Introduction
Breast cancer is the most commonly diagnosed
malignancy among women and a leading cause of cancer-related
mortality worldwide. In the United States, an estimated 266,120 new
cases of breast cancer will be diagnosed and 40,920 patients will
succumb to this disease in 2018 (1). It has been verified that chemotherapy
can improve the survival and quality of life in breast cancer
patients (2). Paclitaxel is a
chemotherapeutic agent widely used in the treatment of breast
cancer. However, ~50% of breast cancer patients fail to respond to
chemotherapeutic agents due to intrinsic or acquired resistance
(3). Resistance to chemotherapy
remains a major obstacle to successful treatment of breast cancer.
Therefore, it is important to investigate the mechanism underlying
the development of drug resistance and develop novel therapeutic
strategies for overcoming resistance to paclitaxel in breast
cancer.
MicroRNAs (miRNAs) are a class of small non-coding
RNA molecules that are ~19-25 nucleotides in length and regulate
the expression of a wide variety of genes, mainly through
degradation of target mRNAs or inhibition of protein translation
(4). Previous studies demonstrated
that miRNAs are implicated in several critical cellular processes,
including proliferation, differentiation, cell-cycle control and
apoptosis (5,6). Recently, a growing volume of evidence
has demonstrated that dysregulation of miRNAs plays an important
role in cancer drug resistance. For example, overexpression of
miR-210 was observed to induce caspase-3-mediated apoptosis and
reverse gemcitabine resistance by regulation of ABCC5 gene
expression in pancreatic cancer (7). It was also observed that loss of
miR-17 and miR-20b enhanced breast cancer resistance to paclitaxel
through upregulating nuclear receptor coactivator 3 levels
(8). Restoration of miR-200c
expression inhibited cell proliferation and increased the
chemosensitivity to cisplatin by targeting AKT2 in osteosarcoma
(9). In female reproductive
cancers, class III β-tubulin was reported to be a target of
miR-200c involved in the chemosensitivity to paclitaxel (10). Our previous study demonstrated that
downregulation of miR-200c was associated with poor response to
neoadjuvant chemotherapy in patients with breast cancer (11). However, the molecular mechanisms
underlying the role of miR-200c in the resistance of breast cancer
to paclitaxel remain largely unexplored.
Sex-determining region Y-box 2 (SOX2) is a member of
the SOX gene family that has been demonstrated to play a critical
role in the regulation of self-renewal and pluripotency in human
embryonic stem cells (12) and
activation of breast cancer stem cells (13). Aberrant expression of SOX2 was
observed in various types of cancer, such as glioblastoma (14), lung cancer (15) and prostate cancer (16). It was demonstrated that
overexpression of SOX2 contributes to resistance of MCF-7 breast
cancer cells to tamoxifen, whereas downregulation of SOX2 enhanced
the sensitivity of MCF-7 cells to tamoxifen (17). In addition, it was found that
knockdown of SOX2 in pancreatic ductal adenocarcinoma cells
increased the response to small-molecule inhibitors targeting MEK
and AKT signaling (18). A recent
study indicated that downregulation of SOX2 reduced invasiveness
and increased sensitivity to paclitaxel by preserving the
epithelial-like properties of breast cancer stem cells (19).
The aims of the present study were to determine
whether miR-200c-3p expression is significantly downregulated in
paclitaxel-resistant breast cancer cells compared with parental
cells, to investigate how the overexpression of miR-200c-3p affects
the chemosensitivity to paclitaxel and cell apoptosis, and to
elucidate the role of SOX2 in this process.
Materials and methods
Cell lines
The human breast cancer cell line MCF-7 was obtained
from the China Center for Type Culture Collection (Shanghai,
China). The paclitaxel-resistant human breast cancer cell line
MCF-7/Tax was established through stepwise selection by increasing
the concentration of paclitaxel (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) over 8 months. MCF-7/Tax cells were cultured in
the continuous presence of 10 µg/ml paclitaxel to maintain the
drug-resistant phenotype (20).
Paclitaxel-resistant human breast cancer MCF-7/Tax cells and
parental MCF-7 cells were maintained in RPMI-1640 medium
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (Invitrogen; Thermo Fisher
Scientific, Inc.) and ampicillin and streptomycin at 37°C in a
humidified atmosphere containing 5% CO2. MCF-7/Tax cells
were further cultured in drug-free medium for >2 weeks prior to
subsequent experiments.
miRNA transfection
Cells were seeded in 6-well plates at a density of
105 cells per well and cultured for 24 h. The cells were
then transfected with miR-200c-3p mimics, miR-200c-3p inhibitor or
negative control using Lipofectamine 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
The miR-200c-3p mimics, miR-200c-3p inhibitor and negative control
were purchased from GenePharma Tech (Shanghai, China).
siRNA transfection
Cells were seeded in 6-well plates and transfected
with SOX2 siRNA or negative control siRNA using Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. siRNAs were supplied by GenePharma Tech.
The sequences used for the siRNAs were as follows: siSOX2,
5′-GGACAUGAUCAGCAUGUAUTT-3′ (sense) and 5′-AUACAUGCUGAUCAUGUCCTT-3′
(antisense); and negative control siRNA,
5′-UUCUCCGAACGUGUCACGUTT-3′ (sense) and 5′-ACGUGACACGUUCGGAGAATT-3′
(antisense). Cells were harvested for further analysis after 48
h.
Cell viability assay
Cell viability was measured using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay according to the manufacturer's protocol. Untransfected or
transfected cells were re-seeded in 96-well plates at a density of
5×103 cells per well. The cells were then treated with
different concentrations of paclitaxel for 24, 48 and 72 h. A total
of 100 µl MTT (Sigma-Aldrich; Merck KGaA) per well was added and
incubated at 37°C for 4 h. Subsequently, 150 µl dimethyl sulfoxide
was added to each well. The absorbance was measured at 570 nm by a
microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Three independent experiments were performed in quadruplicate.
Apoptosis assay
Apoptosis was evaluated by flow cytometry using the
FITC Annexin V Apoptosis Detection kit I (BD Biosciences, Franklin
Lakes, NJ, USA) according to the manufacturer's instructions. The
cells were harvested and washed with phosphate-buffered saline. A
total of 5 µl FITC-labeled enhanced Annexin V, 5 µl propidium
iodide (PI) and 100 µl binding buffer were added. After incubation
in the dark for 15 min at room temperature, the samples were
analyzed by flow cytometry (FACSCalibur, BD Biosciences). All
experiments were performed in triplicate.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA from cultured cells was extracted using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. The concentration and
quality of the RNA was determined using the Nanodrop 1000
Spectrophotometer (Thermo Fisher Scientific, Inc.). Quantitative
analysis of miRNA expression was performed by All-in-One miRNA
RT-qPCR Reagent kit (GeneCopoeia, Guangzhou, China) and U6 snRNA
was used for normalization according to the manufacturer's
instructions. To determine the mRNA levels of SOX2, total RNA was
reverse-transcribed using the PrimeScript 1st Strand cDNA Synthesis
kit (Takara, Dalian, China) with β-actin used as an endogenous
control according to the manufacturer's instructions. PCR was
performed on the StepOnePlus system (ABI 7500; Applied Biosystems;
Thermo Fisher Scientific, Inc.) and each sample was run in
triplicate. The relative expression levels of miRNA or mRNA were
calculated using the comparative Cq method (2−ΔΔCq)
(21). The primers for
amplification were as follows: SOX2 forward,
5′-TGGACAGTTACGCGCACAT-3′ and reverse, 5′-CGAGTAGGACATGCTGTAGGT-3′;
β-actin forward, 5′-TGACGTGGACATCCGCAAAG-3′ and reverse,
5′-CTGGAAGGTGGACAGCGAGG-3′; miR-200c-3p forward,
5′-UAAUACUGCCGGGUAAUGAUGGA-3′; U6 snRNA:
5′-GCTTCGGCAGCACATATACTAAAAT-3′. The universal reverse primer of
miR-200c-3p and U6 snRNA was 5′-GAGACTGCGGATGTATAGAACTTGA-3′.
