Introduction
Hepatocellular carcinoma (HCC) is the fifth most
common malignancy in the world and the second most common cause of
cancer-related death, according to the World Health Organization
data (1,2). Despite the fact that significant
advances have been made in its diagnosis and its treatment options,
which include new chemotherapeutic interventions and targeted
therapies, patients with HCC display poor long-term survival due to
its high rates of intrahepatic and distal metastases (3). Signaling pathways such as the
phosphoinositide 3-kinase (PI3K)/AKT signaling pathway have been
implicated in HCC progression (4).
PI3K/AKT pathway activation contributes to the progression and
development of HCC and regulates the malignant biological functions
of cancer cells (5). Thus, it is
crucial to identify the mechanisms that underlie HCC progression
and metastasis since these data could provide the field with new
targets for cancer therapies.
MicroRNAs (miRNAs) are a group of endogenous,
evolutionarily conserved, noncoding small RNAs that bind to their
complementary sequence within the target mRNA 3′-untranslated
region (UTR) to post-transcriptionally affect gene initiation and
development and gene expression progression. miRNAs then induce
target mRNA degradation or inhibit translation (6,7). A
large amount of evidence indicates that abnormal miRNA expression
plays key roles in many aspects of the progression of HCC (8,9). In
particular, miRNAs display effects on cell proliferation, cell
migration, cell apoptosis, drug resistance, and cell renewal.
Recent studies have used genome-wide methods to show that miRNAs
such as miR-129-2 (10), miR-124
(11), miR-203 (12) and miR-125b (13) are involved in HCC. miR-122 has
important effects on the biology of HCC (14) and is also related to certain liver
diseases. It is worth noting that HCC expresses some small RNAs.
miRNAs have been identified as candidate biomarkers for HCC
diagnosis and treatment and bind to some of the target molecules
involved in various biological processes associated with HCC
development (including cell proliferation, cell differentiation,
cell apoptosis, and metastasis) or act as tumor suppressors
(15,16). Our aim was to identify the functions
and mechanisms of the activities of key miRNAs in liver cancer, as
these data may contribute to the development of methods for
providing more accurate diagnoses or predicting outcomes in
patients with liver cancer. One miRNA, miR-300, is highly expressed
in HCC, but its potential role in the carcinogenesis and tumor
development in liver cancer is unclear.
An increasing amount of evidence indicates that the
poor prognoses and treatment failures of HCC are associated with
the abnormal activation of many signaling pathways. Of these, the
PI3K/AKT signaling pathway is the most important pathway through
which cells regulate malignancy (17). It is worth noting that abnormal Akt
activation is an undesirable prognostic factor in all stages of HCC
and contributes to resistance to single first-generation targeted
therapies. Akt and protein kinase B (57-kDa Ser/Thr kinase) are
activated by extracellular signalling. Akt is often activated in
cancer cells and subsequently activates cell proliferation and
induces immune cell apoptosis (18). However, Akt over-activation
increases intracellular ROS levels by increasing oxygen consumption
and inhibits ROS scavengers downstream of forkhead transcription
factor (FOXO). Akt also induces premature senescence and sensitizes
cells to ROS-mediated apoptosis (19). FOXO1, a member of the FOXO subfamily
that plays an important role in protecting cells from the
microenvironment, is directly phosphorylated by Akt, resulting in
the inhibition of its transcriptional activity. Thus, inhibitors
that target these pathways may be potent therapeutic agents in
cancer.
In the present study, we showed that miR-300 is
highly expressed in HCC tissues and induces the downregulation of
FOXO1, which leads to the downregulation of the AKT/FOXO1 pathways,
resulting in the accelerated proliferation and migration of HCC and
liver cancer cells.
Materials and methods
Clinical tissues
From January 2012 to December 2013, liver tissues
were collected from 36 patients with HCC and normal liver tissues
from 18 patients at the Department of Hepatobiliary Surgery at the
First Affiliated Hospital of Xi'an Jiaotong University. The tissues
were stored at −80°C or embedded in paraffin. No chemotherapy or
radiotherapy was administered to any of the included patients
before surgery. The average age of the patients with HCC was
52.1±9.4 years; the study population included 30 males and 6
females. A total of 26 patients were positive for hepatitis B
virus. According to the TNM classification system (20), 12 patients had stage I disease, 6
patients had stage II disease, 14 patients had stage III disease,
and 4 patients had stage IV disease. Ethical approval for the study
was obtained from the First Affiliated Hospital Ethics Committee of
Xi'an Jiaotong University. All participants provided written
informed consent.
Cell culture and reagents
The human HCC cell lines HCCLM3, HuH-7 and SK-HEP1,
and liver cancer Hep3B, HepG2 and HuH-6 cells were obtained from
the Shanghai Institutes for Biological Sciences (SIBS) of the
Chinese Academy of Sciences (CAS), and the HCC cell lines MHCC97-H
and MHCC97-L were obtained from Fudan University (Shanghai, China).
The cells were cultured in Invitrogen™ RPMI-1640 medium (Thermo
Fisher Scientific, Inc., Waltham, MA, USA) with 10% fetal bovine
serum (FBS; BioWest, Nuaillé, France) and 1%
penicillin-streptomycin (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) at 37°C in a humidified atmosphere of 5%
CO2.
Rapamycin was obtained from Sigma-Aldrich/Merck KGaA
and dissolved in DMSO. The Akt inhibitor AZD5363 (cat. S8019) was
purchased from Selleck Chemicals (Houston, TX, USA).
Phosphatidylinositol 3′-kinase (PI3K) inhibitor (LY294002, cat.
BML-ST420-0025) was purchased from Alexis Biochemicals (San Diego,
CA, USA).
Quantitative reverse-transcriptase
polymerase chain reaction (qRT-PCR)
Total RNA was isolated from HCC tissues or HCC/liver
cancer cells using TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's instructions.
Two micrograms of total RNA were reverse transcribed into cDNA
using a reverse transcription kit (Takara Bio, Inc., Tokyo, Japan).
Then, the cDNA was amplified using a SYBR® Premix Ex
Taq™ II (Perfect Real-Time) Kit (Takara, Bio). The following PCR
primers were used: 5′-GGATGTGCATTCTATGGTGTACC-3′ (forward) and
5′-TTTCGGGATTGCTTATCTCAGAC-3′ (reverse) for FOXO1 (86 bp) and
5′-CTCACCGGATGCACCAATGTT-3′ (forward) and
5′-CGCGTTGCTCACAATGTTCAT-3′ (reverse) for GAPDH (82 bp). qRT-PCR
included an initial denaturation cycle at 95°C for 2 min, followed
by 35 cycles of denaturation at 98°C for 10 sec and annealing and
extension at 60°C for 45 sec. Gene expression levels were
calculated through the ΔΔCt method; U6 (miRNA detection) or GAPDH
(mRNA detection) was used as an internal control. The hsa-miR-300
primer (HmiRQP0377) and snRNA U6 qPCR primer (HmiRQP9001) were
obtained from GeneCopoeia (Guangzhou, China). The results are
expressed as the mean value of three independent experiments.
Cell transfection
miRNA vectors, including precursor miR-300 clones
(miR-300, HmiR0490), and an miR-300 inhibitor (anti-miR-300;
HmiR-AN0377) and miR-300 inhibitor control clone (anti-miR-con,
CmiR-AN0001) were purchased from GeneCopoeia (Guangzhou, China).
Plasmids containing the FOXO1 gene, specific siRNA for FOXO1
(sense, 5′-CCAUGGACAACAACAGUAA-3′; antisense,
5′-CCAUGGACAACAACAGUAA-3′) or scrambled siRNA (sense,
5′-UCACAACCUCCUAGAAAGAGUAGA-3′; antisense,
5′-UACUCUUUCUAGGAGGUUGUGAUU-3′) for FOXO1 were synthesized by
Bioworld Biotech Co., Ltd. (Shanghai, China). Cells were
transfected with the above vectors by Lipofectamine 3000
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. Transfected cells were harvested at 24 h
or the indicated time for qRT-PCR or western blot analysis.
MTT assay
Cells were analyzed with the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
method to detect cell viability. The target cells were transfected
with miR-300, miR-con, anti-miR-300 or anti-miR-con using
Lipofectamine. Briefly, 5,000 cells were seeded in each well and
cultured in a cell incubator for the indicated times (24, 48, 72
and 96 h). Then, 10 µl of MTT (5 mg/ml diluted in 1X PBS) was added
to each well, and the cells were incubated for an additional 4 h at
37°C. After incubation, 100 µl of DMSO was added to the wells to
dissolve the crystals. Then, the absorbance of each well in the
culture plates was measured at 450 nm with a micro-plate reader
(Bio-Rad Laboratories, Hercules, CA, USA) after the plates were
oscillated for 10 min at room temperature (RT).
Preparation of the cell extracts and
western blot analysis
Whole protein was lysed in RIPA buffer supplemented
with protease (Roche, Manheim, Germany) and phosphatase (Roche)
inhibitors, and then the protein concentration was quantified with
a BCA Protein Assay Kit (Qiagen, Valencia, CA, USA), according to
the manufacturer's protocol. The protein samples (40 µg) were
digested, run on 10% SDS-PAGE gels, and then transferred to PVDF
membranes (Millipore, Billerica, MA, USA), which were blocked in 5%
skim milk in 1X TBST for 2 h at RT and incubated in primary
antibodies overnight at 4°C. The membranes were then washed three
times in 1X TBST and incubated with HRP-conjugated secondary
antibodies (goat anti-rabbit IgG-HRP, 1:10,000 dilution; cat. no.
ab6721; goat anti-mouse IgG-HRP, 1:10,000 dilution; cat. no.
ab6789; Abcam, Cambridge, MA, USA) at RT for 2 h. The assay was
performed using an enhanced chemiluminescence kit (Amersham, Little
Chalfont, UK). GAPDH (1:2,000 dilution; cat. no. ab8245; Abcam) was
used as a protein loading control. The FOXO1 primary antibody was
obtained from Abcam (1:1,000 dilution; cat. no. ab52857) and total
AKT (tAKT; 1:500 dilution; cat. no. 4691), phospho-AKT (pAKT, 1:500
dilution; cat. no. 4060), total 4E-BP1 (t4E-BP1, 1:1,000 dilution;
cat. no. 9644), phospho-4E-BP1 (p4E-BP1, 1:500 dilution; cat. no.
