Introduction
Liver cancer is one of the most common types of
cancer worldwide, ranking as the third leading cause of
cancer-associated mortality (1).
Despite the great advances in the use of modern surgical techniques
in combination with chemotherapy, the overall 5-year survival rate
for patients with liver cancer remains poor (2). Therefore, novel strategies for the
anticancer therapy of liver cancer are urgently required.
Phosphatidylinositol-3-phosphate 5-kinase (PIKfyve)
is a lipid kinase that phosphorylates
phosphatidylinositol-3-phosphate (PI3P) to generate
phosphatidylinositol 3,5-bisphosphate [PtdIns(3,5)P2] or
phosphatidylinositol 5-phosphate (PtdIns5P) (3,4).
PtdIns(3,5)P2 and PtdIns5P have been proposed to be involved in
several cellular functions, including vesicular trafficking, ion
channel activation and epidermal growth factor receptor (EGFR)
signaling (5–7). In addition, accumulating evidence has
suggested that PIKfyve is involved in oncogenesis and cancer cell
migration (8,9), and knockdown of PIKfyve resulted in a
significant decrease in cancer cell migration (10). Therefore, the potential role of
PIKfyve inhibition in anticancer therapy was explored in the
present study. Recent studies demonstrated that inhibition of
PIKfyve activity using the inhibitor YM201636 led to a strong
reduction in cell proliferation in multiple cancers (8,11);
however, whether PIKfyve inhibition could be applied for anticancer
therapy of liver cancer remains unknown. Therefore, the aim of the
present study was to investigate the antitumor activity of the
PIKfyve inhibitor, YM201636, in liver cancer.
Materials and methods
Reagents and antibodies
RPMI-1640 and fetal bovine serum (FBS) were
purchased from Gibco (Thermo Fisher Scientific, Inc., Waltham, MA,
USA). MTT and monodansylcadaverine (MDC) were purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). YM201636 (cat. no.
sc-204193) was obtained from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). Monoclonal mouse anti-human EGFR antibody (cat.
no. sc-71034; 1:200), monoclonal mouse anti-human phospho (p)-EGFR
antibody (cat. no. sc-81490; 1:200), monoclonal mouse anti-human
β-actin antibody (cat. no. sc-47778; 1:1,000) were all purchased
from Santa Cruz Biotechnology, Inc. Polyclonal rabbit anti-human
microtubule associated protein 1 light chain 3 (LC3) A/B antibody
(cat. no. 14600-1-AP; 1:1,000) was purchased from ProteinTech
Group, Inc. (Chicago, IL, USA). The horseradish peroxidase
(HRP)-conjugated secondary antibodies including goat anti-rabbit
IgG (cat. no. sc-2054; 1:1,000) and goat anti-mouse IgG (cat. no.
sc-2973; 1:1,000) were also purchased from Santa Cruz
Biotechnology, Inc.
Cell lines and cell culture
All cell lines (HepG2, Huh-7 and H22) used in the
present study were purchased from the Cell Bank of Type Culture
Collection of Chinese Academy of Sciences (Shanghai, China). These
cells were cultured in RPMI-1640 medium supplemented with 10% FBS,
100 U/ml penicillin and 100 µg/ml streptomycin, at 37°C in a
humidified incubator containing 5% CO2.
Transient transfections
HepG2 and Huh-7 cells were transiently transfected
with pcDNA3.1-epidermal growth factor receptor (EGFR) plasmid or
control pcDNA3.1 plasmid (Shanghai GeneChem Co., Ltd., Shanghai,
China) using Lipofectamine® 2000 reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. Briefly, the cells were seeded in 6-well plates at
5×105/well and cultured at 37°C in a humidified
atmosphere with 5% CO2. The subsequent day, when the
cells were ~70% confluent, they were transfected with a mixture of
3 µl Lipofectamine® 2000 and 2 µg plasmids. At 4 h
post-transfection, the cell culture medium was replaced with
RPMI-1640 medium. After 24 h, these transfected cells were
collected to perform MDC staining or western blot analysis.
MTT assay
Cells were seeded in 96-well plates at an initial
density of 4×103 cells/well in 90 µl medium and cultured
overnight. As cells reached 30% confluence, various concentrations
of YM201636 (0.1, 0.2, 0.5, 1, 2 and 5 µM) were added to the cells,
which were then incubated for 24 h. Subsequently, 50 µl 1 mg/ml of
MTT (Sigma-Aldrich; Merck KGaA) was added to each well for 4 h.
Then, the supernatant was removed and 100 µl DMSO was subsequently
added to solubilize the crystal products at room temperature for 10
min. The optical density (OD) was measured at a wavelength of 490
nm using a microplate reader (BioTek Instruments, Inc., Winooski,
VT, USA). The growth inhibition ratio was calculated as follows:
Growth inhibition ratio (%) = (ODcontrol -
ODdrug)/ODcontrol × 100; where
ODcontrol is the OD of the group treated with vehicle.
The experiments were repeated at least 3 times. To verify whether
the mechanism of autophagy promoted cell survival or cell death,
HepG2 and Huh-7 cells were pretreated with 5 mM 3-methyladenine
(3-MA) for 30 min followed by cotreatment with 2 µM YM201636 for 24
h. Then, an MTT assay was performed. The growth inhibition ratio
was calculated as follows: Growth inhibition ratio (%) =
(ODcontrol - ODdrug)/ODcontrol ×
100; where ODcontrol is the OD of the group treated with
vehicle. To examine the effects of EGFR overexpression on cell
growth, HepG2 and Huh-7 cells (80% confluence) seeded in a 6-well
plate were transfected with pcDNA3.1-EGFR or control pcDNA3.1
plasmids using Lipofectamine® 2000. After 24 h, the
cells were harvested and seeded into 96-well plates at a density of
4×103 cells/well for another 24 h. An MTT assay was
performed to determine the OD value of each group. During
incubation, EGF was not added to the culture medium to avoid EGFR
activation.
