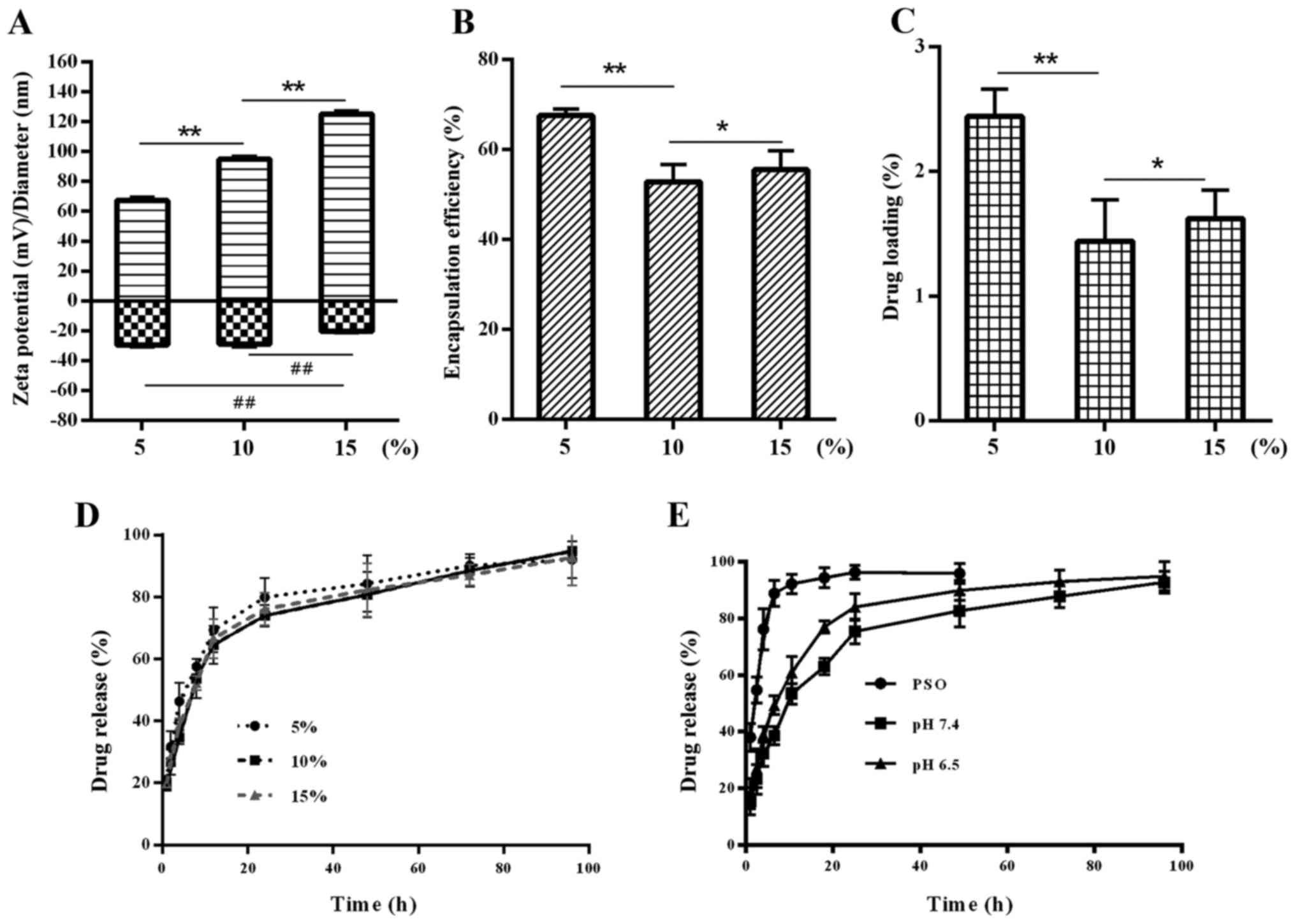
Influence of initial drug amount on NP
properties
Another parameter, which may potentially affect NP
size, is the PSO amount used in the preparation process. At a fixed
100% lipid/polymer mass ratio and a 20% lipid-PEG/lipid mass ratio,
5, 10 and 15% PSO/(lipid + PLGA) were encapsulated.
