Introduction
Accumulating evidence suggests that different cancer
types are hierarchically organized and only a small fraction of all
tumor cells are essential for tumor initiation, invasive growth and
potential dissemination to distant organ sites, through blood or
lymphatic vessels (1). These few
cells, termed ‘cancer stem cells’ (CSCs), possess an enhanced
self-renewal capacity and the ability to differentiate into
multiple lineages (2,3). Although certain properties and
functions of CSCs have been explored, further in-depth research is
impeded by the difficulty of isolating pure CSCs and maintaining
them in vitro (4).
Despite numerous attempts, there remains a lack of
robust methods able to isolate CSCs. Previously, certain studies
have experienced difficulty using markers that originally appeared
to accurately distinguish tumorigenic from non-tumorigenic cells
(5–7). Therefore, the surface markers that
have been used to isolate CSCs may be unreliable.
In an attempt to generate CSCs from normal cancer
cells using small molecules, a homogenously converted cell
population was generated by combined treatment with two small
molecules, CHIR99021 and PD184352 (5). The cells exhibited properties similar
to CSCs, including a self-renewal capacity and stability in
maintaining CSC characteristics. As an inhibitor of glycogen
synthase kinase 3 (GSK3), CHIR99021 (referred to as CHIR) is
implicated in the self-renewal of embryonic stem cells, activating
canonical Wnt signaling (8,9). PD184352 (referred to as PD) is a small
inhibitor of mitogen-activated protein kinase kinase (MEK) that has
been demonstrated to suppress cell proliferation (10). Subsequent studies reported that the
expression levels of certain proteins that notably mediate
migration and invasion change during the process of
epithelial-mesenchymal transition (EMT) (11,12).
This novel method provides a practical strategy to
generate CSCs, which may be subsequently used for small molecule
drug screening in vitro. The mechanisms that mediated the
induction of CSCs remain largely unknown and require further
investigation. The aim of the present study was to investigate the
effect of the inhibition of GSK3 and MEK in CSCs and the underlying
molecular mechanisms.
Materials and methods
Cell culture
Immortalized human mammary epithelial cells (HMLE;
Type Culture Collection of the Chinese Academy of Sciences,
Shanghai, China) were maintained in Dulbecco's modified Eagle's
medium (DMEM; Thermo Fisher Scientific, Inc., Waltham, MA, USA):
F12 media (Thermo Fisher Scientific, Inc.) (1:1) supplemented with
insulin (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), epidermal
growth factor, hydrocortisone (Sigma-Aldrich; Merck KGaA) and 5%
fetal bovine serum (FBS; GE Healthcare Life Sciences, Logan, UT,
USA) at 37°C in a humidified atmosphere containing 5%
CO2. The cells were passaged every 3–5 days. The HMLE
cells were treated with 3 µM PD (Meiyan Biological Technology Co.,
Ltd, Shanghai, China) and 2 µM CHIR (Wako Pure Chemicals
Industries, Ltd., Osaka, Japan). Cultured cells were imaged using
an inverted confocal laser scanning microscope (TCS SP5; Leica
Microsystems GmbH, Wetzlar, Germany) on day 3 at a magnification of
×40-200. In brief, single cells were transferred to 50% Matrigel
and cultured in mammary epithelial cell growth medium (Clonetics;
Lonza Group, Ltd., Basel, Switzerland) supplemented with 10 ng/ml
basic fibroblast growth factor for an additional 7 days at 37°C.
The structures were then imaged using an inverted confocal laser
scanning microscope (TCS SP5; Leica Microsystems GmbH) at a
magnification of ×40-200.
Immunocytochemical analysis
HMLE cells were washed once with PBS (Invitrogen;
Thermo Fisher Scientific, Inc.; without Ca2+ and
Mg2+) and were fixed with a 4% formaldehyde solution
containing 0.15% picric acid (Sigma-Aldrich; Merck KGaA) in PBS for
20 min at 37°C, followed by three washes with PBS. Blocking and
permeabilization were performed using 10% donkey serum (Jackson
ImmunoResearch Laboratories, Inc., West Grove, PA, USA) and 0.3%
Triton X-100 (Sigma-Aldrich; Merck KGaA) solution in PBS for 1 h at
room temperature. All primary antibodies (mouse polyclonal
anti-E-cadherin and anti-vimentin; dilution, 1:1,000; catalog nos.
3195 and 3932, respectively; Cell Signaling Technology, Inc.,
Danvers, MA, USA) were diluted in 1% bovine serum albumin (BSA) and
incubated overnight at 4°C. After 1 h of washing with 0.1% BSA in
PBS, samples were incubated with Alexa 555- or Alexa 488-conjugated
anti-mouse secondary antibodies (dilution, 1:500; cat nos. P0190
and P0188, respectively; Beyotime Institute of Biotechnology,
Haimen, China) for 1 h at room temperature and nuclei were stained
with DAPI (100 µl; dilution, 1:1,000; Sigma-Aldrich; Merck KGaA) at
room temperature for 2–3 min. All images were captured using a
Nikon Eclipse 80i fluorescence microscope equipped with a
PhotometricsCoolSnap HQ2 camera at a magnification of ×40 and
processed with NIS Elements Basic Research Software 6.0 (Nikon
Corporation, Tokyo, Japan).
Cell proliferation assay
The proliferative activity of HMLE cells was
measured using a trypan blue dye exclusion assay. Cells were seeded
in a 96-well plate at a density of 5,000 cells/well. After 12 h, 3
µM PD and 2 µM CHIR were applied to the cells for 24, 48, 72 and 96
h and control cells were treated with the same volume of PBS at
37°C. In order to investigate whether cells treated with CHIR and
PD exhibited CSC properties, cells were treated with serial
dilutions of doxorubicin (100, 200 and 400 ng/ml; Lingnan
Pharmaceutical, Ltd., Guangzhou, China) or paclitaxel (10, 20 and
40 ng/ml; Lingnan Pharmaceutical, Ltd., Guangzhou, China) for 24 h
at 37°C and control cells were treated with dimethyl sulfoxide
(0.2%; Sigma-Aldrich; Merck KGaA). The cells were then trypsinized
and re-suspended in PBS. Trypan blue dye solution (0.4%) was added
to the cell suspension for 2 min at 37°C. After 2 min, the number
of stained (dead) cells and unstained (viable) cells per
mm2 was counted under a phase contrast microscope at a
magnification of ×100.
Cell invasion and migration
assays
For the invasion and migration assays,
1×105 cells/ml HMLE cells were used following treatment
with CHIR and PD. The cell migration and invasion capacity were
determined using a Transwell assay (Corning Incorporated, Corning,
NY, USA). Cells were resuspended in serum-free DMEM medium (Thermo
Fisher Scientific, Inc.). Subsequently, 200 µl cell suspensions
were seeded into the upper chamber with a porous membrane coated
with Matrigel (BD Biosciences, San Jose, CA, USA; for the Transwell
invasion assay) or without coating (for the migration assay).