Western blotting
Protein extracts were obtained using a lysis buffer
and the protein concentration was measured by a bicinchoninic acid
protein assay (Thermo Fisher Scientific, Inc.). Total protein was
separated on SDS-PAGE gels and transferred to polyvinylidene
difluoride membranes (EMD Millipore, Billerica, MA, USA). The
membranes were blocked in TBST solution containing 5% non-fat milk
and incubated with the primary antibody for SOX2 (dilution 1:1,000;
cat. no. 92494; Abcam, Cambridge, UK) and GAPDH (dilution 1:5,000;
cat. no. 201822; Abcam) overnight at 4°C. GAPDH was used as an
internal control. Subsequently, the membranes were incubated with a
horseradish peroxidase-conjugated goat anti-rabbit secondary
antibody (dilution 1:3,000; cat. no. 6721; Abcam) for 1 h at room
temperature. The bands were visualized using the ECL detection
system (Thermo Fisher Scientific, Inc.).
Luciferase reporter assay
The 3′-untranslated region (UTR) of SOX2 containing
the miR-200c-3p binding sites and its corresponding mutated
sequence were cloned into the psi-CHECK2 vector (Promega
Corporation, Madison, WI, USA) downstream of the Renilla
luciferase gene. MCF-7/TAX cells were cotransfected with the
luciferase vectors and miR-200c-3p mimics or negative control in
96-well plates using Lipofectamine 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). After transfection for 48 h, the Renilla
and firefly luciferase activities were measured using the
Dual-Luciferase Reporter assay kit (Promega Corp.) according to the
manufacturer's protocol. The firefly luciferase activity was
normalized to that of Renilla luciferase.
Statistical analysis
The statistical analysis was performed using the
SPSS version 19.0 statistical package (IBM Corp., Armonk, NY, USA).
The data are presented as mean ± standard deviation. Differences
between two groups were analyzed using the Student's t-test. One
way analysis of variance (ANOVA) with Bonferroni post hoc test was
used to analyze the differences among three or more groups.
P<0.05 was considered to indicate statistically significant
differences.
Results
miR-200c-3p expression is
significantly downregulated in paclitaxel-resistant MCF-7/Tax
cells
Paclitaxel-resistant MCF-7/Tax human breast cancer
cells were established through stepwise selection by increasing the
concentration of paclitaxel in MCF-7 cells. The MCF-7 and MCF-7/Tax
cell sensitivity to various concentrations of paclitaxel was
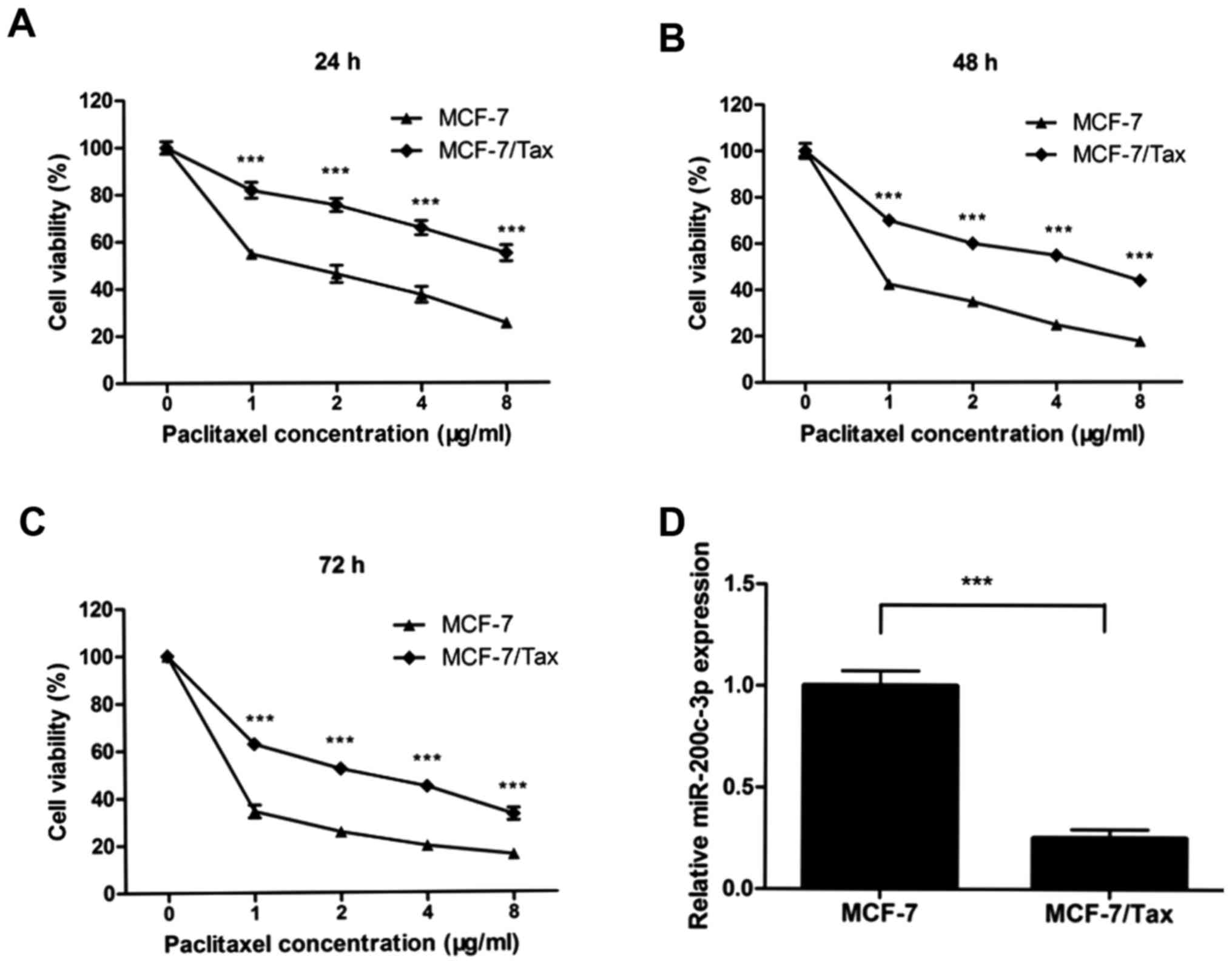
determined with MTT assays. As illustrated in Fig. 1A-C, MCF-7/Tax cells were
significantly resistant to paclitaxel compared with parental MCF-7
cells (all P<0.001). The proliferation of MCF-7/Tax cells was
markedly inhibited in a dose- and time-dependent manner.
