Introduction
Hereditary spherocytosis (HS) is a type of hemolytic
anemia caused by abnormal red blood cell (RBC) membrane proteins,
and approximately 75% patients are autosomal dominant for the
disease (1). HS is found globally
at a different prevalence in different countries. The prevalence of
HS is as high as 1/2000 in northern Europe and North America
(2). Although anemia, jaundice and
splenomegaly are typical clinical manifestations of HS, due to the
different types of membrane protein defects, clinically the disease
is still highly heterogeneous. In approximatelly 20–30% of HS
patients, clinical manifestations are not obvious, except for a
compensatory increase in reticulocytes (RETs) (3). In these patients, hemolytic symptoms
are aggravated when certain factors are induced, the most common of
which are infections or continuous heavy physical activities.
Moreover, severe HS patients often present with severe hemolysis,
and require blood transfusions to maintain a hemoglobin (HGB)
content of >60 g/l. However, this procedure can often lead to
iron overload or other complications in these patients. Although HS
is not an uncommon clinical disease, its definitive diagnosis rate
remains relatively low. The lack of knowledge among Chinese
physicians towards the disease is one of the main reasons of
misdiagnosis and mistherapy of many HS patients. This study reports
a case of severe HS that has been misdiagnosed for up to 9 years.
The aim of the present study was to investigate the factors that
led to the long-term misdiagnosis of the disease, and to provide
the proband with reasonable treatment recommendations based on the
international principles of HS treatment.
Materials and methods
Routine examination
Peripheral venous blood (2 ml) was collected from
the proband and the proband's family into tubes containing the
anticoagulant EDTA-K2. Routine blood tests, including
RBC, HGB, mean corpuscular volume (MCV), mean corpuscular
hemoglobin (MCH), mean sphered cell volume (MSCV), mean
reticulocyte volume (MRV), RET and mean platelet volume (MPV), were
performed using the LH 780 automated hematology analyzer (Beckman
Coulter, Carlsbad, CA, USA). Liver function, especially total
bilirubin (TBiL) and direct bilirubin (DBiL), was assessed using
the 7600 fully automated biochemical analyzer (Hitachi, Ltd.,
Tokyo, Japan). This study procured the informed consents of the
proband and his family members, and the approval of the Ethics
Committee of The First Affiliated Hospital of Guangxi Medical
University (Nanning, China). From all participants, written
informed consent for molecular genetic analysis, data analysis and
publication was obtained.
Flow cytometric analysis of
EMA-labeled red blood cells
EMA working solution, at a final concentration of
0.5 mg/ml, was prepared by diluting EMA powder in
phosphate-buffered saline (PBS; Sigma-Aldrich Co., Dorset, UK). The
EMA solution was stored at −20°C in the dark and thawed at 4°C at
the time of use. PBS-washed RBCs (5 µl) were incubated with 20 µl
EMA working solution for 1 h at room temperature in the dark, and
then washed 3 times with PBS containing 0.5% fetal bovine serum
(FBS; Life Technologies Corp., Shanghai, China). The mixture was
centrifuged for 5 min at 2,500 × g to remove the supernatant, and
the pellet was resuspended in 600 µl PBS for flow cytometric
analysis (Beckman Coulter, High Wycombe, UK) (4). The flow cytometer was calibrated by
recording the fluorescence intensity of 10,000 cells in the FL-1
channel prior to use. Data were analyzed using Flowjo software
(FlowJo, LLC, Ashland, OR, USA).
Sodium dodecyl sulfate-polyacrylamide
gel electrophoresis
RBC membrane proteins were extracted as per
Rungaldier's methods (5), and
3.5–17% sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) was prepared based on the protocol of Fairbanks et
al (6). An RBC membrane protein
precipitate was mixed with RBC membrane lysis solution, containing
mercaptoethanol, and incubated for 1 h in a 37°C waterbath. The
proteins were then separated via SDS-PAGE, along with a broad range
protein ladder (Bio-Rad Laboratories, Hercules, CA, USA) to
indicate the molecular size of the unknown proteins. Upon Coomassie
blue staining, protein bands in the gel were scanned using a Gel
Doc 2000 Imaging System (Bio-Rad Laboratories), and protein defects
were analyzed using Quantity One quantification analysis software
(Bio-Rad Laboratories).
Mass spectrometry
In-gel digestion was performed as per the methods
described by Gharahdaghi et al (7). Briefly, the protein samples from the
proband and healthy controls were separated by SDS-PAGE and stained
with Coomassie blue. Target proteins were then removed from the gel
and placed into 1.5-ml microeppendorf tubes. The protein-containing
gel strips were washed twice by water and decolorized, and
dehydrated in pure acetonitrile (ACN) and dried under vacuum. Gel
strips were digested overnight at room temperature using trypsin
and 30 µl digestion buffer (40 mM NH4HCO3 and
10% ACN), and extracted peptides were centrifuged for 5 min at
10,000 × g. The upper layer of the solution was concentrated by
vacuum drying, and the precipitate was resuspended in 0.1%
trifluoroacetic acid solution. The extracted proteins were analyzed
using the 4800 Plus System Mass spectrometer (AB SCIEX, Boston, MA,
USA).
Gene sequence analysis
Peripheral blood DNA of the proband and his family
members was extracted using a DNA extraction kit (Tiangen Biotech
Co., Ltd., Beijing, China). All relevant primers for the exons,
introns and flanking sequences of the gene were designed by the
Primer Premier 5.0 software, and synthesized by Beijing Genomics
Institute (BGI; Beijing, China). The sequences of target mutations
were as follows: exon 2, forward 5′-ACACATATAAGCGGGGCAAC-3′,
reverse 5′-TTGTACCCACACATACCCATTAAC-3′; exon 40, forward
5′-TGAGTGAATATAGATTTTCCGGC-3′, reverse 5′-TTCTACATTTGGGCCAGTCC-3′;
intron 45, forward 5′-TGGACAGATTCATGTTTTGTGG-3′, reverse
5′-TGGAATGAAAATGTCTCAGCAC-3′. PCR amplification was performed using
an Applied Biosystems 9700 Thermal Cycler (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) in a 50-μl reaction, containing
25 µl PCR Master Mix (Takara Bio, Otsu, Japan), 1 µl each of
forward and reverse primers, 0.2 ng DNA and double-distilled water.