Complete DMEM medium (Thermo Fisher Scientific, Inc.) was added to
the lower wells of the chambers. Once cells were allowed to migrate
for 24 h or invade for 48 h, cells on the upper membrane were
removed using a cotton swab, and those that had migrated or invaded
to the bottom of the inserts were stained with crystal violet (1
mg/ml; Sigma-Aldrich; Merck KGaA) for 15 min at room temperature,
and then the number of migratory and invasive cells were counted in
five randomly selected high-power fields under a light microscope
(magnification, ×40; TS100; Nikon Corporation). The presented data
represent three individual wells.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from HMLE cells following
treatment with CHIR and PD using the RNeasy Plus mini kit with
QiaShredder columns (Qiagen GmbH, Hilden, Germany) according to the
manufacturer's protocol. Total RNA (1 µg) per sample was reverse
transcribed using the iScriptcDNA synthesis kit (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) according to the
manufacturer's protocol and the cDNA was diluted with 100 µl water.
A total of 1/50 of the diluted cDNA was used for qPCR with iQ SYBR
Green Supermix on the CFX96 system (Bio-Rad Laboratories, Inc.).
The PCR conditions were as follows: Initial reaction at 42°C for 1
h was used for cDNA synthesis, followed by denaturation at 94°C for
5 min and 22 cycles of the following reactions: 94°C for 30 sec,
55°C for 30 sec and 72°C for 30 sec. Following the last cycle, the
reaction was amplified at 72°C for 10 min. All qPCR reactions were
performed in triplicate, and expression levels were analyzed using
CFX manager software (version 3.1; Bio-Rad Laboratories, Inc.),
with levels normalized to GAPDH. The expression levels of the genes
were determined using the ΔΔCq method (13). The following primer pairs were used:
E-cadherin forward, 5′-CAATGGTGTCCATGTGAACA-3′ and reverse,
5′-CCTCCTACCCTCCTGTTCG-3′; Vimentin forward,
5′-CGCTTCGCCAACTACAT-3′ and reverse, 5′-AGGGCATCCACTTCACAG-3′;
Twist forward, 5′-AGCAACAGCGAGGAAGAGCC-3′ and reverse,
5′-CACAGCCCGCAGACTTCTTG-3′; α-SMA forward,
5′-TCCCTTGAGAAGAGTTACGAGTTG-3′ and reverse,
5′-ATGATGCTGTTGTAGGTGGTTTC-3′ and GAPDH forward,
5′-AATGGGCAGCCGTTAGGAAA-3′ and reverse,
5′-GCCCAATACGACCAAATCAGAG-3′. Each set of reactions was repeated
using cDNA from at least three independent experiments.
Cell cycle analysis and apoptosis
analysis
HMLE cells were seeded into 6-well plates at a
density of 3×105 cells/well and treated with 3 µM PD and
2 µM CHIR for 48 h at 37°C. Subsequently, cells were collected with
low-speed centrifugation (300 × g, 5 min, 4°C) and cell pellets
were re-suspended in 1 ml PBS solution. Prior to flow cytometry
analysis, cells were washed, centrifuged (300 × g, 5 min, 4°C) and
re-suspended in propidium iodide (PI; Sigma-Aldrich; Merck KGaA)
staining buffer containing 50 µl/ml PI and 250 µl/ml RNase A at 4°C
for 30 min. Finally, the cell mixture was incubated at 4°C for 30
min in the dark environment to detect cell cycle and stained with 5
µl Annexin V-fluorescein isothiocyanate at room temperature for 15
min to detect apoptosis by fluorescence activated cell sorting
(FACS) analysis (Beckman Coulter, Inc., Brea, CA, USA). Apoptotic
cells were analyzed using a flow cytometer and FlowJo software
(version 10; FlowJo LLC, Ashland, OR, USA).
FACS analysis
Cells were washed with PBS and dissociated with
Accutase (Innovative Cell Technologies, Inc., San Diego, CA, USA).
Following harvesting, the cells were washed twice with ice-cold
FACS buffer (Hank's balanced salt solution supplemented with 10 mM
HEPES, 2% FBS and 0.1% sodium azide; Sigma-Aldrich; Merck KGaA).
Non-dissociated cells were removed by passing the cell suspension
through a cell strainer (BD Biosciences) twice. Cells were
incubated with phycoerythrin-conjugated anti-human CD24 antibody
and allophycocyanin-conjugated anti-human CD44 antibody (dilution,
1:50; cat nos. MA5-11833 and MA5-13890, respectively; Thermo Fisher
Scientific, Inc.) for 30 min at 4°C. Following incubation, the
cells were washed twice with 2 ml FACS buffer, fixed for 15 min at
room temperature and suspended in 4% paraformaldehyde solution
(Electron Microscopy Sciences, Hatfield, PA, USA) in PBS. Finally,
>20,000 cells were analyzed using FACS Calibur and CellQuest
software (version 5.1; BD Biosciences). Further analysis was
performed using FlowJo software (version 10; FlowJo LLC).
Expression of CD24 and CD44 was quantified in divided cells and
separated into ‘high’ and ‘low’ expression groups as previous
described (14).
Animal studies
A total of 132 female SCID mice (weight, 16.83±0.65
g; age, 4 weeks old; obtained from the Experimental Animal Centre
of the Third Military Medical University, Chongqing, China) were
maintained under specific pathogen-free conditions with 12-h
light/12-h dark cycles at 26–28°C and 50–65% humidity, with ad
libitum access to food and water. HMLE cells (1×103,
1×104, 1×105 and 1×106 cells in
each group) were subcutaneously injected into the left flank of
SCID mice (n=12 animals/group). Every 7 days post-inoculation, the
length and width of the individual orthotopic tumors were measured
with calipers, and the volume (mm3) was calculated
according to the following formula: 1/2× length ×
width2. The mice were sacrificed at 42 days
post-inoculation. Subcutaneous tumor growth was measured for 42
days following inoculation. Mouse subcutaneous tumors were
harvested and weighed. All animal experiments were ethically
approved by the Research Ethics Committee of Third Military Medical
University. Mice were anaesthetized using 2% pentobarbital sodium
(0.1 ml/100 g; Sigma-Aldrich; Merck KGaA) and cervical vertebrae
were dislocated.
