Introduction
Gastric cancer is the fourth most frequently
diagnosed cancer and the 2nd leading cause of cancer-related deaths
worldwide (1). The morbidity and
mortality rate of gastric cancer have decreased substantially over
the past few decades, and attention has shifted to the key issue of
prevention and treatment of gastric cancer. Increasing evidence
implicates Helicobacter pylori in the development of gastric
cancer. The initial bacterial infection leads to chronic gastritis
and gastric ulcers, which may finally culminate in gastric cancer
(2,3). The current treatments for gastric
cancer include surgery, chemotherapy, radiotherapy, thermal
therapy, immune therapy, and Chinese herbal treatment. However, due
to the difficulty in diagnosing gastric cancer at an early stage,
the prognosis with the current treatment options is often poor
(4,5). Therefore, it is necessary to identify
novel agents that more effectively treat advanced gastric
cancer.
Cinobufagin is a cardiotoxic bufanolide steroid
secreted by the Asiatic toad Bufo gargarizans. Chan Su, a
traditional Chinese medicine, is used to make a variety of valuable
medicinal herbs. Bufalin, resibufogenin, and cinobufagin are the
active ingredients in Chan Su. To date, several reports have
detailed the antitumor effects of cinobufagin in gastric cancer
(6–8). Additionally, research has demonstrated
that cinobufagin may trigger apoptosis and autophagic cell death in
gastric cancer via activation of the reactive oxygen species
(ROS)/JNK/p38 axis. Interestingly, mutation of certain
autophagy-related genes, such as ATG16L1 (autophagy related 16 like
1) and IRGM (immunity related GTPase M), confers susceptibility to
gastric cancer (8). This suggests
that there may be a close relationship between autophagy and the
development of gastric cancer.
Autophagy is a pathophysiological process that
enables the recycling of long-lived proteins or damaged organelles
and is crucial for cell development, differentiation, survival and
homeostasis (9–11). Many investigators have demonstrated
the important role of autophagy in the development and progression
of a wide range of cancers, including gastric cancer. It is
believed that inhibition of autophagy in cancer cells could improve
the toxicity of antitumor drugs and reverse drug resistance
(12,13). In fact, we previously reported that
knockdown of autophagy related 5 (ATG5) in hepatocellular
carcinoma inhibits autophagy and enhances the antitumor effect of
norcantharidin, a traditional Chinese medicine (14).
Previous studies have shown that cinobufagin and
inhibition of autophagy both play anticancer roles in gastric
cancer. Many reports have elucidated the cytotoxic effects of
cinobufagin; however, little is known concerning the mechanism of
cinobufagin-induced cytotoxic activity in gastric cancer (8,13). The
mechanism of autophagy induction in gastric cancer and the
connection between cinobufagin and autophagy are also not well
established. Therefore, the objective of the present study was to
elucidate the relationship of cinobufagin and autophagy in terms of
their anticancer action.
Materials and methods
Reagents and materials
Cinobufagin was obtained from the National Institute
for the Control of Pharmaceutical and Biological Products (Beijing,
China). Cell Proliferation kit I (MTT) was purchased from Roche
Applied Science (Mannheim, Germany). Hank's balanced salt solution
(HBSS) was obtained from Sigma-Aldrich/Merck KGaA (Darmstadt,
Germany). RPMI-1640 media, 10% heat-inactivated fetal bovine serum
(FBS), and pancreatic enzymes were purchased from Gibco; Thermo
Fisher Scientific, Inc. (Waltham, MA, USA). Antibodies, including
β-actin (dilution 1:10,000; cat. no. ab227387), microtubule
associated protein 1 light chain 3 (LC3-II) (dilution 1:3,000; cat.
no. ab51520), caspase-3 (dilution 1:1,000; cat. no. ab2302),
caspase-8 (dilution 1:1,000; cat. no. ab25901), caspase-9 (dilution
1:1,000; cat. no. ab32539), Bax (dilution 1:2,000; cat. no.
ab32503). Bcl-2 (dilution 1:2,000; cat. no. ab182858) and
cytochrome c (dilution 1:5,000; cat. no. ab133504) antibody
were from Abcam (Cambridge, UK). Secondary antibody, included goat
anti-rabbit IgG horseradish peroxidase (dilution 1:4,000; cat. no.
ab6721) and goat anti-mouse IgG horseradish peroxidase (dilution
1:4,000; cat. no. ab205719) were from Abcam (Cambridge, UK).
Antibody against cleaved PARP was purchased from Santa Cruz
Biotechnology, Inc. (dilution 1:500; cat. no. sc-56196; Santa Cruz,
CA, USA). The mitochondrial membrane potential assay kit with JC-1
and RIPA lysis buffer was purchased from Beyotime Institute of
Biotechnology (Suzhou, China). Enhanced chemiluminescence (ECL)
detection kits and protease inhibitor were purchased from Pierce
Biotechnology/Thermo Fisher Scientific, Inc. Horseradish peroxidase
(HRP)-conjugated secondary antibody was obtained from Beijing
Zhongshan Golden Bridge Biotechnology (Beijing, China).
Cell lines and treatment
The human gastric cancer cell line SGC-7901 was
obtained from the Cell Bank of the Chinese Academy of Sciences
(Shanghai, China). Cells were cultured in RPMI-1640 media
supplemented with 10% fetal bovine serum, 50 U/ml penicillin, and
50 U/ml streptomycin and maintained in 5% CO2 at
37°C.
Cells were divided into 8 groups: Sham, cinobufagin,
HBSS, HBSC, 3-MA, 3-MAC, S and SC. To note, the HBSS, 3-MA and S
groups were used as controls. The preliminary results showed no
significant difference of the cell apoptotic ratio and LC3-II
protein expression between the sham, HBSS, 3-MA and S groups.
Therefore, for the simplicity of our results, the groups (sham,
cinobufagin, HBSC, 3-MAC and SC) were used for all subsequent
results of the experiments. i) Sham group: Cells were cultured in
RPMI-1640 media. ii) Cinobufagin group: Cells were cultured in
RPMI-1640 media containing cinobufagin (0, 0.03, 0.06, 0.12 or 0.24
mM) for 24 h (8). iii) HBSS group:
Cells were cultured in HBSS media with Ca2+ and
Mg2+ and supplemented with 10 mM HEPES (1 ml/well) for
0.5 h to induce autophagy (15).
Following incubation in HBSS media, cells were washed twice with
PBS and cultured in RPMI-1640 media for 24 h. iv) HBSC group: Cells
were cultured in HBSS media as described for the HBSS group, and
then cells were washed with PBS and cultured in RPMI-1640 media
containing cinobufagin (0.24 mM) for 24 h. v) 3-MA group: Cells
were cultured in RPMI-1640 media containing 3-MA (10 mM) (16). vi) 3-MAC group: Cells were cultured
in RPMI-1640 media containing 3-MA (10 mM) to inhibit autophagy,
washed twice with PBS, and cultured in RPMI-1640 media containing
cinobufagin (0.24 mM) for 24 h. vii) S group: Cells were
transfected with scrambled siRNA and cultured in RPMI-1640 media.
viii) SC group (ATG5 siRNA+cinobufagin): Cells were
transfected with ATG5 siRNA, cultured in RPMI-1640 media,
washed twice with PBS, and then cultured in RPMI-1640 media
containing cinobufagin (0.24 mM) for 24 h.