Amplification was conducted at 95°C for 5 min, 95°C for 30 sec,
annealing for 30 sec, and 72°C for 40 sec for a total of 35 cycles,
followed by 72°C for 8 min and 4°C hold. PCR products were
recovered, purified from the gel using the Agarose DNA Purification
kit (Solarbio Life Science, Shanghai, China) and sequenced by the
ABI3730×L sequencer (Thermo Fisher Scientific, Inc.). All
variations were classified as pathogenic, potentially pathogenic,
undefined, potentially benign and benign according to the American
College of Medical Genetics and Genomics (ACMG) guidelines and
recommendations (8–10).
Results
Patient and routine blood tests
The proband (male, born in 2006, Han nationality,
Guangxi residence, China) presented with jaundice on the day of
birth, and had 401.5 µmol/l of TBiL. The proband was diagnosed with
neonatal hyperbilirubinemia, but did not undergo exchange
transfusion. At 4 months of age, the proband appeared pale and
dispirited with no obvious presence of inducers. He was diagnosed
with severe anaemia, and received repeated transfusion therapy at a
local hospital at an average of one transfusion per month. At 2
years of age, the proband was admitted to the hospital because of a
fever (up to 38.8°C) that lasted 3 days and was induced by
bronchopneumonia. Moreover, the proband showed symptoms resembling
thalassemia and was diagnosed with thalassemia and G6PD deficiency.
Laboratory test results indicated 2.64×1012/l RBC, 54
g/l haemoglobin (HGB), 68.00 fl MCV, 277.0 g/l mean corpuscular
hemoglobin concentration (MCHC), 47.3 µmol/l TBiL and 15.9 µmol/l
DBiL. The proband was given antitussive and antipyretic medication
but did not show any improvement. Upon antiviral and anti-infection
therapy, the body temperature of the proband returned to normal and
coughing disappeared. Routine blood tests revealed mild anemia,
suggesting improvement of the thalassemia-like illness. At 8 years
of age, the proband was admitted to a Hospital located in Nanning
due to acute upper respiratory tract infections accompanied by
breathing difficulties. Laboratory test results indicated
1.04×1012/l RBC, 30 g/l HGB, 316.70 µmol/l TBiL, 109.52
µmol/l DBiL, 207.2 µmol/l IBiL, 3+ urinary bilirubin (BiL) and 3+
urinary bile, therefore, the proband was diagnosed with chronic
hemolytic anemia and anemic cardiomyopathy. In 2015, the proband
came to our hospital for examination due to ‘9 years of jaundice
and 2 days of fever’, without any history of genetic disorders.
Physical examination showed that the proband was clearly conscious
but low-spirited, and had jaundice in the sclera and on the skin,
along with hepatosplenomegaly. Laboratory test results demonstrated
that the proband was negative for the Coombs test, autoantibody,
and hepatitis B surface antigen, and had a high level of serum
ferritin (3503 ng/ml), normal immunoglobulin levels, normal genetic
testing for thalassemia, normal G6PD activity, and increased
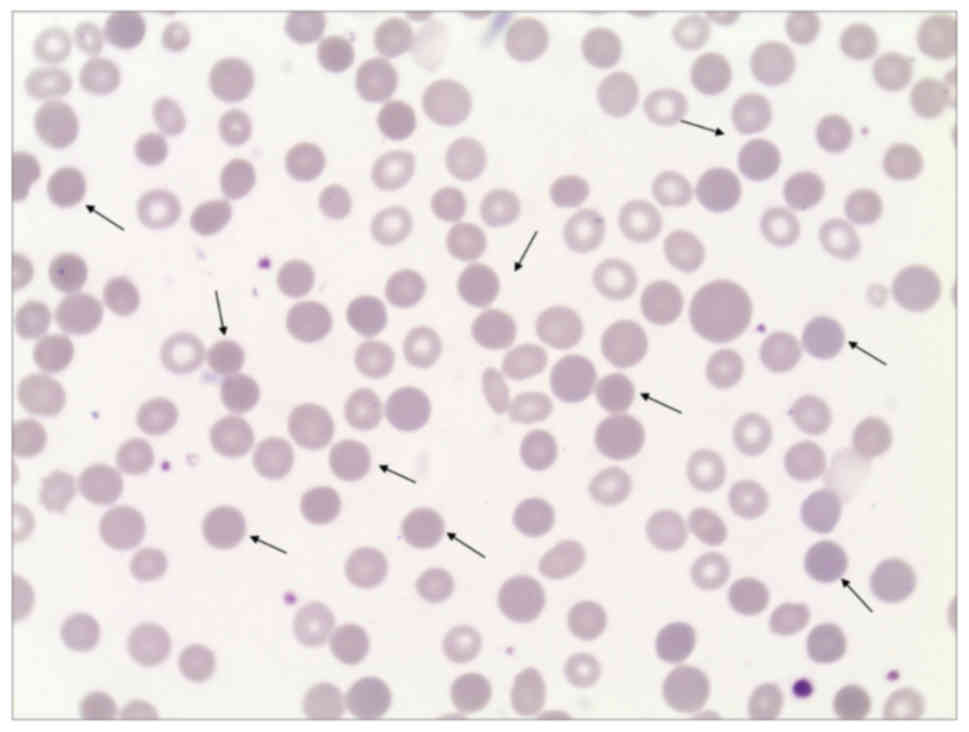
erythrocyte osmotic fragility and spherocytes (25% of all RBCs)
(Fig. 1). In consideration of his
medical history, clinical manifestations and laboratory test
results, the proband was finally diagnosed with HS. Upon completion
of relevant examinations and preoperative preparations, the proband
underwent splenectomy under general anesthesia, and was given
anti-inflammatory, hemostatic and transfusion therapies after the
operation. The proband was discharged from the hospital once his
vital signs had stabilized. Routine blood and biochemical tests
were conducted on the proband's family members and the results are
presented in Table I.
 | Table I.Laboratory test results of the HS
proband and his family members. |
Table I.