Western blot assay
Cells were lysed in a lysis buffer containing
aprotinin, leupeptin and phenylmethanesulfonyl fluoride
(Sigma-Aldrich; Merck KGaA) and phosphatase inhibitor cocktails II
and III (Sigma-Aldrich; Merck KGaA) at 4°C for 30 min. Protein
concentration was quantified using the Bradford method (15). Subsequently, 50 mg total protein
extracts were separated by 10% SDS-PAGE and transferred to
polyvinylidene difluoride membranes (GE Healthcare, Chicago, IL,
USA), followed by blocking for 1 h at room temperature in blocking
buffer (cat no. P0023B; Beyotime Institute of Biotechnology). The
membrane was incubated with the following primary antibodies
overnight at 4°C: Rabbit anti-phosphorylated (p)-MEK1/2, rabbit
anti-p-ERK1/2, rabbit anti-β-catenin, rabbit anti-p-GSK3 and rabbit
anti-anti-β-actin antibody (dilution, 1:1,000; cat nos. 8727, 4376,
8480, 9323 and 4970, respectively; Cell Signaling Technology,
Inc.). Membranes were then washed twice with PBS with Tween-20
(0.1%). Subsequently they were incubated with horseradish
peroxidase-conjugated anti-rabbit secondary antibody (dilution,
1:10,000; cat no. 7074; Cell Signaling Technology, Inc.) for 1 h at
room temperature. Binding of the primary antibody was detected
using an enhanced chemiluminescence kit (GE Healthcare). ImageJ
software version 1.47 (National Institutes of Health, Bethesda, MD,
USA) was used to analyze relative protein band density. Each sample
was analyzed in triplicate.
Statistical analysis
All data are presented as mean ± standard deviation
except where stated otherwise. All statistical analyses were
performed using SPSS 18.0 version (SPSS, Inc. Chicago, IL, USA). An
unpaired Student's t-test was used to compare between the two
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
PD and CHIR induces mesenchymal
morphological transformation and proliferation of HMLE cells
To determine whether PD and CHIR are able to induce
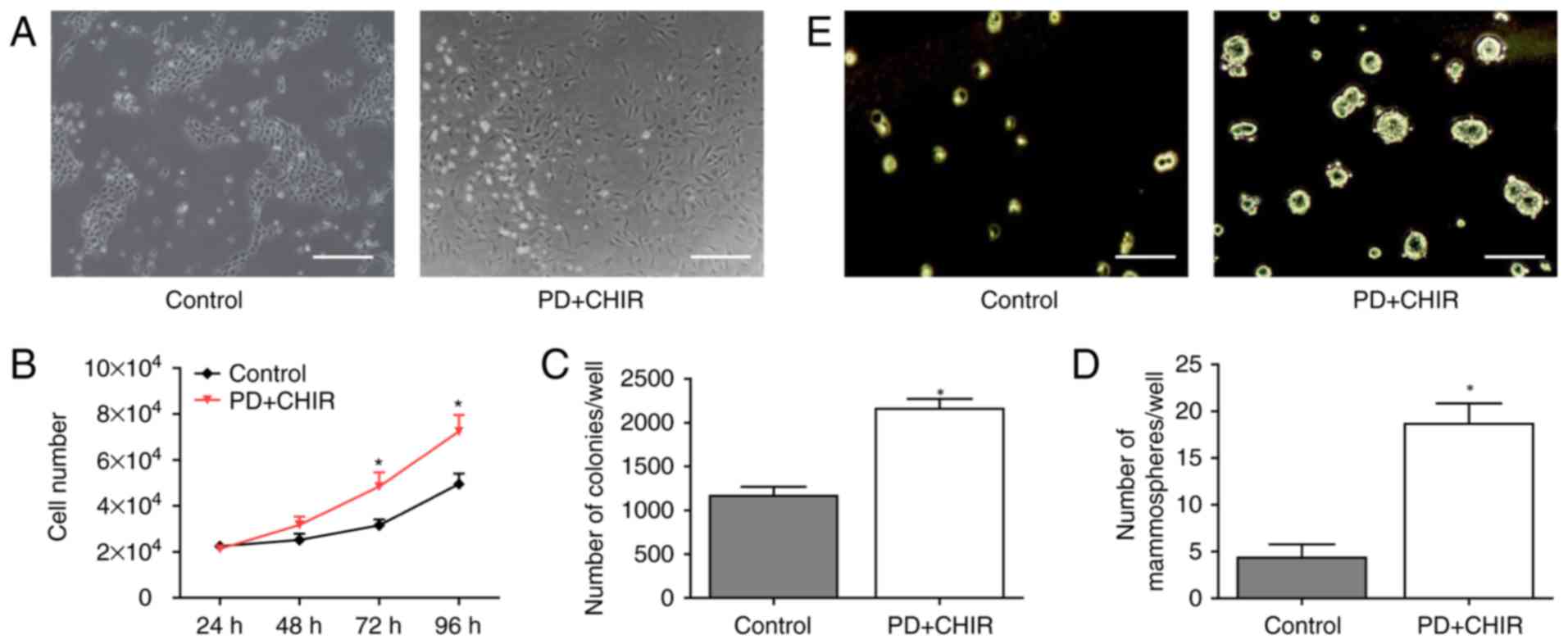
CSCs in vitro, the morphology of HMLE cells was observed
following treatment with PD and CHIR. The untreated cells exhibited
oval or cobblestone shape with tight intercellular junctions
(Fig. 1A; left panel). Following PD
and CHIR treatment for 3 days, the cell contact between cells was
lost and an elongated fibroblastic morphology was observed
(Fig. 1A; right panel). There was
no significant difference in proliferation between the two groups
at 48 h. Subsequently, the treated cells proliferated significantly
faster compared with the control cells at 72 and 96 h (P<0.05;
Fig. 1B), which was consistent with
the results of a previous study that identified that E-cadherin
knockdown generates CSCs (16).
Additionally, there was a significantly increased number of
colonies in the treated cells compared with the control cells
(P<0.05; Fig. 1C). Furthermore,
HMLE cells were seeded at a very low density in plates containing
serum-free medium with or without the compounds, and the number of
newly formed mammospheres was determined. Compared with the control
cells, the treated cells generated a significantly greater number
of mammospheres, and they were larger in size (P<0.05; Fig. 1D). As presented in Fig. 1E, the treated cells were able to
form 20 spheres per 1,000 cells while the control cells formed only
2 spheres per 1,000 cells.