To determine whether miR-200c-3p expression was
implicated in breast cancer cell resistance to paclitaxel,
miR-200c-3p expression was evaluated in MCF-7 and MCF-7/Tax cells
using RT-qPCR analysis. The expression level of miR-200c-3p in
paclitaxel-resistant MCF-7/Tax cells was found to be significantly
downregulated by 4-fold compared with that noted in the parental
MCF-7 cells (P<0.001, Fig. 1D).
These results suggested that reduced miR-200c-3p expression may be
involved in the resistance of breast cancer cells to
paclitaxel.
miR-200c-3p overexpression promotes
paclitaxel sensitivity in paclitaxel-resistant MCF-7/Tax cells
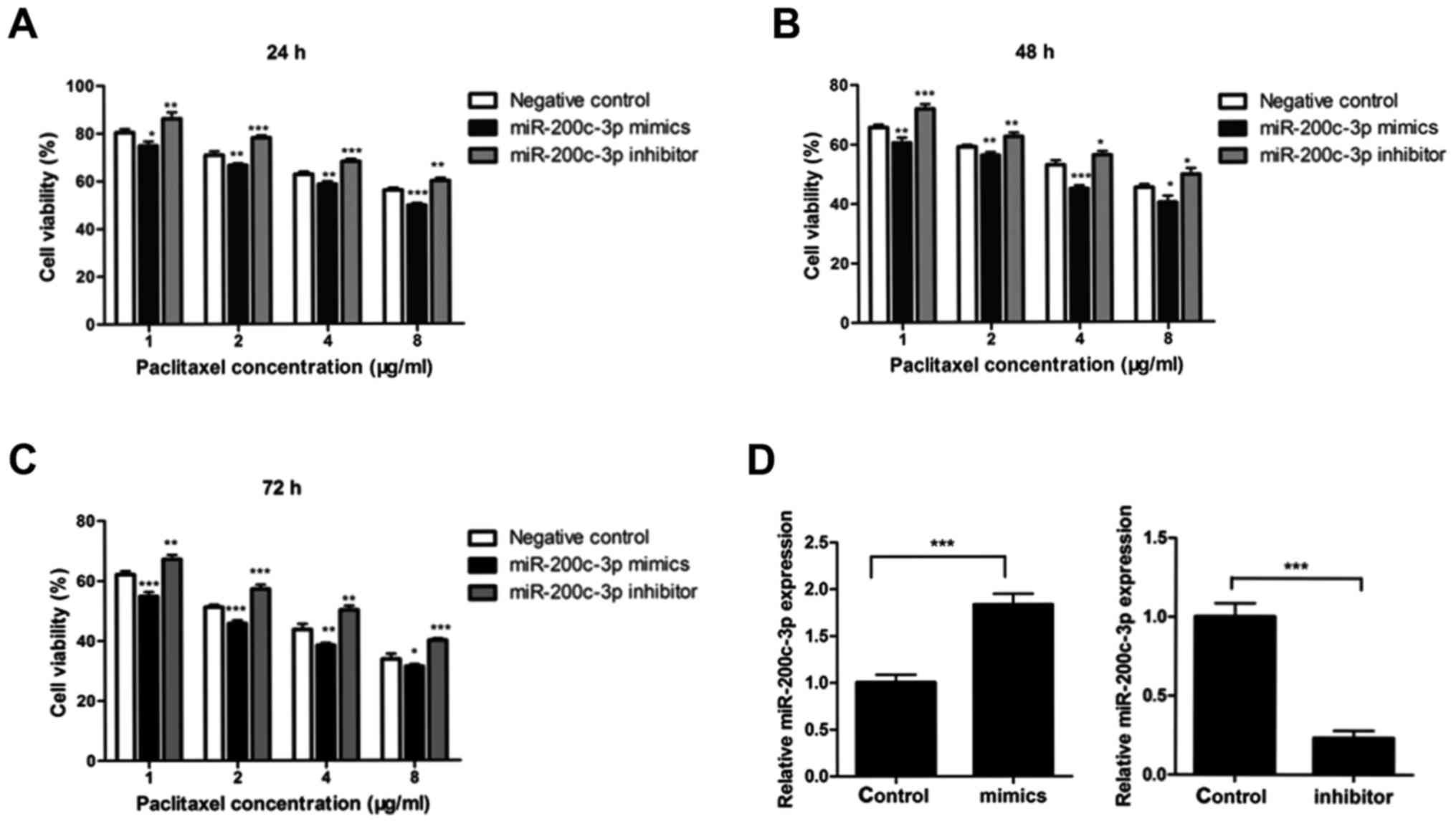
Downregulation of miR-200c-3p expression was
observed in paclitaxel-resistant MCF-7/Tax cells compared with that
noted in the parental MCF-7 cells. To evaluate the effect of
miR-200c-3p on the resistance to paclitaxel, MCF-7/Tax cells were
transfected with miR-200c-3p mimics, miR-200c-3p inhibitor or
negative control for 24 h. The cells were then exposed to various
concentrations of paclitaxel for 24, 48 or 72 h. The cell viability
was determined by MTT assays. As shown in Fig. 2A-C, transfection of miR-200c-3p
mimics into MCF-7/Tax cells significantly increased the
chemosensitivity to paclitaxel. By contrast, downregulation of
miR-200c-3p with transfection of miR-200c inhibitors significantly
decreased the chemosensitivity to paclitaxel in MCF-7/Tax cells
(Fig. 2A-C). miR-200c-3p expression
was significantly increased in MCF-7/Tax cells following
transfection of miR-200c-3p mimics and significantly decreased in
MCF-7/Tax cells transfected with miR-200c-3p inhibitors (Fig. 2D).