Laboratory test results of the HS
proband and his family members.
| Item | Proband | Father | Mother | Normal range |
|---|
| RBC
(×1012/l) | 1.69 | 4.93 | 4.8 | 3.5–5.5 |
| Hb (g/l) | 48.9 | 149.2 | 134.4 | 110.0–160.0 |
| MCV (fl) | 79.69 | 93.77 | 85.81 | 82.00–95.00 |
| MCHC (g/l) | 360.8 | 322.6 | 326.1 | 316.0–354.0 |
| MSCV (fl) | 75.68 | 96.38 | 80.59 | 84.00–104.00 |
| MRV (fl) | 89.30 | 112.18 | 106.68 | 101.00–119.00 |
| Ret (%) | 7 | 1.4 | 0.9 | 0.0–2.0 |
| TBIL (µmol/l) | 178.10 | 15.6 | 10.4 | 3.40–20.50 |
| DBIL (µmol/l) | 59.20 | 5.5 | 3.0 | 0.00–6.8 |
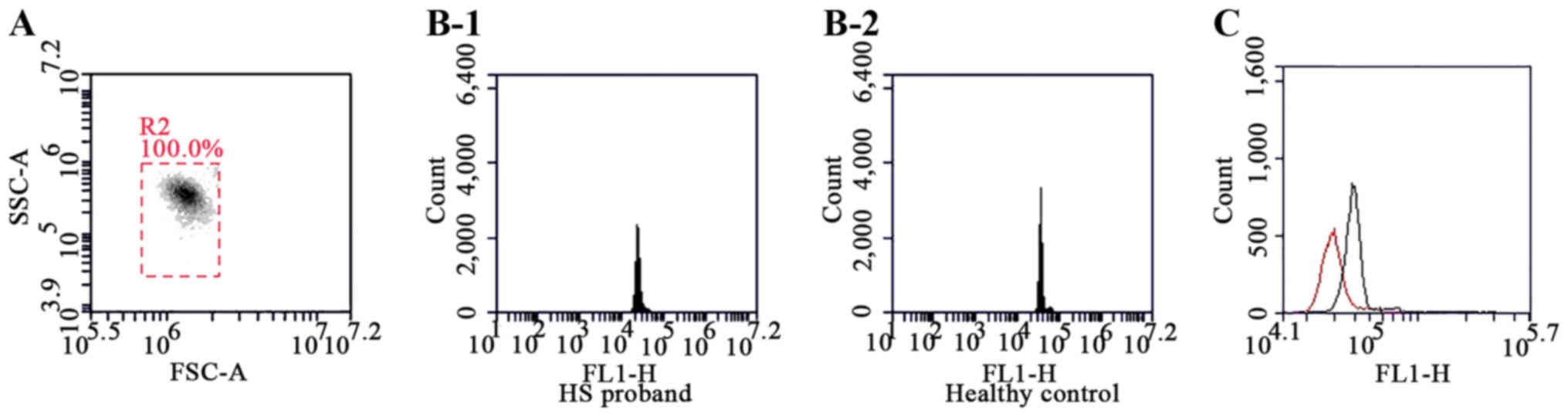
Fluorescence results for red blood
cells from the patient and healthy controls
Blood was collected from 10 healthy individuals that
were age and sex matched with the proband, and was used as negative
controls to reduce the errors between samples (11). A blank control was also included.
Fluorescence of EMA-labeled RBCs was expressed as mean fluorescence
channel units (MFC). The percent reduction in MCF of the proband
was calculated as follows:
MCF(%)=MCFofhealthycontrols-MCFofprobandMCFhealthycontrolsx100%
It was found that the MCF of the proband's blood was
reduced by 35.77% compared with those of the healthy controls
(Fig. 2).
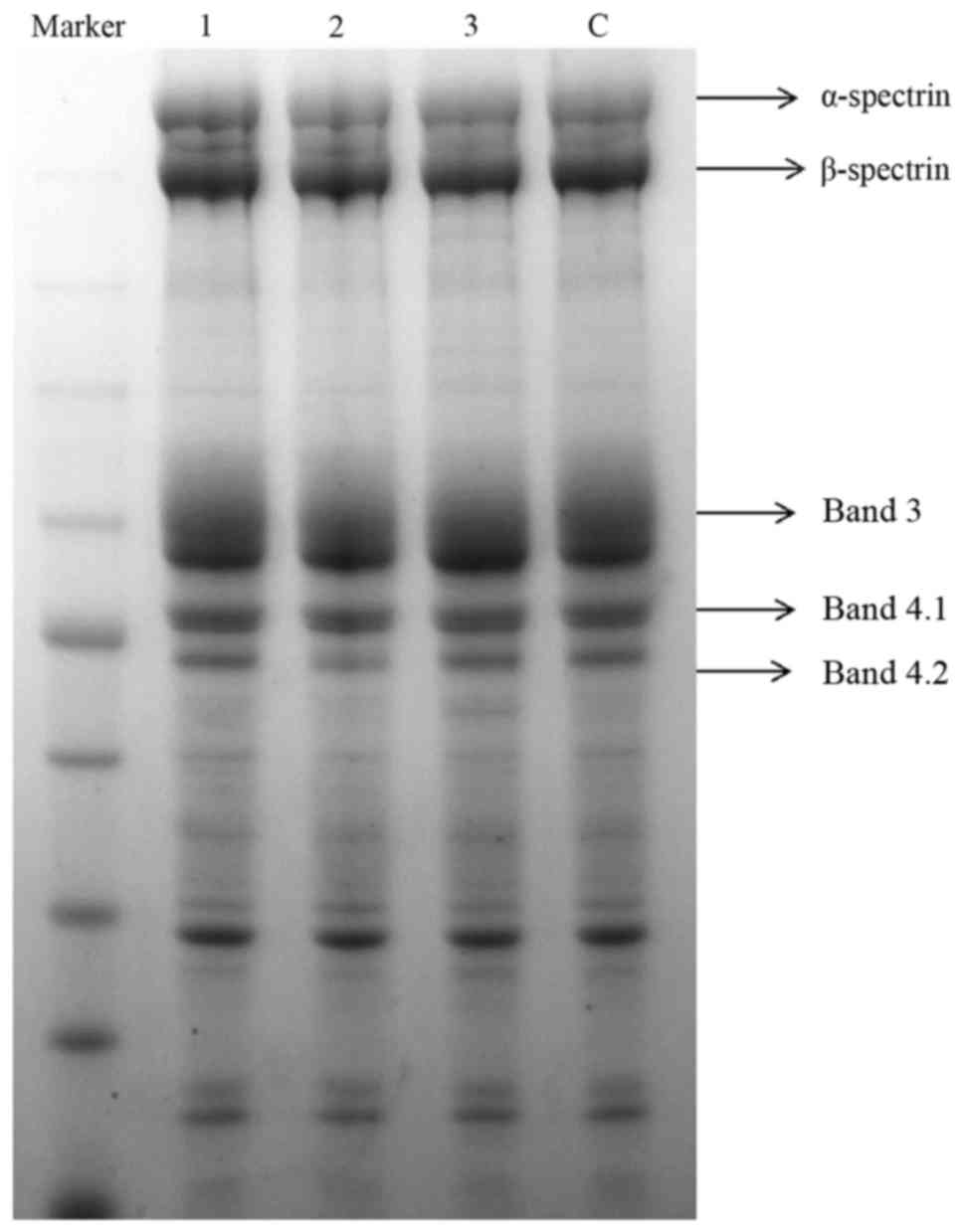
Analysis of red blood cell membrane
proteins
RBC membrane proteins of the proband were evaluated
by SDS-PAGE and Commassie brilliant blue staining. The proband and
his family did not show any significant reduction in membrane
protein levels compared with that of the healthy controls (Fig. 3).