Cell invasion and migration in HMLE
are promoted following treatment with CHIR and PD
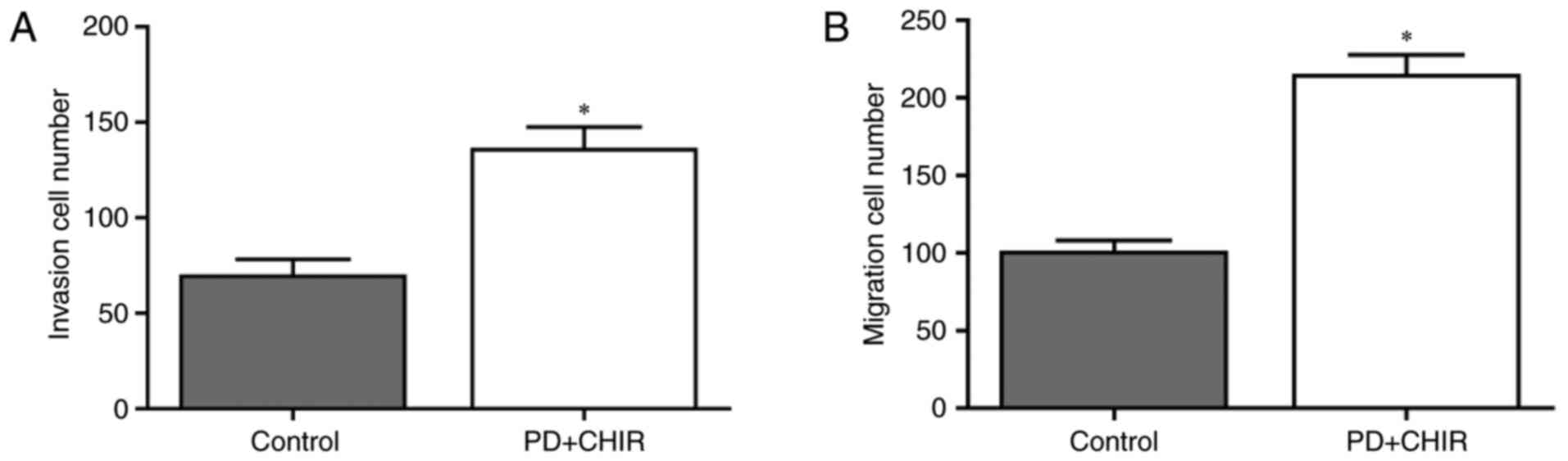
To determine whether treatment with CHIR and PD
promoted migration and invasion potential, HMLE cells were treated
with CHIR and PD, and the invasion and migration of cells were
analyzed. CHIR and PD significantly promoted invasion (P<0.05;
Fig. 2A) and migration (P<0.05;
Fig. 2B) in HMLE cells compared
with the control group. These data suggested that HMLE cells
treated with CHIR and PD exhibited increased motility.
Cell cycle is accelerated and the
apoptosis rate is suppressed in HMLE cells treated with CHIR and
PD
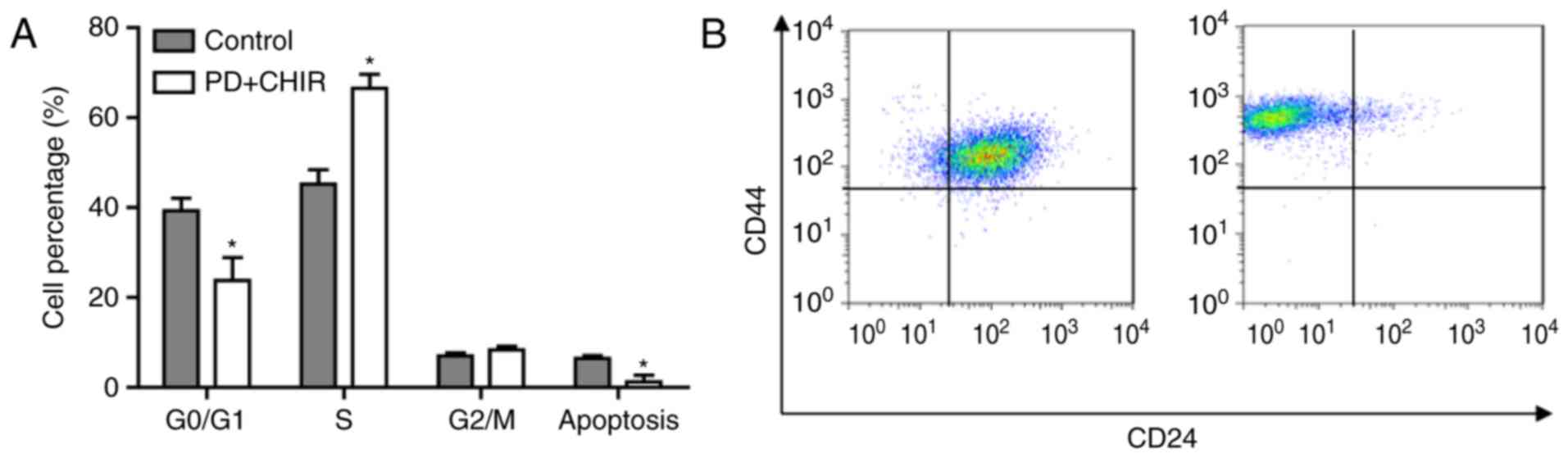
The cell cycle distribution and apoptosis rate in
HMLE cells following treatment with CHIR and PD were determined
using flow cytometry analysis. These data suggested that the cell
cycle prominently shifted from G0/G1 phase to G2/M phase; the
percentage of cells in G0/G1 phase significantly decreased and the
percentage in S phase significantly increased compared with the
control cells (P<0.05). Furthermore, cell apoptosis was
significantly suppressed by treatment with CHIR and PD (P<0.05;
Fig. 3A). Additionally, the present
study observed that the treatment of HMLE cells with the
aforementioned compounds induced a shift in CD44 and CD24
expression, from CD44+/CD24+ to
CD44high/CD24low (Fig. 3B).