2855), total S6K1 (tS6K1; 1:1,000 dilution; cat. no. 2708),
phospho-S6K1 (pS6K1, 1:500 dilution; cat. no. 9208), SNAIL (1:1,000
dilution; cat. no. 3879) and MMP2 (1:1,000 dilutio; cat. no. 40994)
antibodies were purchased from Cell Signaling Technology, Inc.
(Beverly, MA, USA).
Cell migration and invasion
assays
Matrigel-uncoated and -coated Transwell inserts
(8-µm pore size; Corning Inc., Corning, NY, USA) were used to
assess HCC and liver cancer cell migration and invasion in
vitro. Briefly, 2×105 transfected cells were
suspended in 150 µl of serum-free DMEM and placed in the upper
chamber, and 700 µl of 20% FBS containing DMEM was loaded into the
lower chamber. After 24 h of incubation, the cells were fixed in 4%
paraformaldehyde for 20 min and then stained with 0.1% crystalline
violet dye for 15 min. The non-migrating or invading cells were
gently removed using a cotton swab. The mean number of migrated or
invaded cells was determined by averaging the numbers of cells in
10 fields in both inserts with ImageJ software (National Institutes
of Health, Bethesda, MD, USA). All experiments were performed in
duplicate and repeated at least three times.
Wound healing assay
Wound healing assay was performed to examine cell
migration capacity. The cells were seeded in 24-well plates at a
density of 1.2×105 cells/well; they were cultured in
complete medium and grown to 80% fusion. The cells were then
transfected with 50 nM miR-300 or anti-miR-300 and/or treated with
target FOXO1/siFOXO1. After 24 h, the transfection medium was
replaced with fresh medium. When the cells reached 100% confluence,
a sterile micropipette was used to scratch the cell layer. After
the cells were washed with PBS, serum-free medium was added to the
wells. Images were captured immediately after the plates were
scratched and 24 h later to monitor cell migration toward the
injured area with a Zeiss microscope (Carl Zeis, Oberkochen,
Germany). The migratory capability of the cells was quantified via
measurements of the scratch area using ImageJ software (National
Institutes of Health). The experiment was carried out twice.
Anchorage-independent growth ability
(soft agar) assay
Cells (4×103) were suspended in 2 ml of
complete medium and 0.3% agar (Sigma-Aldrich; Merck KGaA). The
agar/cell mixture was placed on top of a bottom layer containing a
1% mixture of complete medium and agar. After the cells were
incubated for 10 days, the sizes of the colonies were measured
using an eye micrometre, and colonies with a diameter >0.1 mm
were counted. The number of colonies of each cell line was manually
counted in triplicate under an inverted microscope (Nikon Eclipse
Ti-S; Nikon, Tokyo, Japan).
Luciferase reporter assay
miR-300 is predicted to interact with the 3′-UTR
sequence of FOXO1 by binding to the corresponding target sequence
or a mutated sequence within the predicted target site. Therefore,
the normal 3′-UTR and a mutated 3′-UTR were inserted into the
pmiR-GLO double luciferase miRNA target expression vector (Promega,
Madison, WI, USA); the resulting plasmids were called FOXO1 WT
3′-UTR and FOXO1 Mut 3′-UTR, respectively. Cells were seeded in
24-well plates and transfected with the FOXO1 WT 3′-UTR or FOXO1
Mut 3′-UTR vector using Lipofectamine. After 48 h, the cells were
harvested and analyzed according to the manufacturer's instructions
(Dual-Luciferase Assay System; Promega). PRL-TK was used to express
Renilla luciferase as an internal control and was
co-transfected according to the correct transfection and harvest
efficiencies.
Tumor sphere culture
Cancer cells were washed and subjected to enzymatic
dissociation to single cells. Afterward, single cancer cells were
re-suspended in tumor sphere media (TSM) consisting of serum-free
DMEM, 20 ng/ml human recombinant epidermal growth factor
(Sigma-Aldrich; Merck KGaA), 10 ng/ml human recombinant basic
fibroblast growth factor-basic (R&D Systems, Minneapolis, MN,
USA) and 10 ng/ml epidermal growth factor (R&D Systems). Cells
were plated at a density of 7.5×104 to 1×105
cells/10-mm dish, and the medium was changed every other day until
the tumor sphere formation was observed.
In vivo experiment
Sixteen male BALB/c nude mice aged 5 weeks (18–20 g)
were purchased from the Shanghai Experimental Animal Center of the
Chinese Academy of Sciences (Shanghai, China). The animals were
maintained under specific pathogen-free (SPF) conditions at Xi'an
Jiaotong University. The specific housing conditions were as
follows: Temperature, 21±2°C; humidity, 30–70%; 12-h light/dark
cycle; the ingested food and water were sterile feed and sterilized
bottled water. All animal studies were performed according to the
guidelines of the Institutional Animal Care and Use Committee of
Xi'an Jiaotong University. Liver cancer cells HepG2 or
HepG2-miR-300 were used for the tumor studies. Six mice were
randomly assigned to each of the following groups. HepG2 and
HepG2-miR-300 cells were harvested (2×106 cells per
well), resuspended in 100 µl of PBS, and then implanted
subcutaneously into the flanks of BALB/c nude mice. The resulting
tumors were designated either as HepG2 or HepG2-miR-300 tumors,
respectively, and were monitored every day and measured every 2
days with a Vernier calliper to record the diameter. Tumor volume
(V) was calculated with the following formula: V = W2 ×
L/2, where L is the length, and W is the width of the tumor. On the
24th day after tumor injection, the experimental animals were
sacrificed via CO2 inhalation according to the animal
care guidelines, and the tumors were removed for subsequent
analyses. This experiment was conducted at the Xi'an Jiaotong
University Experimental Research Laboratory with the consent of the
Experimental Animal Ethics Committee (no. XJTULAC2018-450).
Immunohistochemical (IHC)
analysis
Briefly, 4-µm-thick slices were deparaffinized in
xylene and dehydrated in fractional ethanol. Endogenous peroxidase
activity was blocked by incubating the sections in 3% hydrogen
peroxide at RT for 10 min. The appropriate antibody (1:200) was
used as the primary antibody for the IHC analysis and was detected
with the streptavidin peroxidase conjugation (SP-IHC) method. The
semi-quantitative results were analyzed according to the staining
intensity and the percentage of positively labelled cells as
described by Sinicrope et al (21). Ten independent fields in each
section were analyzed using ImageJ software (National Institutes of
Health) at high magnification (×400) to obtain an average
score.
Statistical analysis
The results are expressed as the mean ± standard
deviation and were calculated using data from at least three
independent repetitions. SPSS 16.0 software (SPSS, Inc., Chicago,
IL, USA) and GraphPad Prism 6.0 (GraphPad Software, Inc., La Jolla,
CA, USA) were used to perform two-tailed Student's t-tests and
Pearson's correlation analyses. One-way and two-way analysis of
variance (ANOVA) were used to test for overall differences.
Differences were defined as significant at P<0.05.
