Introduction
Vascular endothelial growth factor receptor 2
(VEGFR2) is a tyrosine kinase receptor, mainly expressed in
endothelial cells. In several types of solid tumor, VEGFR2 is
critical in tumor angiogenesis, allowing tumor cells to receive
sufficient nutrients and oxygen to grow. The inhibition of VEGFR2
has been shown to cause hypoxia and necrosis of solid tumors
(1). Therefore, VEGFR2 has become a
promising target for tumor treatment (2). Unfortunately, patients can develop
resistance to VEGFR2 inhibitors and this resistance correlates with
poor prognosis. It is suggested that the mechanism underlying this
resistance involves c-Met (1). The
inhibition of VEGFR2 causes tumor hypoxia and activates the hypoxia
inducible factor 1α regulatory pathway, leading to enhanced
expression of c-Met, which can promote tumor metastasis and
establish a vicious circle of hypoxia and malignant metastasis
(1). Therefore, the inhibition of
c-Met may have the potential to overcome the resistance to VEGFR2
inhibition.
Dual c-Met and VEGFR2 kinase inhibitors have
attracted considerable attention in studies (3–5). c-Met
is a tyrosine kinase receptor for hepatocyte growth factor and has
become an important target for the treatment of several types of
solid tumor (6). The activation of
c-Met is associated with several physiological processes, including
cell proliferation, survival, morphogenesis and angiogenesis
(7). Studies have shown the
upregulation of c-Met in various type of cancer, which correlates
with a poor prognosis. Therefore, c-Met inhibitors have been used
to treat patients and have produced beneficial results (8). Previously, it was found that c-Met and
VEGFR2 act synergistically in several types of cancer, and they
share partial signal pathways (9–11).
Therefore, dual c-Met and VEGFR2 kinase inhibitors have the
potential to improve therapeutic outcomes. To date, several dual
c-Met and VEGFR2 inhibitors are available for clinical use
(10), however, their selectivity
is low and they cause several side effects, which are due to their
relatively simple structures of either quinolones/quinazolines or
pyridine. Therefore, the development of a novel structure model for
a dual c-Met and VEGFR2 kinase inhibitor is important for tumor
treatment.
At present, several natural compounds are being used
as chemopreventive agents for cancer, and there is increasing
interest in identifying the antitumor activity of these agents
(12,13). However, no natural products have
been identified with an inhibitory profile towards both c-Met and
VEGFR2. Therefore, the present study aimed to identify natural
products that can be used as leading compounds for dual c-Met and
VEGFR2 kinase inhibitor development through virtual screening
technology against a natural product database. It was found that
chrysoeriol (CAS no. 491-71-4), a natural product, had relative
high binding-affinity to both VEGFR2 (Kd=11 µM)
and c-Met (Kd=12 µM). Pharmacological and
molecular docking experiments were also performed to investigate
the structure activity relationships (SARs) of chrysoeriol and its
analogs. To the best of our knowledge, the present study is the
first to report a natural product with both c-Met and VEGFR2
inhibitory profiles. The SAR results of this lead compound and
analogs have the potential to provide insights into the development
of potent dual c-Met and VEGFR2 inhibitors.
Materials and methods
Software and database use
All calculations were performed using the Dell
PowerEdge R900 workstation (Dell, Inc. Round Rock, TX, USA) under
the Redhat 5.0 platform. The pharmacophore construction and virtual
screening process were performed in Discovery Studio 3.0 (DS 3.0;
Accelrys Software, Inc., San Diego, CA, USA). The binding modes of
the compounds with protein were displayed in Discovery Studio 4.0
(DS 4.0; Accelrys Software, Inc.). The crystal structures of c-Met
(PDB code: 3CTJ) and VEGFR2 (PDB code: 3VHE) were downloaded from
the RCSB Protein Data Bank (PDB) (14–16).
The Traditional Chinese Medicine (TCM) database (http://tcm.cmu.edu.tw/) from ZINC was used for
screening.
Structure-based virtual screening
In the screening process for dual c-Met and VEGFR2,
the crystal structure for c-Met (PDB code: 3CTJ) and the crystal
structure for VEGFR2 (PDB code: 3VHE) were used for the
pharmacophore construction. The pharmacophore construction process
of c-Met was performed as follows. Firstly, 3CTJ was prepared by
using the Protein Wizard in DS 3.0. This protein prepared process
included cleaning water molecules, missing residues and adding
hydrogen atoms. Subsequently, the receptor-ligand based
pharmacophore was constructed using the Receptor-Ligand
Pharmacophore Generation module of DS 3.0. The crystal structure
(PDB code: 3VHE) was used for the pharmacophore construction
process of VEGFR2, and the details of the process were similar to
those for c-Met. Subsequently, the pharmacophore model which met
the interaction model reported in previous articles was selected as
the pharmacophore for c-Met or VEGFR2 (14,16).
These two pharmacophore models were validated with several
compounds, which had been identified as c-Met or VEGFR2 inhibitors.
