Introduction
Based on the invasion of lamina propria, urothelial
bladder cancer is clinically divided into muscle-invasive bladder
cancer (MIBC) and non-muscle-invasive bladder cancer. Particularly
for MIBC, the characteristics including a high occurrence rate of
distant metastases and a low 5-year survival rate demand a high
level of efficacy of the therapeutic regimen (1). Cisplatin (DDP), as the backbone of
combination chemotherapies, is the first-line drug for patients
with advanced bladder cancer (2).
Several large, randomized trials and meta-analyses provide
convincing evidence to support the clinical feasibility and
survival benefit of neoadjuvant DDP-based chemotherapy compared
with surgery alone in patients with MIBC (3–5).
Although bladder cancer is relatively chemosensitive for standard
first-line therapy (such as gemcitabine plus DDP or methotrexate,
vinblastine, doxorubicin and DDP), 30% of MIBC patients still
present with a resistant response to the aforementioned DDP-based
therapeutic regimens and have a very poor prognosis with only a 14
month overall survival period (6,7). There
is no consensus in how to manage MIBC patients who experience the
failure of DDP-based first-line therapy. In addition, DDP-based
regimens are frequently associated with adverse effects, including
renal toxicity and gastrointestinal disorders, which cause
obstacles for therapeutic efficacy, drug continuation and
tolerability (8).
Apoptosis is a controlled type of cell death, which
is characterized by chromatin condensation and membrane budding
(9). DDP primarily induces cancer
cell apoptosis, and failure of the apoptotic pathway may be the
major factor in DDP resistance (10). With respect to the molecular
mechanisms, cross-linked compounds of DNA and DDP contribute to
cytotoxicity by interfering with DNA replication and inducing DNA
damage, which in turn triggers the process of apoptosis (11). Defects in the repair of DNA damage
induced by regimens represent an essential mechanism of cytotoxic
chemotherapy sensitivity (12).
Several studies support the role of various DNA-repair genes, such
as breast cancer susceptibility gene 1, excision repair
cross-complementing group 1, ataxia-telangiectasia mutated kinase,
and retinoblastoma gene 1 as biomarkers of DDP-based chemotherapy
sensitivity (13,14).
Cancerous inhibitor of protein phosphatase 2A
(CIP2A), an oncoprotein with many roles in biological functions, is
overexpressed at a high frquency in a number of tumors, and
performs its oncogenic functions by participating in multiple
pathways, including the phosphoinositide-3-kinase (PI3K)/RAC-α
serine/threonine-protein kinase (AKT), RAS/ERK and the
Wnt/β-catenin pathway (15). Our
previous study revealed the close relationship between the aberrant
expression of CIP2A and uncontrolled cell proliferation in bladder
cancer (16). CIP2A depletion
contributes to cell apoptosis in several cancer types (17). Functional studies also confirmed the
potential role of CIP2A in sensitizing cancer cells to many
chemotherapeutic agents (18).
Exploring novel approaches to bladder cancer treatment to increase
sensitivity to DDP is a critical issue; however, the advent of
targeted therapy is promising.
In this context, the present study was designed to
explore the significance and potential of CIP2A in enhancing the
chemosensitivity of DDP in bladder cancer. In the present study, we
demonstrated that inhibition of CIP2A exacerbated DDP-induced
bladder cancer cell apoptosis by the accumulation of DNA damage.
Furthermore, we further explored the role of the AKT pathway in
DDP-triggered CIP2A inhibition.
Materials and methods
Cell culture and chemicals
Human urothelial bladder cancer cell lines T24 and
J82 were obtained from the Type Culture Collection of the Chinese
Academy of Sciences, (Shanghai, China) and were cultured in
Dulbecco's modified Eagle's medium (HyClone Laboratories; GE
Healthcare Life Sciences, Logan, UT, USA) and supplemented with 10%
fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Cells were cultured at 37°C with 5%
CO2. DDP (Sigma-Aldrich, Merck KGaA, Darmstadt, Germany)
was dissolved in phosphate-buffered saline (PBS) for cell
incubation. The PI3K inhibitor LY294002 was purchased from Selleck
Chemicals (Houston, TX, USA).
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA extraction and reverse-transcription
reactions were performed using an RNA Extraction kit (Takara Bio,
Inc., Otsu, Japan) and PrimeScript™ RT Master Mix (Takara Bio,
Inc.), respectively. The following primers were used for
amplification: CIP2A, forward primer, 5′-CACAAAT-CACCTCGACCCCT-3′
and reverse primer, 5′-CAAAAGCTGAGTGGCGTTCG-3′. β-actin was used as
an internal control: Forward primer 5′-GTGGGGCGCCCCCAGGCACCA-3′ and
reverse primer, 5′-CTCCTTAATGTCACGCACGAT-3′. RT-qPCR was performed
using Power SYBR Green PCR Master Mix (Thermo Fisher Scientific,
Inc.). Analysis of the relative gene expression data was performed
using quantitative PCR and the 2−ΔΔCq method (19).
Western blot analysis
Following cell lysis by ice-cold
radioimmunoprecipitation assay buffer, protein concentrations were
determined. Equal quantities (30 µg) of protein were separated
using 10% SDS-polyacrylamide gels, transferred to a polyvinylidene
difluoride (PVDF) membrane and blocked with 5% bovine serum albumin
(BSA). PVDF membranes were incubated overnight at 4°C with the
following antibodies: CIP2A (dilution 1:500; cat. no. sc-80662;
Santa Cruz Biotechnology, Inc., Dallas, CA, USA), AKT (dilution
1:1,000; cat. no. 9272S; Cell Signaling Technology, Inc., Danvers,
MA, USA), phosphorylated (p)-AKT (dilution 1:1,000; cat. no. 4060S;
Cell Signaling Technology), B cell lymphoma-2 (Bcl-2; dilution
1:1,000; cat. no. sc-492; Santa Cruz Biotechnology, Inc.),
caspase-3 (dilution 1:1,000; cat. no. 9662S; Cell Signaling
Technology), poly(ADP-Ribose) polymerase (PARP; dilution 1:1,000;
cat. no. 5625T; Cell Signaling Technology), γ-H2AX (dilution
1:1,000; cat. no. ab2893; Abcam, Cambridge, UK) and β-actin
(dilution 1:1,000; cat. no. sc-47778; Santa Cruz Biotechnology,
Inc.). After being washed, the membranes were then incubated with
horseradish peroxidase-conjugated secondary antibodies (anti-mouse
IgG, dilution 1:1,000; cat. no. 7076S; Cell Signaling Technology;
anti-rabbit IgG, dilution 1:1,000; cat. no. 7074S; Cell Signaling
Technology) followed by enhanced chemiluminescence detection.