MTT assay
SGC-7901 cells were cultured in RPMI-1640 media on
96-well flat bottom microtiter plates at 1×104 cells/well
overnight, and treated with various concentrations of cinobufagin
(0–0.5 mM) the following day. The cells were then incubated with
MTT (20 µl) solution (5 g/l) for 4 h at 37°C. An automatic
multi-well spectrophotometer was used to calculate the absorbance
value per well at 570 nm. All MTT assays were performed three
times. The cell viability ratio was calculated with the following
formula: Cell viability (%) = average absorbance of the treated
group⁄average absorbance of the sham group × 100%. IC50
values (50% inhibition concentration) were then calculated using
the Statistical Package for the Social Sciences 17.0 (SPSS, Inc.,
Chicago, IL, USA).
RNA extraction and reverse
transcription quantitative real-time PCR
Total RNA was isolated and purified from cells using
TRIzol, and cDNA was synthesized with the PrimeScript RT Master
Mix. Quantitative real-time PCR reactions were carried out using
cDNA (2 µl) as a template in a 20 µl reaction. Primers were
synthesized by Invitrogen; Thermo Fisher Scientific, Inc.
(Shanghai, China). The primers utilized for RT-qPCR reactions were
5′-TTCTCAAAATATACTGTTTC-3′ (sense) and 5′-TATTATGTATCACAAATGG-3′
(antisense) for ATG5; 5′-TCACCCACACTGTGCCCATCTACGA-3′
(sense) and 5′-CAGCGGAACCGCTCATTGCCAATGG-3′ (antisense) for
β-actin, and 5′-CCACTCCTCCACCTTTGAC-3′ (sense) and
5′-AGGGGAGATTCAGTGTGGTG-3′ (antisense) for
glyceraldehyde-3-phosphate dehydrogenase (GADPH). For RT-qPCR, the
following cycles were used: 95°C for 30 sec, 40 cycles of 95°C for
5 sec, and 60°C for 31 sec. The dissociation stage used the
following cycles: 95°C for 15 sec, 60°C for 1 min, and 95°C for 15
sec.
Small interfering RNA (siRNA)
transfection
Small interfering RNAs (siRNAs) against ATG5
and a nonspecific scrambled siRNA were purchased from Ambion
(Austin, TX, USA). All siRNAs were synthesized by Qiagen
(Chatsworth, CA, USA). SGC-7901 cells were cultured in 6-well
plates. Invitrogen™ Lipofectamine 2000 (Thermo Fisher
Scientific, Inc.) was mixed with RPMI-1640 media lacking 10%
heat-inactivated FBS and containing siRNA1, siRNA2, or scrambled
RNA. Mock controls were transfected with Lipofectamine 2000 alone.
Transfections were performed at 37°C in 5% CO2, after
which the medium in each well was replaced by serum-containing
medium for 4–6 h. After 2 h, cells were treated as indicated.
Western blot analysis
Total protein was extracted using Cell Lysis
Reagents (Pierce) and quantified using the BCA method (Pierce;
Thermo Fisher Scientific, Inc.). For cytochrome c analysis,
cell pellets were suspended in HEPES buffer containing 250 mM
sucrose and homogenized. The homogenate was then centrifuged at 800
× g and 4°C for 15 min. The supernatant was centrifuged at 10,000 ×
g for 15 min at 4°C. Mitochondrial pellets and cytosolic fractions
were then collected (17). Proteins
were separated on a 10% SDS-polyacrylamide gel and transferred to a
PVDF membrane. The membrane was blocked and incubated with the
primary antibodies overnight on a shaker in a cold room. Secondary
antibodies (1:4,000) were applied for 1 h before visualization with
an ECL detection kit. The gray value of each protein was measured
by Image-Pro Plus 6.0 (National Institutes of Health, Bethesda, MD,
USA) for statistical analyses.
Immunofluorescence
SGC-7901 cells were fixed on coverslips in 4%
paraformaldehyde for 20 min. Afterwards, cells were blocked in 10%
BSA for 1 h. Primary antibody (LC3-II in 10% BSA, 1:3,000; cat. no.
ab51520; Abcam) was added overnight, followed by incubation with
secondary antibody (1:1,000; cat. no. ab6721; Abcam) for 1 h at
37°C. Cells were then incubated with 4′,6-diamidino-2-phenylindole
(DAPI). Ten visual fields were chosen randomly, optical density was
measured, and semi-quantitative analysis was performed with
Image-Pro Plus 6.0.
Analysis of ROS production
Intracellular ROS levels were detected by flow
cytometry using the dichlorodihydrofluorescein diacetate (DCHF-DA)
assay. SGC-7901 cells were washed with D-Hank's solution three
times, and then cells were cultured in RPMI-1640 media lacking FBS
and containing DCHF-DA (100 µM) at 37°C in the dark for 30 min.
Afterwards, cells were washed with RPMI-1640 media lacking FBS and
treated with pancreatic enzymes. DCF fluorescence was detected at
an excitation wavelength of 488 nm and at an emission wavelength of
525 nm.
Annexin V and propidium iodide (PI)
co-staining assay
Apoptosis was detected using the Annexin V-FITC kit.
SGC-7901 cells were washed twice with ice-cold PBS and then
resuspended in 400 µl of binding buffer at a concentration of 5×105
cells/ml. Next, 5 µl of Annexin V-FITC and 5 µl of PI were added to
the cells. Tubes were gently vortexed and incubated for 15 min in
the dark at 4°C. Samples were analyzed with a FACSCalibur flow
cytometer (BD Biosciences, San Jose, CA, USA) using CellQuest
software (BD Biosciences).
Mitochondrial membrane potential
assay
Mitochondrial membrane potential was monitored by
JC-1. The JC-1 dyeing liquid was added to each well (1 ml/well),
and SGC-7901 cells were incubated at 37°C for 20 min. Afterwards,
cells were washed twice with dyeing buffer (1X) and cultured in
RPMI-1640 media. Finally, cells were observed by fluorescence
microscopy (AX10; Carl Zeiss, Hamburg, Germany).
Statistical analysis
Data are expressed as mean ± standard deviation. The
significance of the differences between groups was determined by
ANOVA and Dunnet's test. Multiple comparisons between the groups
were performed using the S-N-K method after ANOVA. SPSS 17.0
software (SPSS, Inc., Chicago, IL, USA) was used for all
statistical analyses, and P<0.05 was considered statistically
significant.