Results
Aberrant miR-300 expression in human
HCC and liver cancer cells is correlated with a poor prognosis
First, we detected miR-300 levels in HCC and liver
cancer cells (HCCLM3, MHCC-97H, MHCC-97L, HuH-6, HuH-7, Hep3B and
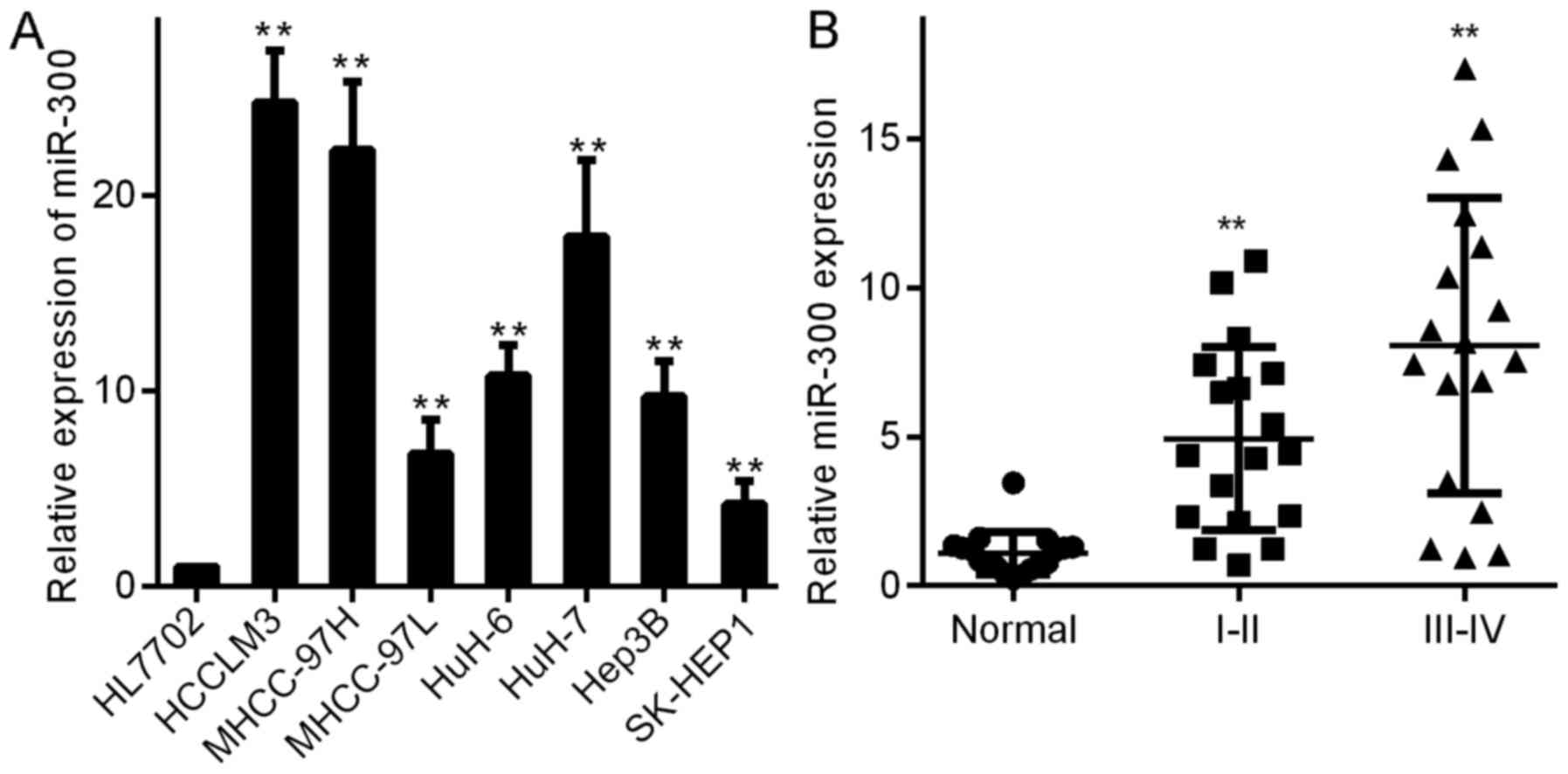
SK-HEP1) and normal liver cells (HL7702). As shown in Fig. 1A, miR-300 was expressed at higher
levels in the liver cancer cells (Fig.
1A) than that in the normal liver cells (HL7702). These results
indicate that miR-300 is associated with liver cancer cell growth.
We measured the expression of miR-300 in 54 HCC tissue samples to
verify that miR-300 is related to the development of HCC. miR-300
was expressed at significantly higher levels in HCC tissues than
that noted in the normal liver tissues (Fig. 1B). Notably, miR-300 levels were
highest in stage III–IV tumors and were higher in stage I–II tumors
than in normal liver tissues (Fig.
1B), demonstrating that aberrant miR-300 expression in human
HCC cells is correlated with a poor prognosis.
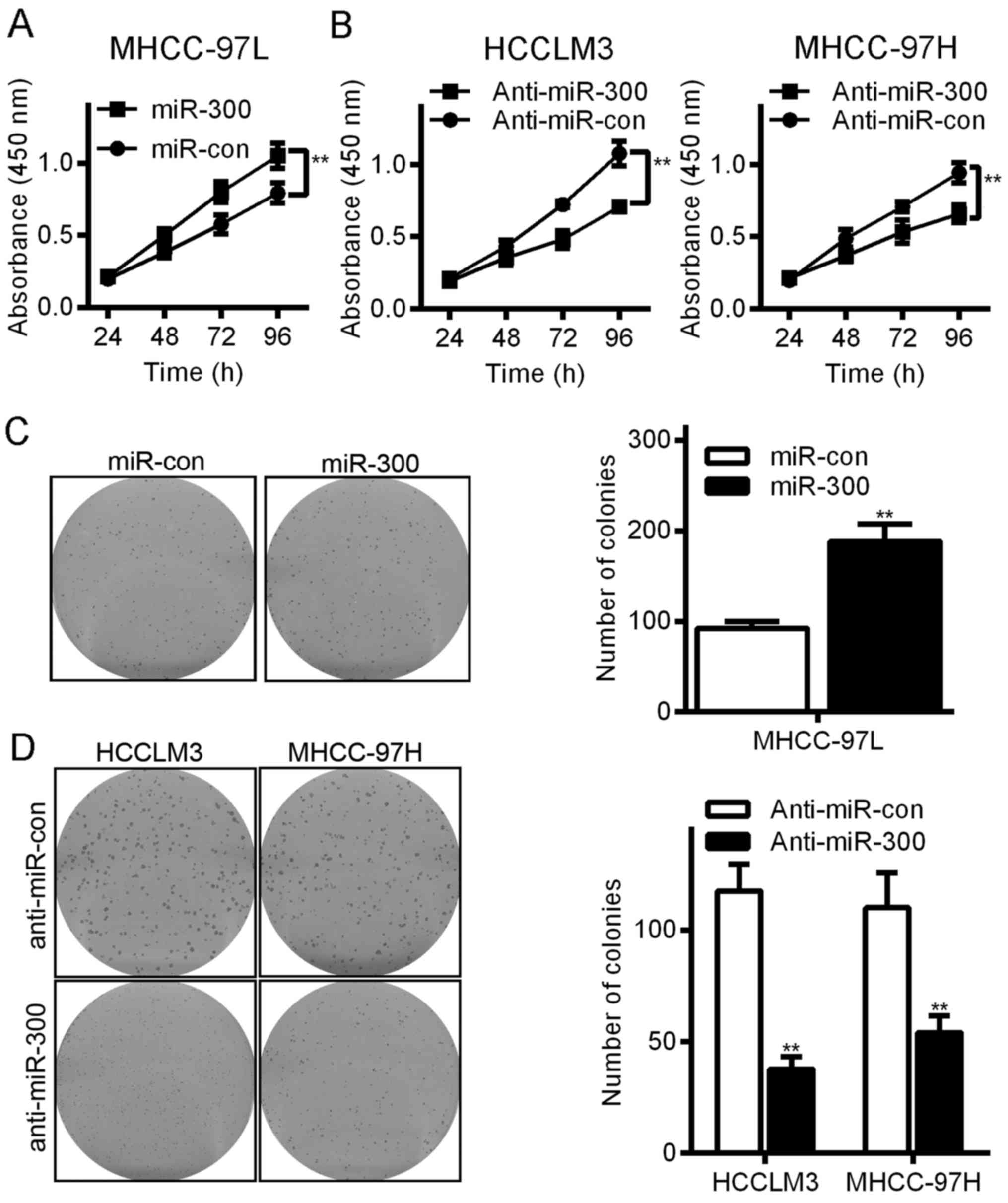
Effects of exogenous miR-300 on colony
formation and cell viability in HCC cells
To better understand the function of miR-300 in HCC,
we used lentiviral vectors to overexpress or silence miR-300 in the
HCC cell lines. The expression levels of miR-300 in the resulting
overexpression and knockdown cell lines were detected with qRT-PCR
(data not shown). First, we used colony formation and MTT assays to
investigate the growth-promoting effect of miR-300 on the HCC cell
lines. The colony formation efficiency of the HCC MHCC-97L cells
was 187.9±19.5 in the miR-300 group and 92.1±7.9 in the control
group (P<0.05, Fig. 2C). In
contrast, downregulating miR-300 reduced the proliferative ability
of the HCCLM3 and MHCC97H cells. The colony formation efficiency of
the HCCLM3 cells was 37.7±5.5 in the anti-miR-300 group and
117.5±12.1 in the anti-miR-con group (P<0.05, Fig. 2D), and the colony formation
efficiency of the MHCC-97H cells was 110.0±15.5 in the anti-miR-300
group and 54.0±7.6 in the anti-miR-con group (P<0.05, Fig. 2D). The MTT assays showed similar
results (Fig. 2A). Thus, miR-300
significantly increased the viability of human HCC cells.