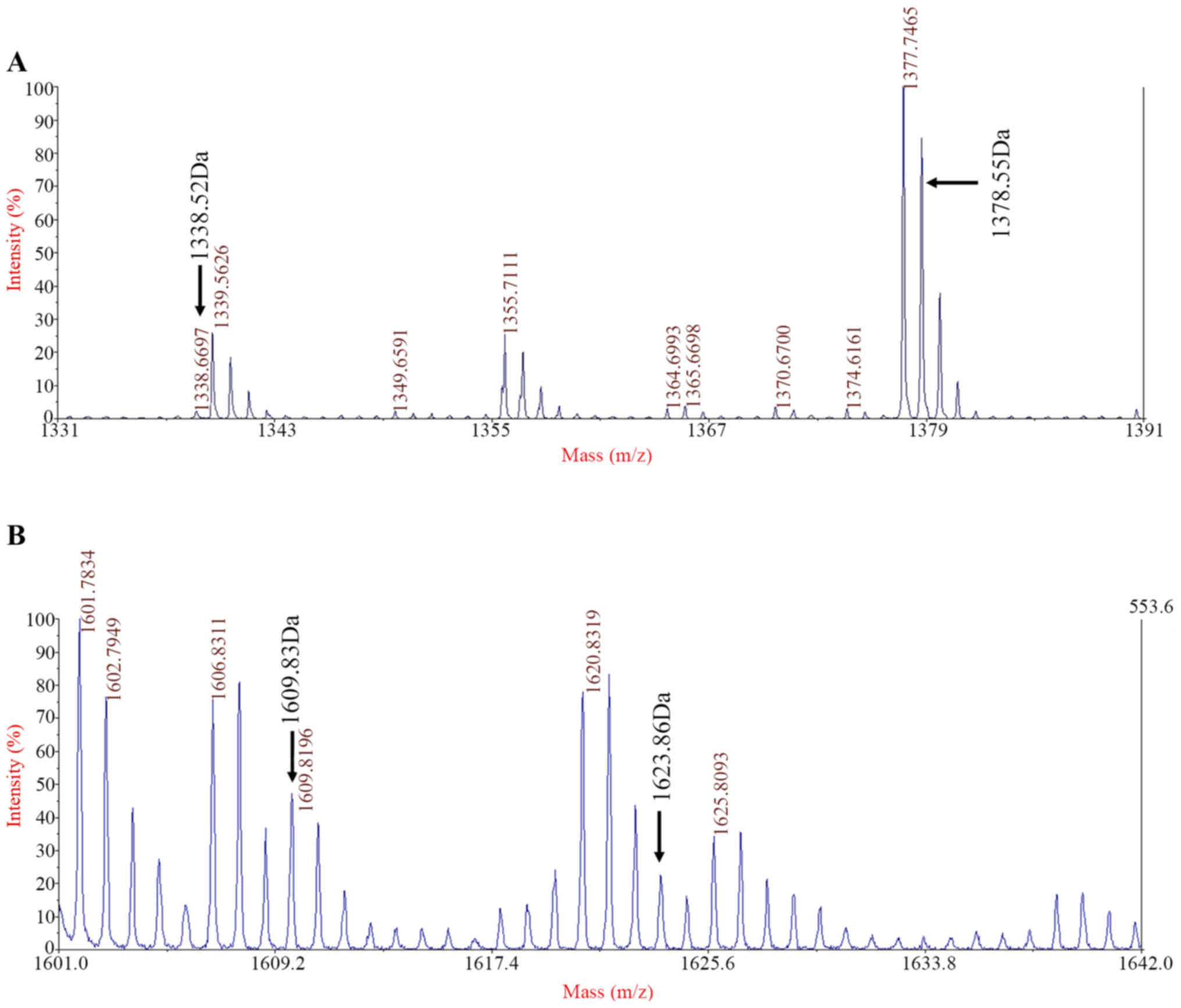
Matrix-assisted laser
desorption/ionization time of flight mass spectrometry (MALDI-TOF
MS)
The α-spectrin peptide chain of the proband was
analyzed using MALDI-TOF MS. When comparing the α-spectrin peptide
chain of the proband with that of reference peptides in the
database it was found that the molecular sizes of RBC α-spectrin in
the proband were 1,338.52 Da (LEDSYHLQVFK) and 1,609.83 Da
(GDCGDTLAATQSLVMK) as opposed to 1,378.55 Da (LEDSYPLQVFK) and
1,623.86 Da (GDCGDTLAATQSLLMK) of normal α-spectrin. This indicated
that the α-spectrin chain of the proband contained two abnormal
fragments (Fig. 4).