Construction of stably transfected
cells
The CIP2A short hairpin RNA (sh-CIP2A) plasmid,
ligated in the pGV101 vector, was purchased from GeneChem
Technologies Co., Ltd. (Shanghai, China). The oligonucleotide
sequence for shRNA was as follows:
5′-GATCCCCCACAGTTTAAGTGGTGGAAACTCGAGTTTCCACCACTTAAACTGTGGTTTTTGGAT-3′.
sh-CIP2A was transfected using Lipofectamine 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. In order to obtain stable cell lines, transfected cells
were selected with 500 µg/ml neomycin for nearly 2 weeks.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
Cells were seeded on a 96-well plate and incubated
with different concentrations of DDP (0, 5, 10, 20 and 40 µg/ml).
At 24 h after incubation, cell viability was determined by MTT
assay (Invitrogen; Thermo Fisher Scientific, Inc.) using a
microplate reader (Multiscan™GO; Thermo Fisher Scientific, Inc.).
Absorbance was determined at a wavelength of 490 nm. All
experiments were performed in triplicate and three wells were used
for each treatment. The drug concentration resulting in 50%
inhibition rate (IC50) was calculated with GraphPad 6.0
software (GraphPad Software, Inc., La Jolla, CA, USA).
Colony forming assay
The effect of DDP on the ability of bladder cancer
cells to form colonies was assessed using a 6-well plate (200
cells/well). One week after the initial incubation of cells,
colonies were incubated with DDP (0 and 5 µg/ml) for 1 week. The
colonies were stained with 0.1% crystal violet.
Annexin V-PI apoptosis assays
After DDP treatment, the harvested cells were
resuspended in Annexin V-binding buffer. Then the cells were
stained with FITC-conjugated Annexin V and PI according to the
manufacturer's protocol (Invitrogen; Thermo Fisher Scientific,
Inc.). After incubation for 30 min at room temperature in the dark,
evaluation of the level of apoptosis was analyzed with a flow
cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Immunofluorescence staining
Cells grown on coverslips in 24-well plates were
fixed with 4% paraformaldehyde and blocked with 5% bovine serum
albumin (BSA). Subsequently, the cells were incubated with
antibodies against γ-H2AX (dilution 1:200; cat. no. ab2893; Abcam;)
overnight at 4°C. The secondary antibody (dilution 1:1,000; cat.
no. A-21441; Invitrogen; Thermo Fisher Scientific, Inc.) was
incubated with the cells at 37°C for 1 h, followed by staining with
4′,6-diamidino-2-phenylindole (DAPI) to visualize the nuclei.
Images were captured using a confocal laser-scanning microscope
(Leica SP8; Leica Microsystems GmbH, Wetzlar, Germany).
Comet assay
Slides were pre-coated with 1% normal melting point
agarose. The mixture of 104 cells and 70 µl 1% low
melting point agarose was rapidly spread onto the pre-coated
slides. Subsequently, the slides were immersed in cold lysis buffer
for at least 1 h and alkaline electrophoresis solution for 20 min,
successively. Following electrophoresis at 25 V and 300 mA for 30
min, the slides were neutralized with Tris-HCl (pH 7.5) and dyed
with ethidium bromide for 20 min. The individual cells were viewed
using an Olympus BX51 UV fluorescence microscope (Olympus Corp.,
Tokyo, Japan).
Immunohistochemical (IHC) and TUNEL
analysis
The slides with paraffin-embedded tissues were
deparaffinized in xylene and rehydrated through graded alcohols.
Subsequently, 3% hydrogen peroxide was used to block the endogenous
peroxidase activity. The slides were incubated with primary
antibodies against CIP2A (dilution 1:50; cat. no. sc-80662; Santa
Cruz Biotechnology, Inc.), γ-H2AX (dilution 1:100; cat. no. ab2893;
Abcam) and p-AKT (dilution 1:200; cat. no. 4060S; Cell Signaling
Technology). After washing 3 times, diaminobenzidine was used for
signal development, and the slides were counterstained with 20%
hematoxylin. Terminal deoxynucleotidyl transferase mediated dUTP
nick end labeling (TUNEL) assay was performed using DeadEnd™
Colorimetric TUNEL system (Promega Corp., Madison, WI, USA),
according to the manufacturer's instructions.
Subcutaneous xenograft models
Animal experiments were performed according to the
protocol approved by the Institutional Animal Care and Use
Committee of Ruijin Hospital Affiliated to the School of Medicine,
Shanghai Jiaotong University. The housing condition for mice is
grade SPF (specific-pathogen-free). The temperature is 26–28°C, and
the light/dark cycle is 10/14-h cycle. Sterilized food and water by
steam under high pressure were provided to the mice. Twenty
4-week-old female BALB/c nude mice (SLAC Laboratory Animal Co.,
Ltd., Shanghai, China) were randomly divided into 4 equal groups
(n=5). sh-CIP2A and sh-Control T24 cells were harvested to prepare
cell suspensions containing 6×106 cells/ml which were
injected into the axillary subcutaneous tissues at 100 µl/mouse,
respectively. On day 21 when the maximum diameter of the tumors
reached 5 mm, intraperitoneal injections of DDP (2 mg/kg, twice
weekly) or an equal volume of 0.9% normal saline were administered
for 3 weeks. To build the tumor growth curve, the tumor volume was
determined using calipers every 7 days and calculated with the
following formula: Tumor volume (mm3)=(Length ×
Width2)/2. On day 42, the mice were euthanized via
dislocation of cervical vertebra, and meanwhile, the xenograft
tumor tissues were removed, weighed and subjected to further IHC
staining analysis. The inhibitory rate of DDP was calculated with
the formula (1-tumor volume of DDP-treated group/tumor volume of
non-DDP-treated group) ×100%.
Statistical analysis
Data are expressed as the mean ± standard deviation
(SD), and bars in the graph represent standard deviation.
Statistical analyses were conducted using SPSS 22.0 software (IBM,
Corp., Armonk, NY, USA). A two-tailed Student's t-test was
conducted to analyze statistical differences between groups.
P-values of <0.05 were considered to indicate statistically
significant differences.