Cells treated with CHIR and PD express
EMT markers
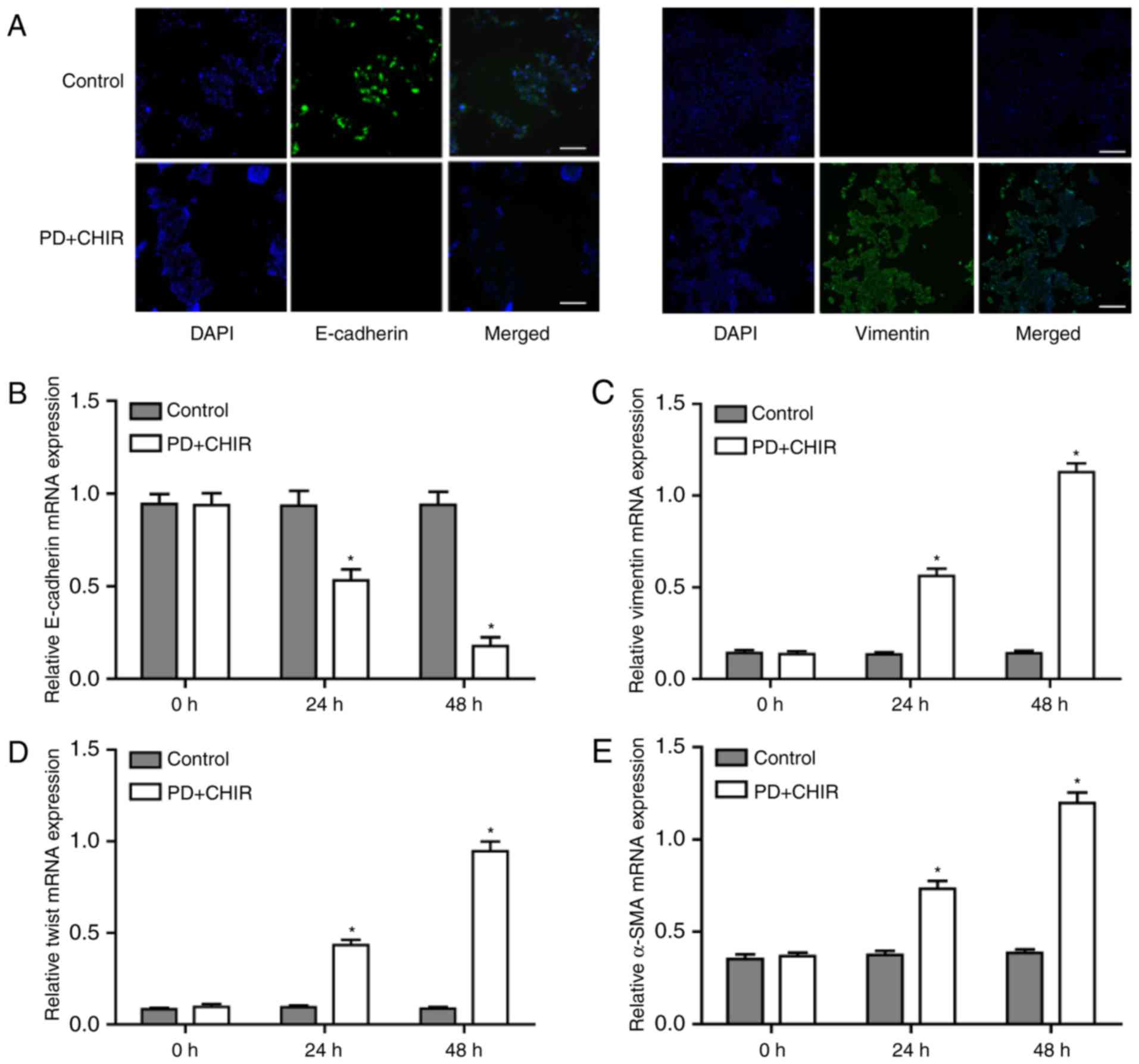
To determine whether the morphological
transformation was associated with the phenotypic switch, the
expression of specific epithelial marker E-cadherin and the stem
cell specific marker Vimentin were determined following treatment
of HMLE cells with PD and CHIR. The mRNA expression levels of
E-cadherin mRNA were significantly decreased following the CHIR and
PD treatments at 24 and 48 h compared with the control cells, and
the expression levels almost undetectable after 48 h of treatment
(P<0.05; Fig. 4A and B; left
panel). By contrast, the mRNA expression levels of vimentin were
low in the control cells, and significantly increased following
treatment with CHIR and PD for 24 and 48 h, with the expression
almost doubling after 48 h (P<0.05; Fig. 4A and C; right panel). Other
mesenchymal markers, Twist and α-smooth muscle actin (α-SMA), were
also examined; the mRNA expression levels of Twist and α-SMA also
exhibit a significant increase at 24 and 48 h post-treatment
compared with the control cells (P<0.05) and the expression
levels at 48 h were double that at 24 h (Fig. 4D and E).
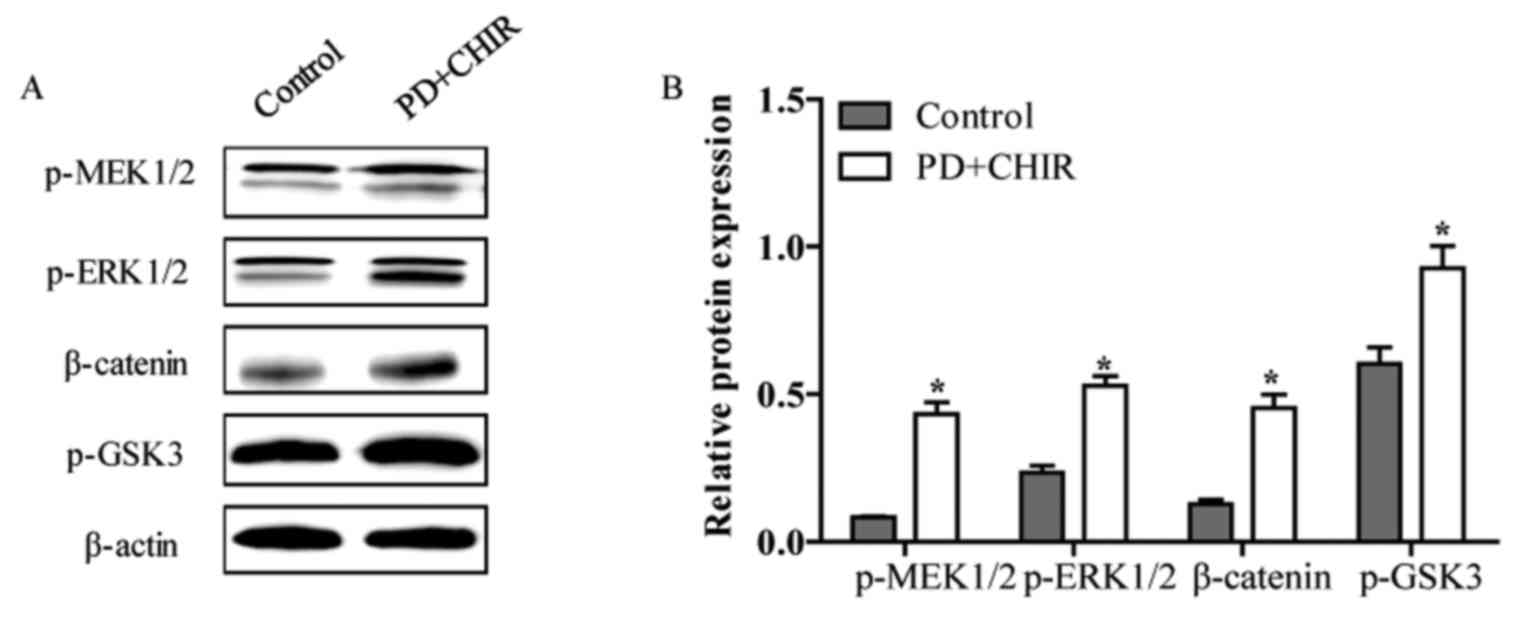
CHIR and PD activate the Wnt and MEK
signaling pathway
The mechanisms of how CHIR and PD accelerated the
cell cycle and reduced cell apoptosis in HMLE cells were examined.