DNA analysis
Human RBC membrane genes SPTA1, SPTB, SLC4A1,
ANKI and EPB42 were amplified by PCR and analyzed by
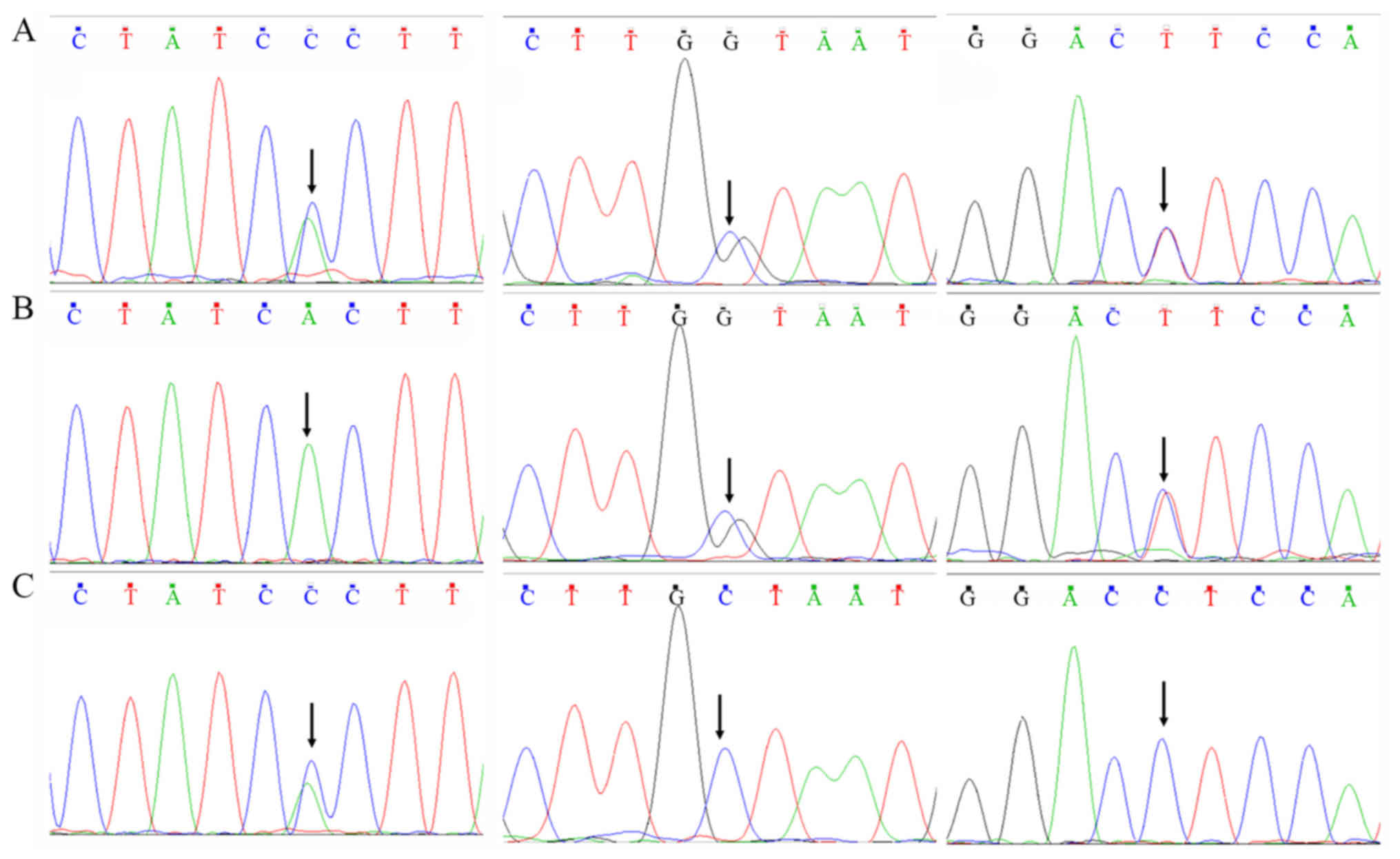
genomic DNA sequencing (Fig. 5).
Comparing the sequencing results to the reference sequences in the
NCBI database showed that the proband carried 3 mutations,
including c.161A>C heterozygous missense mutation in exon 2 of
the SPTA1 gene, leading to the substitution of histidine
(His) by proline (Pro) at position 54 of the α-spectrin peptide;
c.5572C>G heterozygous missense mutation in exon 40 of the
SPTA gene, leading to the substitution of leucine (Leu) by
valine (Val) at position 1858; and a 6531–12C>T heterozygous
mutation in intron 45. Mutation analysis of the proband's family
members showed that the His54Pro mutation was present in the
mother, whereas both Leu1858Val and 6531–12C>T mutations were
present in the father (Fig. 6).
Discussion
Hereditary spherocytosis (HS) has very diverse
clinical manifestations, which typically include anemia, jaundice
and splenomegaly. Common complications include cholecystolithiasis,
hemolytic crisis, hematopoietic crisis and are sometimes combined
with delayed growth, heart diseases and neuromuscular disorders.
Laboratory test results of HS generally show increases in
reticulocytes (RETs) with reduced volume, peripheral spherocytes,
RBC osmotic fragility, mean corpuscular hemoglobin concentration
(MCHC) and red blood cell distribution width (RDW) (3). Therefore, timely and accurate
diagnosis of HS is critical for optimal treatment regimen and the
prognosis of the patient. The Guidelines for the Diagnosis and
Management of HS that was developed by the British Committee for
Standards in Hematology stated that an individual can be
definitively diagnosed with HS without further testing if he/she
has a family history of HS, and presents with typical clinical and
blood characteristics of HS (12).
However, a negative family history was one of the main reasons for
misdiagnosis of the proband. It has been shown that many mild HS
patients have no obvious clinical symptoms, which makes it
especially difficult to diagnose the disease using conventional
methods (13). This was
particularly true in case of the proband's mother, as the size and
morphology of mature RBCs in her peripheral blood were relatively
consistent with almost no signs of abnormality, and HS could only
be identified through a genetic testing approach. Another
possibility would be that HS was recessively inherited through the
family, and individuals with recessive inheritance or serendipitous
mutation have no family history of the disease. Thus, these
possibilities may all have resulted in the prolonged misdiagnosis
and mistherapy of the patient.