Results
Cinobufagin inhibits SGC-7901 cell
proliferation and induces caspase-mediated apoptosis
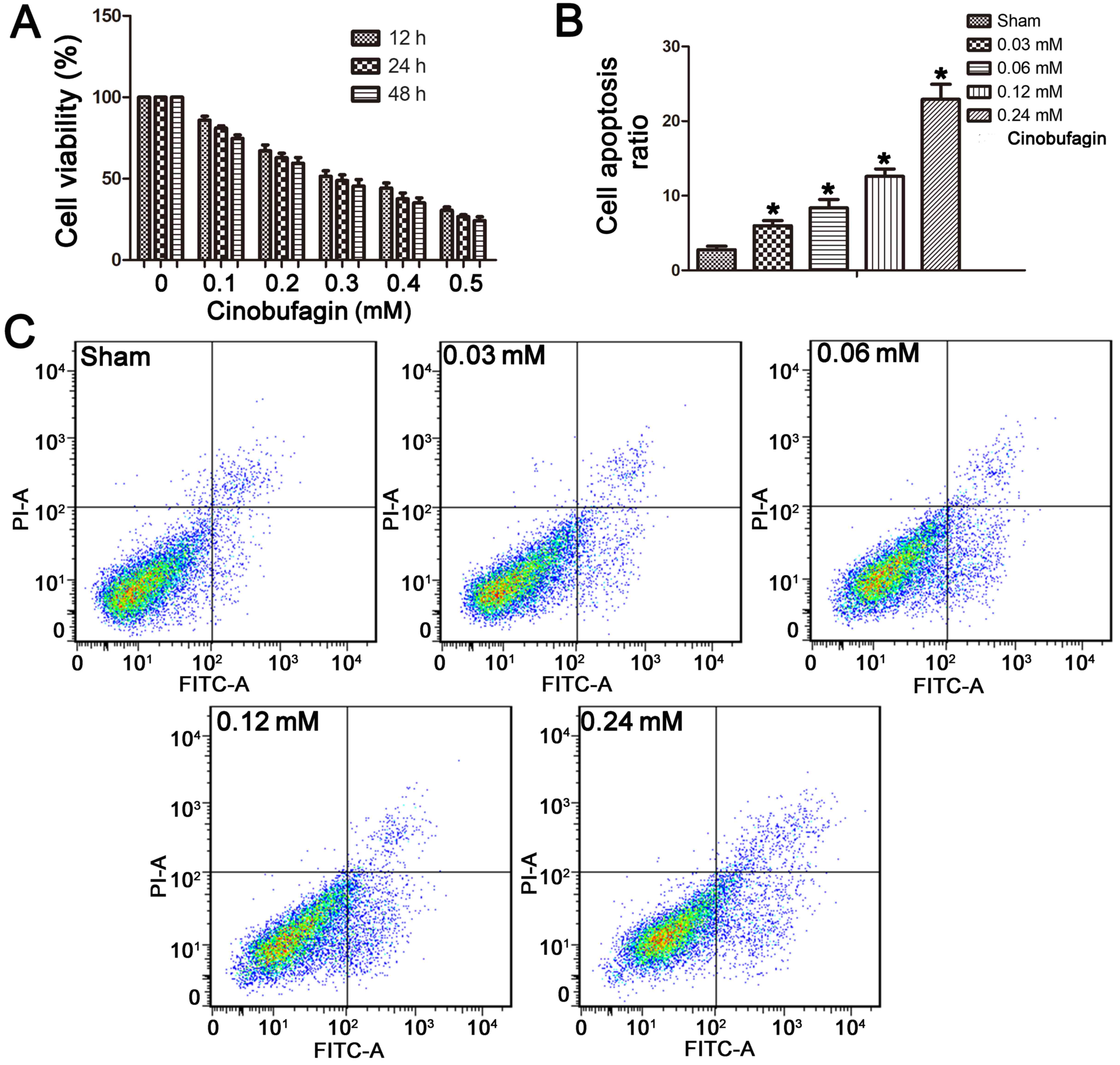
The MTT assay demonstrated that cinobufagin
inhibited the proliferation of the SGC-7901 cells in a dose- and
time-dependent manner after treatment with 0–0.5 mM cinobufagin for
12, 24 or 48 h (Fig. 1A). At
concentrations >0.03 mM, cinobufagin treatment significantly
inhibited cell proliferation. The IC50 value of
cinobufagin treatment at 24 h was 0.24 mM. Thus, a range of
concentrations (0, 0.03, 0.06, 0.12 and 0.24 mM) was applied, and
the concentration of 0.24 mM was used for all subsequent
experiments.
Cinobufagin induces apoptosis in
SGC-7901 cells
Cell apoptosis was detected by co-staining cells
with Annexin V and PI. We found that the apoptotic ratio of
SGC-7901 cells increased with cinobufagin treatment. The basal
apoptotic population of the sham group was 2.77±1.04%. After
treatment with cinobufagin at concentrations of 0.03, 0.06, 0.12,
or 0.24 mM for 24 h, the apoptotic rate increased to 5.99±1.52,
8.35±2.55, 12.59±2.25 and 22.91±4.56%, respectively (Fig. 1B and C). This suggests a
dose-dependent increase in apoptotic ratio in the SGC-7901 cells in
response to cinobufagin treatment.
Autophagy counteracts
cinobufagin-induced apoptosis