Flow cytometry
MDC, a specific marker for autophagic vacuoles, was
used to examine whether YM201636 induced autophagy. HepG2 and Huh-7
cells were grown on coverslips in a 6-well plate overnight at
3×105/well, and then treated with 2 or 5 µM of YM201636
for 24 h. The cells were collected and washed with ice-cold PBS 3
times, then incubated with 50 µM MDC at 37°C for 30 min. The
stained cells were washed, fixed with 4% paraformaldehyde for 10
min at room temperature and analyzed with the imaging flow
cytometer FlowSight® (Amnis®; Merck KGaA). A
laser set at 405 nm was used for excitation. Bright field and MDC
images (green fluorescence) were collected in channels 1 and 8,
respectively. The stained cells (1×104/sample) were
analyzed using IDEAS version 6.0 software (Merck KGaA).
Western blot analysis
The total proteins were isolated from cancer cells
or allograft tumors using radio immunoprecipitation assay buffer
(Beyotime Institute of Biotechnology, Haimen, China) following the
manufacturer's instructions. The protein concentration was
determined using a bicinchoninic acid assay kit (Pierce; Thermo
Fisher Scientific, Inc.). Samples were denatured in 5X SDS-sample
buffer at 95°C for 5 min. Total proteins (40 µg/well) were
separated using SDS-PAGE on 10% gels for EGFR detection or 12% gels
for LC3 detection. Following separation, the protein was
transferred onto polyvinylidene difluoride membranes. Subsequently,
the membranes were blocked using 5% non-fat milk in TBS-Tween
(TBS-T) at room temperature for 1 h. Following blocking, membranes
were incubated with corresponding primary antibodies overnight at
4°C, then washed 3 times with TBST and incubated with the
appropriate HRP-conjugated secondary antibody, and then washed 3
times with TBST. Proteins were detected using the enhanced
chemiluminescence plus reagents (Beyotime Institute of
Biotechnology). The western blots were visualized using a FluroChem
E Imager (Protein Simple, San Jose, CA, USA). Quantity One Software
(Quantity One 462; Bio-Rad Laboratories, Inc., Hercules, CA, USA)
was used to calculate the alteration of corresponding protein
expression.
Evaluation of antitumor effects in
vivo
Male BALB/c mice (weighing 20–25 g; aged 5 weeks)
were purchased from the Laboratory Animal Center of Henan
(Zhengzhou, China). All animal procedures were performed with the
approval of the Institutional Animal Care and Use Committee of