Subsequently, these two pharmacophore models were imported to the
Search 3D database module of DS 3.0 to search the TCM database.
The screening results for dual kinase inhibitors
were subsequently imported into X-score version 1.3 (http://www.sioc-ccbg.ac.cn/?p=42) to predict
their binding energy. According to the values of the binding
energy, the top 10 structures for each model were selected for
further analysis. The compounds that met the following conditions
were considered as potential dual VEGFR2 and c-Met kinase
inhibitors: Compounds must be selected out by both the c-Met and
VEGFR2 pharmacophore models; the compounds must fit into the two
pharmacophores. Additionally, certain measures of the
ligand-receptor interaction in these compounds were analyzed
manually. These measures included clashes with residues in the
active sites of kinase protein; interactions with important
residues (Cys919, Glu 885 and Asp 1086 for VEGFR2; Asp 1222 and Met
1160 for c-Met) and hydrophobic interaction.
Bioassay at the molecular level
The kinase assays of c-Met and VEGFR2 were performed
using the KINOMEscan™ screening platform (17,18).
Binding reactions were assembled by combining kinases, liganded
affinity beads, and test compounds in 1X binding buffer (20%
SeaBlock, 0.17X PBS, 0.05% Tween-20 and 6 mM DTT). The test
compounds were prepared as 40X stocks in 100% DMSO and directly
diluted into the assay. All reactions were performed in
polypropylene 384-well plates in a final volume of 0.02 ml. The
assay plates were incubated at room temperature with shaking for 1
h and the affinity beads were washed with wash buffer (1X PBS and
0.05% Tween-20). The beads were then re-suspended in elution buffer