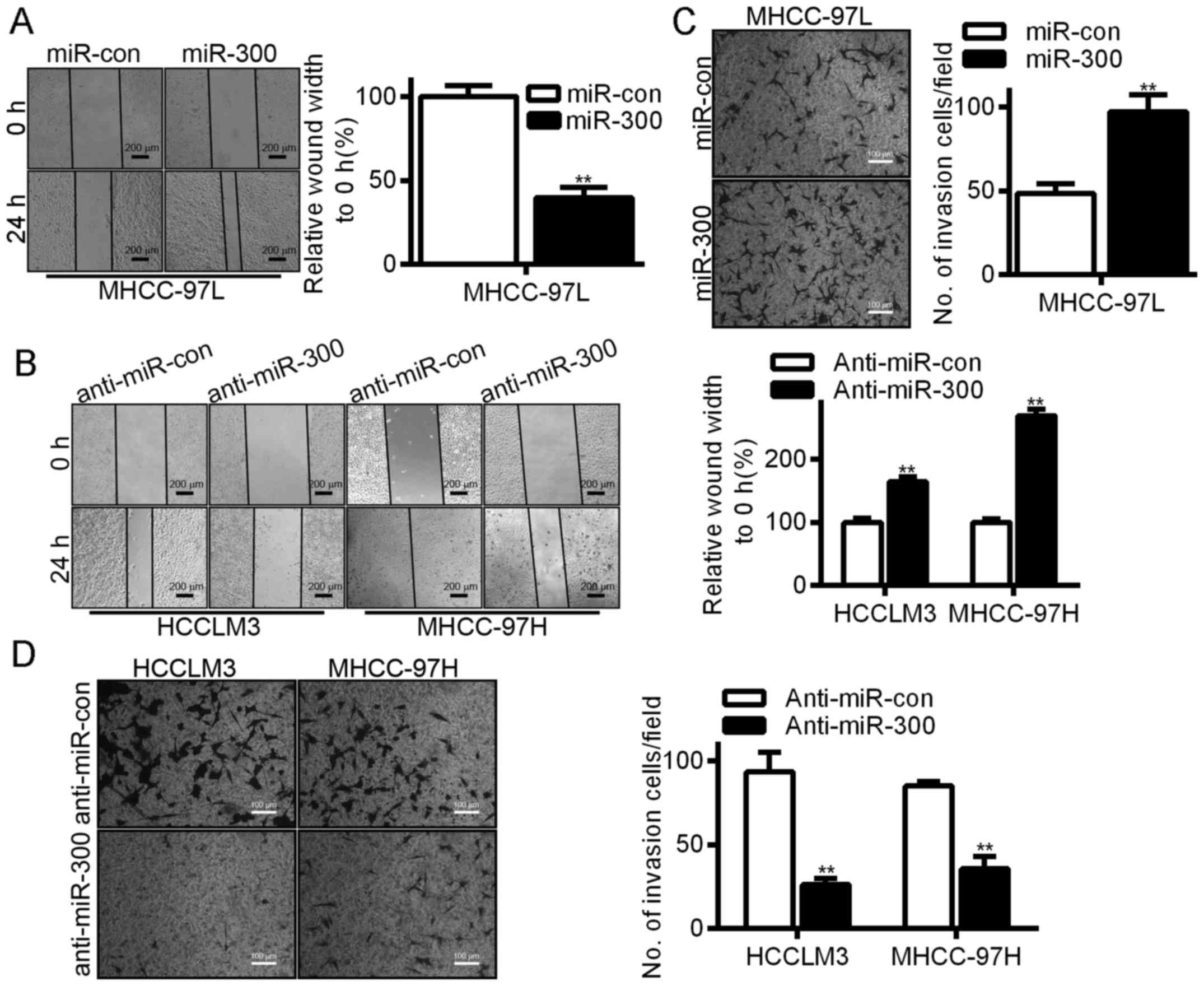
miR-300 promotes migration, invasion
and sphere formation in HCC cells
To further explore the functions of miR-300 in HCC,
we examined the effect of miR-300 on HCC cell migration and
invasiveness. In our wound healing experiments, overexpression of
miR-300 significantly accelerated wound healing in the HCC cells
(Fig. 3A), while silencing of
miR-300 reduced cell mobility (Fig.
3B). We also used a Transwell chamber whose inserts were coated
with or without Matrigel to assess the invasive capacity of the HCC
cells; the experimental results revealed that overexpression of
miR-300 significantly enhanced the rate of HCC cell invasion
(Fig. 3C), while silencing of
miR-300 reduced the rate of HCC cell invasion (Fig. 3D).
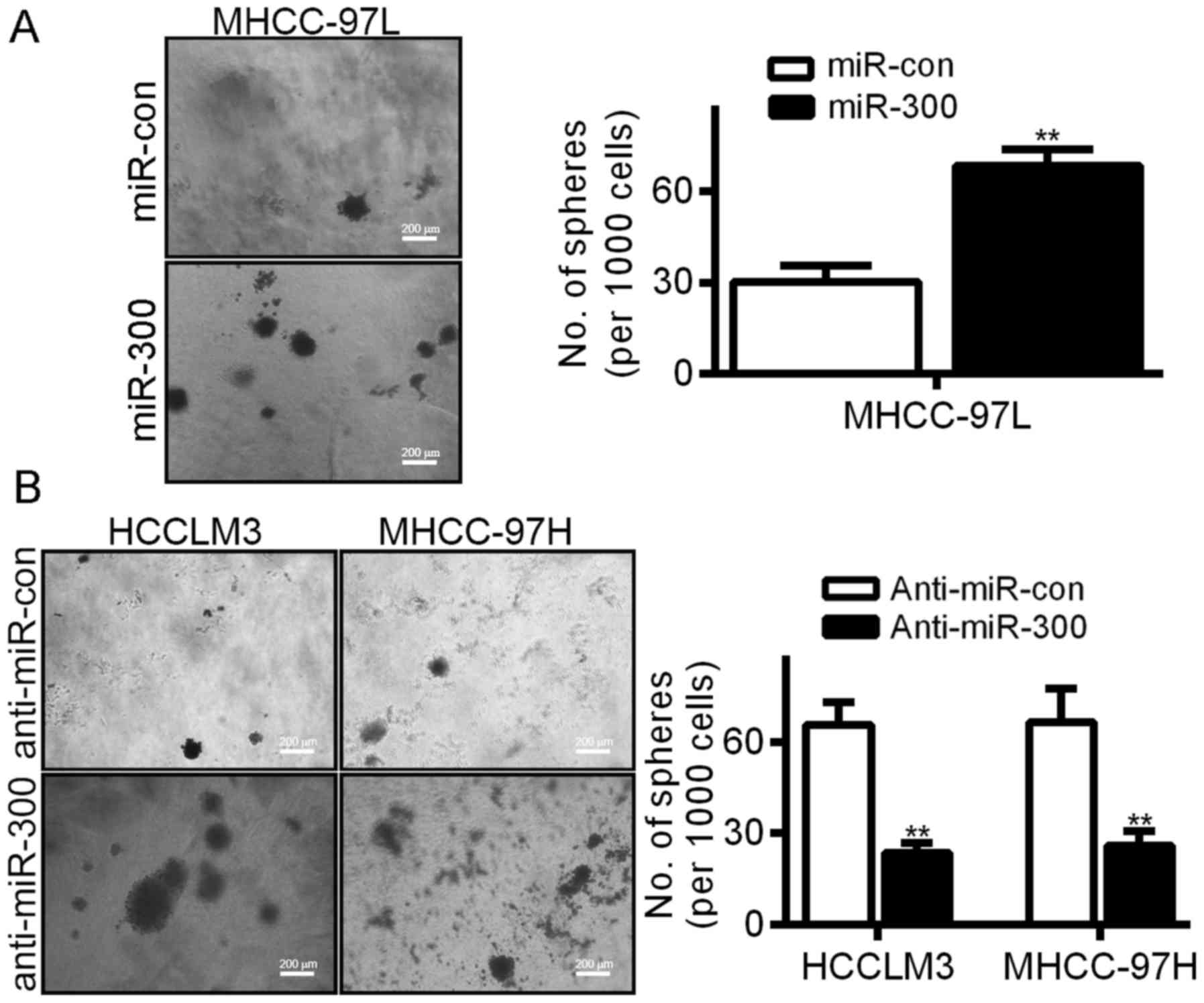
We demonstrated that miR-300 is expressed at higher
levels in HCC tissues (Fig. 1B).
Sphere formation was analyzed to confirm the role of miR-300 in HCC
cells. As shown in Fig. 4A, ectopic
miR-300 expression promoted an increase in the number of HCC cell
spheres (Fig. 4A), while silencing
of miR-300 reduced sphere formation (Fig. 4B).
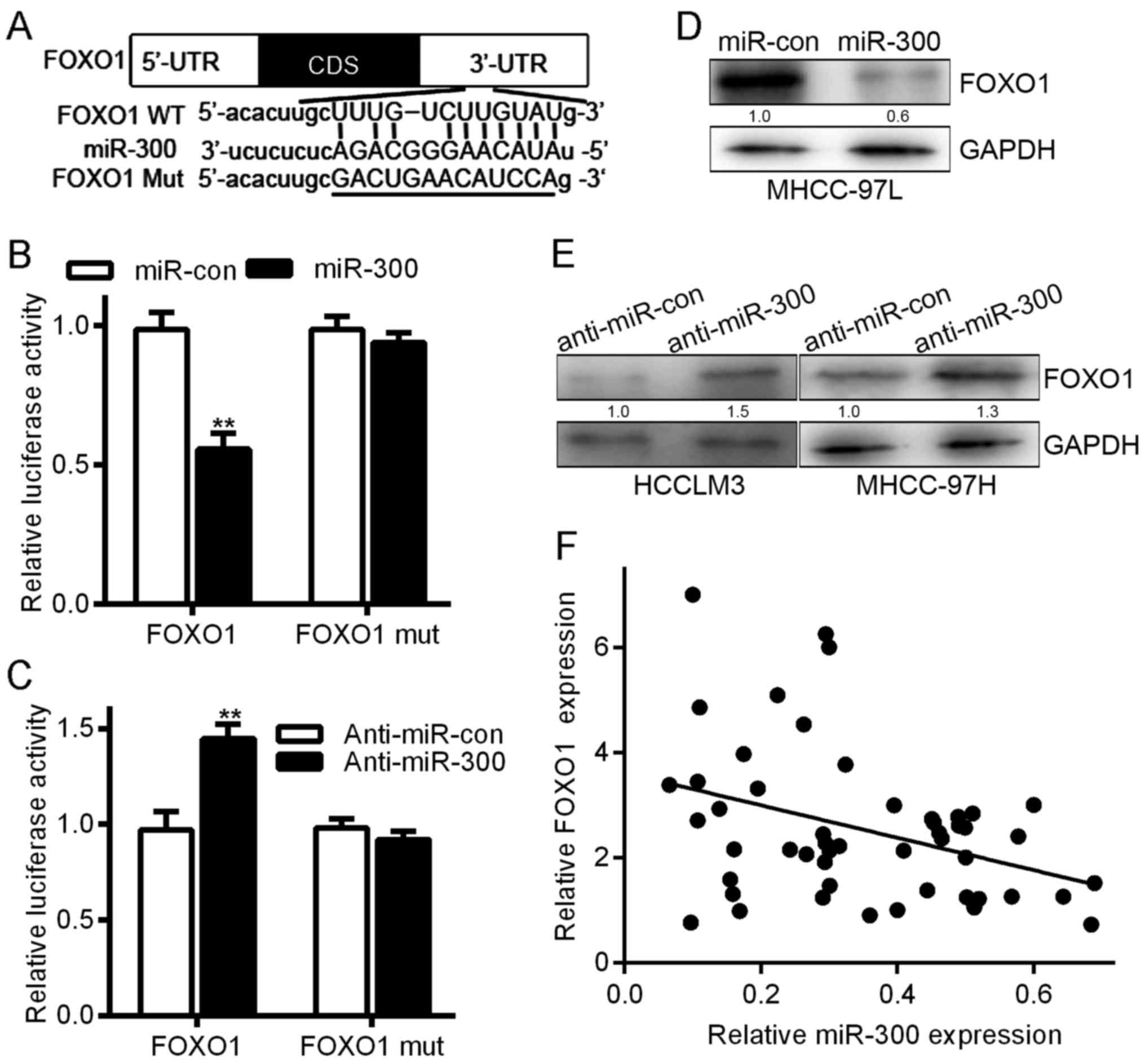
FOXO1 is a potential target of
miR-300, and FOXO1 levels are inversely correlated with miR-300
levels in HCC tissues
The TargetScan and RNAhybrid algorithms predicted
that FOXO1 is a direct target of miR-300 (Fig. 5A). To verify that FOXO1 is a target
of miR-300, we analyzed the association between miR-300 and FOXO1.