Henan University (Kaifeng, China). For the development of solid
tumors, mice were subcutaneously injected with 2×106 H22
cells. At 1 day after inoculation, the mice were randomly divided
into the control and YM201636 groups, which were treated with 5%
DMSO or 2 mg/kg YM201636, respectively, via oral administration
once daily for 7 consecutive days. The mice were anesthetized using
ether for ~30 sec via the respiratory route, and the heart rate and
respiratory rate were monitored to ensure the animals were simply
anesthetized. Then the mice were euthanized by cervical
dislocation, and solid tumors were isolated and weighed. Meanwhile,
the heart, liver, kidney, lung, and spleen of the mice were
collected and weighed on the last day. The organ weight index was
investigated for systemic toxicity evaluation as follows: Organ
index (%) = (organ weight/bodyweight) × 100.
Statistical analysis
Statistical analyses were performed using GraphPad
Prism 5 software for Windows (GraphPad Software, Inc., La Jolla,
CA, USA). All data are expressed as the mean ± standard error. A
two-tailed unpaired t-test was used for the comparison of the mean
values between two groups. One-way analysis of variance (ANOVA)
followed by a Dunnett's test or two-way ANOVA followed by a
Bonferroni post hoc test was used for multiple comparisons.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effects of YM201636 on HepG2 and Huh-7
cell viability
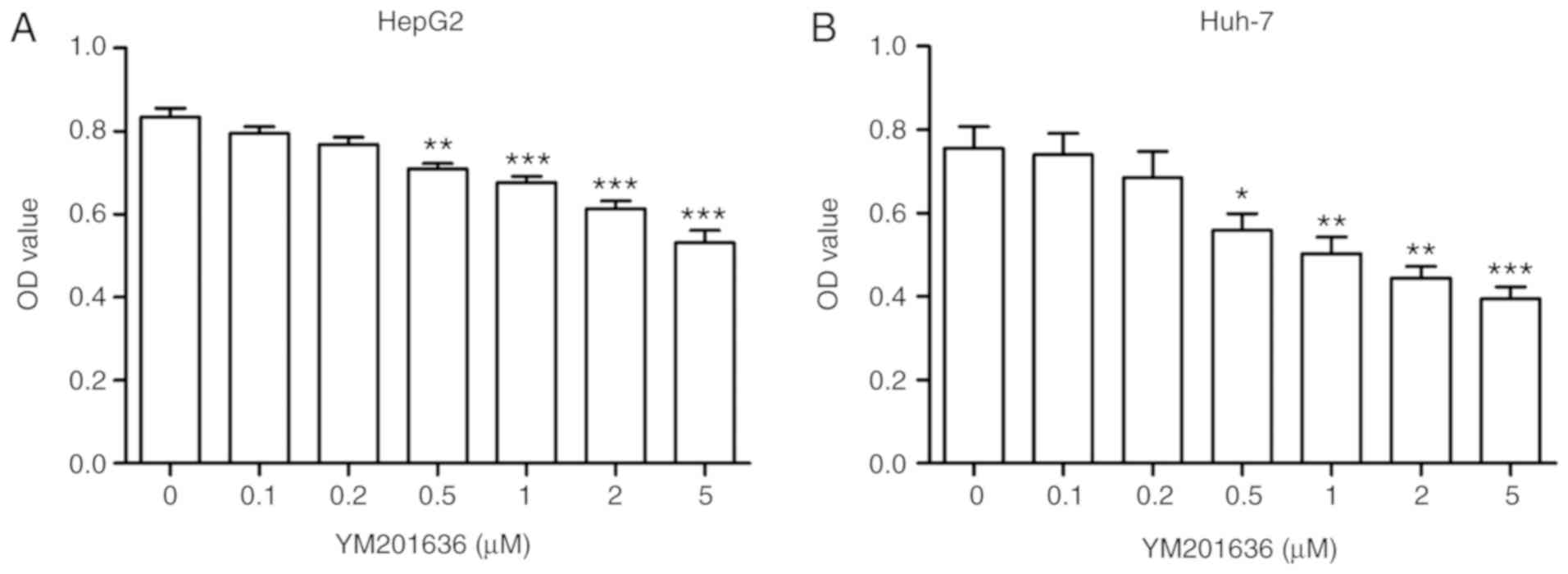
In order to determine whether the PIKfyve inhibitor
YM201636 affects the cell viability of hepatoma, HepG2 and Huh-7
cells were cultured and incubated in the presence of increasing
concentrations of the drug for 24 h. Cell viability rates were then
detected using an MTT assay. Following incubation of HepG2 and
Huh-7 cells with 0.1,0.2, 0.5, 1, 2 and 5 µM YM201636 for 24 h,
YM201636 reduced the HepG2 and Huh-7 cell viability in a
dose-dependent manner (Fig. 1). The
data indicated that YM201636 may inhibit the cell growth of liver
cancer cell lines.
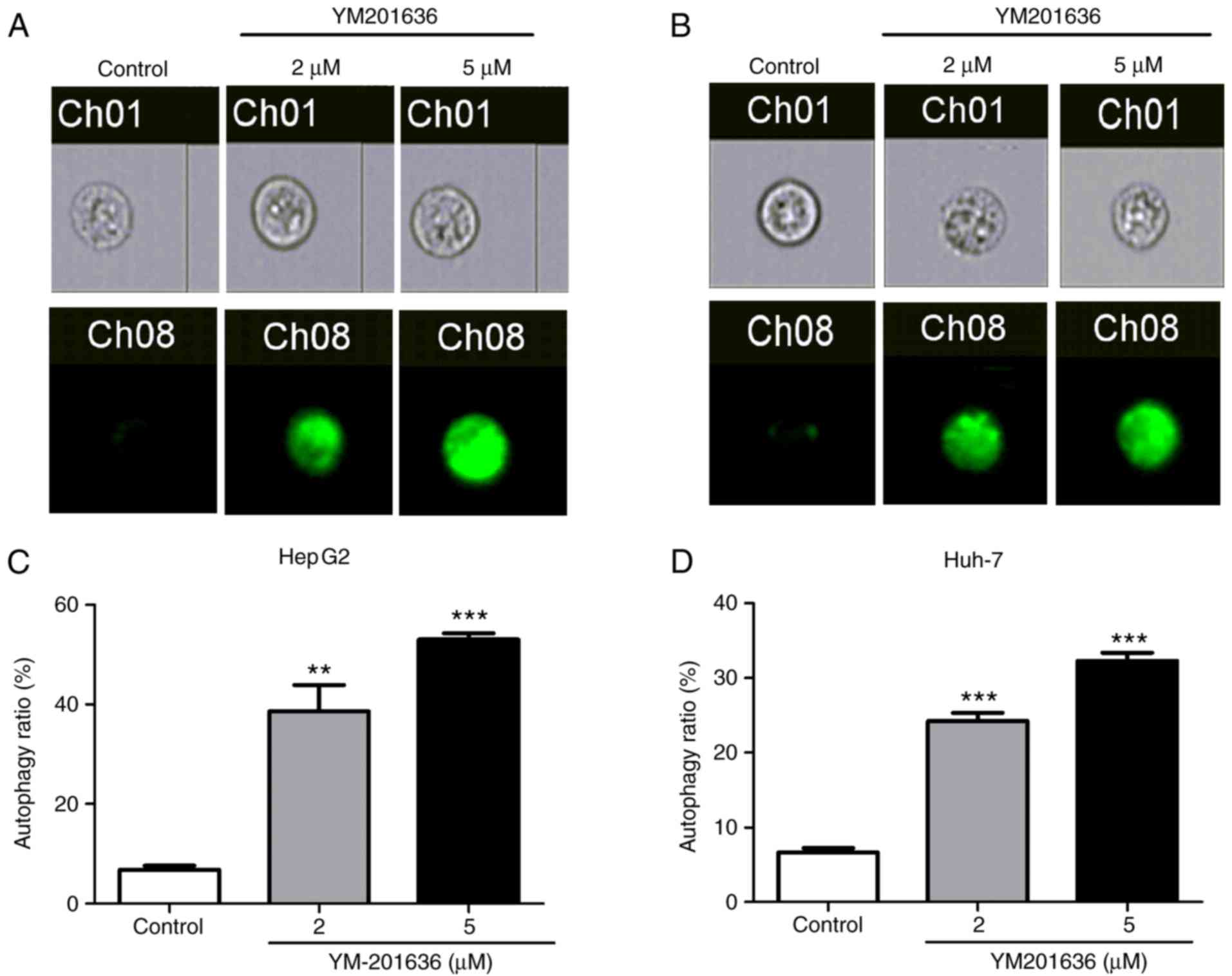
YM201636 induces autophagy in liver
cancer cell lines
Previous studies have demonstrated that knockdown or
inhibition of PIKfyve induces secretory autophagy in multiple cell
lines (12,13). Therefore, the present study
investigated whether decreased cell viability was associated with
autophagy in liver cancer cell lines using an MDC staining assay.