The relative protein levels of p-MEK1/2, p-ERK1/2, β-catenin and
p-GSK3 were determined by western blot analysis. The relative
protein levels of p-MEK1/2, p-ERK1/2, β-catenin and p-GSK3 were
significantly increased in HMLE cells following treatment with CHIR
and PD compared with the control cells (P<0.05; Fig. 5A and B). This suggested that CHIR
and PD-induced proliferation, invasion and migration and the
inhibition of cell apoptosis may be associated with the activation
of the Wnt and MEK signaling pathway.
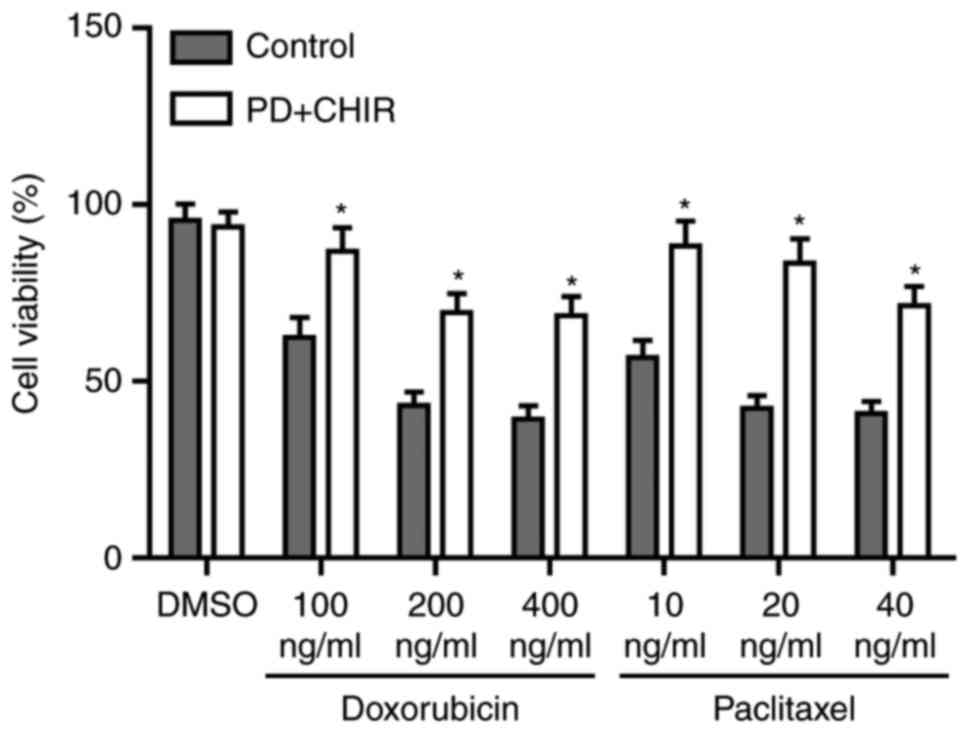
Cells treated with CHIR and PD
demonstrate the properties of CSCs
To investigate whether cells treated with CHIR and
PD exhibit CSC properties, cells were treated with serial dilutions
doxorubicin or paclitaxel for 24 h. The percentage of drug
resistant cells was significantly increased by CHIR and PD compared
with untreated control cells (P<0.05; Fig. 6). To determine the tumor initiation
ability of the induced HMLE cells, SCID mice were injected with
103, 104, 105 and 106
cells. Tumors formed in all of the mice injected with CHIR and
PD-treated cells, whereas 104 of the control cells were
required to initiate tumor formation and only 75% of mice formed
tumors when injected 105 with control cells (Table I).
 | Table I.Tumor incidence in mice injected with
increasing dilutions of cells. |
Table I.
Tumor incidence in mice injected with
increasing dilutions of cells.
| Cells injected | Control | PD+CHIR |
|---|
|
1×106 | 4/4 | 4/4 |
|
1×105 | 3/4 | 4/4 |
|
1×104 | 1/4 | 4/4 |
|
1×103 | 0/4 | 4/4 |
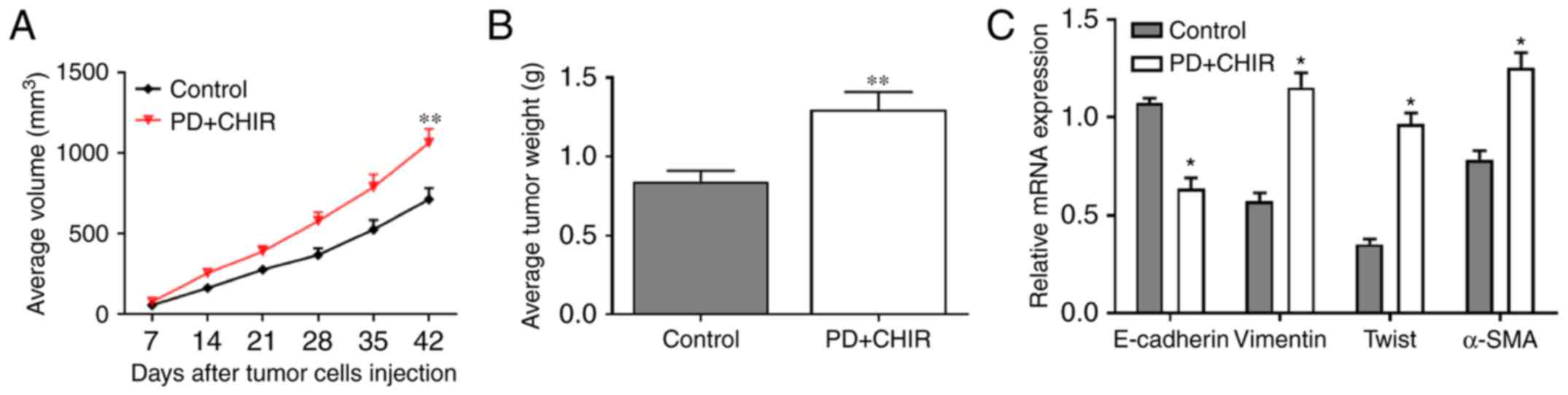
Cells treated with CHIR and PD
promotes tumor growth in vivo
To further investigate the effects of CHIR and PD
in vivo, 1×103 HMLE cells were injected into SCID
mice. To evaluate tumor growth, the length and width of
subcutaneous tumors were measured every 7 days post-inoculation.