Results
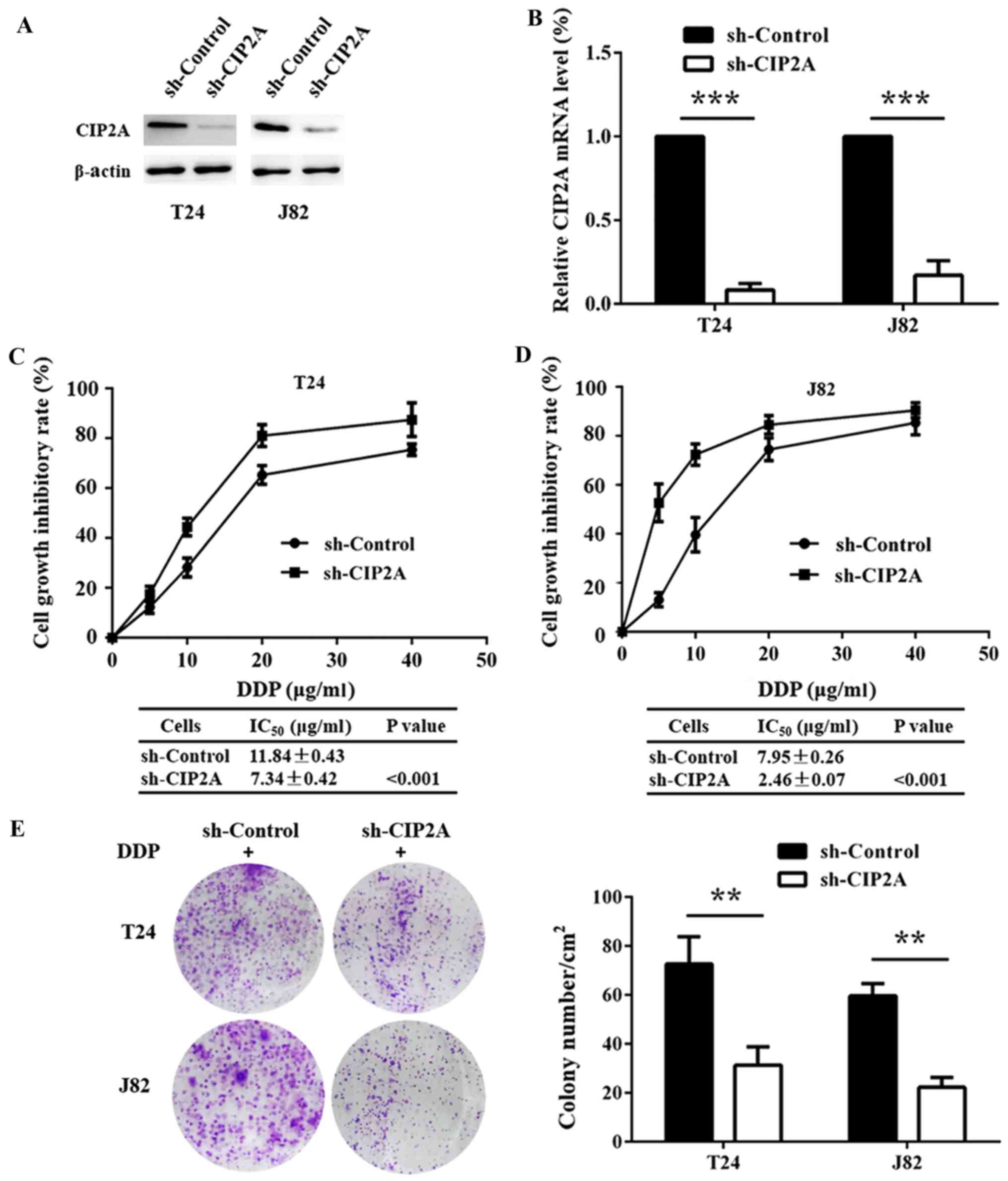
CIP2A depletion increases drug
sensitivity to DDP in bladder cancer cells
CIP2A-stable knockdown T24 and J82 cells were
successfully established to elucidate the role of CIP2A in
chemoresistance to DDP. CIP2A expression was markedly decreased at
both the mRNA and protein levels (Fig.
1A and B). Knockdown of CIP2A led to a significant decrease in
the IC50 of DDP compared with sh-Control cells (Fig. 1C and D). According to the results of
the colony formation experiment following DDP treatment, we
observed that inhibition of CIP2A expression significantly
decreased the number of colonies formed (Fig. 1E). These results revealed a causal
link between CIP2A depletion and the increased chemosensitivity to
DDP.
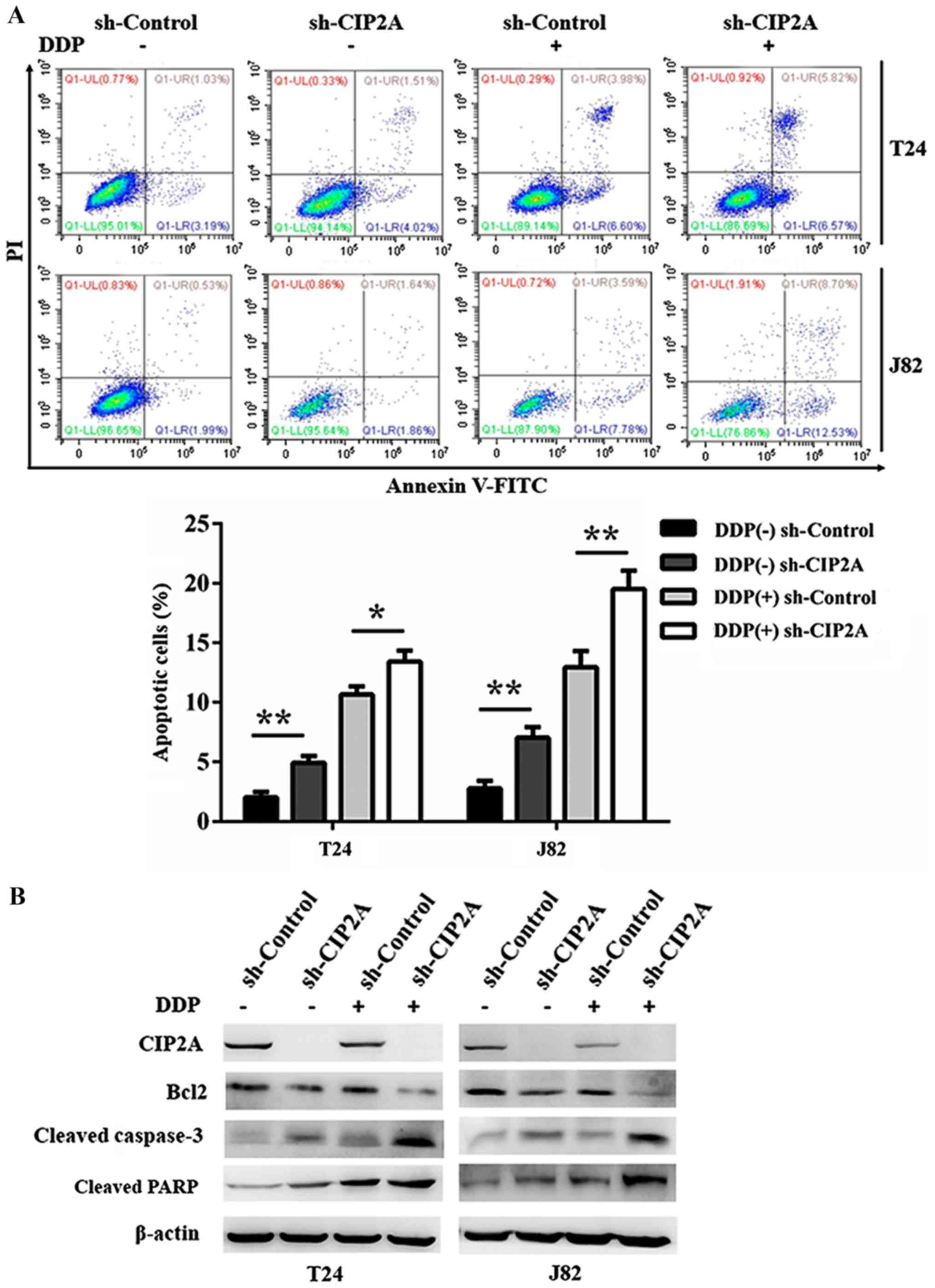
Knockdown of CIP2A induces increased
apoptosis in bladder cancer cells upon DDP treatment
Given that apoptosis is one of the major mechanisms
responsible for the impairment of proliferation induced by DDP
(8), we subsequently investigated
the role of CIP2A in DDP-induced apoptosis. Flow cytometry results
revealed that upon DDP treatment, the apoptotic rates of sh-CIP2A
cells were markedly higher compared with the sh-Control cells
(Fig. 2A). Consistently, following
DDP treatment, decreased Bcl2 and increased cleaved caspase-3 and
cleaved PARP were detected in sh-CIP2A T24 and J82 cells (Fig. 2B). Therefore, our results indicated
that knockdown of CIP2A promoted DDP-induced apoptosis in bladder
cancer cells in vitro.