Following treatment with 2 or 5 µM YM201636 for 24 h, HepG2 and
Huh-7 cells exhibited strong MDC staining (Fig. 2), with enhanced fluorescence in
cells treated with 5 µM YM201636. Additionally, the percentage of
autophagic cells in the population was increased in HepG2 and Huh-7
cells treated with YM201636 for 24 h (Fig. 2). To further confirm whether
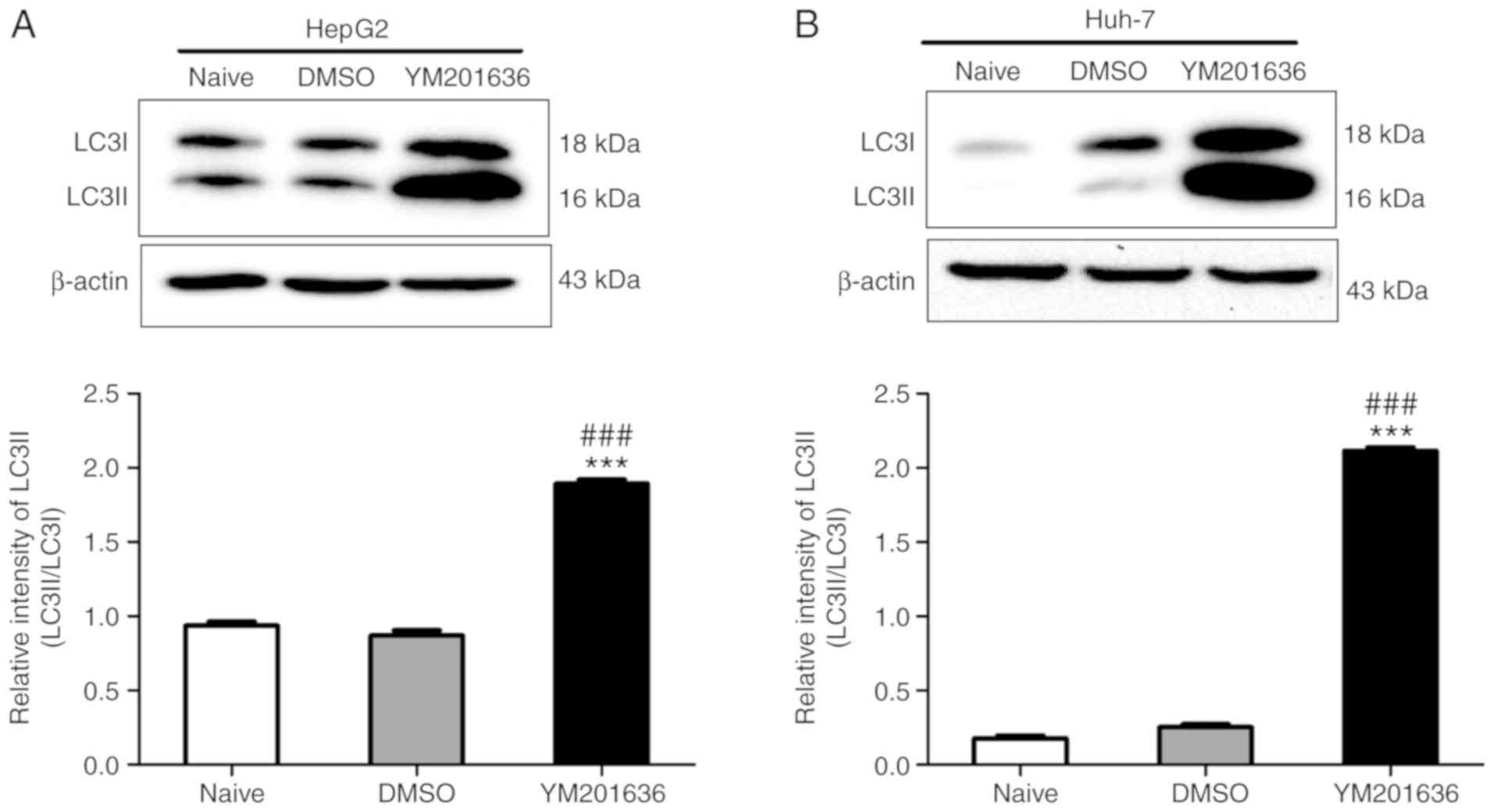
YM201636 induces autophagy in liver cancer cell lines, the ratio of
LC3I to LC3II was determined, an established autophagosome-related
marker (14,15). The results demonstrated that
YM201636 significantly promoted the conversion of LC3I to LC3II in
HepG2 and Huh-7 cells (Fig. 3). To
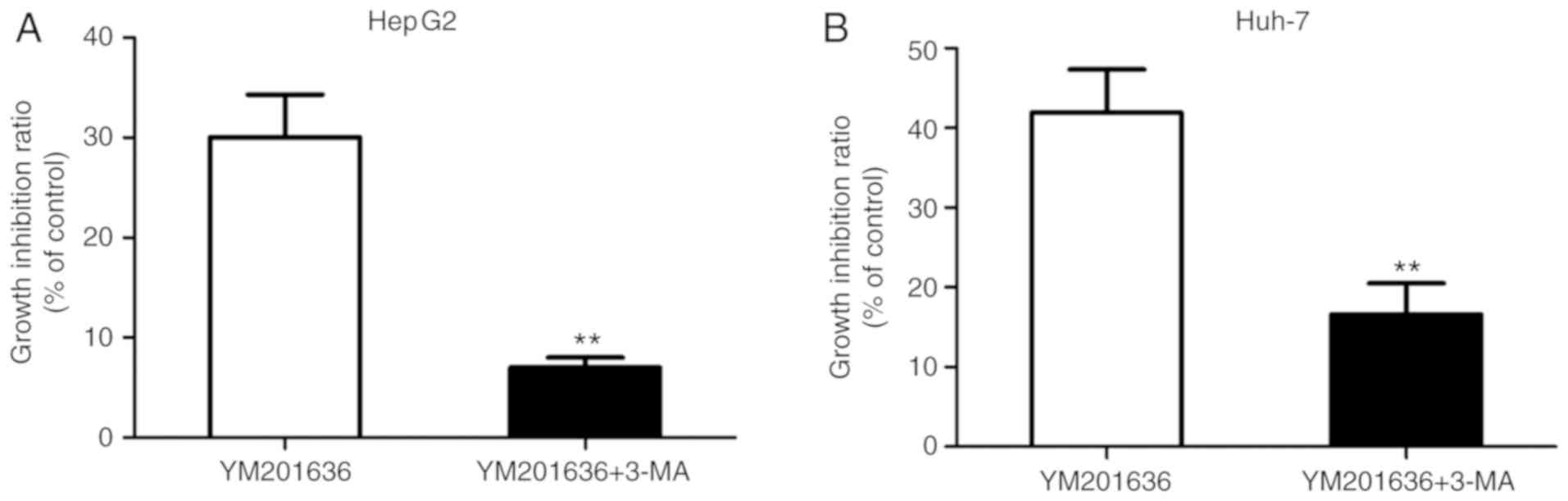
verify whether the mechanism of autophagy promoted cell survival or
cell death, the autophagy inhibitor 3-MA was applied to inhibit
YM201636 induced-autophagy in HepG2 and Huh-7 cells; 3-MA inhibits