(1X PBS, 0.05% Tween-20 and 0.5 µM non-biotinylated affinity
ligand) and incubated at room temperature with shaking for min. The
kinase concentration in the eluates was measured by quantitative
polymerase chain reaction analysis as previously described
(17,18). In the preliminary experiments, the
compounds were screened at the concentration 10 µM, and the results
for primary screen binding interactions are reported as ‘% Ctrl’,
where lower numbers indicated stronger hits. In the subsequent
Kd-determining assay, the compounds were screened at a
series of concentrations under the condition of 10 µM kinase
protein. All the kinase assays were repeated three times to confirm
results.
Statistical analysis
All statistical analyses were performed in SPSS
version 21.0 (IBM SPSS, Armonk, NY, USA). Student's t-test was used
to evaluate the difference between groups. All tests were two-sided
and P<0.05 was considered to indicate a statistically
significant difference.
Structure preparation for molecular
docking
The crystal structures of c-Met (PDB code: 3CTJ) and
VEGFR2 (PDB code: 3VHE) were used for molecular docking. The
proteins of 3CTJ or 3VHE were processed in DS 3.0. First, the water
molecules and other nonsense small molecules were cleaned. Second,
the residues and loop segments were deleted and all hydrogen atoms
were added.
Molecular docking screening
The LibDock module in DS 3.0 was used for docking
screening. The prepared c-Met and VEGFR2 structures were imported
into DS 3.0, and the small molecules were docked into the
corresponding protein, as required. The docking results were
analyzed using DS 4.0 and the results were visualized using PyMol