Identification of a suitable laboratory test has
important implication in the improvement of HS diagnosis. Since
anemia, jaundice and splenomegaly are all clinical manifestations
of G6PD deficiency, thalassemia, and AIHA, and given that
thalassemia is highly prevalent in regions of the Guangxi Province,
it is easy to confuse HS with these diseases. When a patient is
suspected of having hemolytic anemia, examination of peripheral RBC
morphology, RBC protein electrophoresis, genetic analysis,
detection of G6PD levels, and a Coombs test can all help
distinguish HS from these diseases (14). In particular, the concurrent use of
genetic analysis and RBC protein electrophoresis is a conventional
means for the diagnosis of thalassemia in Guangxi regions. In
addition, routine blood testing is also a common conventional
measure for HS screening. In a study by Broséus et al
(15) it was reported that MCV
<80 f1, MCHC >354 g/l and RDW >14% in routine peripheral
blood tests showed a 63% sensitivity and a 100% specificity for the
diagnosis of HS. Subsequently, Liao et al (16) found that both sensitivity and
specificity of MCHC were undesirable for the diagnosis of HS in
patients with increased RET levels, and that increased blood TBiL
levels could interfere with the detection of HGB and result in a
false increase in MCHC (17). On
the other hand, MSCV is a valuable indicator for HS screening
(15). A study in 57 HS patients
and 109 thalassemia patients by Liao et al (16) showed that MSCV<MCV could
effectively distinguish HS from thalassemia. Furthermore, Tao et
al (18) reported that the
combination of MSCV<MCV, flow cytometry and osmotic fragility
test demonstrated 89.28% sensitivity and 96.14% specificity for the
diagnosis of H. MRV is a novel indicator for HS screening. The
value of MRV in the screening of HS was first reported by Lazarova
et al (19) in 2014, where
they demonstrated that MRV ≤96.72 fl showed 100% sensitivity and
88% specificity for the diagnosis of HS. Moreover, in a study
performed on 53 HS patients, 109 thalassemia patients, 56 G6PD
deficiency patients, and 52 AIHA patients by Xu et al
(20) it was demonstrated that MRV
≤95.77 fl showed 86.8% sensitivity and 91.2% specificity for the
diagnosis of HS. The limitations of basic hospitals and
insufficient knowledge of clinicians towards HS were the main
reasons for misdiagnosis of the proband. The proband was first
diagnosed with thalassemia and G6PD deficiency by a local hospital.
At the first time of diagnosis, HGB electrophoresis and G6PD
detection were not conducted. Thus, the proband was diagnosed with
hemolytic anemia at another hospital in Nanning, China. It is
important to note that at the time of his second diagnosis, the RBC
parameters of the proband were typical, where pre-treatment values
of MCV, MCHC, MRV and MSCV all met the diagnostic criteria for HS.
Therefore, diagnosis of the patient would not have been difficult
if the clinician had performed reasonable comprehensive analyses
and further diagnostic testing.
The undesirable sensitivity and specificity of
traditional HS screening methods are also a reason of prolonged
misdiagnosis or missed diagnosis of several HS patients (21). SDS-PAGE is mainly used for the
detection of changes in the relative levels of RBC membrane
proteins in HS patients compared to healthy controls. The SDS-PAGE
approach can be used to detect the presence of abnormal proteins in
roughly 80% of HS patients, however, the type of protein defect
cannot be confirmed in approximately 10% of HS patients (22). In this study, membrane protein
defects in the proband were not detected by SDS-PAGE because the
excess amount of ankyrin proteins in the RETs interfered with the
gradient of the gel and the separation of RBC membrane
proteins.
Detection of EMA-labeled RBCs by flow cytometry is a
novel method of preliminary screening for HS, especially in
atypical patients (23). EMA is a
fluorescent probe that covalently binds to Lys-430 on the first
extracellular ring of band 3 protein and spectrin (24). Upon EMA labeling, the MCF of RBCs is
reduced, and >21% reduction in MCF was found to show 97%
sensitivity and 100% specificity for the diagnosis of HS (25). This approach is fast, requires a
small amount of blood, and is therefore, a superior auxiliary
method for the clinical diagnosis of HS.
HS is caused by the deficiency or functional change
of RBC membrane proteins resulting from genetic mutations. As the
shape-changing ability of RBCs is reduced, and upon entering the
microcirculation of the spleen, these cells are easily captured for
phagocytosis. The main RBC membrane protein-encoding genes include
SPTA1, SPTB, ANK1, SLC4A1 and EPB42, and the
corresponding proteins they encode include α-spectrin, β-spectrin,
ankyrin, band 3 and 4.2 protein, respectively. Previous studies
have shown that the severity of HS is associated with the type of
membrane protein defect in the patient (26). Specifically, patients with defects
in α-spectrin present with severe anemia, while those with defects
in ankyrin, band 3 and 4.2 protein present with mild to moderate
anemia (27).
It is found that approximately 5% of HS patients
have a defect in α-spectrin and they acquired the disease by
recessive inheritance (28).
Generally, a single heterozygous mutation in the SPTA1 gene
will not lead to protein defects or the appearance of obvious
clinical symptoms in the patients, because overexpression of
α-spectrin will maintain protein levels. Only homozygous or
compound heterozygous mutations will result in disease (29). The proband was identified as a
carrier for compound heterozygous mutations in the SPTA1
gene. In addition, 3 site mutations were found in the proband,
including c.161A>C in exon 2, c.5572C>G in exon 40, and
6531–12C>T in intron 45. In particular, c.161A>C was a newly
identified mutation that had not yet been reported in the dbSNP
database, and hence there is no information available regarding the
mutation in the 1000 Genomes dataset, such as its distribution
frequency and the mechanism of pathogenesis. Harmfulness prediction
was made through the web PolyPhen-2 (genetics.bwh.harvard.edu/pph2/) (30) and Mutation Taster (www.mutationtaster.org/). The results showed that
c.161A>C is pathogenic or potentially pathogenic according to
the ACMG guidelines and recommendations. We speculated that the
mutation may affect the level of mRNA expression or the structural
stability of proteins. In addition, c.5572C>G and 6531–12C>T
together result in a polymorphic ‘LELY’ allele that is highly
frequent but lowly expressed (31).