Progressive solid tumor types were observed in all mice. The volume
of the tumors generated from CHIR and PD-treated HMLE cells was
significantly higher compared with those produced from control HMLE
cells (P<0.01; Fig. 7A). The
mean tumor weights (mean ± standard deviation) of subcutaneous
tumors were as follows: CHIR and PD group, 1.316±0.235 g, and the
control group, 0.823±0.124 g. The weight of tumors from the CHIR
and PD group were significantly higher compared with the control
group (P<0.01; Fig. 7B).
Additionally, the relative mRNA expression of vimentin, Twist and
α-SMA were significantly higher in the tumor tissue of the CHIR and
PD group compared with the control group, and the relative mRNA
expression of E-cadherin was significantly lower in the CHIR and PD
group compared with that in the control group (P<0.05; Fig. 7C). The data suggested that the
treatment of HMLE cells with CHIR and PD promoted the growth of
HMLE-engrafted tumor types in vivo.
Discussion
To understand the effects of CSCs on tumor
initiation and therapeutic resistance (17,18),
improved methods of isolation and cell expansion in vitro
need to be developed. Although CSCs may be distinguished via
certain cell surface markers in a variety of tumor types, cancer
cells that are negative for markers may also exhibit a
proliferative CSC phenotype (19).
The results of previous studies have indicated that cautions should
be taken when using surface markers to identify CSCs due to the
phenotypic plasticity of tumor cells (19,20).
CHIR is implicated in the self-renewal of HMLE cells, activating
canonical Wnt signaling (8,9). PD is a small inhibitor of MEK that has
been demonstrated to suppress HMLE cell proliferation (10). However, in the present study,
non-stem cancer cells were co-treated with an GSK3 inhibitor (CHIR)
and an MEK inhibitor (PD). The treated cells exhibited increased
expression of CSC markers, increased tumorigenicity and therapeutic
resistance. The observations of previous studies indicated, at
least in part, the potential to rapidly and effectively generate
CSCs by stimulation cells with specific molecules (21–23).
Previous studies have suggested that GSK3 serves an
important function in various cellular processes, including
mediating signaling downstream of Wnt, fibroblast growth factor
(FGF) and Hedgehog during the progression of cancer (24,25).
Upregulation of Wnt ligands, induced by blocking GSK3, bind to the
frizzled/low-density-lipoprotein-related protein co-receptor
complex, which results in the stabilization and nuclear
translocation of β-catenin. β-catenin functions as a powerful
trans-activator of T-cell factor/lymphoid enhancer factor
transcription factors, which regulate important downstream target
genes that promote cell proliferation, differentiation and tissue
development. The MEK signaling pathway comprises several key
signaling components and phosphorylation events that have important
roles in tumorigenesis (26,27).
These activated kinases regulate cell growth, differentiation,
proliferation and migration (28).
Notably, in the present study, cancer cells treated with CHIR and
PD to inhibit MEK signaling proliferated faster compared with the
control cells. One potential explanation for this is that CHIR not
only inhibits the Wnt signaling pathway, but also engages in cross
talk with FGF signaling to enhance the expansion (29). In addition, previous studies have
reported that there is bidirectional crosstalk between ERK and Wnt
signaling pathways (29,30). Following the inhibition of MEK using
CHIR, the accumulation of β-catenin resulted in the activation of
ERK and stimulated the upregulation of oncogenes, including c-myc
and Ras (31,32).
EMT serves a critical role in the acquisition of
malignant traits by carcinoma cells, particularly during the
process of metastasis (32,33). During EMT, epithelial cells lose
cell-cell junctions and polarity, resulting in cells exhibiting an
enhanced migratory, mesenchymal cell phenotype (34). In the present study, treatment with
CHIR and PD induced the specific molecular changes that are markers
of EMT, including the downregulation of E-cadherin expression, and
the upregulation of Twist and vimentin expression. In addition,
studies revealed mammosphere cultures formed and the tumors
observed in SCID mice prevented the success of the transition and
capturing of CSCs (35,36). These results demonstrated that EMT
is an important feature of CSCs. However, Andarawewa et al
(37) reported that MEK/ERK
activation induced by ionizing radiation co-operated with
transforming growth factor-β1 to induce EMT in normal mammary
epithelial cells, and the inhibition of MAPK/ERK activity notably
prevented the downregulation of E-cadherin protein levels and
restored cell-cell interaction. Sineva and Pospelov (38) additionally reported that GSK3
inhibition affected the proliferation rate of stem cells by
increasing the accumulation of cells in the G1 phase. Notably, in
the present study, the treated cancer cells exhibited the increased
expression of CSC surface markers, various CSC properties and an
increase in proliferation compared with the untreated control
cells. One of the potential mechanisms involved in the observed
aforementioned effects is that the compounds reduce
β-catenin/E-cadherin-mediated adhesion, rather than the
β-catenin-dependent transcription of EMT- or cell cycle-associated
genes. However, the underlying molecular mechanisms of these
effects remain poorly understood.
Collectively, the results of the present study
demonstrated that the co-stimulation of cancer cells with PD and
CHIR efficiently generated CSCs. Notably, these results also
indicated that the process of inducing differentiated breast
epithelial tumor cells is sufficient to promote the initiation and
establishment of a tumor. The mechanisms underlying the observed
effects remain largely unknown and require further
investigation.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZM and SL designed the present study. SL, LG, WQ and
CL performed the experiments. SL analyzed the data. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were ethically approved by
the Research Ethics Committee of Third Military Medical University
(Chongqing, China).
Patient consent for publication
Not applicable.
Competing interest
The authors declare that they have no competing
interests.
References
|
1
|
Reya T, Morrison SJ, Clarke MF and
Weissman IL: Stem cells, cancer, and cancer stem cells. Nature.
414:105–111. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Visvader JE and Lindeman GJ: Cancer stem
cells in solid tumours: Accumulating evidence and unresolved
questions. Nat Rev Cancer. 8:755–768. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dalerba P, Cho RW and Clarke MF: Cancer
stem cells: Models and concepts. Annu Rev Med. 58:267–284. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rowehl RA, Crawford H, Dufour A, Ju J and
Botchkina GI: Genomic analysis of prostate cancer stem cells
isolated from a highly metastatic cell line. Cancer Genomics
Proteomics. 5:301–310. 2008.PubMed/NCBI
|
|
5
|
Abraham BK, Justenhoven C, Pesch B, Harth
V, Weirich G, Baisch C, Rabstein S, Ko YD, Brüning T, Fischer HP,
et al: Investigation of genetic variants of genes of the
hemochromatosis pathway and their role in breast cancer. Cancer
Epidemiol Biomarkers Prev. 14:1102–1107. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hill RP and Perris R: ‘Destemming’ cancer
stem cells. J Natl Cancer Inst. 99:1435–1440. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhu QS, Rosenblatt K, Huang KL, Lahat G,
Brobey R, Bolshakov S, Nguyen T, Ding Z, Belousov R, Bill K, et al:
Vimentin is a novel AKT1 target mediating motility and invasion.