Fig. 2A shows that
the NPs increased in size when the amount of PSO was increased. The
ζ potential decreased at 15% PSO (P<0.01). An increase in PSO
did not change the EE and DL. A significant increase in EE and DL
was observed in the samples containing 5% PSO as compared with
those containing 10 or 15% (Fig. 2B and
C). Drug release was also comparable, but no significant
differences in the in vitro drug release rates of different
samples were observed (Fig. 2D).
The results indicated that, in the present study, the volume of the
core polymer and the surface area of the NPs determined the DL,
which is in agreement with previous reports from the literature
(21,22). Hence, the mass ratio of PSO to
lipid/PLGA of 5% was chosen for subsequent experiments.
In vitro drug release profile
In vitro release of PSO from P-LPNs was
monitored over 96 h using the dialysis-bag method. P-LPNs were
thereby compared to free PSO in the dialysis bag. A number of
studies have shown that tumor tissue represents an acidic
microenvironment due to insufficient oxygen supply and thus
anaerobic energy provision. Generally the pH in tumors lies between
6.0 and 6.9, while in non-neoplastic tissue it is maintained at a
pH value of between 7.35 and 7.45 (23,24).
Thus, PBS at pH 7.4 and 6.5 was used as the dialysis medium to
study the sustained release from NPs at these different pH
conditions. As shown in Fig. 2E,
free PSO in solution was basically removed from the dialysis bag
within the first 6 h, while the P-LPNs showed delayed release
characteristics. The release characteristics were improved due to
the nano-formulation, and the cumulative release of PSO exceeded
90% after 96 h.
The half-lives of PSO release in P-LPNs were 11.9
and 7.5 h at pH 7.4 and 6.5, respectively (Fig. 2E). In the first 10 h, cumulative
release reached ~53% at pH 7.4 and 61% at pH 6.5 for the P-LPNs.
Drug release at pH 6.5 was thus faster than at pH 7.4, probably due
to a more rapid degradation of surface lipids in a more acidic
environment. These findings are in accordance with data showing
that internalization of drug-loaded nanocarriers into cells is
stimulated by degradation in the acidic environment of lysosomes
(25).
In vitro stability of LPNs. In order to investigate
the long-term stability and protein binding of LPNs, a formulation
of 100% lipid/PLGA mass ratio, 20% lipid-PEG/lipid mass ratio and
5% PSO/(lipid + PLGA) mass ratio were chosen. Lipid-polymer hybrid
NPs were incubated in PBS, RPMI-1640 medium, RPMI-1640 medium
containing 10% FBS, 10% (v/v) human plasma in PBS or 100% human
plasma at 37°C for 120 h. Fig. 3A
shows that size and polydispersity of NPs incubated in either PBS
or RPMI-1640 medium did not change significantly. This indicates
that a monolayer comprised of PEGylated lipid molecules stabilizes
the polymer core and prevents NP aggregation over the course of 120
h.