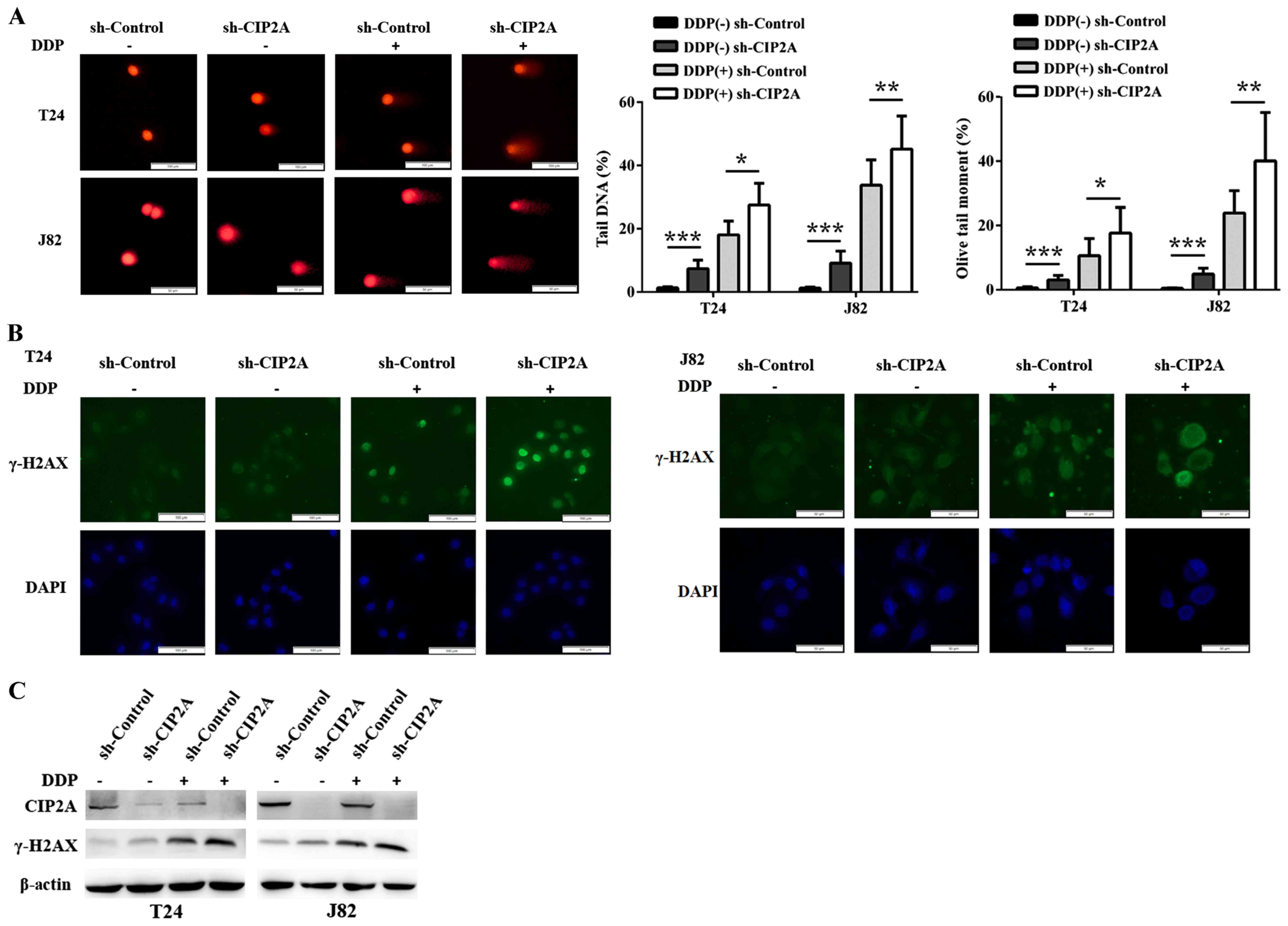
Knockdown of CIP2A in bladder cancer
cells enhances DDP-induced DNA damage
DDP can lead to the intracellular accumulation of
DNA double-strand breaks (DSBs) and γ-H2AX protein, which are
responsible for cell apoptosis (20). Comet assays confirmed that upon
treatment with DDP, both tail DNA content and DNA migration
distance of cells with CIP2A downregulation increased significantly
compared with the control group (Fig.
3A). In addition, the immunofluorescence images intuitively
illustrated that low expression of CIP2A accelerated DDP-induced
foci formation of γ-H2AX in T24 and J82 cells (Fig. 3B). Consistently, DDP markedly
enhanced the accumulation of γ-H2AX at the protein level in
sh-CIP2A cells (Fig. 3C),
indicating that CIP2A depletion may greatly increase the genotoxic
effect of DDP in aggravating DNA damage in bladder cancer
cells.
DDP attenuates CIP2A expression via
the AKT pathway in bladder cancer cells
Based on the aforementioned results, we inferred
that CIP2A may serve as a molecular target of DDP and play a
crucial role in apoptosis, once DDP enters the cells. Following
cell exposure for 48 h to the indicated DDP concentrations, CIP2A
was negatively modulated at both the mRNA and protein levels in a
dose-dependent manner (Fig. 4A and
B). In addition, the decrease of CIP2A protein expression was
accompanied by a concomitant reduction in p-AKT levels and
corresponding change of apoptosis-related indicators (Fig. 4B). To determine the causal
relationship between AKT phosphorylation and CIP2A expression, we
tested the level of p-AKT in the sh-Control and sh-CIP2A bladder
cancer cells. The western blotting results revealed that there was
no significant difference between the two cell lines (Fig. 4C). However, the expression of CIP2A
was downregulated when treated with LY294002, an inhibitor to
phosphorylation of AKT (Fig. 4D).
These results provided evidence that CIP2A is a downstream effector
in the AKT signaling pathway, through which DDP inhibits CIP2A
expression in bladder cancer cells.