The 6531–12C>T mutation can cause 50% of the transcription
codons to be skipped during translation of exon 46, resulting in
the deficiency of 6 amino acids that link α-spectrin with
β-spectrin. The c.5572C>G mutation can lead to amino acid
changes and linkage disequilibrium with 6531–12C>T. In general,
a single heterozygous mutation (mother) or αLELY(father)
is sufficient to ensure the correct structural expression of the
α-spectrin protein and prevents the development of corresponding
pathological changes in the carrier. Even if an individual develops
relevant pathological changes, the damage is very minimal (32). Simultaneous inheritance of both
alleles in the proband resulted in significant anemia due to the
vicious interaction between the two mutations, in which the
presence of αLELY exacerbated the pathogenesis of
c.161A>C, and the presence of c.161A>C exacerbated the trans
effect of αLELY on the structure of α-spectrin (33).
In conclusion, although HS is not an uncommon or
rare disorder, its sporadic nature and the absence of reliable
laboratory indicators for definitive diagnosis of HS prompt the
development of a comprehensive assessment involving multiple
laboratory indicators that will allow for accurate diagnosis of the
disease. EMA-labeled RBC detection by flow cytometry, SDS-PAGE, and
an osmotic fragility test can be used concurrently to determine the
fragility, and the presence of membrane protein defects in RBCs.
Furthermore, performance of a blood smear and routine laboratory
tests, such as RBC, HBG, MCV, MSCV, RET%, MRV, TBiL and DBiL, can
effectively distinguish HS from thalassemia, G6PD deficiency,
iron-deficiency anemia and AIHA.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (no. 81360263).
Availability of data and materials
The datasets supporting the conclusions of this
article are included within the article.
Authors' contributions
SM drafted the initial manuscript and organized the
data and contributed to the design of the report and to the
critical writing of the manuscript. HW is a physician involved in
the care of the patient in the Pediatric Hematology Clinic. YQ is
the principal physician caring for the hematological issues of this
patient. XD, LL and ZD, research scientists at Clinical Laboratory,
conceived of and established the flow-cytometric studies and the
gene sequencing aspects that are central to this report. FL, a
senior hematologist and scientist, has been involved as a
consultant on the clinical case and supervises the scientists in
aspects of hematology and genetics. He provided critical advice in
case management, contributed to the design and critically reviewed
the manuscript. All authors read and approved the manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Written informed consent was obtained from all
participants in this study. The study protocol was approved by the
Ethics Committee of the First Affiliated Hospital at the Guangxi
Medical University in China.
Patient consent for publication
This study procured the informed consents of the
proband and his family members. From all participants, written
informed consent for molecular genetic analysis, data analysis and
publication was obtained.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
EMA
|
eosin-5-maleimide
|
|
DBiL
|
direct bilirubin
|
|
HGB
|
haemoglobin
|
|
HS
|
hereditary spherocytosis
|
|
IBiL
|
indirect bilirubin
|
|
MALDI-TOF
|
matrix-assisted laser
desorption/ionization time of flight
|
|
MCF
|
mean channel florescence
|
|
MCHC
|
mean corpuscular hemoglobin
concentration
|
|
MCV
|
mean corpuscular volume
|
|
MFC
|
mean fluorescence channel units
|
|
MPV
|
mean platelet volume
|
|
MRV
|
mean reticulocyte volume
|
|
MSCV
|
mean sphered corpuscular volume
|
|
RETs
|
reticulocytes
|
|
RBCs
|
reb blood cells
|
|
RDW
|
red blood cell distribution width
|
|
SDS-PAGE
|
sodium dodecyl sulfate-polyacrylamide
gel electrophoresis
|
|
TBiL
|
total bilirubin
|
References
|
1
|
Yawata Y, Kanzaki A, Yawata A, Doerfler W,
Ozcan R and Eber SW: Characteristic features of the genotype and
phenotype of hereditary spherocytosis in the Japanese population.
Int J Hematol. 71:118–135. 2000.PubMed/NCBI
|
|
2
|
Bolton-Maggs PH, Langer JC, Iolascon A,
Tittensor P and King MJ; General Haematology Task Force of the
British Committee for Standards in Haematology, : Guidelines for
the diagnosis and management of hereditary spherocytosis − 2011
update. Br J Haematol. 156:37–49. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
King MJ, Garçon L, Hoyer JD, Iolascon A,
Picard V, Stewart G, Bianchi P, Lee SH and Zanella A; International
Council for Standardization in Haematology, : ICSH guidelines for
the laboratory diagnosis of nonimmune hereditary red cell membrane
disorders. Int J Lab Hematol. 37:304–325. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ciepiela O, Kotuła I, Górska E,
Stelmaszczyk-Emmel A, Popko K, Szmydki-Baran A, Adamowicz-Salach A
and Demkow U: Delay in the measurement of eosin-5′-maleimide (EMA)
binding does not affect the test result for the diagnosis of
hereditary spherocytosis. Clin Chem Lab Med. 51:817–823. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rungaldier S, Oberwagner W, Salzer U,
Csaszar E and Prohaska R: Stomatin interacts with GLUT1/SLC2A1,
band 3/SLC4A1, and aquaporin-1 in human erythrocyte membrane
domains. Biochim Biophys Acta. 1828:956–966. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fairbanks G, Steck TL and Wallach DF:
Electrophoretic analysis of the major polypeptides of the human
erythrocyte membrane. Biochemistry. 10:2606–2617. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gharahdaghi F, Weinberg CR, Meagher DA,
Imai BS and Mische SM: Mass spectrometric identification of
proteins from silver-stained polyacrylamide gel: A method for the
removal of silver ions to enhance sensitivity. Electrophoresis.
20:601–605. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Richards CS, Bale S, Bellissimo DB, Das S,
Grody WW, Hegde MR, Lyon E and Ward BE; Molecular Subcommittee of
the ACMG Laboratory Quality Assurance Committee, : ACMG
recommendations for standards for interpretation and reporting of
sequence variations: Revisions 2007. Genet Med. 10:294–300. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al ACMG
Laboratory Quality Assurance Committee, : Standards and guidelines
for the interpretation of sequence variants: a joint consensus
recommendation of the American College of Medical Genetics and
Genomics and the Association for Molecular Pathology. Genet Med.