(version 1.7.x; Schrödinger, New York, NY, USA).
Results
Structure-based virtual screening
To determine potential dual c-Met and VEGFR2 kinase
inhibitors, structure-based virtual screening was performed in DS
3.0 against the TCM database, which contained almost 30,000 natural
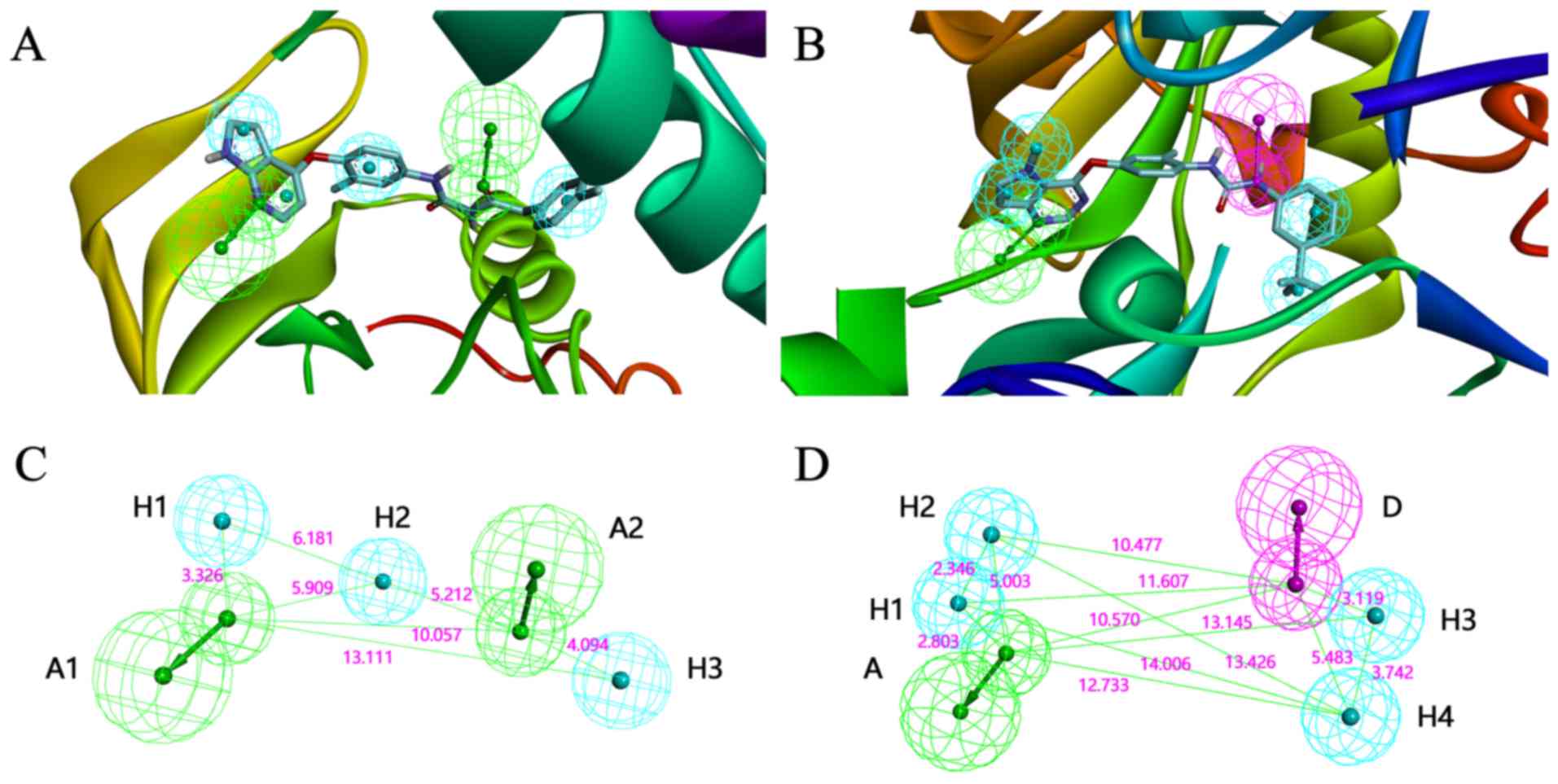
compounds. In the pharmacophore construction process, it was found
that the structure and the pharmacophore features of 3CTJ (Fig. 1A) were similar to those of 3VHE
(Fig 1B). Therefore, it was
hypothesized that the compounds meeting the pharmacophore features
of 3CTJ and 3VHE may inhibit the activity of c-Met and VEGFR2.
Therefore, two pharmacophore models were constructed based on the
crystal structure of 3CTJ and 3VHE, and virtual screening for each
model was performed, respectively (Fig.
1C and D).
Subsequently, the two models described above were
evaluated in the prepared TCM database in DS 3.0, and >200
molecules were retained. All of these molecules were subjected to
protein-ligand binding energy prediction using XSCORE software.
These compounds were then ranked based on the values of predicted
binding energy. The highest 20 ranked compounds for each
pharmacophore model were used for further analysis. Following a
series of analytical processes, eight natural compounds were
selected as the candidates (Table
I). These candidates were purchased for the subsequent
pharmacological experiments.
 | Table I.Predictions of binding energy with
candidate compounds. |
Table I.
Predictions of binding energy with
candidate compounds.
| Compound (CAS
no.) | Binding energy with
VEGFR2 (kcal/mol) | Binding energy with
c-Met (kcal/mol) | Fit value of VEGFR2