Co-transfection with miR-300 significantly decreased FOXO1
luciferase reporter activity (Fig.
5B, one-way ANOVA, P<0.05), while co-transfection with a
miR-300 inhibitor had the opposite effect (Fig. 5C, one-way ANOVA, P<0.05). When
the reporter gene carrying the mutated FOXO1 was co-transfected
into the cells, the effect on luciferase activity was eliminated
(Fig. 5B and C, one-way ANOVA
test). miR-300 overexpression downregulated FOXO1 protein levels in
MHCC-97L cells. Conversely, miR-300 downregulation increased FOXO1
levels in HCCLM3 and MHCC-97H cells (Fig. 5D and E). Moreover, a linear
correlation analysis showed that FOXO1 expression was inversely
proportional to miR-300 expression (Fig. 5F). Collectively, these data suggest
that miR-300 exerts its effects on HCC by directly suppressing
FOXO1.
The AKT/FOXO1 and AKT/mTOR pathways
contribute to the maintenance of a miR-300-mediated malignant
phenotype in HCC cells
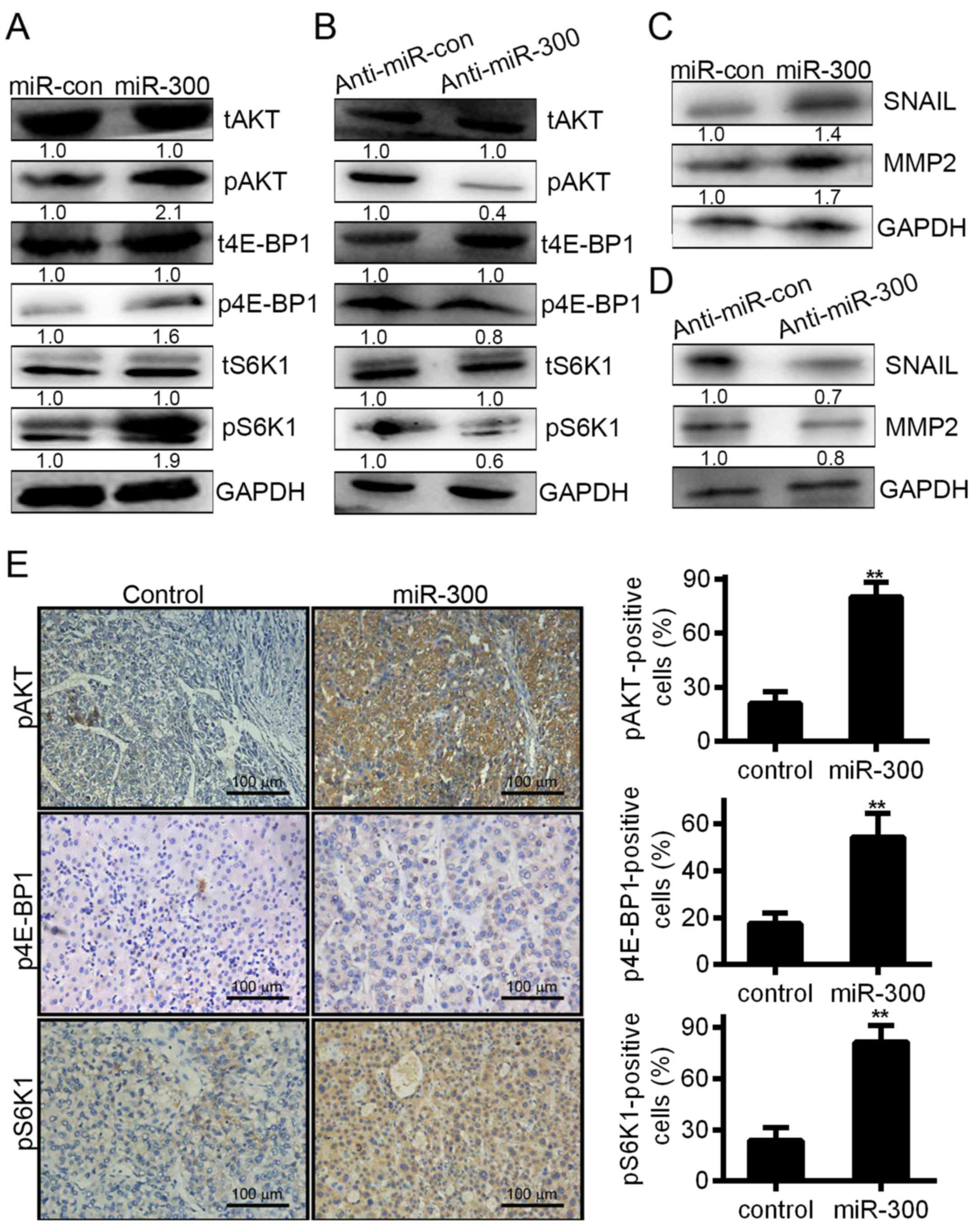
Next, we detected the effect of the miR-300-mediated
inhibition of FOXO1 on the maintenance of malignant phenotypes in
HCC cells. Notably, overexpression of miR-300 significantly
increased the expression levels of phosphorylated AKT, leading to
an increase in the expression levels of phosphorylated S6K and
4E-BP1 (Fig. 6A), while inhibiting
miR-300 in HCCLM3 cells strongly inhibited AKT, S6K and 4E-BP1
expression (Fig. 6B), suggesting
that miR-300 activated the AKT/FOXO1 and AKT/mTOR signaling
pathways. Accordingly, SNAIL and MMP2 were found to promote tumor
invasion and metastasis in HCC (22). As shown, the increased expression of
the MMP2 and SNAIL proteins (Fig.
6C) may have been caused by miR-300 overexpression; when
miR-300 was knocked down in HCCLM3 cells, MMP2 and SNAIL protein
expression was downregulated (Fig.