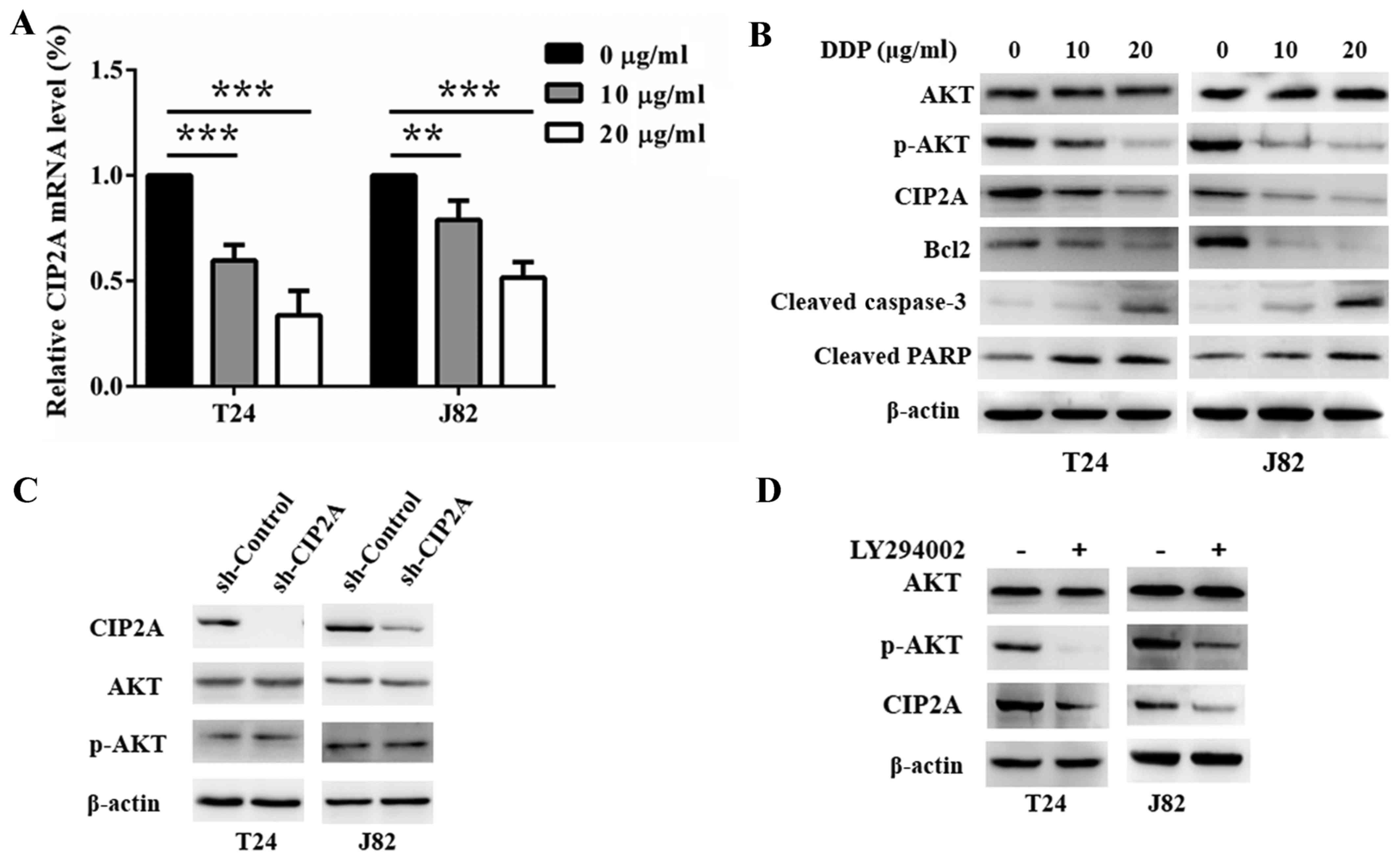 | Figure 4.DDP attenuates the expression of
CIP2A via the AKT pathway in bladder cancer cells. Upon DDP
treatment at different concentrations (0, 10, 20 µg/ml) in T24 and
J82 cells, CIP2A mRNA levels were determined by (A) RT-qPCR, and
(B) the protein levels of AKT, p-AKT, CIP2A, Bcl2, cleaved
caspase-3, and cleaved PARP were determined by western blotting.
(C) The protein levels of AKT, p-AKT and CIP2A in sh-Control and
sh-CIP2A T24 and J82 cells were detected by western blotting. (D)
Following 20 µmol/l LY294002 treatment for 24 h, western blotting
was used to test the protein levels of AKT, p-AKT and CIP2A in T24
and J82 cells. Every experiment was conducted at least 3 times.
**P<0.01, ***P<0.001. DDP, cisplatin; sh, short hairpin;
CIP2A, cancerous inhibitor of protein phosphatase 2A. |
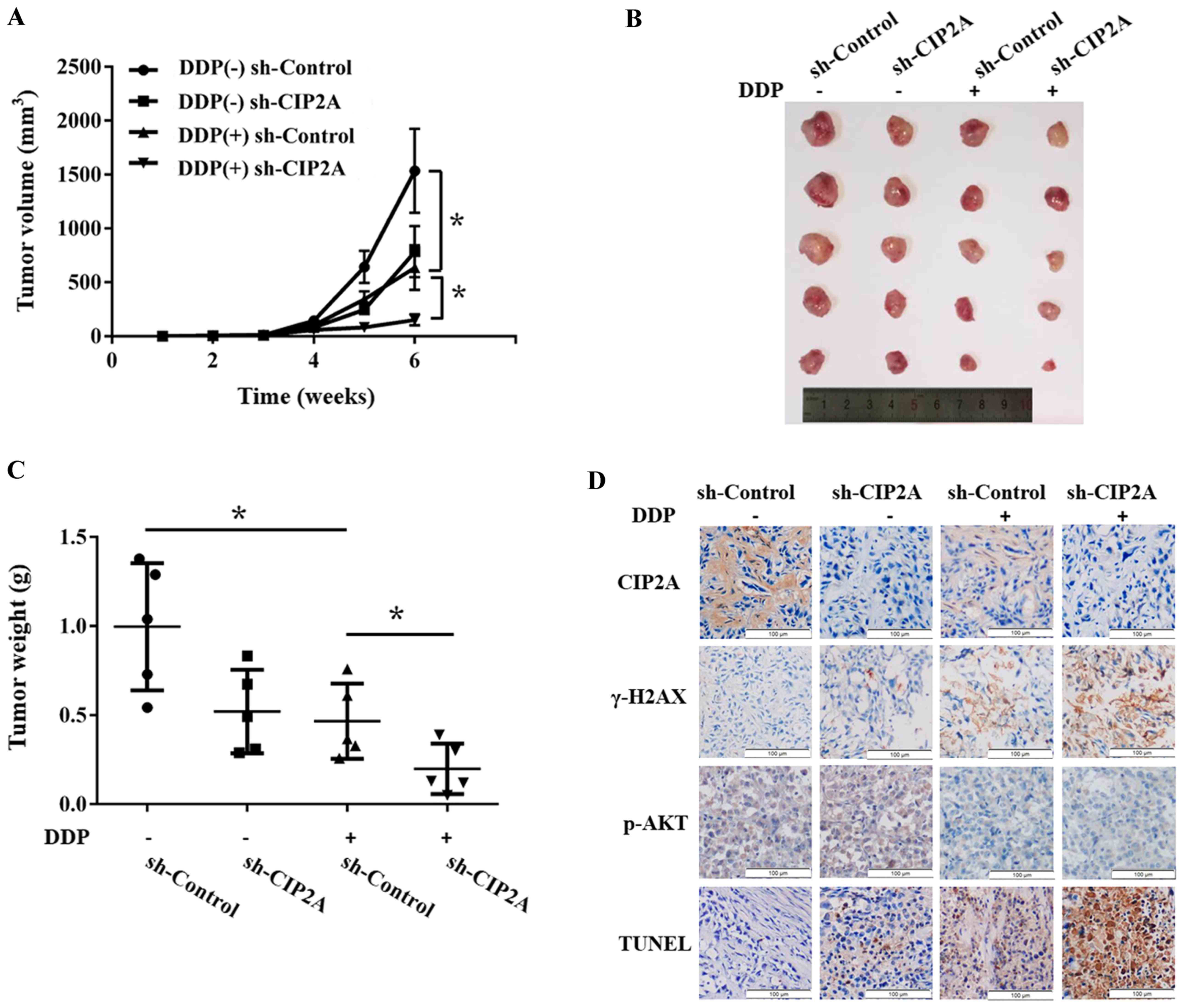
Inhibition of CIP2A expression
enhances DDP sensitivity in nude mouse xenografts
A nude mouse xenograft model was established using
sh-Control and sh-CIP2A T24 cells to verify our previous
conclusions in vivo. After intraperitoneal administration of
DDP for 3 weeks, tumors in the sh-CIP2A group grew at a slower rate
(Fig. 5A and B), accompanied by
significant reductions in weight (Fig.