17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li MM, Datto M, Duncavage EJ, Kulkarni S,
Lindeman NI, Roy S, Tsimberidou AM, Vnencak-Jones CL, Wolff DJ,
Younes A, et al: Standards and guidelines for the interpretation
and reporting of sequence variants in cancer: A joint consensus
recommendation of the Association for Molecular Pathology, American
Society of Clinical Oncology, and College of American Pathologists.
J Mol Diagn. 19:4–23. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hunt L, Greenwood D, Heimpel H, Noel N,
Whiteway A and King MJ: Toward the harmonization of result
presentation for the eosin-5′-maleimide binding test in the
diagnosis of hereditary spherocytosis. Cytometry B Clin Cytom.
88:50–57. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Manciu S, Matei E and Trandafir B:
Hereditary spherocytosis - diagnosis, surgical treatment and
outcomes. A literature review. Chirurgia (Bucur). 112:110–116.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Barcellini W, Bianchi P, Fermo E,
Imperiali FG, Marcello AP, Vercellati C, Zaninoni A and Zanella A:
Hereditary red cell membrane defects: Diagnostic and clinical
aspects. Blood Transfus. 9:274–277. 2011.PubMed/NCBI
|
|
14
|
Guillaud C, Loustau V and Michel M:
Hemolytic anemia in adults: Main causes and diagnostic procedures.
Expert Rev Hematol. 5:229–241. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Broséus J, Visomblain B, Guy J, Maynadié M
and Girodon F: Evaluation of mean sphered corpuscular volume for
predicting hereditary spherocytosis. Int J Lab Hematol. 32:519–523.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liao L, Deng ZF, Qiu YL, Chen P, Chen WQ
and Lin FQ: Values of mean cell volume and mean sphered cell volume
can differentiate hereditary spherocytosis and thalassemia.
Hematology. 19:393–396. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kar R and Sharma CB: Bilirubin peak can be
mistaken as Hb Bart's or Hb H on High-performance liquid
chromatography. Hemoglobin. 35:171–174. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tao YF, Deng ZF, Liao L, Qiu YL, Chen WQ
and Lin FQ: Comparison and evaluation of three screening tests of
hereditary spherocytosis in Chinese patients. Ann Hematol.
94:747–751. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lazarova E, Pradier O, Cotton F and Gulbis
B: Automated reticulocyte parameters for hereditary spherocytosis
screening. Ann Hematol. 93:1809–1818. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu Y, Yang W, Liao L, Deng Z, Qiu Y, Chen
W and Lin F: Mean reticulocyte volume: A specific parameter to
screen for hereditary spherocytosis. Eur J Haematol. 96:170–174.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Deng Z, Liao L, Yang W and Lin F:
Misdiagnosis of two cases of hereditary spherocytosis in a family
and review of published reports. Clin Chim Acta. 441:6–9. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
King MJ and Zanella A: Hereditary red cell
membrane disorders and laboratory diagnostic testing. Int J Lab
Hematol. 35:237–243. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang J, Zheng B, Zhao Y, Chen X, Liu Y, Bo
L, Zheng Y, Zhang F, Ru K and Wang H: Flow cytometric test using
eosin-5′-maleimide (EMA) labelling of red blood for diagnosis of
hereditary spherocytosis. Zhonghua Xue Ye Xue Za Zhi. 36:598–601.
2015.(In Chinese). PubMed/NCBI
|
|
24
|
Suemori S, Wada H, Nakanishi H, Tsujioka
T, Sugihara T and Tohyama K: Analysis of hereditary elliptocytosis
with decreased binding of eosin-5-maleimide to red blood cells.
BioMed Res Int. 2015:4518612015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Joshi P, Aggarwal A, Jamwal M, Sachdeva
MU, Bansal D, Malhotra P, Sharma P and Das R: A comparative
evaluation of Eosin-5′-maleimide flow cytometry reveals a high
diagnostic efficacy for hereditary spherocytosis. Int J Lab
Hematol. 38:520–526. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Perrotta S, Della Ragione F, Rossi F,
Avvisati RA, Di Pinto D, De Mieri G, Scianguetta S, Mancusi S, De
Falco L, Marano V, et al: Beta-spectrinBari: A truncated beta-chain
responsible for dominant hereditary spherocytosis. Haematologica.
94:1753–1757. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Serpillon S, Floyd BC, Gupte RS, George S,
Kozicky M, Neito V, Recchia F, Stanley W, Wolin MS and Gupte SA:
Superoxide production by NAD(P)H oxidase and mitochondria is
increased in genetically obese and hyperglycemic rat heart and
aorta before the development of cardiac dysfunction. The role of
glucose-6-phosphate dehydrogenase-derived NADPH. Am J Physiol Heart
Circ Physiol. 297:H153–H162. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Perrotta S, Gallagher PG and Mohandas N:
Hereditary spherocytosis. Lancet. 372:1411–1426. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Da Costa L, Galimand J, Fenneteau O and
Mohandas N: Hereditary spherocytosis, elliptocytosis, and other red
cell membrane disorders. Blood Rev. 27:167–178. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Adzhubei I, Jordan DM and Sunyaev SR:
Predicting functional effect of human missense mutations using
PolyPhen-2. Curr Protoc Hum Genet Chapter. 7:Unit7.20. 2013.
|
|
31
|
Maréchal J, Wilmotte R, Kanzaki A, Dhermy
D, Garbarz M, Galand C, Tang TK, Yawata Y and Delaunay J: Ethnic
distribution of allele alpha LELY, a low-expression allele of
red-cell spectrin alpha-gene. Br J Haematol. 90:553–556. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Randon J, Boulanger L, Marechal J, Garbarz
M, Vallier A, Ribeiro L, Tamagnini G, Dhermy D and Delaunay J: A
variant of spectrin low-expression allele alpha LELY carrying a
hereditary elliptocytosis mutation in codon 28. Br J Haematol.
88:534–540. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tolpinrud W, Maksimova YD, Forget BG and
Gallagher PG: Nonsense mutations of the alpha-spectrin gene in
hereditary pyropoikilocytosis. Haematologica. 93:1752–1754. 2008.
View Article : Google Scholar : PubMed/NCBI
|