miR-200c-3p overexpression promotes
apoptosis of paclitaxel-resistant MCF-7/Tax cells
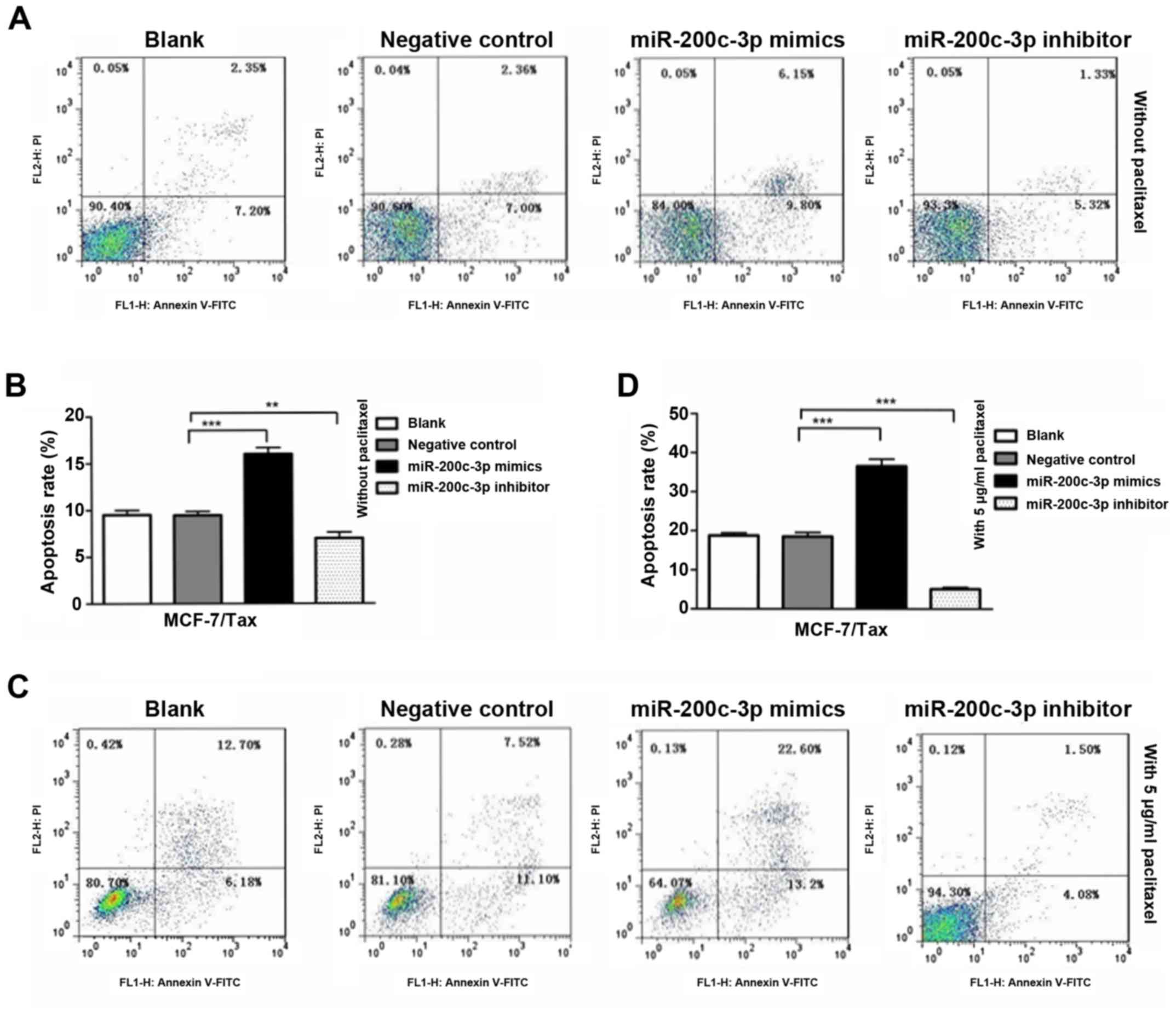
The potential role of miR-200c-3p in the apoptosis
of MCF-7/Tax cells was evaluated following transfection of the
cells with miR-200c-3p mimics, miR-200c-3p inhibitor or negative
control. The cell apoptosis was analyzed using flow cytometry. As
shown in Fig. 3A and B,
overexpression of miR-200c-3p in MCF-7/Tax cells significantly
promoted cell apoptosis compared with the negative control
(P<0.001). By contrast, inhibition of miR-200c-3p in MCF-7/Tax
cells significantly decreased apoptosis compared with the negative
control (P<0.01). In addition, a marked increase in the
apoptotic rate was observed in the MCF-7/Tax cells transfected with
miR-200c-3p mimics following treatment with 5 µg/ml
paclitaxel for 48 h compared with the negative control, whereas a
significant decrease in the apoptotic rate was detected in
MCF-7/Tax cells transfected with miR-200c-3p inhibitor following
paclitaxel treatment compared with the negative control
(P<0.001, Fig. 3C and D). These
findings indicate that overexpression of miR-200c-3p promoted
MCF-7/Tax cell apoptosis, with or without paclitaxel treatment,
whereas the downregulation of miR-200c-3p exerted the opposite
effect.
Prediction and validation of SOX2 as a
target of miR-200c-3p
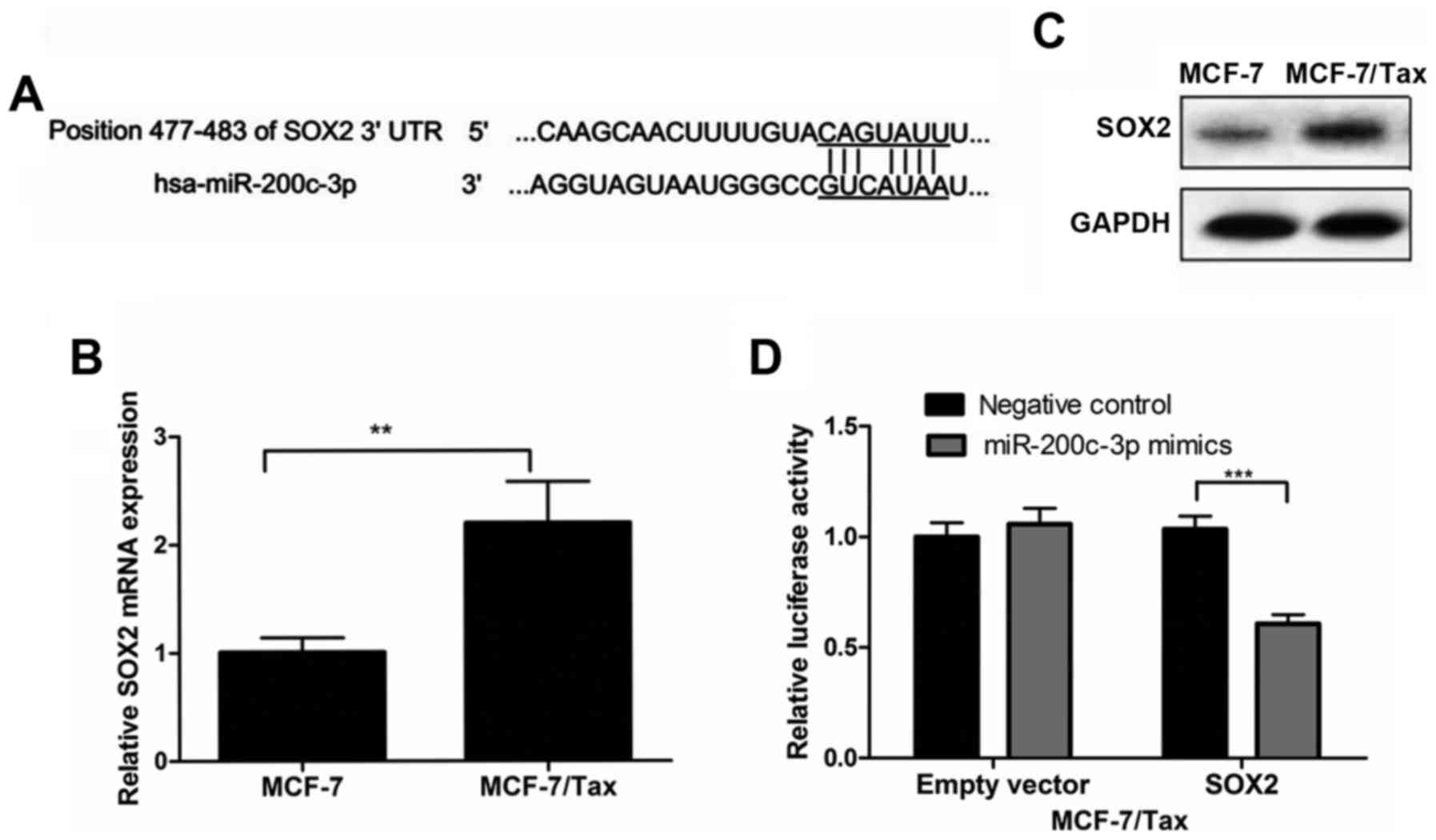
To further explore the molecular mechanism of action
of miR-200c-3p in the resistance of breast cancer cells to
paclitaxel, target gene prediction of miR-200c-3p was performed
using the TargetScan database, one of the most widely used miRNA
target prediction algorithms. Bioinformatic analysis indicated that
the SOX2 gene included the putative miR-200c-3p targeting sequence.