5C). Further analysis yielded the additional information that
the inhibitory rates of the sh-CIP2A group and sh-Control group
were 79.7 and 54.9% respectively, revealing the increased DDP
sensitivity caused by CIP2A knockdown. Compared with the DDP(+)
sh-Control group, the TUNEL assays revealed that the percentage of
apoptotic cells was markedly higher in the DDP(+) sh-CIP2A group.
In addition, the IHC assay illustrated a stronger positive stain of
γ-H2AX (Fig. 5D). According to the
IHC staining results, the positive rates of both p-AKT and CIP2A in
tumors undergoing a DDP therapeutic regimen were markedly decreased
(Fig. 5D). These results supported
the hypothesis that inhibition of CIP2A expression enhanced DDP
sensitivity in vivo.
Discussion
DDP is the backbone of several mainstream
chemotherapeutic regimens for advanced bladder cancer, in
particular in patients with distant metastasis (1). Two large, randomized trials clearly
supported the idea that DDP provides 5-year cancer-specific
survival benefits compared with surgery alone in patients with MIBC
(3,4). However, relatively poor response and
chemotherapy resistance frequently lead to treatment failure
(21). Thus, it is urgent to
discover novel approaches to increasing sensitivity to DDP, which
remains poorly understood. In the present study, we focused our
research on the involvement of CIP2A in DDP-induced DNA damage and
cell apoptosis to enhance the chemosensitivity of DDP.
CIP2A is a human oncoprotein, which has been
confirmed to be overexpressed in bladder cancer (22). Huang et al (23) confirmed that the levels of specific
CIP2A protein increase with increasing tumor grade and stage of
bladder cancer. Furthermore, several recent studies also have
provided evidence of the potent role of CIP2A in bladder cancer
cell proliferation and epithelial mesenchymal transition (16,24).
Thus, it is plausible to infer the potential role that CIP2A serves
as a prognostic indicator and therapeutic target for bladder
cancer.
The intrastrand cross-links of purine bases and DDP
are responsible for the process of DSBs, blocking cell division and
resulting in apoptotic cell death, which contributes to DDP
cytotoxicity (11). Hence, DNA
damage-induced apoptosis is a promising target for increasing the
sensitivity of DDP. Several molecular mechanisms leading to
apoptosis have been found to be implicated in DDP treatment of
human cancers, such as the MAPK (25), JNK (26) and AKT pathway (27). In a screen of diagnostic chronic
myeloid leukemia samples, samples with low CIP2A levels were
characterized by upregulation of pro-apoptotic BCL-2 family members
(28). Furthermore, a series of
studies confirmed that CIP2A is implicated in apoptosis with a
potent chemo-sensitizing potential (29,30).
The view of previous researchers is further validated by our
results that CIP2A knockdown enhanced sensitivity to DDP by
promoting DDP-induced bladder cancer cell apoptosis.
In molecular terms, aggravating DNA damage is an
important target to improve the efficacy of DDP-based neoadjuvant
therapy. The most revealing evidence supports the development of
hypersensitivity to DDP by accumulation of DNA fragments in various
malignancies (31,32). Basu et al (35) confirmed that checkpoint kinase Chk1
inhibition explains the cell-killing activity of DNA-damaging
agents (33–35). Increased Chk1 activity promotes
CIP2A transcription, and CIP2A downregulation is essential for
maximal inhibition of cancer cell viability in response to Chk1
inhibition (36). We have
considered the close association between CIP2A and DNA damage. Our
data indicated that CIP2A depletion induced DNA damage and
exacerbated DDP-induced formation of γ-H2AX foci, confirming the
hypothesis and elucidating the role of CIP2A in the process of
chemotherapeutic drug-induced DNA damage. DNA repair is one of the
main mechanisms underlying DDP resistance. Extensive progress has
been made in the field of DNA-repair related genes as prognostic
markers for DDP treatment in bladder cancer (14). In addition, Myant et al
(37) demonstrated that CIP2A is
indispensable for the efficient recovery and regeneration of
intestinal tissue in response to DNA damage by promoting MYC Ser62
phosphorylation, implying the potential ability of CIP2A to resist
DNA damage. Thus, the association between CIP2A and DNA repair
requires further investigation.
AKT belongs to a family of serine/threonine kinases
which impacts on multiple cellular processes, including cell
proliferation and survival (38).
AKT activation is achieved by regulation of its phosphorylation
status, predominantly on two highly conserved residues, Thr308 and
Ser473 (39). It is accepted that
AKT activation by Ser473 phosphorylation prevents apoptosis
following ionizing radiation-induced DNA damage (40). Our results also revealed the
inhibition of AKT Ser473 phosphorylation by DDP, confirming the
viewpoint that DDP-induced DNA damage is AKT-dependent once again
(41). Furthermore, in the present
study, our data revealed that CIP2A downregulation was triggered by
AKT inactivation, indicating that CIP2A acted downstream of AKT.
However, several studies hold different opinions that CIP2A
controls the activity of AKT by promoting its Ser473
phosphorylation (42,43). However, with regard to the
controversial issue, Wiegering et al (44) reported that CIP2A depletion did not
affect AKT Ser473 phosphorylation in various colon cancer cell
lines, which is consistent with our results. Therefore, further
studies are needed to clarify the relationship between the CIP2A
and AKT pathway.