6D). In addition, the phosphorylation of AKT, S6K and 4E-BP1
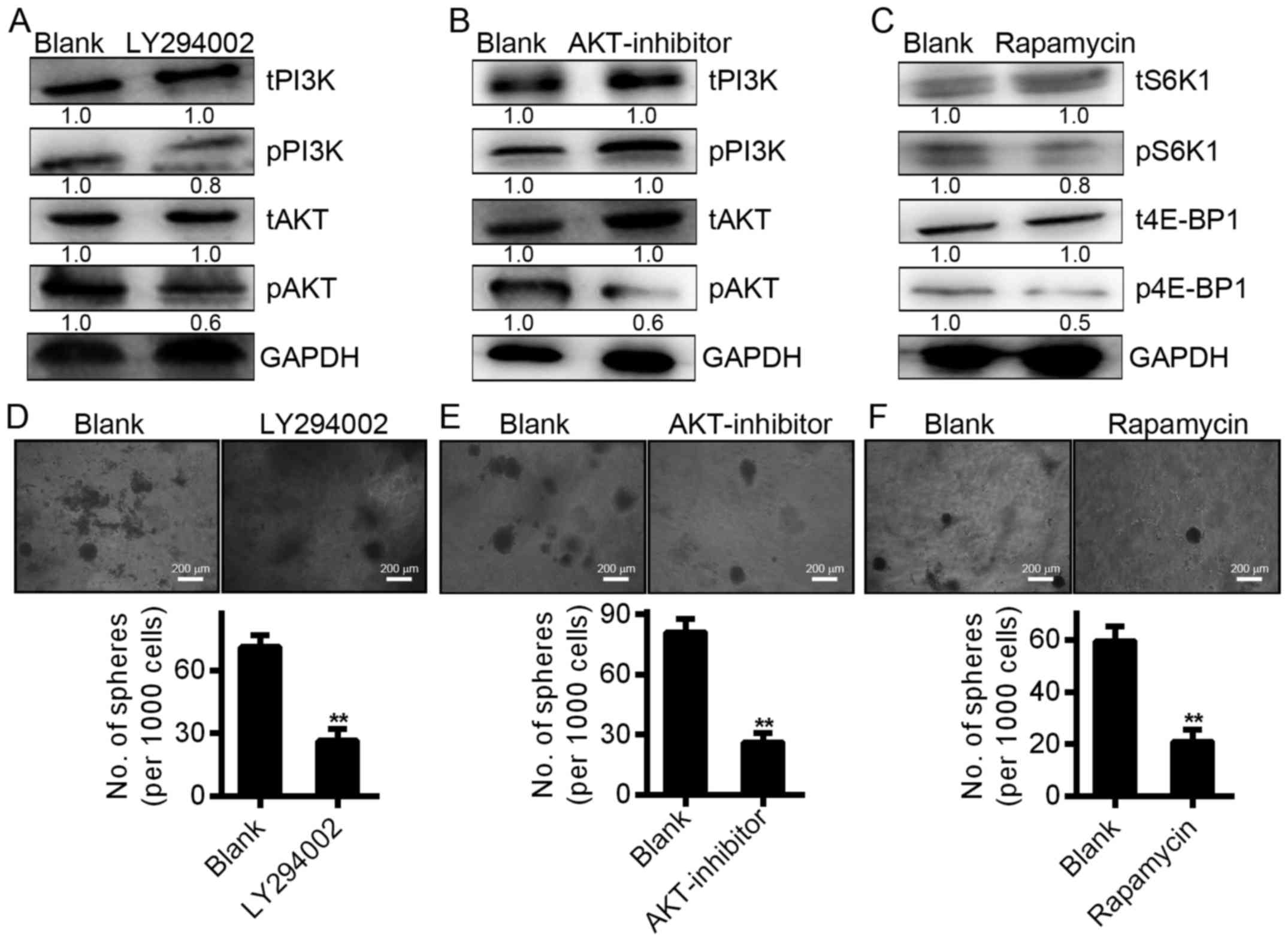
was increased in tumor cells overexpressing miR-300 (Fig. 6E). To confirm that AKT/FOXO1 and
AKT/mTOR pathway activation is involved in the effects of miR-300,
we used specific inhibitors of each signaling pathway in our
subsequent experiments. The results indicated that LY294002 (20
µmol/l), a P13K inhibitor, significantly reduced the
phosphorylation of P13K and Akt (Fig.
7A), while the AKT inhibitor (6 µmol/l) reduced the
phosphorylation of Akt but not P13K (Fig. 7B). Rapamycin (2 µmol/l), an
inhibitor of the mTOR pathway, reduced the phosphorylation of S6K
and 4E-BP1 (Fig. 7C). In addition,
all three types of inhibitors reduced the growth of
miR-300-overexpressing cells (Fig.
7D-F). These results indicate that miR-300 promotes a
proliferative phenotype in HCC cells by activating the AKT/FOXO1
and AKT/mTOR signaling pathways.
Suppression of FOXO1 in liver cancer
cells is essential for miR-300-induced viability
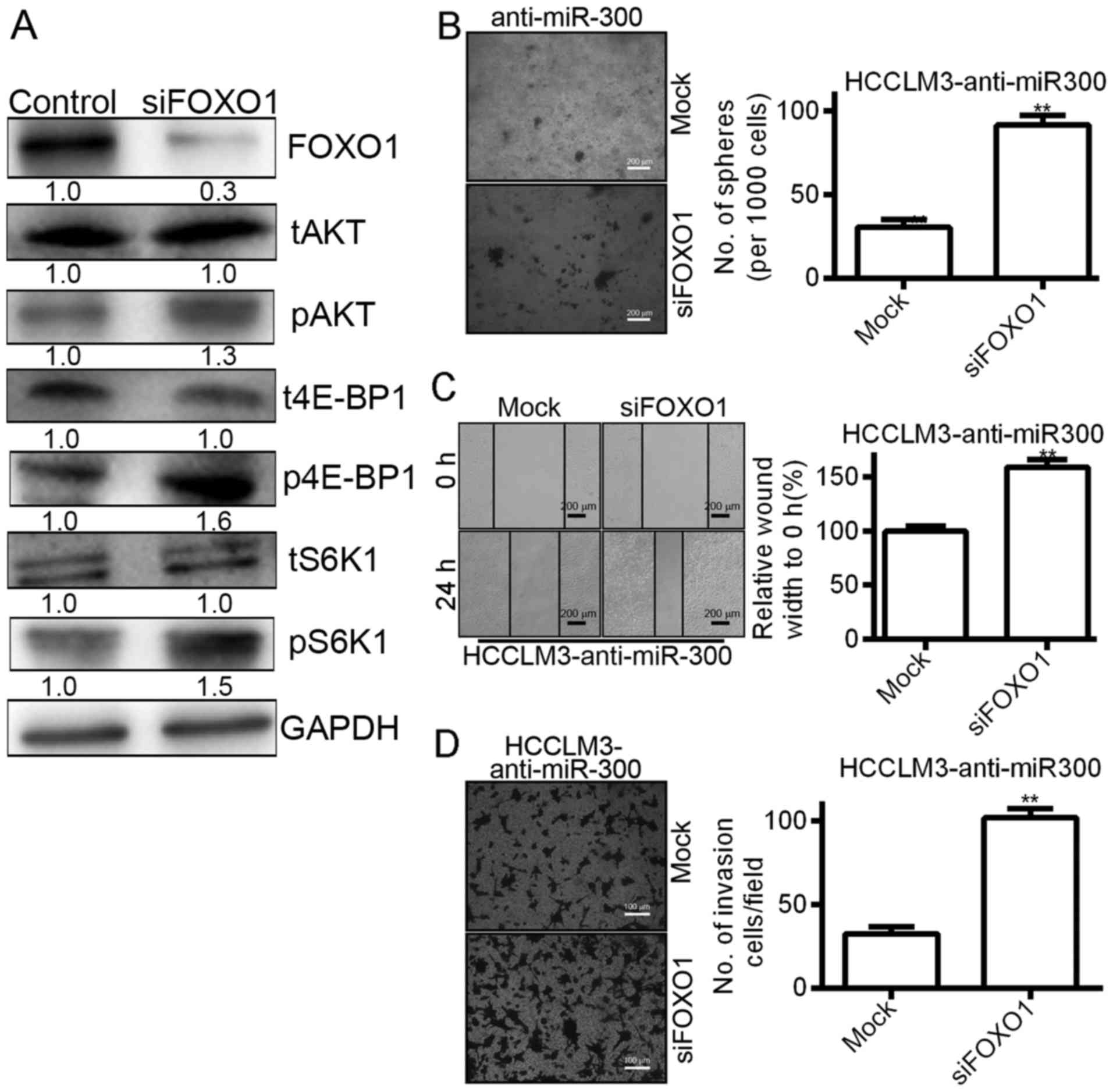
To investigate the indispensable role of FOXO1 in
miR-300-mediated biological functions, we silenced FOXO1 in
HCCLM3-anti-miR300 cells (Fig. 8A).
The results as shown in Fig. 8A
indicated that ectopic knockdown of FOXO1 increased the
phosphorylation of S6K and 4E-BP1 in HCCLM3-anti-miR-300 cells
(Fig. 8A). In subsequent functional
experiments, silencing of FOXO1 in anti-miR-300 cells significantly
induced cell viability and clone formation in soft agar (Fig. 8B) and wound healing assays (Fig. 8C). In addition, silencing of FOXO1
promoted the invasiveness of HCCLM3 cells transfected with miR-300
(Fig. 8D). These data indicate that
FOXO1 plays a key role in miR-300-induced viability, migration and
invasion.