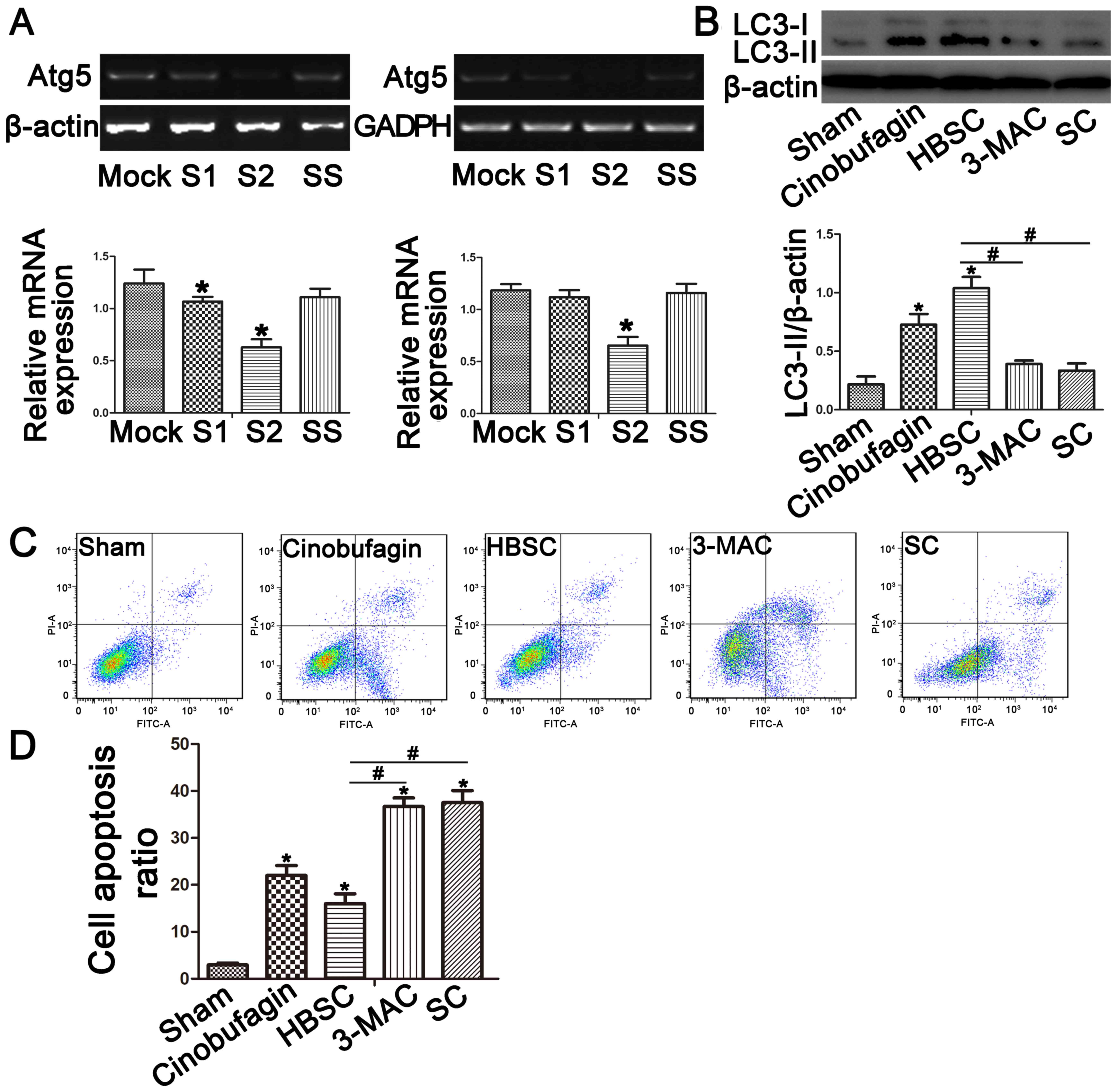
To reduce autophagy activity, ATG5 expression
was knocked down by siRNA in SGC-7901 cells. RT-qPCR confirmed
decreased ATG5 gene expression in the SC group compared with
the sham group (Fig. 2A;
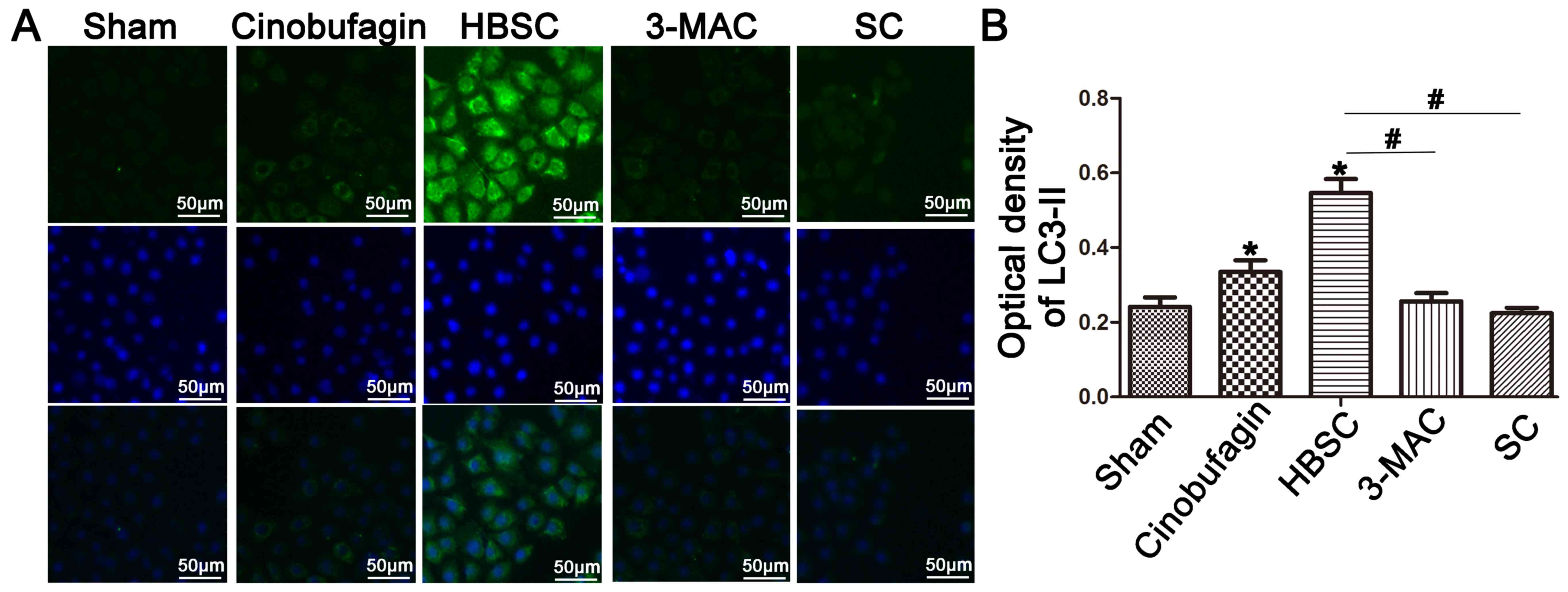
P<0.05). The autophagosome marker LC3-II was then used to
monitor induction of autophagy by western blotting (Fig. 2B) and immunostaining (Fig. 3). When autophagy was inhibited by
treatment with 3-MA or ATG5 siRNA, LC3-II expression was
reduced in the 3-MAC and SC groups compared with the sham group
(Figs. 2B and 3; P<0.05). In contrast, western
blotting and immunostaining revealed that SGC-7901 cells treated
with HBSS displayed increased LC3-II protein levels (Figs. 2B and 3; P<0.05). To compare subsequent
apoptosis ratios, SGC-7901 cells were co-stained with Annexin V and
PI. Staining demonstrated that inhibition of autophagy increased
the apoptosis rates (Fig. 2C and D;
P<0.05), whereas autophagy activation in the HBSC group reduced
early and late apoptosis rates compared with the sham group
(Fig. 2C and D; P<0.05).
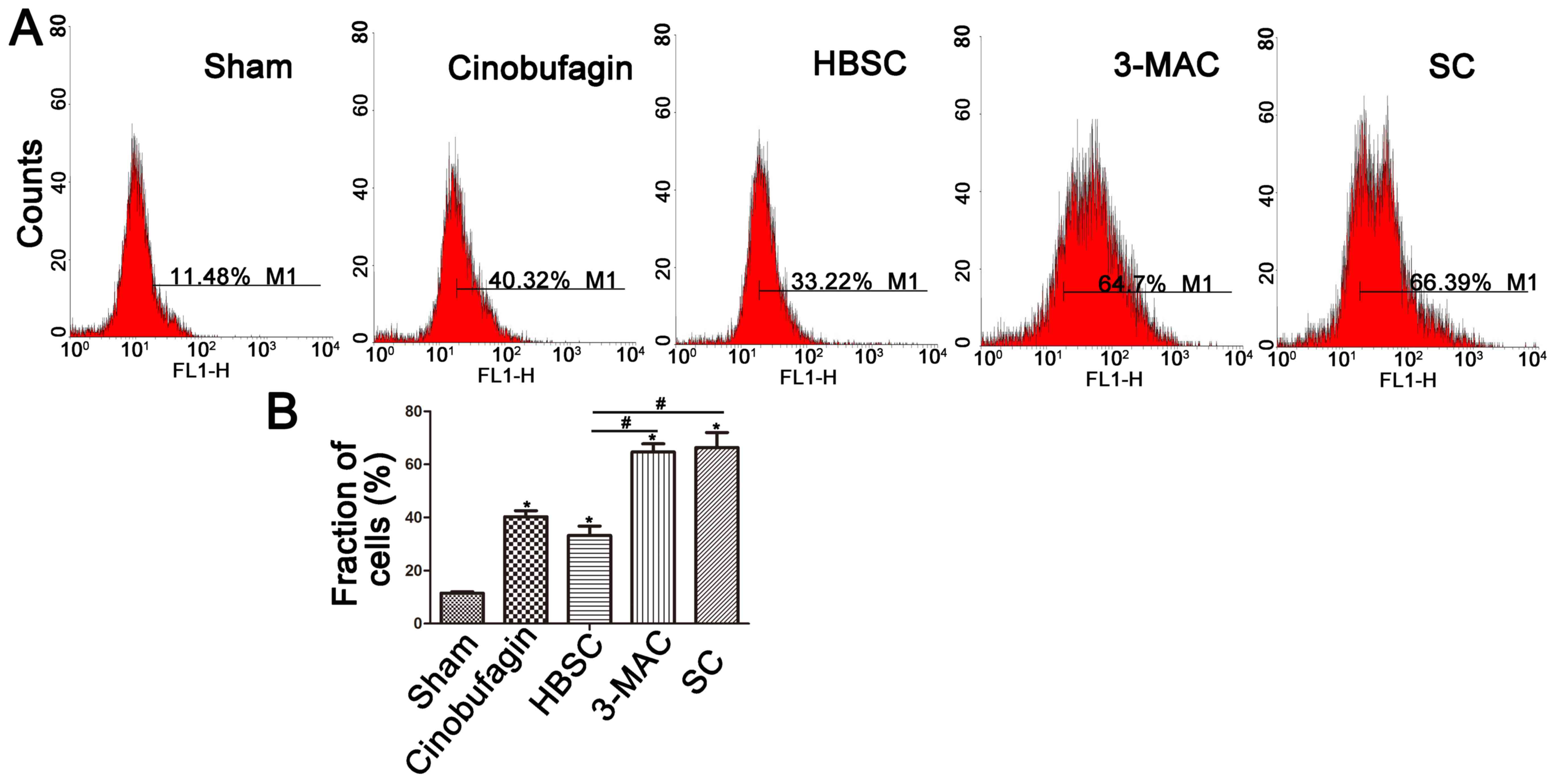
ROS generation is induced by
cinobufagin and is further enhanced by inhibition of autophagy
ROS levels were then detected by DCFH-DA assay.