The predicted interaction between miR-200c-3p and its target site
in the SOX2 3′ UTR is illustrated in Fig. 4A.
The expression of SOX2 at the mRNA level was
evaluated in MCF-7/Tax and MCF-7 cells using RT-qPCR analysis. The
results demonstrated that SOX2 mRNA expression in MCF-7/Tax cells
was significantly increased by 2-fold compared with parental MCF-7
cells (P<0.01, Fig. 4B).
Consistent with SOX2 mRNA expression, the SOX2 protein expression
in MCF-7/Tax cells, as determined by western blotting, was also
increased compared with parental MCF-7 cells (Fig. 4C).
To determine whether SOX2 is a direct target of
miR-200c-3p, a dual luciferase reporter assay was performed.
MCF-7/Tax cells were co-transfected with the SOX2 reporter or empty
vector and miR-200c-3p mimic or miR-negative control. At 48 h after
transfection, the Renilla and firefly luciferase activities
were measured using the Dual-Luciferase Reporter assay kit. As
shown in Fig. 4D, the luciferase
activity was markedly suppressed in the presence of miR-200c-3p
mimics, whereas no reduction in luciferase activity was observed
with the empty vector control or in the presence of miR-negative
control.
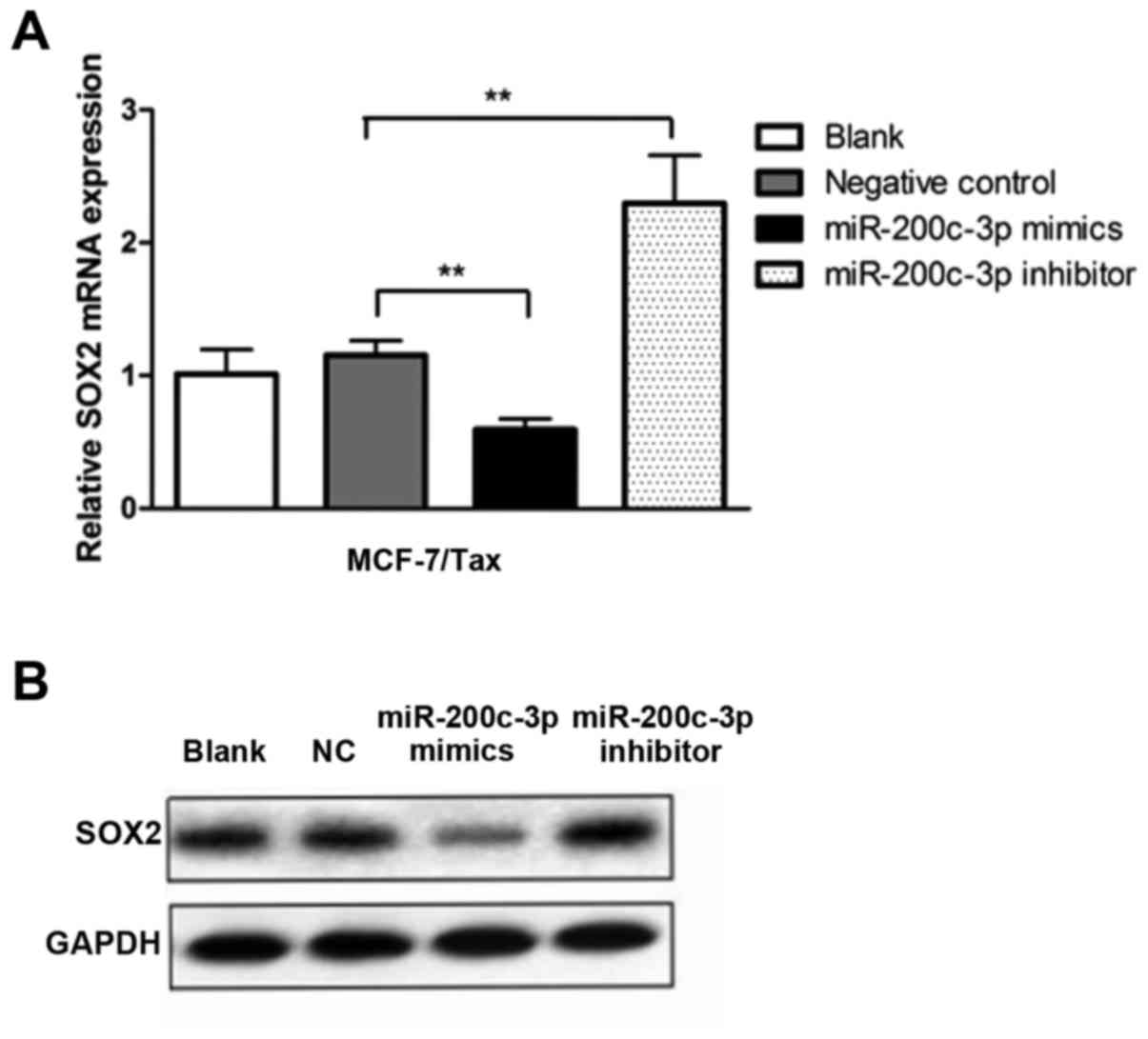
Upregulation of miR-200c-3p in
MCF-7/Tax cells suppresses SOX2 expression
To further verify the effect of miR-200c-3p on SOX2
gene expression in paclitaxel-resistant breast cancer cells, the
SOX2 mRNA and protein expression in MCF-7/Tax cells was determined
following transfection with miR-200c-3p mimics, miR-200c-3p
inhibitor or negative control. It was observed that miR-200c-3p
overexpression in the MCF-7/Tax cells resulted in a significant
decrease in SOX2 mRNA compared with the negative control
(P<0.01, Fig. 5A). By contrast,
downregulation of miR-200c-3p significantly increased the
expression of SOX2 mRNA in the MCF-7/Tax cells compared with the
negative control (P<0.01, Fig.