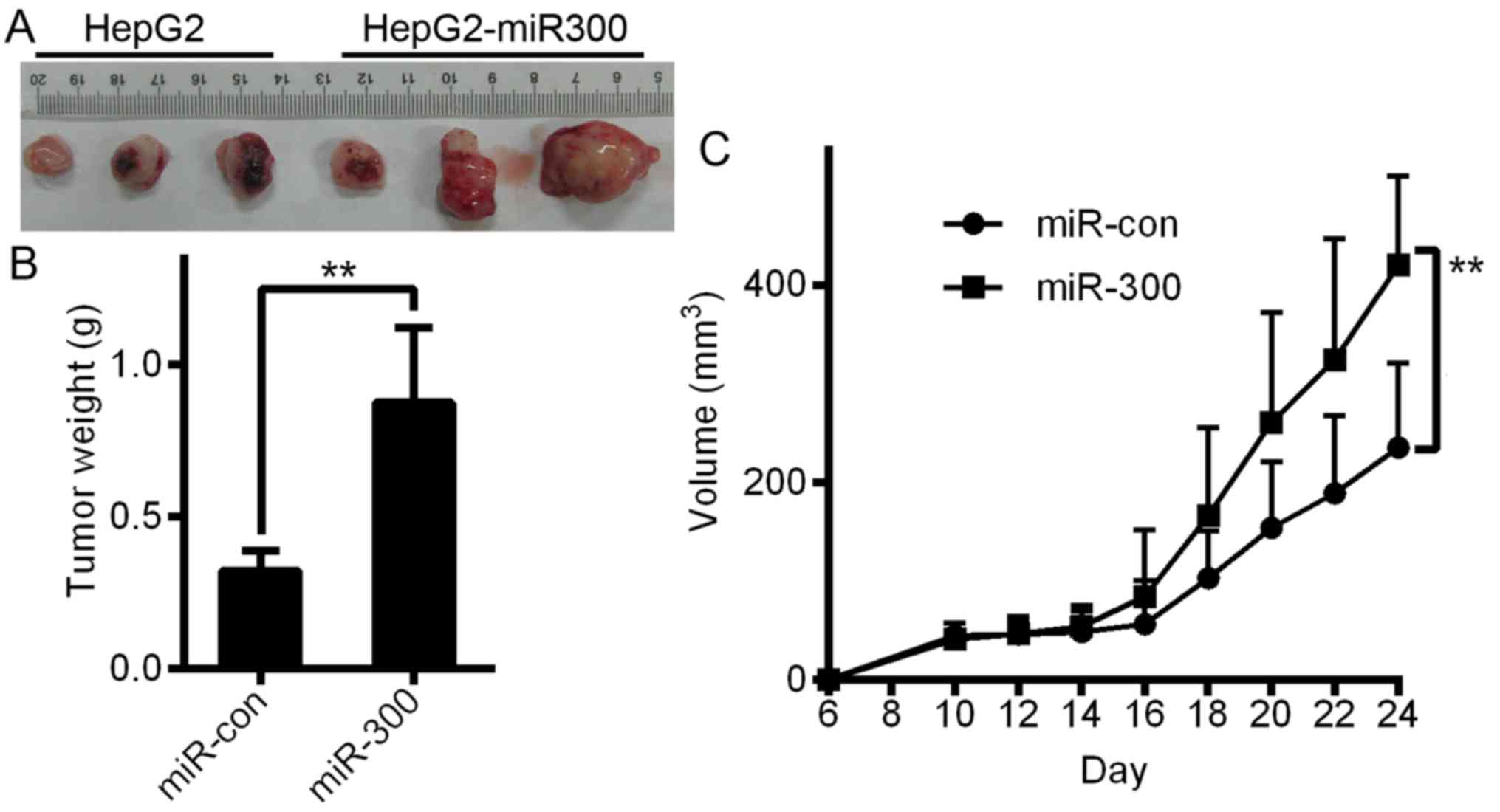
miR-300-overexpressing cells and control cells were
subcutaneously inoculated into thymic Balb/C mice to evaluate the
function of miR-300 to confirm whether its growth-promoting effect
in vitro was related to liver cancer growth in vivo.
As shown in Fig. 9A and B, in the
in vivo assays, miR-300 overexpression significantly
contributed to tumor growth and increases in tumor weight (Fig. 9B) and tumor volume (Fig. 9C). Based on these results, we
concluded that miR-300 promotes the viability of liver cancer cells
both in vivo and in vitro.
Discussion
Many studies have indicated that aberrant miRNA
expression contributes to the progression and development of
cancers, including HCC (23).
miRNAs have become novel prognostic biomarkers for and effective
therapeutic targets in HCC (24).
This study provides initial data showing that miR-300 expression is
increased in HCC cell lines and tissues and that in HCC,
invasiveness and the risk of relapse are associated with a higher
level of miR-300 expression. These results indicated that miR-300
plays an important role in promoting tumor functions in HCC.
More recently, many studies have demonstrated that
miRNAs play important roles in maintaining normal cellular
functions and that disorders of miRNA expression can lead to cancer
progression (25). In this study,
we investigated the expression and function of miR-300 in HCC and
liver cancer cells. We also explored the molecular mechanisms
underlying the effects of miR-300 to gain a better understanding of
its roles, and we found that the AKT/FOXO1 and AKT/mTOR signaling
pathways contributed to the malignant phenotype observed in
miR-300-overexpressing HCC cells and that the specific mechanism
responsible for this effect may involve the inhibition of FOXO1
expression. It is noteworthy that there was a strong correlation
between high miR-300 expression and low FOXO1 expression and the
malignant nature of HCC in both implanted tumors and clinical HCC
samples. These data collectively demonstrate that miR-300 affects
the development of HCC.
FOXO1 is a member of the FOXO subfamily of
transcription factors and acts as a tumor suppressor and regulates
genes involved in apoptotic responses, cell cycle checkpoints and
cellular metabolism. A number of studies indicate that FOXO1
expression is decreased in various types of cancer tissues compared
with normal tissues. However, the mechanism of aberrant FOXO1
expression is poorly understood. Recent studies have shown that
post-transcriptional regulation plays a key role in the
downregulation of FOXO1 and the modulation of its activity. miR-300
has been shown to play an anti-apoptotic role in bladder and
prostate cancer by targeting the 3′-UTR of FOXO1 to reduce its
expression. The transcription factor FOXO1 is expressed in a
variety of species and belongs to a subfamily of Forkhead protein
family class O transcription factors. FOXO1 exerts its
tumor-suppressive function by modulating the transcription of
important regulators of the cell cycle and apoptosis (26–28),
and previous studies support the notion that FOXO1 functions as a
tumor suppressor by demonstrating its role in the regulation of
cell cycle progression and cell differentiation, metabolism and
survival (28). Furthermore, lower
levels of FOXO1 expression have been observed in many tumor types,
such as Hodgkin lymphoma (29) and
breast cancer (30). FOXO1
transcriptional activity is regulated by the PI3K/AKT signaling
pathway (31), and FOXO1 is
downregulated in prostate cancer by several molecular mechanisms.
As previously reported, FOXO1 activity is inhibited by Akt
signaling pathway hyperactivity, which occurs in up to 50% of all
prostate cancers, primarily as a result of a PTEN deletion
(32). Furthermore, the locus of
FOXO1 on chromosome 13q14 is deleted in patients with prostate
cancer (33). Our results indicated
that FOXO1 is regulated by an alternate mechanism in which its
3′-UTR is bound by miR-300.
FOXO1 belongs to a subfamily of forkhead
transcription factors that contain a conserved forkhead DNA-binding
domain (34). FOXO subfamily
members participate in various signaling pathways and regulate many
biochemical processes, such as cell cycle progression, cell
differentiation and the cell apoptosis (35–37).
FOXO1 is a tumor suppressor, and FOXO1 downregulation has been
implicated in tumor function (29,33).
Since FOXO is a downstream target of the serine/threonine protein
kinase B (PKB)/Akt signaling pathway, FOXO1 inhibition results from
high expression of Akt, whose activation induces the
phosphorylation of FOXO1, which causes the tumor suppressor to
translocate from the nucleus to the cytoplasm, where it is
subsequently degraded.
Ichiyama et al reported that FOXO1 is one of
the targets of the microRNA-183-96-182 cluster. miR-183C drives
Th17 pathogenicity in autoimmune diseases by inhibiting FOXO1
(38). Furthermore, two miRNAs,
miR-96 and miR-370, target FOXO1 and regulate its expression in
prostate cancer cells. Suppressing these miRNAs was found to
increase FOXO1 protein levels and decrease cell viability (39,40).
Our study showed that FOXO1 is a direct target of miR-300, which
downregulated FOXO1 expression in vitro, as shown by the
luciferase experiment. These results indicated that in addition to
the known mechanisms that regulate FOXO1 expression, other
mechanisms (e.g., the epigenetic regulation of miRNAs) regulate
FOXO1 expression. The regulation of FOXO1 by specific miRNAs plays
an important role in tumor progression.
It is important to determine whether miR-300 and
FOXO1 are useful as biomarkers for diagnostic assays. Our results
indicated that high miR-300 expression levels and low FOXO1
expression levels are associated with poor clinical features in
patients with HCC. Furthermore, the results demonstrated that high
miR-300 expression, FOXO1 downregulation, and a combination of both
are significantly associated with a poor prognosis in patients with
HCC. Our results also indicated that miR-300 and FOXO1 are
promising prognostic indicators in patients with HCC.
To test our hypothesis independently of patient
samples, we decided to perform cell-based assay. Based on our
results, liver cancer HepG2 cells exhibited a lower miRNA-300
level. In the last few decades, the HepG2 cell line was considered
HCC. However, the HepG2 cell line has been reported to be
misidentified. López-Terrada et al, who initially isolated
these cells, recently corrected their report and claimed that HepG2
cells should in fact be considered a hepatoblastoma cell line
(41). To exclude the misleading
effect of cytological background on the conclusion, we used
MHCC-97L cells as a parallel group. Our results of HepG2 and
MHCC-97L cells in vitro were similar (data not shown).
Therefore, we selected the human liver cancer HepG2 cell line for
the in vivo experiment, although it has hepatoblastoma
characteristics. The aforementioned misidentification issue is
unlikely to affect the outcome of our study.
In conclusion, our results indicate that FOXO1 is
downregulated by miR-300 in HCC cells and that FOXO1 mediates
miR-300-induced cell viability. In addition, pAKT, p4E-BP1, pS6K1,
SNAIL and MMP2 expression was found to be dysregulated in
miR-300-overexpressing HCC cells, indicating that miR-300 levels
are correlated with PI3K/AKT signaling pathway activity. In this
study, the results of the gain-of-expression and functional
loss-of-expression experiments confirmed that overexpression of
miR-300 promoted HCC cell migration and invasion, whereas knockdown
of miR-300 inhibited these metastatic behaviors both in
vitro and in vivo.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Scientific
and Technological Project Foundation of Xi'an City
[2017113SFYX007(6)], the Scientific and Technological Development
Research Project Foundation of Shaanxi Province (2016SF-121) and
the Fundamental Research Funds for the Central Universities of
Xi'an Jiaotong University.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
YC, XL, LF and XC conceived and designed the study.