Intracellular ROS generation was increased in the cinobufagin group
and was further enhanced by inhibition of autophagy. However, ROS
levels were dowregulated when autophagy was induced (Fig. 4A and B).
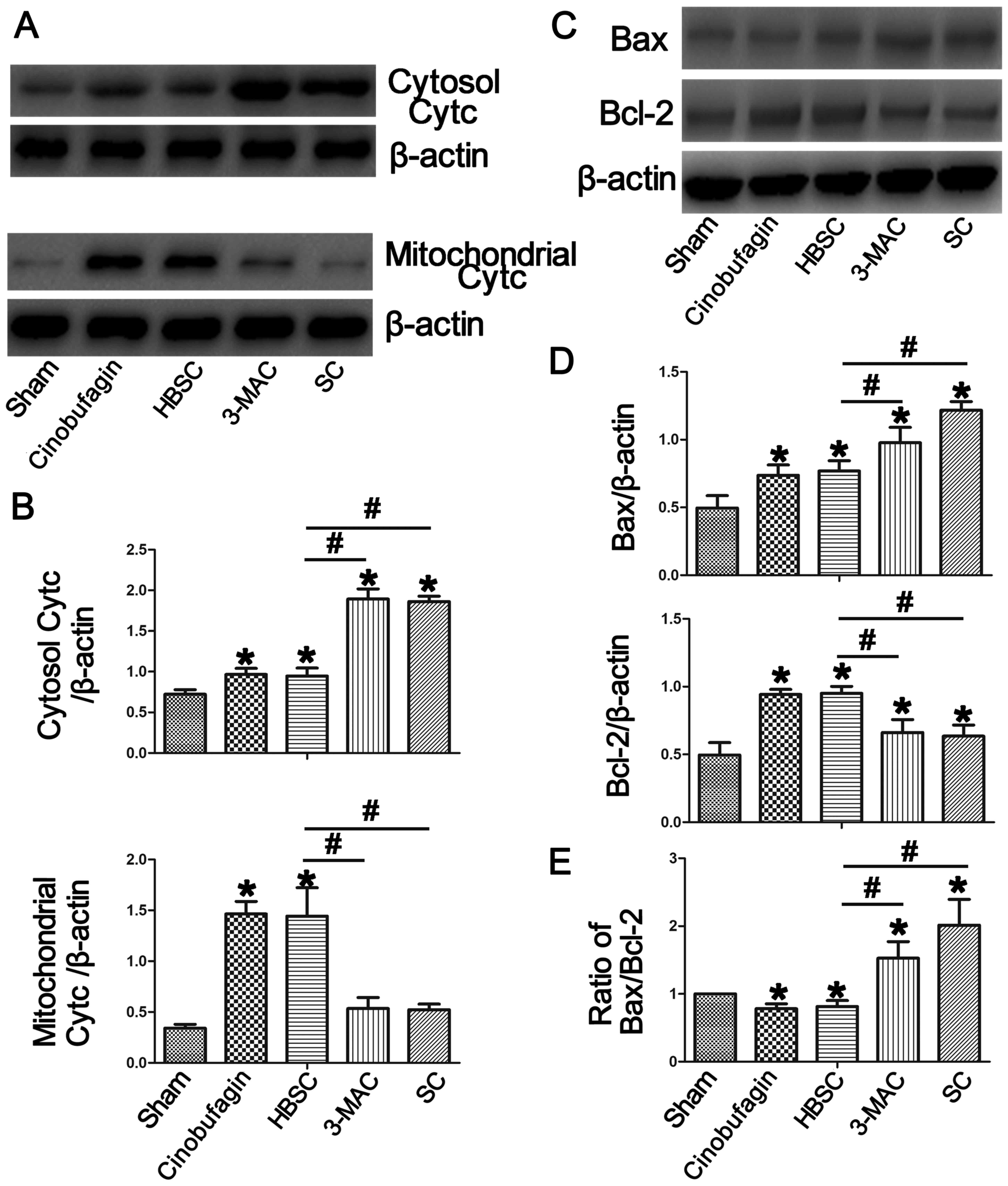
Inhibition of autophagy induces Bax
and cytochrome c, but reduces Bcl-2
To further investigate the mechanism of
cinobufagin-induced apoptosis, we detected the levels of apoptotic
proteins by western blotting. As shown in Fig. 5, downregulation of autophagy
increased Bax (Bcl-2 associated X, apoptosis regulator) and
cytosolic cytochrome c protein levels compared with the sham
group, which was reversed when autophagy was induced in the HBSC
group (P<0.05). In the same way, mitochondrial cytochrome
c and Bcl-2 (Bcl-2, apoptosis regulator) were upregulated in
the HBSC group compared with the sham, 3-MAC and SC groups
(P<0.05).