5A). As shown in Fig. 5B,
overexpression of miR-200c-3p in MCF-7/Tax cells also suppressed
the expression of the SOX2 protein, whereas downregulation of
miR-200c-3p exerted the opposite effect.
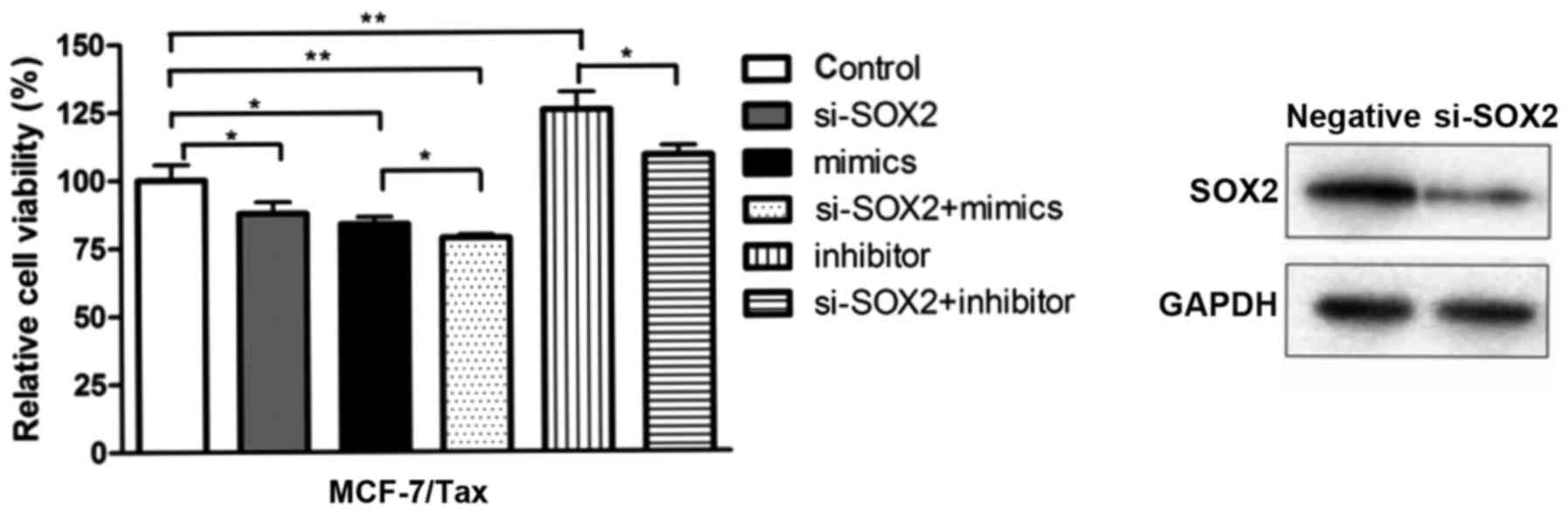
SOX2 knockdown increases
chemosensitivity to paclitaxel in MCF-7/Tax cells
To investigate whether SOX2 is implicated in the
resistance of breast cancer cells to paclitaxel, the
paclitaxel-resistant MCF-7/Tax cells were transfected with
siRNA-SOX2, miR-200c-3p mimics, miR-200c-3p inhibitor, siRNA-SOX2
combined with miR-200c-3p mimics, siRNA-SOX2 combined with
miR-200c-3p inhibitor or negative control. Subsequently, the
transfected cells were exposed to 5 µg/ml paclitaxel for 48 h and
cell viability was determined with MTT assays. The transfection
efficiency of MCF-7/Tax cells transfected with siRNA-SOX2 was
detected by western blot analysis (Fig.
6). As shown in Fig. 6,
transfection with miR-200c-3p mimics and siRNA-SOX2 reduced cell
viability more prominently compared with transfection with
miR-200c-3p mimics alone (P<0.05). In addition, treatment with
miR-200c-3p inhibitor and siRNA-SOX2 also reduced cell viability
compared with treatment with miR-200c-3p inhibitor alone
(P<0.05). It was observed that SOX2 knockdown was able to
increase the sensitivity of MCF-7/Tax cells to paclitaxel compared
with the control.
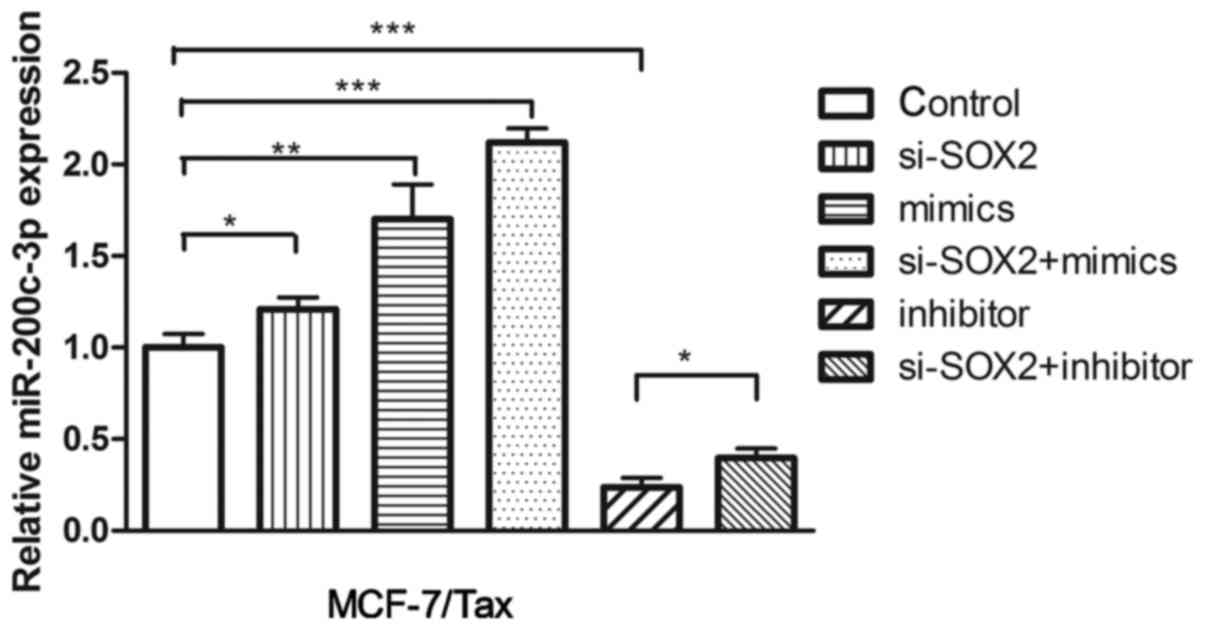
SOX2 knockdown in MCF-7/Tax cells
upregulates miR-200c-3p expression
To investigate the potential effect of SOX2 on
miR-200c-3p expression, siRNA interference was used to knock down
SOX2 in MCF-7/Tax cells. The expression level of miR-200c-3p was
determined by RT-qPCR. As shown in Fig.
7, SOX2 knockdown resulted in significantly increased levels of
miR-200c-3p expression in MCF-7/Tax cells compared with the control
(P<0.05). In addition, co-treatment with miR-200c-3p mimics and
siRNA-SOX2 significantly increased miR-200c-3p expression in
MCF-7/Tax cells (P<0.001, Fig.