YC, CZ, GQ, GWa and GWe performed the experiments. YC, XL, LF and
XC analyzed the data. YC and XL wrote the paper. LF and XC reviewed
and edited the manuscript. All authors read and approved the
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Ethical approval for the study using human tissues
was obtained from the First Affiliated Hospital Ethics Committee of
Xi'an Jiaotong University. All participants provided written
informed consent. The animal experiment was conducted at the Xi'an
Jiaotong University Experimental Research Laboratory with the
consent of the Experimental Animal Ethics Committee (No.
XJTULAC2018-450).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing
interests related to this study.
References
|
1
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: Epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Maluccio M and Covey A: Recent progress in
understanding, diagnosing, and treating hepatocellular carcinoma.
CA Cancer J Clin. 62:394–399. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Altomare DA and Testa JR: Perturbations of
the AKT signaling pathway in human cancer. Oncogene. 24:7455–7464.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cui SX, Shi WN, Song ZY, Wang SQ, Yu XF,
Gao ZH and Qu XJ: Des-gamma-carboxy prothrombin antagonizes the
effects of Sorafenib on human hepatocellular carcinoma through
activation of the Raf/MEK/ERK and PI3K/Akt/mTOR signaling pathways.
Oncotarget. 7:36767–36782. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
He L and Hannon GJ: MicroRNAs: Small RNAs
with a big role in gene regulation. Nat Rev Genet. 5:522–531. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu Z, Dou C, Yao B, Xu M, Ding L, Wang Y,
Jia Y, Li Q, Zhang H, Tu K, et al: Methylation-mediated repression
of microRNA-129-2 suppresses cell aggressiveness by inhibiting high
mobility group box 1 in human hepatocellular carcinoma. Oncotarget.
7:36909–36923. 2016.PubMed/NCBI
|
|
9
|
Yang W, Dou C, Wang Y, Jia Y, Li C, Zheng
X and Tu K: MicroRNA-92a contributes to tumor growth of human
hepatocellular carcinoma by targeting FBXW7. Oncol Rep.
34:2576–2584. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lu CY, Lin KY, Tien MT, Wu CT, Uen YH and
Tseng TL: Frequent DNA methylation of MiR-129-2 and its potential
clinical implication in hepatocellular carcinoma. Genes Chromosomes
Cancer. 52:636–643. 2013.PubMed/NCBI
|
|
11
|
Furuta M, Kozaki KI, Tanaka S, Arii S,
Imoto I and Inazawa J: miR-124 and miR-203 are epigenetically
silenced tumor-suppressive microRNAs in hepatocellular carcinoma.
Carcinogenesis. 31:766–776. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu Y, Ren F, Rong M, Luo Y, Dang Y and
Chen G: Association between underexpression of microrna-203 and
clinicopathological significance in hepatocellular carcinoma
tissues. Cancer Cell Int. 15:622015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Alpini G, Glaser SS, Zhang JP, Francis H,
Han Y, Gong J, Stokes A, Francis T, Hughart N, Hubble L, et al:
Regulation of placenta growth factor by microRNA-125b in
hepatocellular cancer. J Hepatol. 55:1339–1345. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Coulouarn C, Factor VM, Andersen JB,
Durkin ME and Thorgeirsson SS: Loss of miR-122 expression in liver
cancer correlates with suppression of the hepatic phenotype and
gain of metastatic properties. Oncogene. 28:3526–3536. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gramantieri L, Ferracin M, Fornari F,
Veronese A, Sabbioni S, Liu CG, Calin GA, Giovannini C, Ferrazzi E,
Grazi GL, et al: Cyclin G1 is a target of miR-122a, a microRNA
frequently down-regulated in human hepatocellular carcinoma. Cancer
Res. 67:6092–6099. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lin CJ, Gong HY, Tseng HC, Wang WL and Wu
JL: miR-122 targets an anti-apoptotic gene, Bcl-w, in human
hepatocellular carcinoma cell lines. Biochem Biophys Res Commun.
375:315–320. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Greer EL and Brunet A: FOXO transcription
factors at the interface between longevity and tumor suppression.
Oncogene. 24:7410–7425. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hartmann W, Küchler J, Koch A, Friedrichs
N, Waha A, Endl E, Czerwitzki J, Metzger D, Steiner S, Wurst P, et
al: Activation of phosphatidylinositol-3′-kinase/AKT signaling is
essential in hepatoblastoma survival. Clin Cancer Res.
15:4538–4545. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nogueira V, Park Y, Chen CC, Xu PZ, Chen
ML, Tonic I, Unterman T and Hay N: Akt determines replicative
senescence and oxidative or oncogenic premature senescence and
sensitizes cells to oxidative apoptosis. Cancer Cell. 14:458–470.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gospodarowicz MK, Brierley JD and
Wittekind C: TNM classifcation of malignant tumours. 8th edition.
John Wiley & Sons; Oxford, UK: 2017
|
|
21
|
Sinicrope FA, Ruan SB, Cleary KR, Stephens
LC, Lee JJ and Levin B: Bcl-2 and p53 oncoprotein expression during
colorectal tumorigenesis. Cancer Res. 55:237–241. 1995.PubMed/NCBI
|
|
22
|
Miyoshi A, Kitajima Y, Sumi K, Sato K,
Hagiwara A, Koga Y and Miyazaki K: Snail and SIP1 increase cancer
invasion by upregulating MMP family in hepatocellular carcinoma
cells. Br J Cancer. 90:1265–1273. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xiang ZL, Zhao XM, Zhang L, Yang P, Fan J,
Tang ZY and Zeng ZC: MicroRNA-34a expression levels in serum and
intratumoral tissue can predict bone metastasis in patients with
hepatocellular carcinoma. Oncotarget. 7:87246–87256. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dou C, Wang Y, Li C, Liu Z, Jia Y, Li Q,
Yang W, Yao Y, Liu Q and Tu K: MicroRNA-212 suppresses tumor growth
of human hepatocellular carcinoma by targeting FOXA1. Oncotarget.
6:13216–13228. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Calin GA and Croce CM: MicroRNA-cancer
connection: The beginning of a new tale. Cancer Res. 66:7390–7394.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Machida S, Spangenburg EE and Booth FW:
Forkhead transcription factor FoxO1 transduces insulin-like growth
factor's signal to p27Kip1 in primary skeletal muscle satellite
cells. J Cell Physiol. 196:523–531. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim SJ, Winter K, Nian C, Tsuneoka M, Koda
Y and McIntosh CH: Glucose-dependent insulinotropic polypeptide
(GIP) stimulation of pancreatic beta-cell survival is dependent
upon phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB)
signaling, inactivation of the forkhead transcription factor Foxo1,
and down-regulation of bax expression. J Biol Chem.
280:22297–22307. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Grinius L, Kessler C, Schroeder J and
Handwerger S: Forkhead transcription factor FOXO1A is critical for
induction of human decidualization. J Endocrinol. 189:179–187.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xie L, Ushmorov A, Leithäuser F, Guan H,
Steidl C, Färbinger J, Pelzer C, Vogel MJ, Maier HJ, Gascoyne RD,
et al: FOXO1 is a tumor suppressor in classical Hodgkin lymphoma.
Blood. 119:3503–3511. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu Y, Elshimali Y, Sarkissyan M, Mohamed
H, Clayton S and Vadgama JV: Expression of FOXO1 is associated with
GATA3 and Annexin-1 and predicts disease-free survival in breast
cancer. Am J Cancer Res. 2:104–115. 2012.PubMed/NCBI
|
|
31
|
Rena G, Guo S, Cichy SC, Unterman TG and
Cohen P: Phosphorylation of the transcription factor forkhead
family member FKHR by protein kinase B. J Biol Chem.
274:17179–17183. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Morgan TM, Koreckij TD and Corey E:
Targeted therapy for advanced prostate cancer: Inhibition of the
PI3K/Akt/mTOR pathway. Curr Cancer Drug Targets. 9:237–249. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dong XY, Chen C, Sun X, Guo P, Vessella
RL, Wang RX, Chung LW, Zhou W and Dong JT: FOXO1A is a candidate
for the 13q14 tumor suppressor gene inhibiting androgen receptor
signaling in prostate cancer. Cancer Res. 66:6998–7006. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Calnan DR and Brunet A: The FoxO code.
Oncogene. 27:2276–2288. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Huang H and Tindall DJ: FOXO factors: A
matter of life and death. Future Oncol. 2:83–89. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Furukawa-Hibi Y, Yoshida-Araki K, Ohta T,
Ikeda K and Motoyama N: FOXO forkhead transcription factors induce
G2-M checkpoint in response to oxidative stress. J Biol
Chem. 277:26729–26732. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Arden KC: FoxOs in tumor suppression and
stem cell maintenance. Cell. 128:235–237. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ichiyama K, Gonzalez-Martin A, Kim BS, Jin
HY, Jin W, Xu W, Sabouri-Ghomi M, Xu S, Zheng P, Xiao C and Dong C:
The MicroRNA-183-96-182 cluster promotes T helper 17 cell
pathogenicity by negatively regulating transcription factor Foxo1
expression. Immunity. 44:1284–1298. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Haflidadóttir BS, Larne O, Martin M,
Persson M, Edsjö A, Bjartell A and Ceder Y: Upregulation of miR-96
enhances cellular proliferation of prostate cancer cells through
FOXO1. PLoS One. 8:e724002013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wu Z, Sun H, Zeng W, He J and Mao X:
Upregulation of MircoRNA-370 induces proliferation in human
prostate cancer cells by downregulating the transcription factor
FOXO1. PLoS One. 7:e458252012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
López-Terrada D, Cheung SW, Finegold MJ
and Knowles BB: Hep G2 is a hepatoblastoma-derived cell line. Hum
Pathol. 40:1512–1515. 2009. View Article : Google Scholar
|