(3VHE) | Fit value of c-Met
(3CTJ) |
|---|
| Ref | −9.36 | −9.58 |
|
|
| 491-71-4 | −7.89 | −8.48 | 4.14170 | 4.27562 |
| 572-31-6 | −8.11 | −7.58 | 4.13896 | 4.23563 |
| 79995-67-8 | −7.58 | −8.81 | 4.05977 | 4.16375 |
| 34444-37-6 | −6.94 | −7.51 | 3.98707 | 4.13593 |
| 63644-62-2 | −6.73 | −8.11 | 3.89755 | 4.12793 |
| 37831-70-2 | −6.15 | −6.65 | 3.68365 | 3.99841 |
| 61276-17-3 | −6.54 | −9.28 | 3.64415 | 3.74544 |
| 61303-13-7 | −6.36 | −8.22 | 3.63678 | 4.27562 |
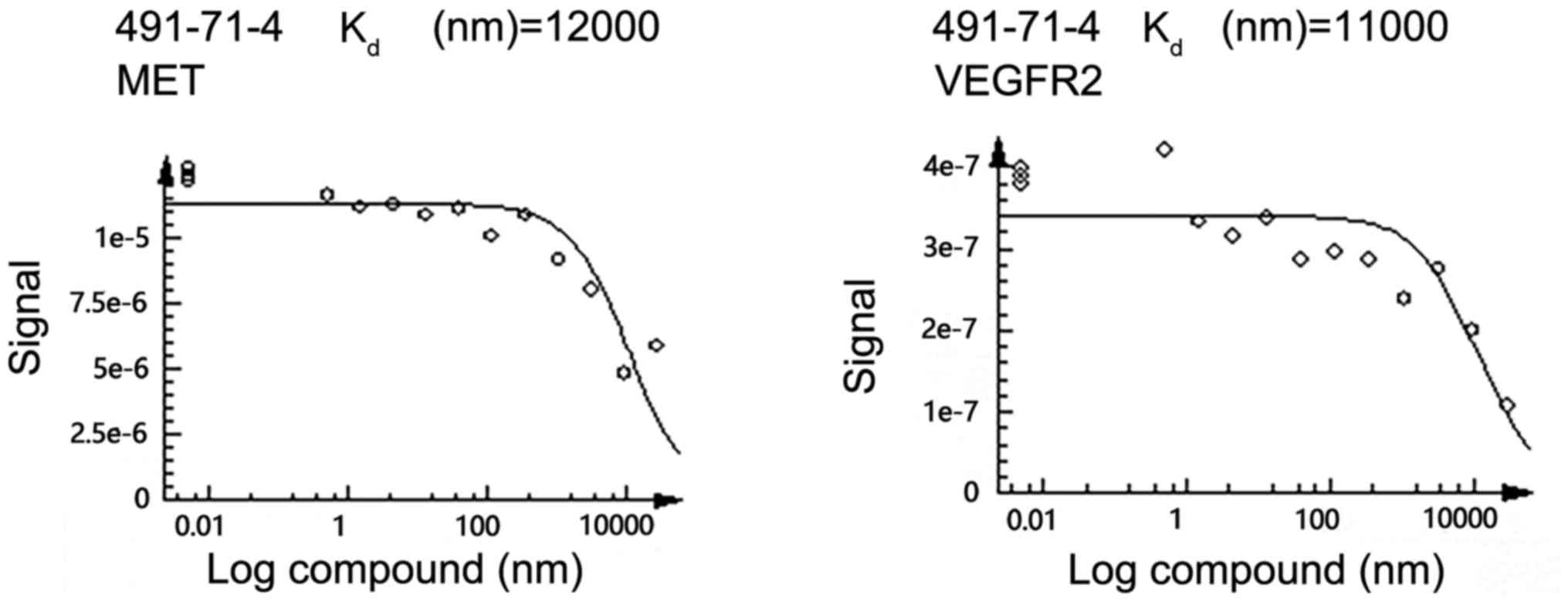
Bioassay validation
Preliminary screening using the KINOMEscan system
was used for primary screening, the results of which are shown in
Table II. Among the candidates,
chrysoeriol (CAS no. 491-71-4) showed desirable binding to both
c-Met and VEGFR2. Therefore, chrysoeriol was selected as a
potential dual VEGFR2 and c-Met kinase inhibitor for further
assessment using a dose-response experiment. The dose-response
curves are shown in Fig. 2. The
average Kd value for chrysoeriol to VEGFR2 and c-Met
were 11 and 12 µm, respectively. These results indicated that
chrysoeriol was a promising leading compound for further structure
optimization.
| Table II.Preliminary experiment results of
binding of candidate compounds. |
Table II.
Preliminary experiment results of
binding of candidate compounds.
| Compound (CAS
no.) | c-MET (% ctrl at 10
µM) | VEGFR2 (% ctrl at 10
µM) |
|---|
| 491-71-4 | 47 | 55 |
| 572-31-6 | 97 | 100 |
| 79995-67-8 | 74 | 100 |
| 34444-37-6 | 87 | 100 |
| 63644-62-2 | 88 | 100 |
| 37831-70-2 | 88 | 100 |
| 61276-17-3 | 84 | 100 |
| 61303-13-7 | 84 | 100 |
Molecular docking
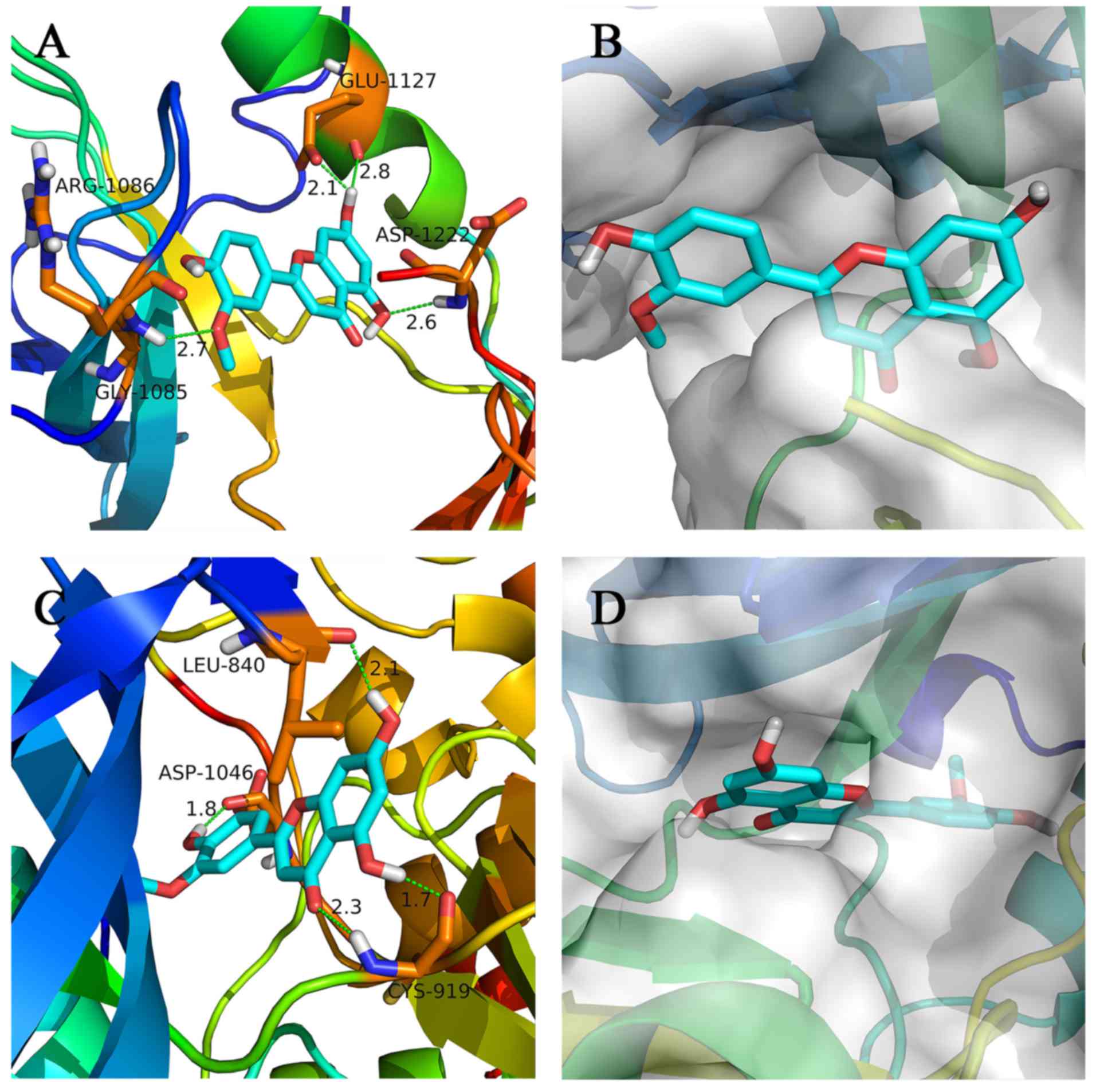
To elucidate the interaction of chrysoeriol with