autophagy by blocking autophagosome formation via the inhibition of
type III phosphatidylinositol 3-kinases (PI3Ks) (16,17).
Co-treatment with 5 mM 3-MA for 24 h attenuated the inhibitory
effects of YM201636 on liver cancer cell lines (Fig. 4). Collectively, these results
suggested that YM201636 inhibits the growth of HepG2 and Huh-7
cells via the induction of autophagy.
YM201636 induced-autophagy depends on
EGFR overexpression
It has been previously reported that PIKfyve
inhibition blocks the lysosomal degradation of EGFR, resulting in
increased expression levels of EGFR in MCF-10A cells (18), and overexpression of inactive EGFR
has been demonstrated to be associated with autophagy (19). Therefore, the present study
investigated whether YM201636-induced autophagy is dependent upon
EGFR overexpression. To test this hypothesis, the total protein
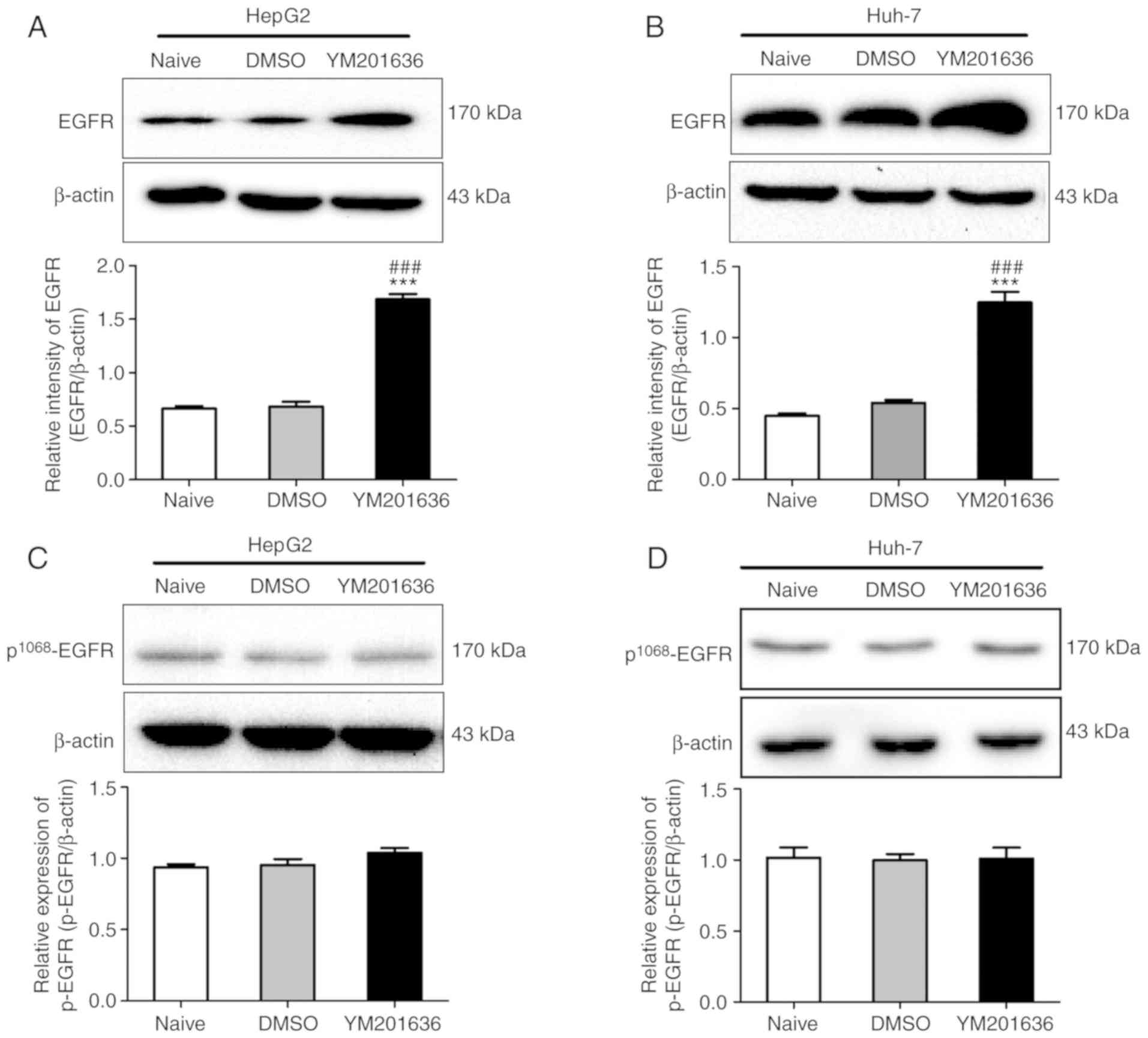
expression of EGFR was determined in HepG2 and Huh-7 cells treated