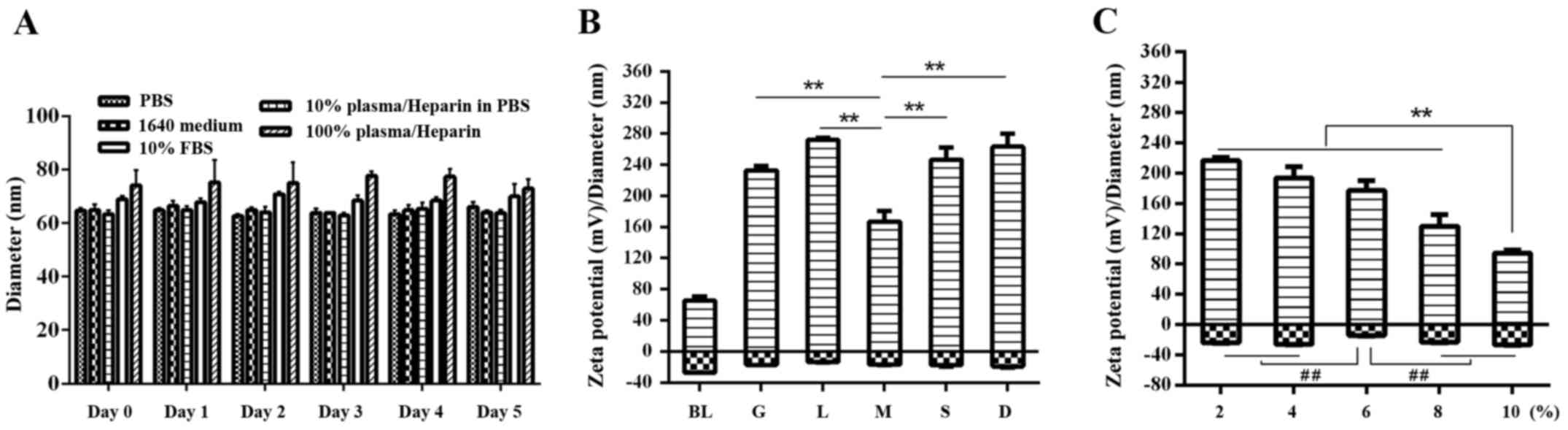 | Figure 3.Post-formulation stability of P-LPNs.
(A) In vitro stability of P-LPNs over 5 days. NPs were
incubated in PBS or RPMI-1640 medium. 10% (v/v) fetal bovine serum,
10% (v/v) human heparin plasma in PBS and 100% human heparin plasma
were also used. Incubation proceeded for up to 5 days at 37°C. An
aliquot of the NP suspension was used to measure NP size using
Malvern dynamic light scattering. (B) Post-formulation stability of
the P-LPNs following freeze-drying and lyophilization with five
cryroprotectants (5%, w/v), glucose, lactose, mannitol, sucrose and
D-trehalose. (C) Post-formulation stability of the P-LPNs assessed
following lyophilization in mannitol at concentrations of 2, 4, 6,
8 and 10% (w/v). **P<0.01 indicates statistically significant
differences between 5% mannitol and the other lyoprotectants,
including glucose, lactose, mannitol, sucrose and D-trehalose.
##P<0.01 indicates a statistically significant
difference in the ζ potential. BL, before lyophilization; G,
glucose; L, lactose; M, mannitol; S, sucrose; D, D-trehalose; PBS,
phosphate-buffered saline; FBS, fetal bovine serum; NP,
nanoparticle; P-LPN, psoralen-loaded lipid-polymer hybrid
nanoparticles. |
In 10% FBS and in 10 or 100% human plasma, the
change in NP size was used as a criterion for evaluation of protein
adsorption and bio-fouling. As shown in Fig. 3A, the hybrid NPs were stable in 10%
FBS, as they retained a size of 64±4 nm (mean ± SEM polydispersity
= 0.22±0.11; n=3), over 120 h. A small increase in size was
observed in 100% human serum as compared with that in 10% human
serum, but without statistical significance. These results
suggested that PEGylation of the NP prevents protein adsorption on
the NP surface. The formulation has appropriate stability, which
was consistent with the results of a previous study (20). In addition to improving the
solubility and stability of the nanocarriers in aqueous solution,
PEGylation has been reported to minimize opsonisation during
circulation in the blood stream and the reticuloendothelial systems
in the liver, spleen, and kidneys, thus increasing NP circulation
time (26,27).
Optimization of the lyoprotectant
formulation for LPNs
Five different lyoprotectants (5%, w/v), glucose,
lactose, mannitol, sucrose and D-trehalose, were used during the
lyophilisation procedure to identify the one that was superior.