Oncogene. 30:457–470. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
An WF, Germain AR, Bishop JA, Nag PP,
Metkar S, Ketterman J, Walk M, Weiwer M, Liu X, Patnaik D, et al:
Discovery of potent and highly selective inhibitors of GSK3b.
2010.
|
|
9
|
Reya T and Clevers H: Wnt signalling in
stem cells and cancer. Nature. 434:843–850. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pellicano F, Simara P, Sinclair A,
Helgason GV, Copland M, Grant S and Holyoake TL: The MEK inhibitor
PD184352 enhances BMS-214662-induced apoptosis in CD34+
CML stem/progenitor cells. Leukemia. 25:1159–1167. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li L, Bennett SA and Wang L: Role of
E-cadherin and other cell adhesion molecules in survival and
differentiation of human pluripotent stem cells. Cell Adh Migr.
6:59–70. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Satelli A and Li S: Vimentin in cancer and
its potential as a molecular target for cancer therapy. Cell Mol
Life Sci. 68:3033–3046. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kawano K, Efferson CL, Peoples GE, Carter
D, Tsuda N, Murray JL and Ioannides CG: Sensitivity of
undifferentiated, high-TCR density CD8+ cells to
methylene groups appended to tumor antigen determines their
differentiation or death. Cancer Res. 65:2930–2937. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kruger NJ: The Bradford method for protein
quantitationMethods in Molecular Biology: Basic Protein and Peptide
Potocols. Chapter 32. Walker JM: Humana Press; Totowa, NJ: pp.
9–15. 1994
|
|
16
|
Gupta PB, Onder TT, Jiang G, Tao K,
Kuperwasser C, Weinberg RA and Lander ES: Identification of
selective inhibitors of cancer stem cells by high-throughput
screening. Cell. 138:645–659. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vinogradov S and Wei X: Cancer stem cells
and drug resistance: The potential of nanomedicine. Nanomedicine
(Lond). 7:597–615. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Facompre N, Nakagawa H, Herlyn M and Basu
D: Stem-like cells and therapy resistance in squamous cell
carcinomas. Adv Pharmacol. 65:235–265. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huang SD, Yuan Y, Tang H, Liu XH, Fu CG,
Cheng HZ, Bi JW, Yu YW, Gong DJ, Zhang W, et al: Tumor cells
positive and negative for the common cancer stem cell markers are
capable of initiating tumor growth and generating both progenies.
PLoS One. 8:e545792013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jaggupilli A and Elkord E: Significance of
CD44 and CD24 as cancer stem cell markers: An enduring ambiguity.
Clin Dev Immunol. 2012:7080362012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li Y, Wang L, Pappan L, Galliher-Beckley A
and Shi J: IL-1β promotes stemness and invasiveness of
colon cancer cells through Zeb1 activation. Mol Cancer. 11:872012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang ZL, Fan ZQ, Jiang HD and Qu JM:
Selective Cox-2 inhibitor celecoxib induces epithelial-mesenchymal
transition in human lung cancer cells via activating MEK-ERK
signaling. Carcinogenesis. 34:638–646. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xie G, Yao Q, Liu Y, Du S, Liu A, Guo Z,
Sun A, Ruan J, Chen L, Ye C and Yuan Y: IL-6-induced
epithelial-mesenchymal transition promotes the generation of breast
cancer stem-like cells analogous to mammosphere cultures. Int J
Oncol. 40:1171–1179. 2012.PubMed/NCBI
|
|
24
|
Hur EM and Zhou FQ: GSK3 signalling in
neural development. Nat Rev Neurosci. 11:539–551. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fisar Z and Hroudová J: Intracellular
signalling pathways and mood disorders. Folia Biol (Praha).
56:135–148. 2010.PubMed/NCBI
|
|
26
|
Zou Y, Liu FY, Wu J, Wan L, Fang SF, Zhang
ZY, Luo Y, Chen MH, Huang MZ, He M and Huang OP: Mutational
analysis of the RAS/RAF/MEK/ERK signaling pathway in 260 Han
Chinese patients with cervical carcinoma. Oncol Lett. 14:2427–2431.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xiong Y, Huang F, Li X, Chen Z, Feng D,
Jiang H, Chen W and Zhang X: CCL21/CCR7 interaction promotes
cellular migration and invasion via modulation of the MEK/ERK1/2
signaling pathway and correlates with lymphatic metastatic spread
and poor prognosis in urinary bladder cancer. Int J Oncol.
51:75–90. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Santarpia L, Lippman SM and El-Naggar AK:
Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy.
Expert Opin Ther Targets. 16:103–119. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Singh AM, Reynolds D, Cliff T, Ohtsuka S,
Mattheyses AL, Sun Y, Menendez L, Kulik M and Dalton S: Signaling
network crosstalk in human pluripotent cells: A Smad2/3-regulated
switch that controls the balance between self-renewal and
differentiation. Cell Stem Cell. 10:312–326. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gong K, Zhou F, Huang H, Gong Y and Zhang
L: Suppression of GSK3β by ERK mediates
lipopolysaccharide induced cell migration in macrophage through
β-catenin signaling. Protein Cell. 3:762–768. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sears R, Nuckolls F, Haura E, Taya Y,
Tamai K and Nevins JR: Multiple Ras-dependent phosphorylation
pathways regulate Myc protein stability. Genes Dev. 14:2501–2514.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dave B, Mittal V, Tan NM and Chang JC:
Epithelial-mesenchymal transition, cancer stem cells and treatment
resistance. Breast Cancer Res. 14:2022012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang Y and Zhou BP: Epithelial-mesenchymal
transition in breast cancer progression and metastasis. Chin J
Cancer. 30:603–611. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xiang J, Fu X, Ran W and Wang Z: Grhl2
reduces invasion and migration through inhibition of TGFβ-induced
EMT in gastric cancer. Oncogenesis. 6:e2842017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Phillips TM, McBride WH and Pajonk F: The
response of CD24(−/low)/CD44+ breast cancer-initiating
cells to radiation. J Natl Cancer Inst. 98:1777–1785. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Andarawewa KL, Erickson AC, Chou WS,
Costes SV, Gascard P, Mott JD, Bissell MJ and Barcellos-Hoff MH:
Ionizing radiation predisposes nonmalignant human mammary
epithelial cells to undergo transforming growth factor beta induced
epithelial to mesenchymal transition. Cancer Res. 67:8662–8670.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sineva GS and Pospelov VA: Inhibition of
GSK3beta enhances both adhesive and signalling activities of
beta-catenin in mouse embryonic stem cells. Biol Cell. 102:549–564.
2012. View Article : Google Scholar
|