7). Treatment with siRNA-SOX2 and miR-200c-3p inhibitor
significantly upregulated miR-200c-3p expression in MCF-7/Tax cells
compared following treatment with the miR-200c inhibitor alone
(P<0.05, Fig. 7).
Discussion
Although notable improvements have been made in the
treatment of breast cancer over the past decades, drug resistance
remains a major obstacle to successful treatment of breast cancer
patients. Recent studies have demonstrated that aberrant miR-200c
expression may play an important role in cancer chemotherapeutic
resistance. Decreased miR-200c expression was previously observed
in cisplatin-resistant breast cancer cells compared to parental
MCF-7 breast cancer cells (22). In
addition, Zhu et al reported that miR-200c expression was
downregulated in vincristine-resistant gastric cancer cells
compared with parental SGC7901 gastric cancer cells (23). miR-200c expression was also
downregulated in cisplatin-resistant lung cancer cells compared
with parental A549 lung cancer cells (23). In our previous study, we
demonstrated that the miR-200c expression level in
doxorubicin-resistant breast cancer cells was markedly
downregulated compared with that in MCF-7 breast cancer cells
(11). In the present study,
miR-200c-3p expression in paclitaxel-resistant breast cancer cells
was found to be significantly downregulated compared with parental
MCF-7 breast cancer cells. In addition, overexpression of
miR-200c-3p in MCF-7/Tax cells enhanced the chemosensitivity to
paclitaxel, while downregulation of miR-200c-3p decreased the
chemosensitivity to paclitaxel in MCF-7/Tax cells. Furthermore, it
was demonstrated that overexpression of miR-200c-3p promoted
MCF-7/Tax cell apoptosis, with or without paclitaxel treatment.
These findings indicate that downregulation of miR-200c-3p
expression is involved in the resistance of breast cancer cells to
paclitaxel, which was consistent with previous reports. However,
Hamano et al reported that miR-200c expression was increased
in cisplatin-resistant esophageal cancer cells and overexpression
of miR-200c induced chemoresistance in esophageal cancer (24). The inconsistent correlation between
miR-200c expression and chemoresistance may be due, in part, to the
differences among tumor types.
SOX2 has been demonstrated to play an important role
in the maintenance of breast cancer stem cells (13). A previous study indicated that SOX2
may be implicated in drug resistance, and downregulation of SOX2
may enhance chemosensitivity of breast cancer cells to paclitaxel
(19). Overexpression of SOX2 was
demonstrated to enhance the resistance of PC-3 prostate cancer
cells to paclitaxel by promoting cell proliferation and preventing
cell apoptosis (16). The miR-200
family was found to be downregulated in human breast cancer stem
cells as well as mammary stem cells, suggesting an association
between the miR-200 family and stemness (25). In the present study, SOX2 mRNA and
SOX2 protein expression in paclitaxel-resistant MCF-7/Tax cells
were found to be markedly increased compared with their parental
MCF-7 cells, while miR-200c-3p expression in MCF-7/Tax cells was
downregulated compared with MCF-7 cells. Moreover, upregulation of
miR-200c-3p in MCF-7/Tax cells suppressed the expression of SOX2 at
the mRNA and protein levels, whereas the downregulation of
miR-200c-3p exerted the opposite effect. The luciferase reporter
assay revealed that SOX2 was a direct target of miR-200c-3p in
breast cancer. It was demonstrated that a single miRNA could
regulate tens or hundreds of target genes, and one gene could also
be regulated by multiple miRNAs. Antiapoptotic factors, such as the
B-cell lymphoma-2 (BCL-2) and X-linked inhibitor of apoptosis
protein (XIAP) genes, were found to be upregulated in
vincristine-resistant gastric cancer cells and cisplatin-resistant
lung cancer cells (23). BCL-2 and
XIAP were identified as target genes of miR-200c and were involved
in drug resistance (23). In
clear-cell renal cell carcinoma, miR-200c was reported to enhance
the efficiency of sorafenib and imatinib by targeting heme
oxygenase-1 (HO-1) (26). The
findings of the study suggest that downregulation of miR-200c-3p
contributes to paclitaxel resistance in breast cancer cells, at
least partially through targeting the SOX2 gene.
It was demonstrated that overexpression of the
miR-200 family upregulated E-cadherin expression, whereas
inhibition of the miR-200 family reduced E-cadherin expression
(27,28). The miR-200 family was demonstrated
to play a key role in epithelial-to-mesenchymal transition (EMT) by
targeting the E-cadherin transcriptional repressors ZEB1 and ZEB2
(29,30). Interestingly, ZEB1 and ZEB2 were
also found to repress miR-200 family transcription by binding to
their regulatory E-boxes (29,30).
Therefore, ZEB1/ZEB2 and the miR-200 family formed a
double-negative feedback loop involved in tumor progression and
metastasis (29,30). In the present study, the knockdown
of SOX2 using SOX2 siRNA markedly increased miR-200c-3p expression
in paclitaxel-resistant MCF-7/Tax cells. Furthermore, knockdown of
SOX2 in MCF-7/Tax cells increased chemosensitivity to paclitaxel. A
previous study reported that miR-200 expression was regulated by
SOX2 through contributing to the cell cycle exit and neuronal
differentiation of neural stem/progenitor cells (31). It was indicated that the
transcription of the mmu-miR-200c/141 gene cluster was activated by
SOX2 through binding to their promoters (31). In colorectal carcinoma, it was
demonstrated that the miR-200c-SOX2 negative feedback loop
mechanism was involved in the regulation of cell stemness,
proliferation and metastasis (32).
It was found that SOX2 could bind to specific promoter
transcription factor binding sites of miR-200c and inhibit the
transcription (32).
In summary, we herein demonstrated that miR-200c-3p
plays a crucial role in the resistance of breast cancer cells to
paclitaxel, possibly partially through targeting SOX2. Furthermore,
the evidence provided supports the role of SOX2 in the regulation
of miR-200c-3p expression. Taken together, the findings of the
present study point to the miR-200c-3p/SOX2 loop as a promising
future therapeutic target for overcoming paclitaxel resistance in
breast cancer patients.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
Natural Science Foundation of Zhejiang Province (LQ13H160016) and
the Medical Science and Technology Program of Zhejiang Province
(2013KYA026 and 2015DTA004) and the National Nature Science
Foundation of China (81672597).
Availability of data and materials
All the datasets generated/analyzed in the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JC, XW and ZC conceived and designed the study. JC,
WT, HH, FC and JH performed the experiments. JC and WT wrote the
manuscript. HH, FC, JH, XW and ZC reviewed and edited the
manuscript. All authors read and approved the manuscript and agree
to be accountable for all aspects of the research in ensuring that
the accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests to disclose.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hassan MS, Ansari J, Spooner D and Hussain
SA: Chemotherapy for breast cancer (Review). Oncol Rep.