Fig. 3B shows that particle size
increased by more than two times and ζ potential sharply increased
in all samples due to the addition of lyoprotectant. Mannitol
resulted in the lowest size following re-dissolution (167±13.2 nm)
and was therefore chosen as the optimal lyoprotectant for further
experiments. When the concentration of mannitol was increased from
2% to 10% (w/v), a more than two-fold decrease in particle size was
observed (down to 100 nm). A concentration of 10% mannitol resulted
in the smallest size following reconstitution (94.4±4.1 nm), which
thus most closely matched the original particle size (Fig. 3C). The ζ potential showed no
significant difference at concentrations of 2, 4, 8 and 10%, but
was lower at 6% (P<0.01).
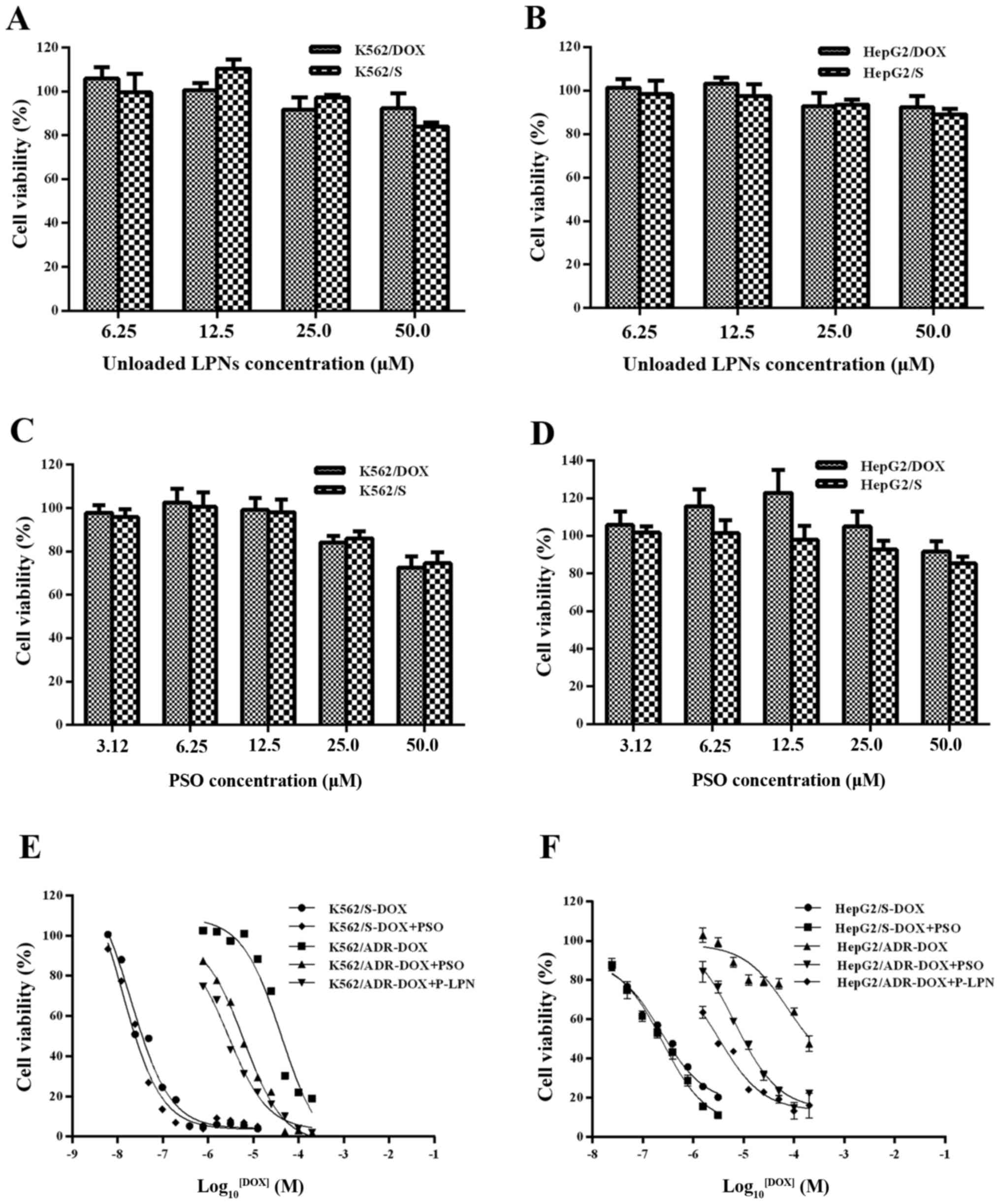
In vitro cytotoxicity of LPNs
In vitro cytotoxicity of drug-free LPNs, free
PSO and P-LPN was assessed in the K562 and the HepG2 cell lines
over 48 h, using the CCK-8 method. The unloaded LPNs at
concentrations between 6.25 and 50 µM (Fig. 4A and B) and the modulator PSO alone
at concentrations between 3.12 and 25 µM (Fig. 4C and D) did not exhibit any
cytotoxic effect. The DOX IC50 was 19.8 nM in the drug-sensitive
K562 cells, 41.2 µM in the DOX-resistant K562 cells, 230 nM in the
sensitive HepG2 cells and 74.9 µM in the DOX-resistant HepG2 cells
(Table II). The resistance indices
of DOX-resistant K562 and HepG2 cells were 2,086 and 326,
respectively, which demonstrated that the resistant cells were
highly resistant to DOX. Co-administration of DOX and P-LPN at an
identical concentration of 20 µM PSO exhibited significantly higher
cytotoxicity compared with that of free DOX or (DOX + PSO) at all
drug concentrations (Fig. 4E and
F). This demonstrated that co-administration of DOX and P-LPNs
was superior to the same regimen in solution in all cases. The NP
formulation showed a 14- and 23-fold stronger effect in inhibiting