c-Met and VEGFR2, molecular docking was performed. The binding
modes of chrysoeriol with c-Met and VEGFR2 were analyzed using
PyMol (version 1.7.x; Schrödinger) and DS 4.0, and the results are
shown in Fig 3A-D. The conformation
of chrysoeriol showed a good fit with the c-Met and VEGFR2 active
shape. In addition, chrysoeriol showed interaction with the
critical amino acid residue of the c-Met and VEGFR2 kinase
proteins. The H-bonds and hydrophobic contacts were shown to be
important for the interactions between chrysoeriol and kinase
proteins. The binding modes were beneficial to discern the critical
groups and are useful as templates for further structure
optimization.
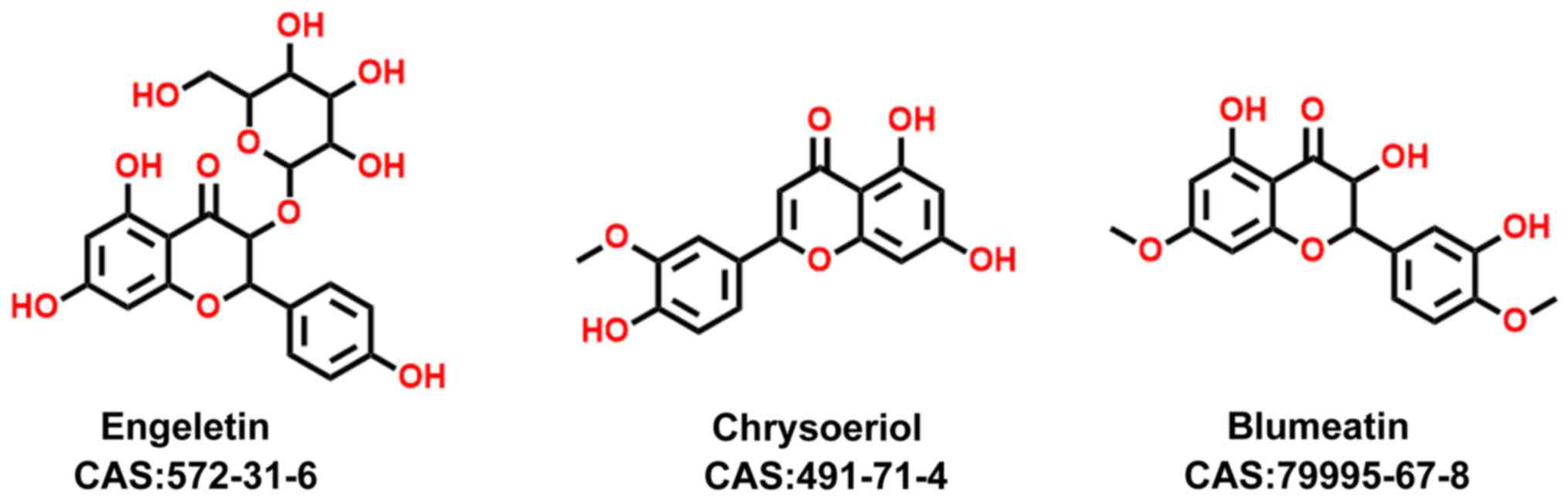
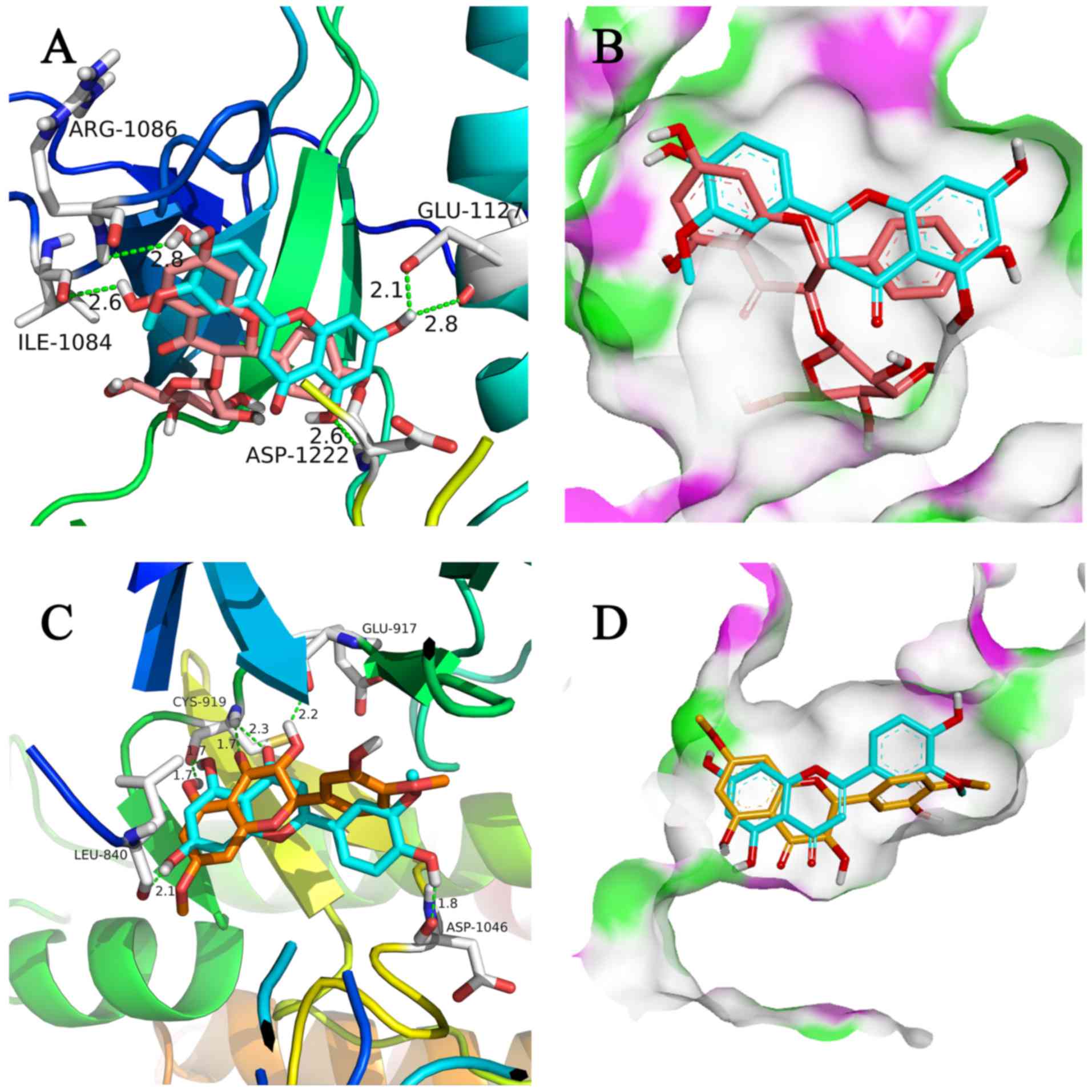
It was found that three compounds with similar
structures showed distinct binding affinities toward c-Met and
VEGFR2 (Fig. 4). Upon dual kinase
inhibitor screening, engeletin (CAS no. 572-31-6) showed no
inhibitory activity towards c-Met or VEGFR2, whereas chrysoeriol
showed almost 50% binding to c-Met and VEGFR2 with 10 µM kinase
protein (P<0.01). Blumeatin B (CAS no. 79995-67-8) showed weaker
binding to c-Met than engeletin, and showed negligible binding to
VEGFR2 (P<0.01). To understand the SARs of these three
compounds, molecular docking was performed, the results of which
are shown in Fig. 5A-D. Based on
the results shown in Fig. 5A and B,
the modes of interaction of chrysoeriol and engeletin with c-Met
and VEGFR2 were investigated. Engeletin is a classical flavanonol
compound, and chrysoeriol is a flavanonol analogue. The
interactions among these three compounds, c-Met and VEGFR2 and SARs
are discussed below.