24:1121–1131. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
O'Driscoll L and Clynes M: Biomarkers and
multiple drug resistance in breast cancer. Curr Cancer Drug
Targets. 6:365–384. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ng EK, Wong CL, Ma ES and Kwong A:
MicroRNAs as new players for diagnosis, prognosis, and therapeutic
targets in breast cancer. J Oncol. 2009:3054202009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lim LP, Lau NC, Garrett-Engele P, Grimson
A, Schelter JM, Castle J, Bartel DP, Linsley PS and Johnson JM:
Microarray analysis shows that some microRNAs downregulate large
numbers of target mRNAs. Nature. 433:769–773. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Amponsah PS, Fan P, Bauer N, Zhao Z,
Gladkich J, Fellenberg J and Herr I: microRNA-210 overexpression
inhibits tumor growth and potentially reverses gemcitabine
resistance in pancreatic cancer. Cancer Lett. 388:107–117. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ao X, Nie P, Wu B, Xu W, Zhang T, Wang S,
Chang H and Zou Z: Decreased expression of microRNA-17 and
microRNA-20b promotes breast cancer resistance to taxol therapy by
upregulation of NCOA3. Cell Death Dis. 7:e24632016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu Y, Zhu ST, Wang X, Deng J, Li WH,
Zhang P and Liu BS: MiR-200c regulates tumor growth and
chemosensitivity to cisplatin in osteosarcoma by targeting AKT2.
Sci Rep. 7:135982017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cochrane DR, Howe EN, Spoelstra NS and
Richer JK: Loss of miR-200c: A marker of aggressiveness and
chemoresistance in female reproductive cancers. J Oncol.
2010:8217172010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen J, Tian W, Cai H, He H and Deng Y:
Down-regulation of microRNA-200c is associated with drug resistance
in human breast cancer. Med Oncol. 29:2527–2534. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fong H, Hohenstein KA and Donovan PJ:
Regulation of self-renewal and pluripotency by Sox2 in human
embryonic stem cells. Stem Cells. 26:1931–1938. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Leis O, Eguiara A, Lopez-Arribillaga E,
Alberdi MJ, Hernandez-Garcia S, Elorriaga K, Pandiella A, Rezola R
and Martin AG: Sox2 expression in breast tumours and activation in
breast cancer stem cells. Oncogene. 31:1354–1365. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Alonso MM, Diez-Valle R, Manterola L,
Rubio A, Liu D, Cortes-Santiago N, Urquiza L, Jauregi P, de Munain
Lopez A, Sampron N, et al: Genetic and epigenetic modifications of
Sox2 contribute to the invasive phenotype of malignant gliomas.
PLoS One. 6:e267402011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rudin CM, Durinck S, Stawiski EW, Poirier
JT, Modrusan Z, Shames DS, Bergbower EA, Guan Y, Shin J, Guillory
J, et al: Comprehensive genomic analysis identifies SOX2 as a
frequently amplified gene in small-cell lung cancer. Nat Genet.
44:1111–1116. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li D, Zhao LN, Zheng XL, Lin P, Lin F, Li
Y, Zou HF, Cui RJ, Chen H and Yu XG: Sox2 is involved in paclitaxel
resistance of the prostate cancer cell line PC-3 via the PI3K/Akt
pathway. Mol Med Rep. 10:3169–3176. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Piva M, Domenici G, Iriondo O, Rábano M,
Simões BM, Comaills V, Barredo I, López-Ruiz JA, Zabalza I, Kypta R
and Vivanco MD: Sox2 promotes tamoxifen resistance in breast cancer
cells. EMBO Mol Med. 6:66–79. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wuebben EL, Wilder PJ, Cox JL, Grunkemeyer
JA, Caffrey T, Hollingsworth MA and Rizzino A: SOX2 functions as a
molecular rheostat to control the growth, tumorigenicity and drug
responses of pancreatic ductal adenocarcinoma cells. Oncotarget.
7:34890–34906. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mukherjee P, Gupta A, Chattopadhyay D and
Chatterji U: Modulation of SOX2 expression delineates an end-point
for paclitaxel-effectiveness in breast cancer stem cells. Sci Rep.
7:91702017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang Q, Wang Y, Lu X, Zhao Z, Zhu L, Chen
S, Wu Q, Chen C and Wang Z: MiR-125b regulates
epithelial-mesenchymal transition via targeting Sema4C in
paclitaxel-resistant breast cancer cells. Oncotarget. 6:3268–3279.
2015.PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pogribny IP, Filkowski JN, Tryndyak VP,
Golubov A, Shpyleva SI and Kovalchuk O: Alterations of microRNAs
and their targets are associated with acquired resistance of MCF-7
breast cancer cells to cisplatin. Int J Cancer. 127:1785–1794.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhu W, Xu H, Zhu D, Zhi H, Wang T, Wang J,
Jiang B, Shu Y and Liu P: miR-200bc/429 cluster modulates multidrug
resistance of human cancer cell lines by targeting BCL2 and XIAP.
Cancer Chemother Pharmacol. 69:723–731. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hamano R, Miyata H, Yamasaki M, Kurokawa
Y, Hara J, Moon JH, Nakajima K, Takiguchi S, Fujiwara Y, Mori M and
Doki Y: Overexpression of miR-200c induces chemoresistance in
esophageal cancers mediated through activation of the Akt signaling
pathway. Clin Cancer Res. 17:3029–3038. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shimono Y, Zabala M, Cho RW, Lobo N,
Dalerba P, Qian D, Diehn M, Liu H, Panula SP, Chiao E, et al:
Downregulation of miRNA-200c links breast cancer stem cells with
normal stem cells. Cell. 138:592–603. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gao C, Peng FH and Peng LK: MiR-200c
sensitizes clear-cell renal cell carcinoma cells to sorafenib and
imatinib by targeting heme oxygenase-1. Neoplasma. 61:680–689.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Park SM, Gaur AB, Lengyel E and Peter ME:
The miR-200 family determines the epithelial phenotype of cancer
cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes
Dev. 22:894–907. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Korpal M, Lee ES, Hu G and Kang Y: The
miR-200 family inhibits epithelial-mesenchymal transition and
cancer cell migration by direct targeting of E-cadherin
transcriptional repressors ZEB1 and ZEB2. J Biol Chem.
283:14910–14914. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Brabletz S and Brabletz T: The ZEB/miR-200
feedback loop-a motor of cellular plasticity in development and
cancer. EMBO Rep. 11:670–677. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hill L, Browne G and Tulchinsky E:
ZEB/miR-200 feedback loop: At the crossroads of signal transduction
in cancer. Int J Cancer. 132:745–754. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Peng C, Li N, Ng YK, Zhang J, Meier F,
Theis FJ, Merkenschlager M, Chen W, Wurst W and Prakash N: A
unilateral negative feedback loop between miR-200 microRNAs and
Sox2/E2F3 controls neural progenitor cell-cycle exit and
differentiation. J Neurosci. 32:13292–13308. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lu YX, Yuan L, Xue XL, Zhou M, Liu Y,
Zhang C, Li JP, Zheng L, Hong M and Li XN: Regulation of colorectal
carcinoma stemness, growth, and metastasis by an
miR-200c-Sox2-negative feedback loop mechanism. Clin Cancer Res.
20:2631–2642. 2014. View Article : Google Scholar : PubMed/NCBI
|