cell growth in resistant K562 and HepG2 cells compared with free
DOX (19.8 and 230 nM, respectively), and a 2.2- and 2.1-fold
stronger effect than free DOX plus PSO (6.2 and 6.8 µM). These
changes were statistically significant (P<0.05).
| Table II.IC50 value for free DOX
alone or in combination with free PSO or LPNs in K562 and HepG2
cells. |
Table II.
IC50 value for free DOX
alone or in combination with free PSO or LPNs in K562 and HepG2
cells.
|
| IC50,
nmol/la |
|---|
|
|
|
|---|
| Sample | K562/S | K562/DOX | HepG2/S | HepG2/DOX |
|---|
| DOX in
solution | 19.8±0.31 | 41.200±0.26 | 229.8±0.57 | 74.930±0.82 |
| DOX + PSO | 12.2±0.43 |
6.190±0.57 | 245.1±0.73 |
6.777±0.46 |
| DOX + P-LPNs | / |
2.860±0.12 | / |
3.254±0.69 |
| Resistance
indexb |
| 2086 |
| 326 |
In the present study, a formulation of lipid-polymer
hybrid nanocarriers with a lipid shell and a PLGA core for
sustained and controlled release of PSO was developed. Parameters
that could have an effect on the preparation of NPs were
investigated to optimize the formulation. Next, the physical
properties and cytotoxicity were studied. P-LPNs were prepared
using the ESE method. It was found that the amount of lipid served
an important role in particle size and stability. A WPC/WPLGA of 1
proved optimal. PEGylated surface lipids had a minor effect on
in vitro drug release, while high PEG surface density
increased particle size and ζ potential. The optimal
lipid-PEG/lipid mass ratio was determined to be 20%. When the
PSO/(lipid + PLGA) mass ratio was increased, size increased, and ζ
potential decreased at 15% PSO. However, there were no apparent
changes in EE and DL. Optimized NP formulations were evaluated for
physical stability in five different media in the absence and
presence of human plasma. There was no marked change in NP size
following a 5-day incubation period, which suggested that the
formulation had sufficient stability in PBS, medium and plasma.