Discussion
It is well established that a number of solid tumors
obtain oxygen and nutrients by angiogenesis through various key
kinase proteins expressed in several tissues. As inhibitors that
target only VEGFR2 can result in a feedback increase of c-Met,
which is associated with distant metastasis, dual c-Met and VEGFR2
kinase inhibitors are considered as more efficient therapeutics.
However, no natural products have been identified to inhibit both
c-Met and VEGFR2 kinase activity. Therefore, the present study
aimed to identify natural products as leading compounds for the
development of dual c-Met and VEGFR2 kinase inhibitors.
In the present study, potential dual c-Met and
VEGFR2 kinase inhibitors were examined using virtual screening
technology. In the screening process, eight compounds were selected
as candidates. The results from enzymatic assays showed that the
binding-affinities of chrysoeriol to c-Met and VEGFR2 were
relatively high with Kd values of 12 and 11 µm,
respectively. The results from the docking experiments revealed
that chrysoeriol was able to bind efficiently to the active binding
cavities of c-Met and VEGFR2. Taken together, these results
indicated that chrysoeriol was a potential dual c-Met and VEGFR2
kinase inhibitor. Engeletin and blumeatin B, which have structures
resembling that of chrysoeriol, showed markedly lower binding
affinities. To understand their SARs, molecular docking was
performed in DS 3.0 and the results are shown in Fig. 5. There is a glucoside structure on
the side chain, and polarity was higher than the substituent group
located in the same position of chrysoeriol. For chrysoeriol, two
hydroxyl groups in the A-ring formed a hydrogen bond with Glu1127
and Asp1222, respectively, and the hydroxyl group in the B-ring
formed a hydrogen bond with Arg1086. However, for engeletin, only
one hydroxyl group appeared to interact with c-Met (Fig. 5A). This observation allowed
identification of the critical binding groups of chrysoeriol. In
addition, the active cavity of c-Met was large, therefore,
molecules with a relatively large size, particularly those with
large groups at the location of flavanonol, may have favorable
biological activity.
Similar to chrysoeriol, blumeatin B is a flavanonol
compound. However, for blumeatin B, the hydroxyl group in C-3
formed a torsional tension with the B-ring, which was not conducive
to the formation of a planar structure of A, B and C rings
(Fig. 5C and D). Therefore, it was
difficult to form hydrogen bonds with ASP1046, which may account
for the relatively weak binding of blumeatin B to c-Met and
VEGFR2.
Chrysoeriol, a flavanonol compound, was able to
inhibit the activity of c-Met and VEGFR2, and may serve as the
leading compound for novel drug development. The medicinal value of
the flavanonol compounds has been shown in several drugs and their
safety is widely recognized. Therefore, chrysoeriol and analogs may
offer potential in the chemotherapeutic treatment of cancer. The
results of the present study provide a novel model for the
development of a dual c-Met and VEGFR2 inhibitor to address the
drug resistance of VEGFR2 inhibitors, and the SAR analysis may
guide further structure optimizations.
Current knowledge of the biofunctions of chryoseriol
is limited. To develop chrysoeriol into a drug for clinical use,
further investigations of its cytotoxicity, pharmacokinetics,
absorption, distribution, metabolism and excretion, and
target-signaling pathway are required. The novel findings of the
present study require integration into ongoing investigations on
the structural optimization of chryoseriol, and subsequent cellular
experiments and pharmacology experiments to determine the
biological and pharmacological characterizations.
Acknowledgements
We are very grateful to Professor Mingfeng Bai for
his guidance in writing and language revision.
Funding
The present study was supported by grant from the
National Science Foundation of China (grant no. 81001092), the
Natural Science Foundation of Liaoning Province (grant no.
20170541008), the Fund for Long-term Training of Young Teachers in
Shenyang Pharmaceutical University (grant no. ZQN2015002), the
Training Program Foundation for the Distinguished Young Scholars of
University in Liaoning Province (grant no. LJQ2015109) and the
CSCO-Merck Serono Cancer Research Fund (grant no.