with YM201636. Western blotting demonstrated that the total protein
expression levels of EGFR were significantly upregulated in HepG2
and Huh-7 cells treated with 2 µM YM201636 for 24 h (Fig. 5). To investigate whether the
increased EGFR was activated in HepG2 and Huh-7 cells treated with
YM201636, the phosphorylation levels of EGFR at Tyr1068,
an indicator of EGFR activation, were examined. As shown in
Fig. 5, the phosphorylation levels
of EGFR at Tyr1068 were notably unaffected in HepG2 and
Huh-7 cells treated with YM201636. The results suggested that the
increased total protein constituted inactive EGFR.
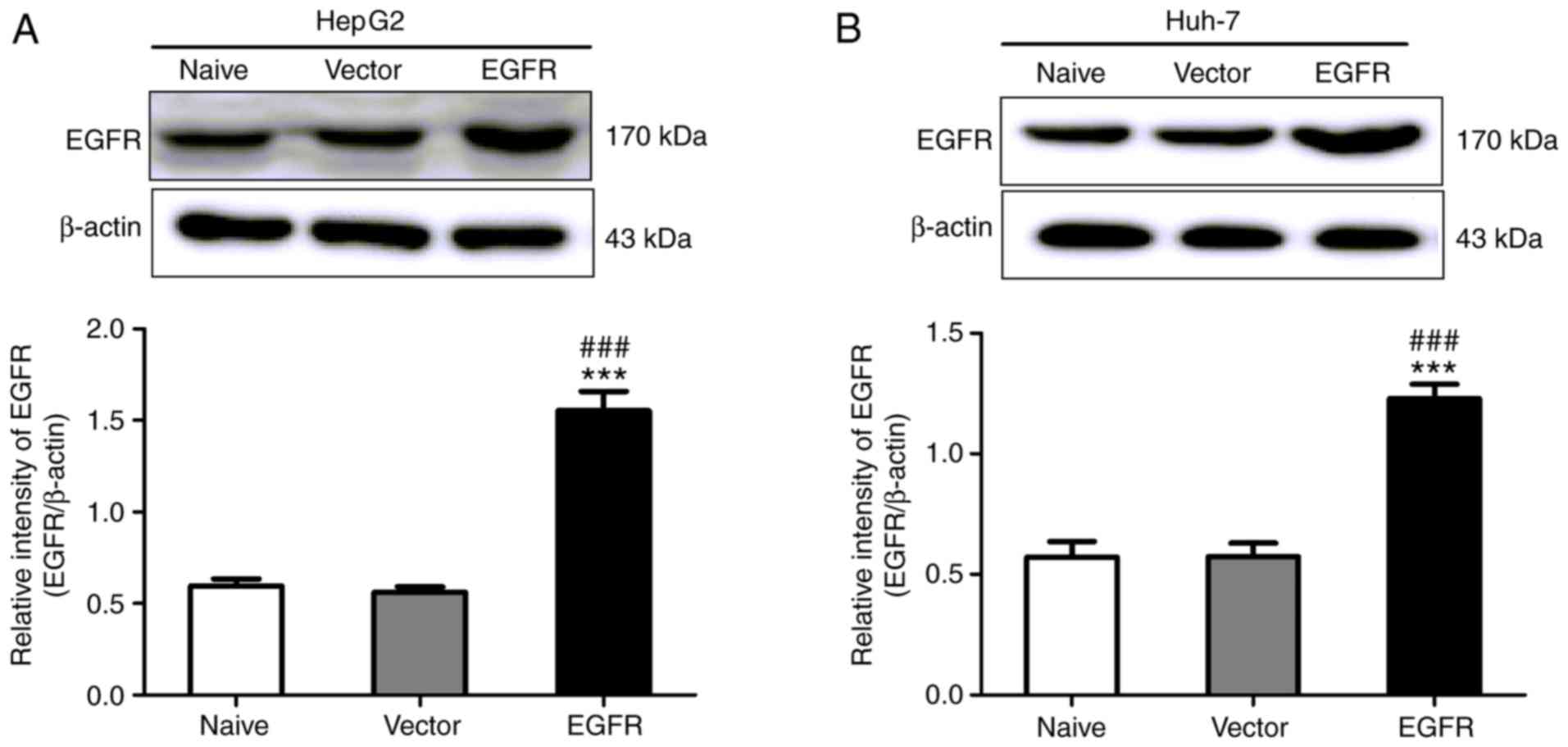
To further investigate the hypothesis that inactive
EGFR mediates autophagy in HepG2 and Huh-7 cells, HepG2 and Huh-7
cells were transfected with the pcDNA3.1-EGFR plasmid with no EGF
treatment. At 24 h after transfection, the expression of EGFR was
increased significantly (Fig. 6).