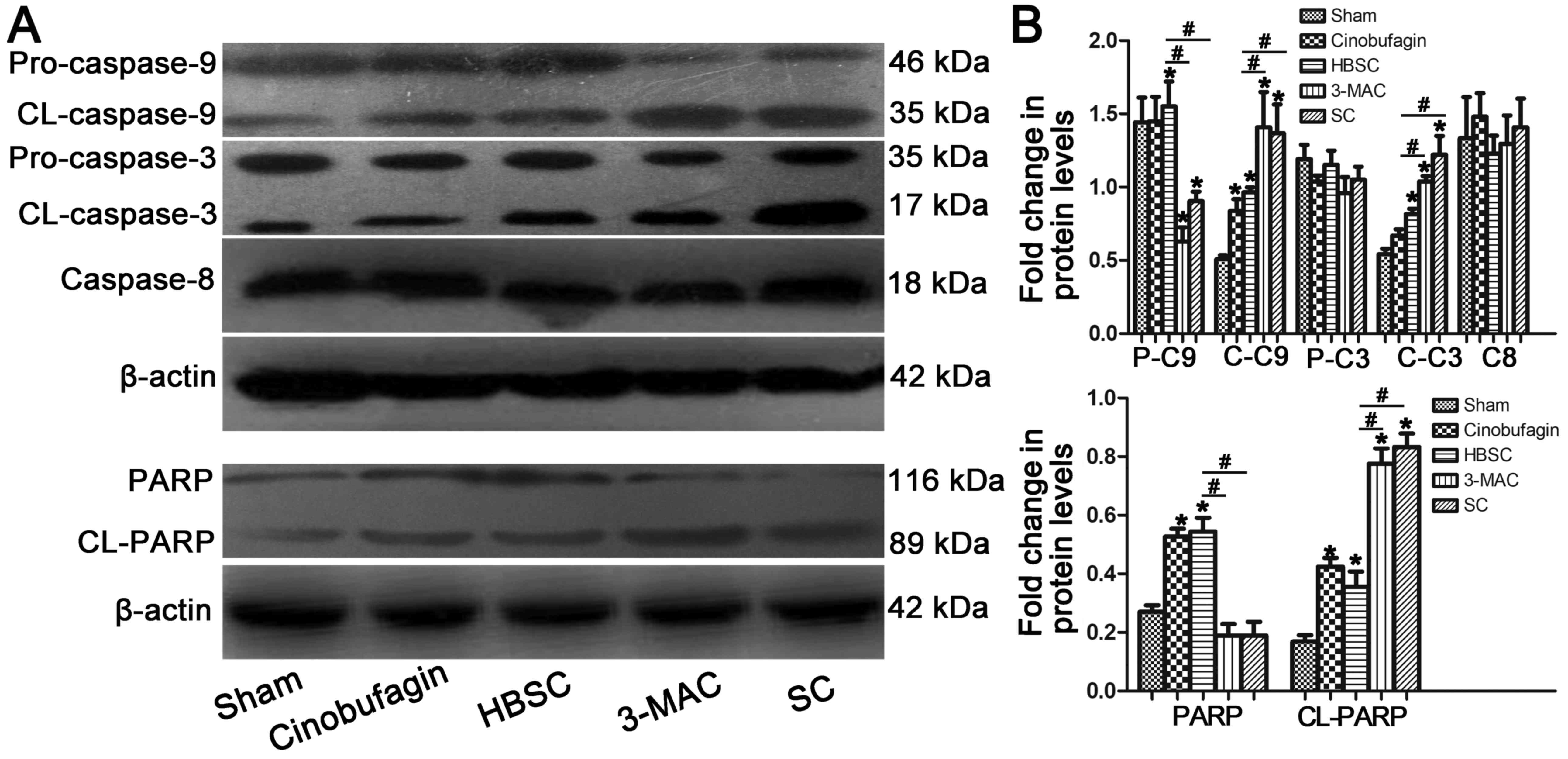
Inhibition of autophagy induces
pro-apoptotic protein expression
Elevated cleaved caspase-3 and cleaved caspase-9
levels were detected when autophagy was inhibited in SGC-7901
cells; however, this increase was abrogated in the HBSC group. No
significant changes in caspase-8 protein expression were observed
between any of the groups. Inhibition of autophagy in SGC-7901
cells also promoted the cleavage of PARP [poly(ADP-ribose)
polymerase] protein from the full-length form to the cleaved form
(Fig. 6; P<0.05).
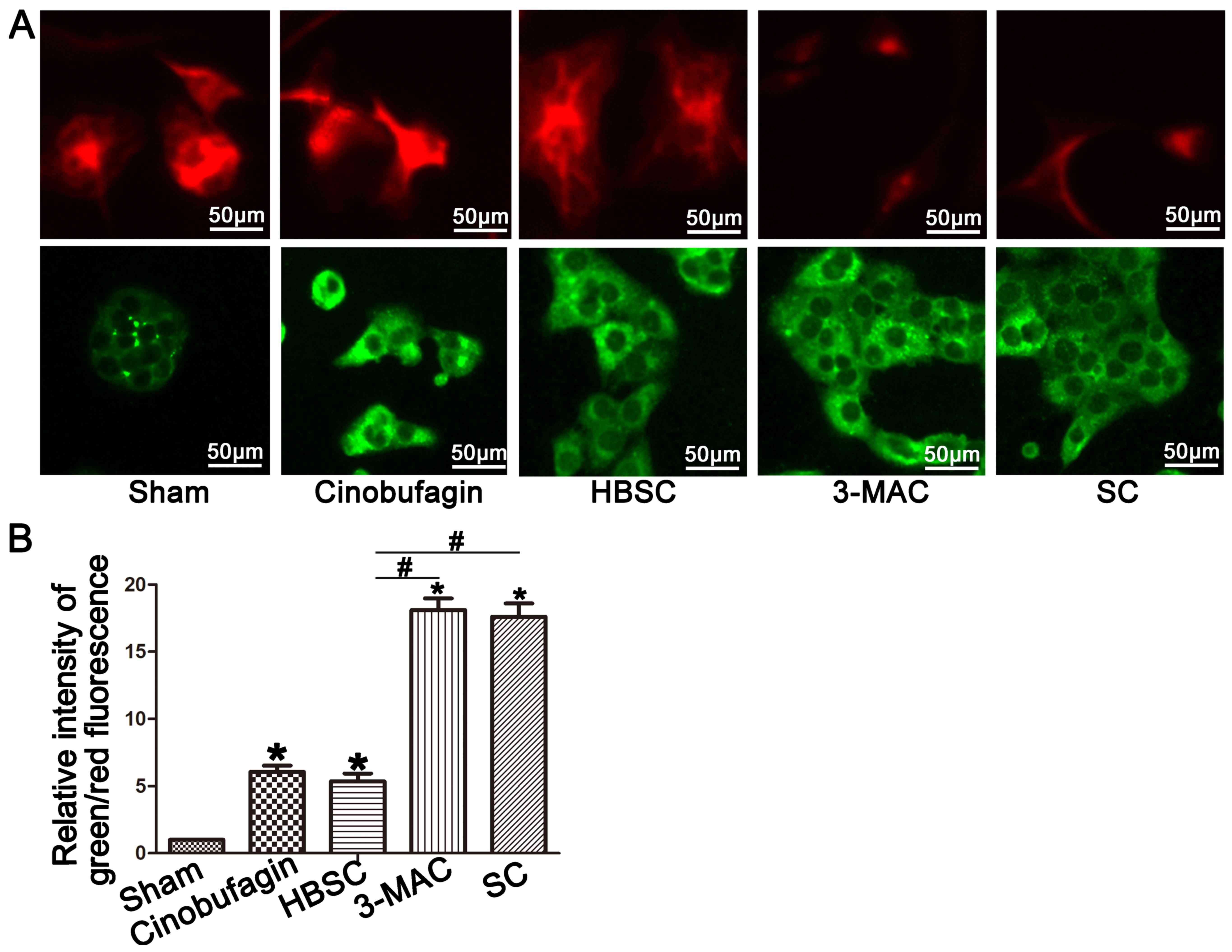
Mitochondrial membrane potential is
disrupted in the 3-MAC and SC groups
Elevated green fluorescence indicates increased
mitochondrial membrane potential disruption and mitochondrial
depolarization when observed with JC-1. We found increased red
fluorescence in the sham and HBSC groups but enhanced green
fluorescence in the 3-MAC and SC groups (Fig. 7A and B; P<0.05).