Mannitol at a concentration of 10% (w/v) was found to be the best
lyoprotectant. Unloaded LPNs were well tolerated by the human model
cell lines in vitro. Compared with free PSO, P-LPNs showed
enhanced toxicity of the chemotherapeutic drug DOX in DOX-resistant
cells. In summary, LPNs hold promise as a drug delivery system for
effective chemotherapy. Further studies on the in vivo
efficacy of these formulations will be required prior to broader
use in a clinical context.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81273707), the Ministry of
Education in the New Century Excellent Talents (grant no.
NECT-12-0677), the Natural Science Foundation of Guangdong (grant
nos. S2013010012880 and 2016A030311037), the Science and Technology
Program of Guangzhou (grant nos. 2014J4500005 and 201704030141),
the Science Program of the Department of Education of Guangdong
(grant nos. 2013KJCX0021 and 2015KGJHZ012), the Science and
Technology Program of Guangdong (grant no. 2015A050502027), and the
Special Project of International Scientific and Technological
Cooperation in Guangzhou Development District (grant no.
2017GH16).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
SPCC and YC designed the research and took
responsible for all aspects of the study. YY and TC performed the
majority of the research work and wrote the initial draft of the
paper. LB assisted in some of the experiments. PC analyzed the
majority of the data and revised the manuscript critically for
important intellectual content. RC contributed to refining the
ideas, performing additional analyses and finalizing the paper. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rogueda PG and Traini D: The nanoscale in
pulmonary delivery. Part 1: Deposition, fate, toxicology and
effects. Expert Opin Drug Deliv. 4:595–606. 2007.
|
|
2
|
Cooper DL, Conder CM and Harirforoosh S:
Nanoparticles in drug delivery: Mechanism of action, formulation
and clinical application towards reduction in drug-associated
nephrotoxicity. Expert Opin Drug Deliv. 11:1661–1680. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yuan Y, Cai T, Xia X, Zhang R, Chiba P and
Cai Y: Nanoparticle delivery of anticancer drugs overcomes
multidrug resistance in breast cancer. Drug Deliv. 23:3350–3357.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jia F, Liu X, Li L, Mallapragada S,
Narasimhan B and Wang Q: Multifunctional nanoparticles for targeted
delivery of immune activating and cancer therapeutic agents. J
Control Release. 172:1020–1034. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu Y, Pan J and Feng SS: Nanoparticles of
lipid monolayer shell and biodegradable polymer core for controlled
release of paclitaxel: Effects of surfactants on particles size,
characteristics and in vitro performance. Int J Pharm. 395:243–250.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mandal B, Bhattacharjee H, Mittal N, Sah
H, Balabathula P, Thoma LA and Wood GC: Core-shell-type
lipid-polymer hybrid nanoparticles as a drug delivery platform.
Nanomedicine (Lond). 9:474–491. 2013. View Article : Google Scholar
|
|
7
|
Vyas S, Rai S, Paliwal R, Gupta PN, Khatri
K, Goyal AK and Vaidya B: Solid lipid nanoparticles (SLNs) as a
rising tool in drug delivery science: One step up in
nanotechnology. Curr Nanosci. 4:30–44. 2008. View Article : Google Scholar
|
|
8
|
Cheow WS and Hadinoto K: Factors affecting
drug encapsulation and stability of lipid-polymer hybrid
nanoparticles. Colloids Surf B Biointerfaces. 85:214–220. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mukherjee B, Satapathy BS, Mondal L, Dey
NS and Maji R: Potentials and challenges of active targeting at the
tumor cells by engineered polymeric nanoparticles. Curr Pharm
Biotechnol. 14:1250–1263. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thevenot J, Troutier AL, David L, Delair T
and Ladavière C: Steric stabilization of lipid/polymer particle
assemblies by poly(ethylene glycol)-lipids. Biomacromolecules.