Acquiring the appropriate biomarkers, which could
help clinicians to identify patients who will really benefit from
the DDP-based chemotherapy is a major issue. Multiple molecular
biomarker candidates have been studied, including regulators of
apoptosis, cell survival and cellular mechanisms of drug uptake and
transport (12). However, none have
shown clinical utility. Based on the evidence of the critical role
of CIP2A in sensitizing bladder cancer cells to DDP in the present
study, CIP2A may be an innovative molecular biomarker, which can be
used to predict a response to DDP, which could lead to optimizing
the individualized therapeutic regimen.
In summary, we have provided evidence that DDP
triggered the inhibition of CIP2A expression via promoting the AKT
inactivation. In addition, CIP2A knockdown increased DDP-induced
bladder cancer cell apoptosis by accelerating DNA damage, revealing
CIP2A as a promoter of sensitivity to DDP. Further investigation of
the AKT/CIP2A pathway should clarify the detailed mechanism and
could give new insights leading to a better curative effect of
DDP-based therapy in bladder cancer treatment.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (no. 81472379 to ZS) and
Zhejiang Provincial Natural Science Foundation of China (no.
LY16H160016 to SC). The present study was supported by the Shanghai
Key Laboratory of Reproductive Medicine, School of Medicine,
Shanghai Jiaotong University.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Ethics approval and consent to
participate
Animal experiments were performed according to the
protocol approved by the Institutional Animal Care and Use
Committee (IACUC) of Ruijin Hospital Affiliated to the School of
Medicine, Shanghai Jiaotong University.
Authors' contributions
ZS and SZ conceived and designed the experiments.
FG, XiaoW, SC and TX performed the experiments. XianW, YS and FD
coordinated the research and analyzed the data. FG and XiaoW wrote
the manuscript. All authors read and approved the manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
DDP
|
cisplatin
|
|
CIP2A
|
cancerous inhibitor of protein
phosphatase 2A
|
|
MIBC
|
muscle-invasive bladder cancer
|
References
|
1
|
Trenta P, Calabro F, Cerbone L and
Sternberg CN: Chemotherapy for Muscle-invasive bladder cancer. Curr
Treat Options Oncol. 17:62016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Massari F, Santoni M, Ciccarese C,
Brunelli M, Conti A, Santini D, Montironi R, Cascinu S and Tortora
G: Emerging concepts on drug resistance in bladder cancer:
Implications for future strategies. Crit Rev Oncol Hematol.
1:81–90. 2015. View Article : Google Scholar
|
|
3
|
Grossman HB, Natale RB, Tangen CM,
Speights VO, Vogelzang NJ, Trump DL, deVere White RW, Sarosdy MF,
Wood DJ, Raghavan D and Crawford ED: Neoadjuvant chemotherapy plus
cystectomy compared with cystectomy alone for locally advanced
bladder cancer. N Engl J Med. 9:859–866. 2003. View Article : Google Scholar
|
|
4
|
International Collaboration of Trialists;
Medical Research Council Advanced Bladder Cancer Working Party (now
the National Cancer Research Institute Bladder Cancer Clinical
Studies Group); European Organisation for Research and Treatment of
Cancer Genito-Urinary Tract Cancer Group; Australian Bladder Cancer
Study Group; National Cancer Institute of Canada Clinical Trials
Group; Finnbladder; Norwegian Bladder Cancer Study Group; Club
Urologico Espanol de Tratamiento Oncologico Group, . Griffiths G,
Hall R, et al: International phase III trial assessing neoadjuvant
cisplatin, methotrexate, and vinblastine chemotherapy for
muscle-invasive bladder cancer: Long-term results of the BA06 30894
trial. J Clin Oncol. 16:2171–2177. 2011.
|
|
5
|
Advanced Bladder Cancer Meta-analysis
Collaboration. Neoadjuvant chemotherapy in invasive bladder cancer:
A systematic review and meta-analysis. Lancet. 361:1927–1934. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yafi FA, North S and Kassouf W: First- and
second-line therapy for metastatic urothelial carcinoma of the
bladder. Curr Oncol. 1:e25–e34. 2011.
|
|
7
|
von der Maase H, Sengelov L, Roberts JT,
Ricci S, Dogliotti L, Oliver T, Moore MJ, Zimmermann A and Arning
M: Long-term survival results of a randomized trial comparing
gemcitabine plus cisplatin, with methotrexate, vinblastine,
doxorubicin, plus cisplatin in patients with bladder cancer. J Clin
Oncol. 23:4602–4608. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cummings BS, Lasker JM and Lash LH:
Expression of glutathione-dependent enzymes and cytochrome P450s in
freshly isolated and primary cultures of proximal tubular cells
from human kidney. J Pharmacol Exp Ther. 293:677–685.
2000.PubMed/NCBI
|
|
10
|
Galluzzi L, Vitale I, Michels J, Brenner
C, Szabadkai G, Harel-Bellan A, Castedo M and Kroemer G: Systems
biology of cisplatin resistance: Past, present and future. Cell
Death Dis. 5:e12572014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Galluzzi L, Senovilla L, Vitale I, Michels
J, Martins I, Kepp O, Castedo M and Kroemer G: Molecular mechanisms
of cisplatin resistance. Oncogene. 31:1869–1883. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Buttigliero C, Tucci M, Vignani F,
Scagliotti GV and Di Maio M: Molecular biomarkers to predict
response to neoadjuvant chemotherapy for bladder cancer. Cancer
Treat Rev. 54:1–9. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Quinn JE, Kennedy RD, Mullan PB, Gilmore
PM, Carty M, Johnston PG and Harkin DP: BRCA1 functions as a
differential modulator of chemotherapy-induced apoptosis. Cancer
Res. 63:6221–6228. 2003.PubMed/NCBI
|
|
14
|
Hoffmann AC, Wild P, Leicht C, Bertz S,
Danenberg KD, Danenberg PV, Stöhr R, Stöckle M, Lehmann J, Schuler
M and Hartmann A: MDR1 and ERCC1 expression predict outcome of
patients with locally advanced bladder cancer receiving adjuvant
chemotherapy. Neoplasia. 8:628–636. 2010. View Article : Google Scholar
|
|
15
|
De P, Carlson J, Leyland-Jones B and Dey
N: Oncogenic nexus of cancerous inhibitor of protein phosphatase 2A
(CIP2A): An oncoprotein with many hands. Oncotarget. 5:4581–4602.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gao F, Xu T, Wang X, Zhong S, Chen S,
Zhang M, Zhang X, Shen Y, Wang X, Xu C and Shen Z: CIP2A mediates
fibronectin-induced bladder cancer cell proliferation by
stabilizing β-catenin. J Exp Clin Cancer Res. 36:702017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang CY, Chao TT, Chang FY, Chen YL, Tsai
YT, Lin HI, Huang YC, Shiau CW, Yu CJ and Chen KF: CIP2A mediates
erlotinib-induced apoptosis in non-small cell lung cancer cells
without EGFR mutation. Lung Cancer. 85:152–160. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu P, Yao J, He J, Zhao L, Wang X, Li Z
and Qian J: CIP2A down regulation enhances the sensitivity of
pancreatic cancer cells to gemcitabine. Oncotarget. 7:14831–14840.