Discussion
In summary, we demonstrated that cinobufagin induced
SGC-7901 cell apoptosis, and inhibition of autophagy cooperatively
enhanced this effect. These processes may occur partly through ROS
generation and activation of the mitochondrial programmed cell
death pathway.
Gastric cancer is one of the leading causes of
cancer mortality in the world, and is also one of the most common
types of tumors diagnosed in China (1,2).
Gastric cancer involves multiple pathways and molecular
alterations. As a result, the mechanism and treatment of gastric
cancer have been the major focus of cancer research. Due to the
high recurrence rates of gastric cancer, it is necessary to
identify novel and promising therapeutics.
As medical technology advances, an increasing number
of Chinese patent medicines targeting gastric cancer have been
filed. Cinobufagin is extracted from Chan Su and was used in
Chinese medicine to treat tumors for many years. Cinobufagin has
been demonstrated to induce apoptosis in human leukemia,
hepatocellular carcinoma and prostate cancer (7,8).
Currently, cinobufagin is widely used in the treatment of tumor
diseases.
Cinobufagin possesses potent anticancer activity by
triggering apoptosis in various types of cancers, including
gastric, breast, and hepatic cancer cells. The mechanism of
cinobufagin-induced apoptosis involves both the FAS- and
mitochondrial-mediated apoptotic pathways (7,18).
Additional research has found that cinobufagin induces cytotoxicity
in gastric cancer cells by apoptosis, and here we report cell
apoptosis of the human gastric cancer cell line SGC-7901 in
response to cinobufagin. The basal apoptotic population of the sham
group was 2.77±1.04%. After treatment with increasing
concentrations of cinobufagin (0.03, 0.06, 0.12 and 0.24 mM) for 24
h, the apoptotic rate increased to 5.99±1.52, 8.35±2.55, 12.59±2.25
and 22.91±4.56%, respectively. Thus, we confirmed that cinobufagin
increases the apoptosis ratio of gastric cancer cells in a
dose-dependent manner.
One study found that inhibition of autophagy by
miR-181a overexpression potentiated the toxicity of cisplatin and
reversed drug resistance (13). An
increasing number of studies have demonstrated the key role of
autophagy in gastric cancer. Autophagy is a process by which
cytoplasmic macromolecules or organelles are degraded in lysosomes
to maintain cellular homeostasis. Autophagy is an adaptive response
to stress and can promote survival; however, it also appears to
promote cell death and morbidity (11,19,20).
Generally speaking, autophagy is Janus-faced. Some studies have
shown that autophagy leads to autophagic cell death under certain
conditions, but others have demonstrated that it keeps cells alive
under stressful ‘life-threatening’ conditions. One such study
reported that autophagy was activated as a protective mechanism
against matrine-induced apoptosis. They found that the inhibition
of autophagy enhanced the antitumor potential of matrine in gastric
cancer (20). Similarly, we
demonstrated that cinobufagin and the inhibition of autophagy,
alone or in combination, induced apoptosis in the human gastric
cancer cell line SGC-7901.
Apoptosis is a process of programmed cell death that
occurs in multicellular organisms and is characterized by blebbing,
cell shrinkage, nuclear fragmentation, chromatin condensation,
chromosomal DNA fragmentation, and global mRNA decay (21). To the best of our knowledge,
apoptosis can be induced by two alternative signaling pathways: The
death receptor pathway and the mitochondrial pathway. The action of
many anticancer drugs is mediated through the mitochondrial pathway
(18,22–24).
Additionally, many reports show that cinobufagin is effective in
the prevention and treatment of cancer by triggering apoptosis and
autophagic cell death via activation of the ROS/JNK/p38 axis
(8). Moreover, mitophagy, which is
the process of mitochondrial degradation by autophagy, plays a
crucial role in cell death and apoptosis (25). Therefore, we focused on the
mitochondrial apoptotic pathway when investigating the relationship
of cinobufagin, autophagy and apoptosis.
The anti-apoptotic protein Bcl-2 and the
pro-apoptotic protein Bax are crucial initiators of the
mitochondrial death cascade. Therefore, we first compared Bcl-2 and
Bax protein levels in our study. We found decreased expression of
Bax and increased expression of Bcl-2 protein when autophagy was
induced in the HBSC group. In contrast, downregulation of autophagy
by 3-MA or ATG5 siRNA prevented this enhanced expression.
The release of cytochrome c from mitochondria into the
cytosol is the pivotal factor in the mitochondrial programmed cell
death pathway. When autophagy was inhibited, we found that
cytosolic cytochrome c protein was enhanced and
mitochondrial cytochrome c protein was accordingly
decreased, confirming our initial findings. Caspase-9 is activated
after cytochrome c is released into the cytosol and
undergoes subsequent incorporation into a complex containing
cytochrome c, apoptotic protease activation factor 1
(APAF1), and procaspase-9. Caspase-9 can activate downstream
caspases, including caspase-7 and caspase-3, that eventually lead
to apoptosis (26,27). Therefore, we also detected caspase-3
and caspase-9 protein expression. In this study, increased cleaved
caspase-3 and cleaved caspase-9 protein expression was observed
when autophagy was inhibited in the 3-MAC and SC groups. In
contrast, both cleaved caspase-3 and cleaved caspase-9 protein
expression was decreased when autophagy was induced in the sham and
HBSC groups.
The PARP protein, which plays an important role in
the apoptotic pathway, is cleaved by activated caspase-3.
Therefore, we compared PARP protein levels between the cell groups
in the present study. We found that PARP protein was increasingly
cleaved from the full-length 116 kDa form to the cleaved 89 kDa
form in the 3-MAC and SC groups. This effect was reversed in the
HBSC group. Once the mitochondrial programmed cell death pathway is
activated, the mitochondrial permeability transition pore opens and
membrane potential decreases (23,24).
JC-1 dye is used to monitor mitochondrial membrane potential and
exhibits enhanced green fluorescence when membrane potential is
disrupted. In this study, we observed more red fluorescence in the
sham and HBSC groups, but elevated green fluorescence in the 3-MAC
and SC groups. This suggests that mitochondrial membrane potential
is disrupted by autophagy inhibition in combination with
cinobufagin treatment and could indicate increased cell death.
ROS are chemically reactive oxygen-containing
molecules, and examples include peroxides, superoxide, hydroxyl
radicals, and singlet oxygen (28).
Generation of ROS induced by chemotherapeutic drugs has been
observed to trigger the mitochondrial apoptosis pathway (29). Consistent with this observation, we
found that cinobufagin induced ROS production in SGC-7901 cells.
Inhibition of autophagy further enhanced ROS production and was
attenuated by the induction of autophagy. Altogether, these studies
suggest that the inhibition of autophagy enhances
cinobufagin-induced apoptosis, which may occur, in part, through
the mitochondrial programmed cell death pathway.