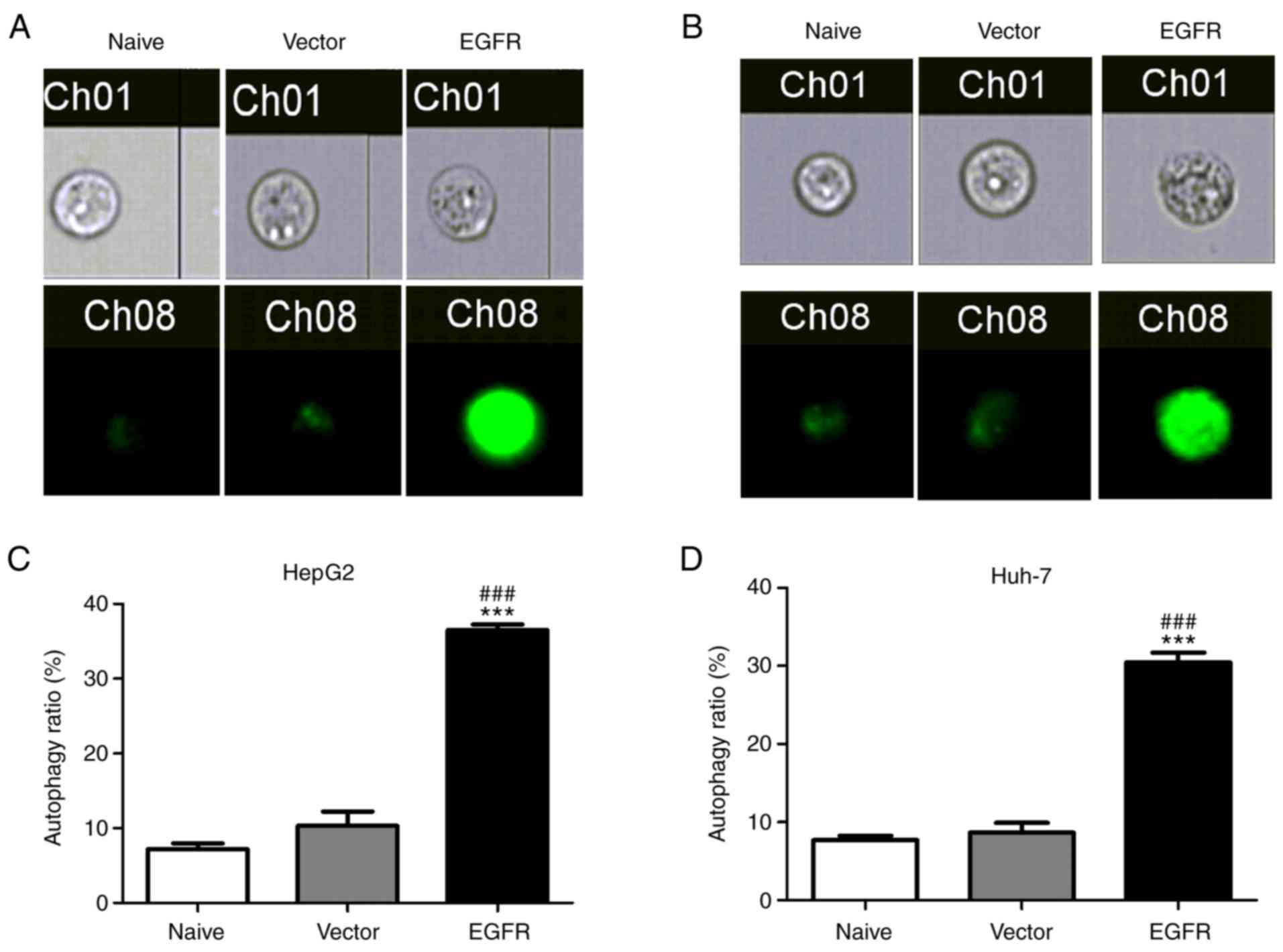
Subsequently, whether overexpression of EGFR was able to affect
autophagy in liver cancer cells was examined. As demonstrated in
Fig. 7, HepG2 and Huh-7 cells
transfected with EGFR exhibited strong MDC staining and an increase
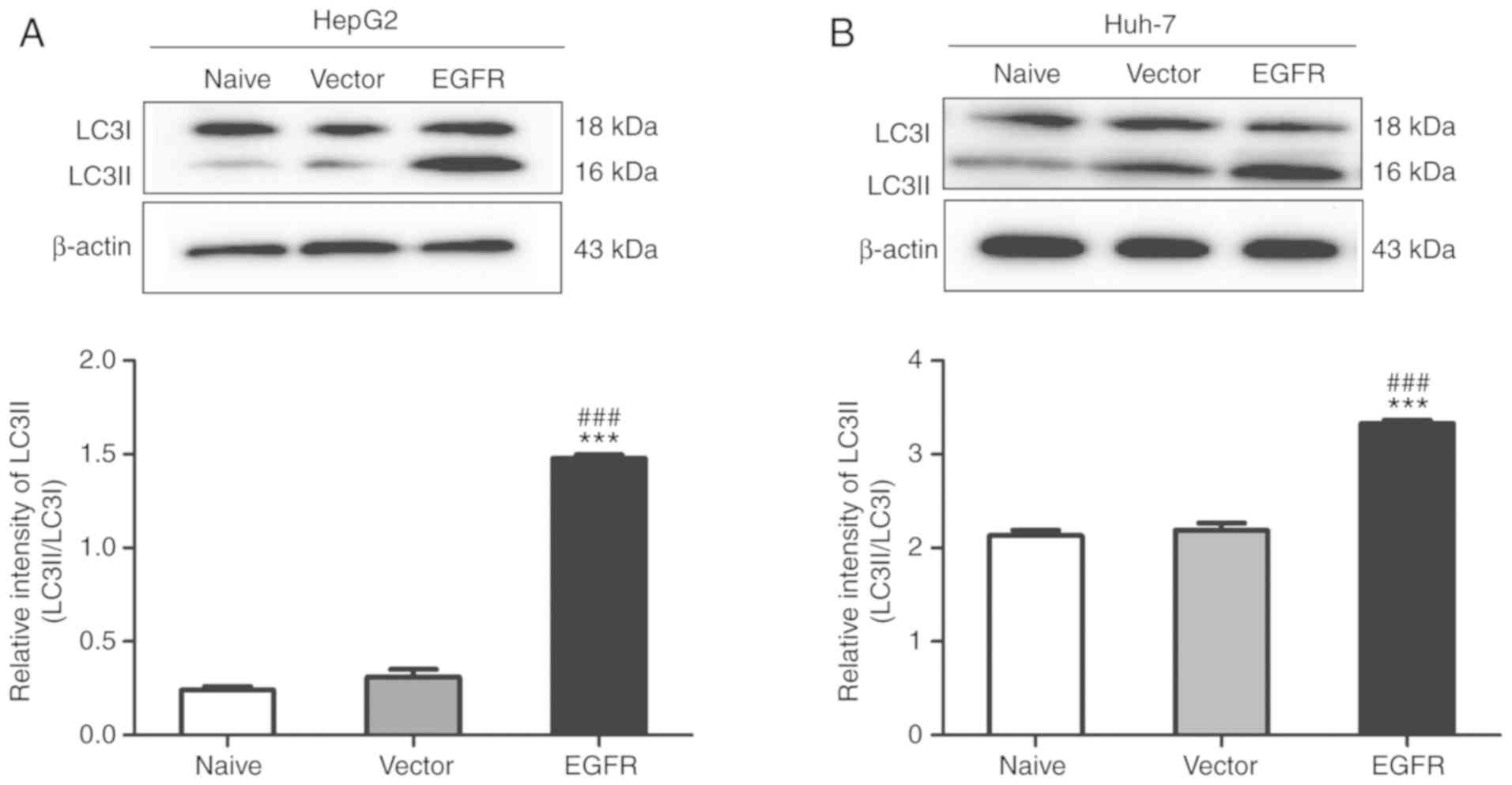
in the proportion of autophagic cells. Additionally, overexpression
of EGFR also promoted the transformation of LC3I into LC3II in