Y-MX2016-013).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
SL, JNC and HWH were mainly responsible for
completing the experiments and writing the manuscript. XYZ, HLJ,
WL, PLW and JW were mainly responsible for analyzing the data. FNL
was responsible for the main idea of this article. All authors read
and approved the manuscript and agree to be accountable for all
aspects of the research in ensuring that the accuracy or integrity
of any part of the work are appropriately investigated and
resolved.
Ethics approval and consent to
participate
This article does not involve ethical conflicts.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interest.
References
|
1
|
Vaupel P: The role of hypoxia-induced
factors in tumor progression. The Oncologist. Suppl 5:S10–S17.
2004. View Article : Google Scholar
|
|
2
|
Carmeliet P: Angiogenesis in health and
disease. Nat Med. 9:653–660. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Qian F, Engst S, Yamaguchi K, Yu P, Won
KA, Mock L, Lou T, Tan J, Li C, Tam D, et al: Inhibition of tumor
cell growth, invasion, and metastasis by EXEL-2880 (XL880,
GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine
kinases. Cancer Res. 69:8009–8016. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Matsumoto S, Miyamoto N, Hirayama T, Oki
H, Okada K, Tawada M, Iwata H, Nakamura K, Yamasaki S, Miki H, et
al: Structure-based design, synthesis, and evaluation of
imidazo[1,2-b]pyridazine and imidazo[1,2-a]pyridine derivatives as
novel dual c-Met and VEGFR2 kinase inhibitors. Bioorg Med Chem.
21:7686–7698. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhan Z, Ai J, Liu Q, Ji Y, Chen T, Xu Y,
Geng M and Duan W: Discovery of anilinopyrimidines as dual
inhibitors of c-met and VEGFR-2: Synthesis, SAR, and cellular
activity. ACS Med Chem Lett. 5:673–678. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mizuno S and Nakamura T: Hepatocyte growth
factor: A regenerative drug for acute hepatitis and liver
cirrhosis. Regen Med. 2:161–170. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Molnarfi N, Benkhoucha M, Funakoshi H,
Nakamura T and Lalive PH: Hepatocyte growth factor: A regulator of
inflammation and autoimmunity. Autoimmun Rev. 14:293–303. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yap TA and de Bono JS: Targeting the
HGF/c-Met axis: State of play. Mol Cancer Ther. 9:1077–1079. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang X, Zhang Y, Yang Y, Lim S, Cao Z, Rak
J and Cao Y: Vascular endothelial growth factor-dependent
spatiotemporal dual roles of placental growth factor in modulation
of angiogenesis and tumor growth. Proc Natl Acad Sci USA.
110:13932–13937. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang J, Jiang X, Jiang Y, Guo M, Zhang S,
Li J, He J, Liu J, Wang J and Ouyang L: Recent advances in the
development of dual VEGFR and c-Met small molecule inhibitors as
anticancer drugs. Eur J Med Chem. 108:495–504. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
De Falco S, Gigante B and Persico MG:
Structure and function of placental growth factor. Trends
Cardiovasc Med. 12:241–246. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nishimoto RN: OFIRMEV: An old drug becomes
new again. Anesth Prog. 61:99–102. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen H, Wu J, Gao Y, Chen H and Zhou J:
Scaffold repurposing of old drugs towards new cancer drug
discovery. Curr Top Med Chem. 16:2107–2114. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cai ZW, Wei D, Schroeder GM, Cornelius LA,
Kim K, Chen XT, Schmidt RJ, Williams DK, Tokarski JS, An Y, et al:
Discovery of orally active pyrrolopyridine- and aminopyridine-based
met kinase inhibitors. Bioorg Med Chem Lett. 18:3224–3229. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cui JJ, McTigue M, Nambu M, Tran-Dubé M,
Pairish M, Shen H, Jia L, Cheng H, Hoffman J, Le P, et al:
Discovery of a novel class of exquisitely selective
mesenchymal-epithelial transition factor (c-MET) protein kinase
inhibitors and identification of the clinical candidate
2-(4-(1-(quinolin-6-ylmethyl)-1H-[1,2,3]triazolo[4,5-b]pyrazin-6-yl)-1H-pyrazol-1-yl)ethanol
(PF-04217903) for the treatment of cancer. J Med Chem.
55:8091–8109. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Oguro Y, Miyamoto N, Okada K, Takagi T,
Iwata H, Awazu Y, Miki H, Hori A, Kamiyama K and Imamura S: Design,
synthesis, and evaluation of
5-methyl-4-phenoxy-5H-pyrrolo[3,2-d]pyrimidine derivatives: Novel
VEGFR2 kinase inhibitors binding to inactive kinase conformation.
Bioorg Med Chem. 18:7260–7273. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fabian MA, Biggs WH III, Treiber DK,
Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P,
Edeen PT, Floyd M, et al: A small molecule-kinase interaction map
for clinical kinase inhibitors. Nat Biotechnol. 23:329–336. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Karaman MW, Herrgard S, Treiber DK,
Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI,
Edeen PT, et al: A quantitative analysis of kinase inhibitor
selectivity. Nat Biotechnol. 26:127–132. 2008. View Article : Google Scholar : PubMed/NCBI
|