In summary, we demonstrated that cinobufagin induces
SGC-7901 cell apoptosis, which is further enhanced by the
inhibition of autophagy. Collectively, this study and previous
reports support a model where cinobufagin treatment and autophagy
inhibition cooperatively induce SGC-7901 cell apoptosis by
enhancing ROS generation and activating the mitochondrial apoptotic
pathway. However, the definite relationship between autophagy and
mitochondrial apoptotic pathways, and the function of autophagy in
the apoptosis pathways, requires further study.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Science Foundation of China (81670570), Key Research and
Development Program of Jiangsu Province (BE2016789) and the Xuzhou
City Subject Foundation (KC16SH029).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YW, XX and TZ designed the research; YW XX, BL and
QZ performed the experiments; YW, XX, MW, HZ, HG and QT analyzed
the data; YW, XX, BL, HG, QT and XL interpreted the results of the
experiments; YW, XX, TZ, MW, HZ and QZ prepared the figures; YW,
XX, MW, HZ, HG and XL revised the manuscript; YW, XX, BL, QT and TZ
approved the final version of the manuscript. All authors read and
approved the manuscript and agree to be accountable for all aspects
of the research in ensuring that the accuracy or integrity of any
part of the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rugge M, Genta RM, Di Mario F, El-Omar EM,
El-Serag HB, Fassan M, Hunt RH, Kuipers EJ, Malfertheiner P, Sugano
K, et al: Gastric cancer as preventable disease. Clin Gastroenterol
Hepatol. 15:1833–1843. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Greenlee RT, Hill-Harmon MB, Murray T and
Thun M: Cancer statistics, 2001. CA Cancer J Clin. 51:15–36. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brenner H, Rothenbacher D and Arndt V:
Epidemiology of stomach cancer. Methods Mol Biol. 472:467–477.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lim SC, Parajuli KR, Duong HQ, Choi JE and
Han SI: Cholesterol induces autophagic and apoptotic death in
gastric carcinoma cells. Int J Oncol. 44:805–811. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang Y, Tang X, Liu X, Li F and Lin X:
Simultaneous determination of three bufadienolides in rat plasma
after intravenous administration of bufadienolides extract by ultra
performance liquid chromatography electrospray ionization tandem
mass spectrometry. Anal Chim Acta. 610:224–231. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mommersteeg MC, Yu J, Peppelenbosch MP and
Fuhler GM: Genetic host factors in helicobacter pylori-induced
carcinogenesis: Emerging new paradigms. Biochim Biophys Acta.
1869:42–52. 2018.
|
|
8
|
Ma K, Zhang C, Huang MY, Li WY and Hu GQ:
Cinobufagin induces autophagy-mediated cell death in human
osteosarcoma U2OS cells through the ROS/JNK/p38 signaling pathway.
Oncol Rep. 36:90–98. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rautou PE, Mansouri A, Lebrec D, Durand F,
Valla D and Moreau R: Autophagy in liver diseases. J Hepatol.
53:1123–1134. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nivon M, Richet E, Codogno P, Arrigo AP
and Kretz-Remy C: Autophagy activation by NFκB is essential for
cell survival after heat shock. Autophagy. 5:766–783. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kumar A, Singh UK and Chaudhary A:
Targeting autophagy to overcome drug resistance in cancer therapy.
Future Med Chem. 7:1535–1542. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhao J, Nie Y, Wang H and Lin Y: MiR-181a
suppresses autophagy and sensitizes gastric cancer cells to
cisplatin. Gene. 576:828–833. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xiong X, Wu M, Zhang H, Li J, Lu B, Guo Y,
Zhou T, Guo H, Peng R, Li X, et al: Atg5 siRNA inhibits autophagy
and enhances norcantharidin-induced apoptosis in hepatocellular
carcinoma. Int J Oncol. 47:1321–1328. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Settembre C, Di Malta C, Polito VA, Garcia
Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D,
Colella P, et al: TFEB links autophagy to lysosomal biogenesis.
Science. 332:1429–1433. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Carchman EH, Rao J, Loughran PA, Rosengart
MR and Zuckerbraun BS: Heme oxygenase-1-mediated autophagy protects
against hepatocyte cell death and hepatic injury from
infection/sepsis in mice. Hepatology. 53:2053–2062. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lewis JS, Meeke K, Osipo C, Ross EA,
Kidawi N, Li T, Bell E, Chandel NS and Jordan VC: Intrinsic
mechanism of estradiol-induced apoptosis in breast cancer cells
resistant to estrogen deprivation. J Natl Cancer Inst.
97:1746–1759. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qi F, Inagaki Y, Gao B, Cui X, Xu H,
Kokudo N, Li A and Tang W: Bufalin and cinobufagin induce apoptosis
of human hepatocellular carcinoma cells via Fas- and
mitochondria-mediated pathways. Cancer Sci. 102:951–958. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Reed M, Morris SH, Jang S, Mukherjee S,
Yue Z and Lukacs NW: Autophagy-inducing protein beclin-1 in
dendritic cells regulates CD4 T cell responses and disease severity
during respiratory syncytial virus infection. J Immunol.
191:2526–2537. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen YY, Sun LQ, Wang BA, Zou XM, Mu YM
and Lu JM: Palmitate induces autophagy in pancreatic β-cells via
endoplasmic reticulum stress and its downstream JNK pathway. Int J
Mol Med. 32:1401–1406. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xie SQ, Zhang YH, Li Q, Xu FH, Miao JW,
Zhao J and Wang CJ: 3-Nitro-naphthalimide and nitrogen mustard
conjugate NNM-25 induces hepatocellular carcinoma apoptosis via
PARP-1/p53 pathway. Apoptosis. 17:725–734. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shen B, He PJ and Shao CL: Norcantharidin
induced DU145 cell apoptosis through ROS-mediated mitochondrial
dysfunction and energy depletion. PLoS One. 8:e846102013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang JY, Lin MT, Tung HY, Tang SL, Yi T,
Zhang YZ, Tang YN, Zhao ZZ and Chen HB: Bruceine D induces
apoptosis in human chronic myeloid leukemia K562 cells via
mitochondrial pathway. Am J Cancer Res. 6:819–826. 2016.PubMed/NCBI
|
|
24
|
Lin M, Tang S, Zhang C, Chen H, Huang W,
Liu Y and Zhang J: Euphorbia factor L2 induces apoptosis in A549
cells through the mitochondrial pathway. Acta Pharm Sin B. 7:59–64.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim I and Lemasters JJ: Mitophagy
selectively degrades individual damaged mitochondria after
photoirradiation. Antioxid Redox Signal. 14:1919–1928. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang JY, Yi T, Liu J, Zhao ZZ and Chen
HB: Quercetin induces apoptosis via the mitochondrial pathway in KB
and KBv200 cells. J Agric Food Chem. 61:2188–2195. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang XH, Jia DZ, Liang YJ, Yan SL, Ding Y,
Chen LM, Shi Z, Zeng MS, Liu GF and Fu LW: Lgf-YL-9 induces
apoptosis in human epidermoid carcinoma KB cells and multidrug
resistant KBv200 cells via reactive oxygen species-independent
mitochondrial pathway. Cancer Lett. 249:256–270. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kang JY, Eggert M, Mouli S, Aljuffali I,
Fu X, Nie B, Sheil A, Waddey K, Oldham CD, May SW, et al:
Pharmacokinetics, antitumor and cardioprotective effects of
liposome-encapsulated phenylaminoethyl selenide in human prostate
cancer rodent models. Pharm Res. 32:852–862. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ott M, Gogvadze V, Orrenius S and
Zhivotovsky B: Mitochondria, oxidative stress and cell death.
Apoptosis. 12:913–922. 2007. View Article : Google Scholar : PubMed/NCBI
|