2016.PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 4:402–408. 2001.
View Article : Google Scholar
|
|
20
|
Rajeswaran A, Trojan A, Burnand B and
Giannelli M: Efficacy and side effects of cisplatin- and
carboplatin-based doublet chemotherapeutic regimens versus
non-platinum-based doublet chemotherapeutic regimens as first line
treatment of metastatic non-small cell lung carcinoma: A systematic
review of randomized controlled trials. Lung Cancer. 59:1–11. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
David KA, Milowsky MI, Ritchey J, Carroll
PR and Nanus DM: Low incidence of perioperative chemotherapy for
stage III bladder cancer 1998 to 2003: A report from the National
Cancer Data Base. J Urol. 178:451–454. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xue Y, Wu G, Wang X, Zou X, Zhang G, Xiao
R, Yuan Y, Long D, Yang J, Wu Y, et al: CIP2A is a predictor of
survival and a novel therapeutic target in bladder urothelial cell
carcinoma. Med Oncol. 30:4062013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Huang LP, Savoly D, Sidi AA, Adelson ME,
Mordechai E and Trama JP: CIP2A protein expression in high-grade,
high-stage bladder cancer. Cancer Med. 1:76–81. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pang X, Fu X, Chen S, Zhu X, Qi H, Li Y,
Li F and Tan W: Overexpression of CIP2A promotes bladder cancer
progression by regulating EMT. Clin Transl Oncol. 18:289–295. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Basu A and Tu H: Activation of ERK during
DNA damage-induced apoptosis involves protein kinase Cdelta.
Biochem Biophys Res Commun. 334:1068–1073. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jones EV, Dickman MJ and Whitmarsh AJ:
Regulation of p73-mediated apoptosis by c-Jun N-terminal kinase.
Biochem J. 405:617–623. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hayakawa J, Mittal S, Wang Y, Korkmaz KS,
Adamson E, English C, Ohmichi M, McClelland M and Mercola D:
Identification of promoters bound by c-Jun/ATF2 during rapid
large-scale gene activation following genotoxic stress. Mol Cell.
16:521–535. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lucas CM, Milani M, Butterworth M, Carmell
N, Scott LJ, Clark RE, Cohen GM and Varadarajan S: High CIP2A
levels correlate with an antiapoptotic phenotype that can be
overcome by targeting BCL-XL in chronic myeloid leukemia. Leukemia.
30:1273–1281. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Janghorban M, Farrell AS, Allen-Petersen
BL, Pelz C, Daniel CJ, Oddo J, Langer EM, Christensen DJ and Sears
RC: Targeting c-MYC by antagonizing PP2A inhibitors in breast
cancer. Proc Natl Acad Sci USA. 111:9157–9162. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chao TT, Wang CY, Chen YL, Lai CC, Chang
FY, Tsai YT, Chao CH, Shiau CW, Huang YC, Yu CJ and Chen KF:
Afatinib induces apoptosis in NSCLC without EGFR mutation through
Elk-1-mediated suppression of CIP2A. Oncotarget. 6:2164–2179. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
McLaughlin M, Barker HE, Khan AA, Pedersen
M, Dillon M, Mansfield DC, Patel R, Kyula JN, Bhide SA, Newbold KL,
et al: HSP90 inhibition sensitizes head and neck cancer to
platin-based chemoradiotherapy by modulation of the DNA damage
response resulting in chromosomal fragmentation. BMC Cancer.
17:862017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kritsch D, Hoffmann F, Steinbach D, Jansen
L, Photini Mary S, Gajda M, Mosig AS, Sonnemann J, Peters S,
Melnikova M, et al: Tribbles 2 mediates cisplatin sensitivity and
DNA damage response in epithelial ovarian cancer. Int J Cancer.
141:1600–1614. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Smith J, Tho LM, Xu N and Gillespie DA:
The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and
cancer. Adv Cancer Res. 108:73–112. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ma CX, Janetka JW and Piwnica-Worms H:
Death by releasing the breaks: CHK1 inhibitors as cancer
therapeutics. Trends Mol Med. 17:88–96. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Basu B, Yap TA, Molife LR and de Bono JS:
Targeting the DNA damage response in oncology: Past, present and
future perspectives. Curr Opin Oncol. 24:316–324. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Khanna A, Kauko O, Böckelman C, Laine A,
Schreck I, Partanen JI, Szwajda A, Bormann S, Bilgen T, Helenius M,
et al: Chk1 targeting reactivates PP2A tumor suppressor activity in
cancer cells. Cancer Res. 73:6757–6769. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Myant K, Qiao X, Halonen T, Come C, Laine
A, Janghorban M, Partanen JI, Cassidy J, Ogg EL, Cammareri P, et
al: Serine 62-phosphorylated MYC associates with nuclear lamins and
its regulation by CIP2A is essential for regenerative
proliferation. Cell Rep. 12:1019–1031. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Manning BD and Cantley LC: AKT/PKB
signaling: Navigating downstream. Cell. 129:1261–1274. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liao Y and Hung MC: Physiological
regulation of Akt activity and stability. Am J Transl Res. 2:19–42.
2010.PubMed/NCBI
|
|
40
|
Bozulic L, Surucu B, Hynx D and Hemmings
BA: PKBalpha/Akt1 acts downstream of DNA-PK in the DNA
double-strand break response and promotes survival. Mol Cell.
30:203–213. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hayakawa J, Ohmichi M, Kurachi H, Kanda Y,
Hisamoto K, Nishio Y, Adachi K, Tasaka K, Kanzaki T and Murata Y:
Inhibition of BAD phosphorylation either at serine 112 via
extracellular signal-regulated protein kinase cascade or at serine
136 via Akt cascade sensitizes human ovarian cancer cells to
cisplatin. Cancer Res. 60:5988–5994. 2000.PubMed/NCBI
|
|
42
|
Tseng LM, Liu CY, Chang KC, Chu PY, Shiau
CW and Chen KF: CIP2A is a target of bortezomib in human triple
negative breast cancer cells. Breast Cancer Res. 14:R682012.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chen KF, Liu CY, Lin YC, Yu HC, Liu TH,
Hou DR, Chen PJ and Cheng AL: CIP2A mediates effects of bortezomib
on phospho-Akt and apoptosis in hepatocellular carcinoma cells.
Oncogene. 29:6257–6266. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wiegering A, Pfann C, Uthe FW, Otto C,
Rycak L, Mäder U, Gasser M, Waaga-Gasser AM, Eilers M and Germer
CT: CIP2A influences survival in colon cancer and is critical for
maintaining Myc expression. PLoS One. 10:e752922013. View Article : Google Scholar
|