8:3651–3660. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang L, Zhu D, Dong X, Sun H, Song C,
Wang C and Kong D: Folate-modified lipid-polymer hybrid
nanoparticles for targeted paclitaxel delivery. Int J Nanomedicine.
10:2101–2114. 2015.PubMed/NCBI
|
|
12
|
Feng S-S and Chien S: Chemotherapeutic
engineering: Application and further development of chemical
engineering principles for chemotherapy of cancer and other
diseases. Chem Eng Sci. 58:4087–4114. 2003. View Article : Google Scholar
|
|
13
|
Chan JM, Zhang L, Yuet KP, Liao G, Rhee
JW, Langer R and Farokhzad OC: PLGA-lecithin-PEG core-shell
nanoparticles for controlled drug delivery. Biomaterials.
30:1627–1634. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hsieh MJ, Chen MK, Yu YY, Sheu GT and
Chiou HL: Psoralen reverses docetaxel-induced multidrug resistance
in A549/D16 human lung cancer cells lines. Phytomedicine.
21:970–977. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jiang J, Wang X, Cheng K, Zhao W, Hua Y,
Xu C and Yang Z: Psoralen reverses the P-glycoprotein-mediated
multidrug resistance in human breast cancer MCF-7/ADR cells. Mol
Med Rep. 13:4745–4750. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cai J, Chen S, Zhang W, Hu S, Lu J, Xing J
and Dong Y: Paeonol reverses paclitaxel resistance in human breast
cancer cells by regulating the expression of transgelin 2.
Phytomedicine. 21:984–991. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jambhrunkar S, Qu Z, Popat A, Karmakar S,
Xu C and Yu C: Modulating in vitro release and solubility of
griseofulvin using functionalized mesoporous silica nanoparticles.
J Colloid Interface Sci. 434:218–225. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jeong H, Samdani KJ, Yoo DH, Lee DW, Kim
NH, Yoo IS and Lee JH: Resveratrol cross-linked chitosan loaded
with phospholipid for controlled release and antioxidant activity.
Int J Biol Macromol. 93:757–766. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tahir N, Madni A, Balasubramanian V,
Rehman M, Correia A, Kashif PM, Mäkilä E, Salonen J and Santos HA:
Development and optimization of methotrexate-loaded lipid-polymer
hybrid nanoparticles for controlled drug delivery applications. Int
J Pharm. 533:156–168. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang L, Chan JM, Gu FX, Rhee JW, Wang AZ,
Radovic-Moreno AF, Alexis F, Langer R and Farokhzad OC:
Self-assembled lipid - polymer hybrid nanoparticles: A robust drug
delivery platform. ACS Nano. 2:1696–1702. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mu L and Feng SS: Vitamin E TPGS used as
emulsifier in the solvent evaporation/extraction technique for
fabrication of polymeric nanospheres for controlled release of
paclitaxel (Taxol). J Control Release. 80:129–144. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ruan G and Feng SS: Preparation and
characterizations of PLA-PEG-PLA micro-spheres for controlled
release of paclitaxel. Biomaterials. 24:5037–5044. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Abaza M and Luqmani YA: The influence of
pH and hypoxia on tumor metastasis. Expert Rev Anticancer Ther.
13:1229–1242. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schornack PA and Gillies RJ: Contributions
of cell metabolism and H+ diffusion to the acidic pH of
tumors. Neoplasia. 5:135–145. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Saraswathy M and Gong S: Different
strategies to overcome multidrug resistance in cancer. Biotechnol
Adv. 31:1397–1407. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Åslund AKO, Sulheim E, Snipstad S, von
Haartman E, Baghirov H, Starr N, Kvåle Løvmo M, Lelú S, Scurr D,
Davies CL, et al: Quantification and qualitative effects of
different PEGylations on Poly(butyl cyanoacrylate) Nanoparticles.
Mol Pharm. 14:2560–2569. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Harris JM and Chess RB: Effect of
pegylation on pharmaceuticals. Nat Rev Drug Discov. 2:214–221.
2003. View
Article : Google Scholar : PubMed/NCBI
|