HepG2 and Huh-7 cells (Fig. 8).
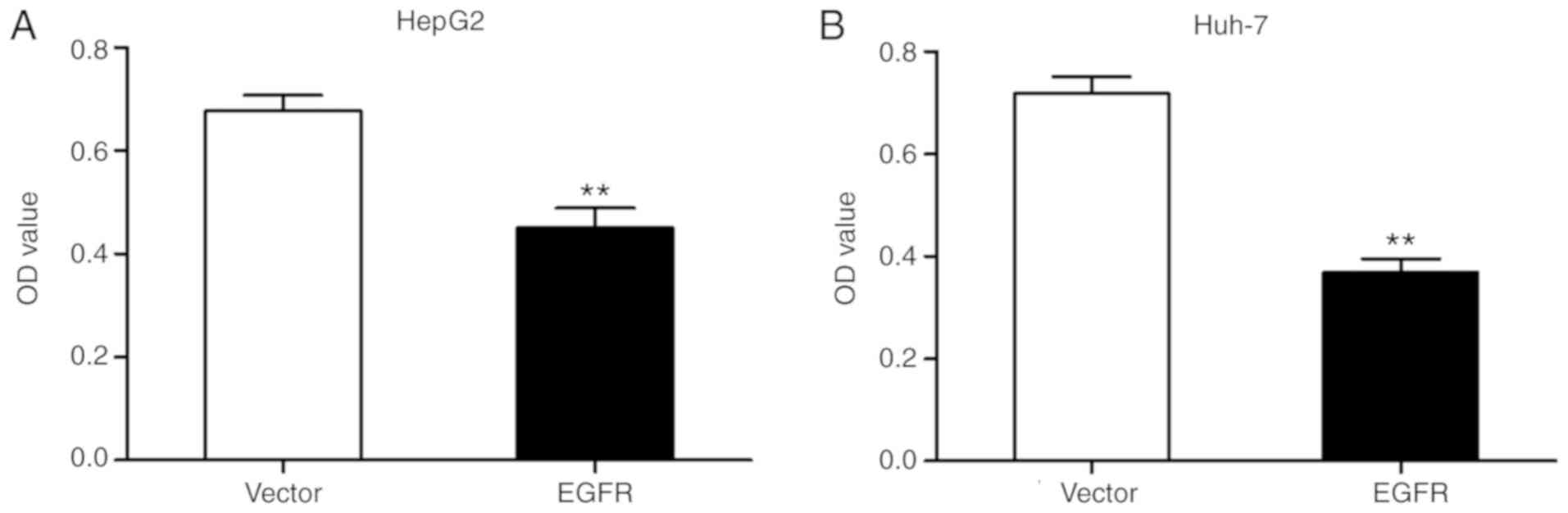
Furthermore, the effects of EGFR overexpression on liver cancer
cell growth were determined. Overexpression of EGFR in HepG2 and
Huh-7 cells inhibited cell proliferation (Fig. 9). The results indicated that
YM201636 induced-autophagy was dependent on EGFR overexpression in
HepG2 and Huh-7 cells.
YM201636 inhibits the in situ growth
of hepatoma in vivo
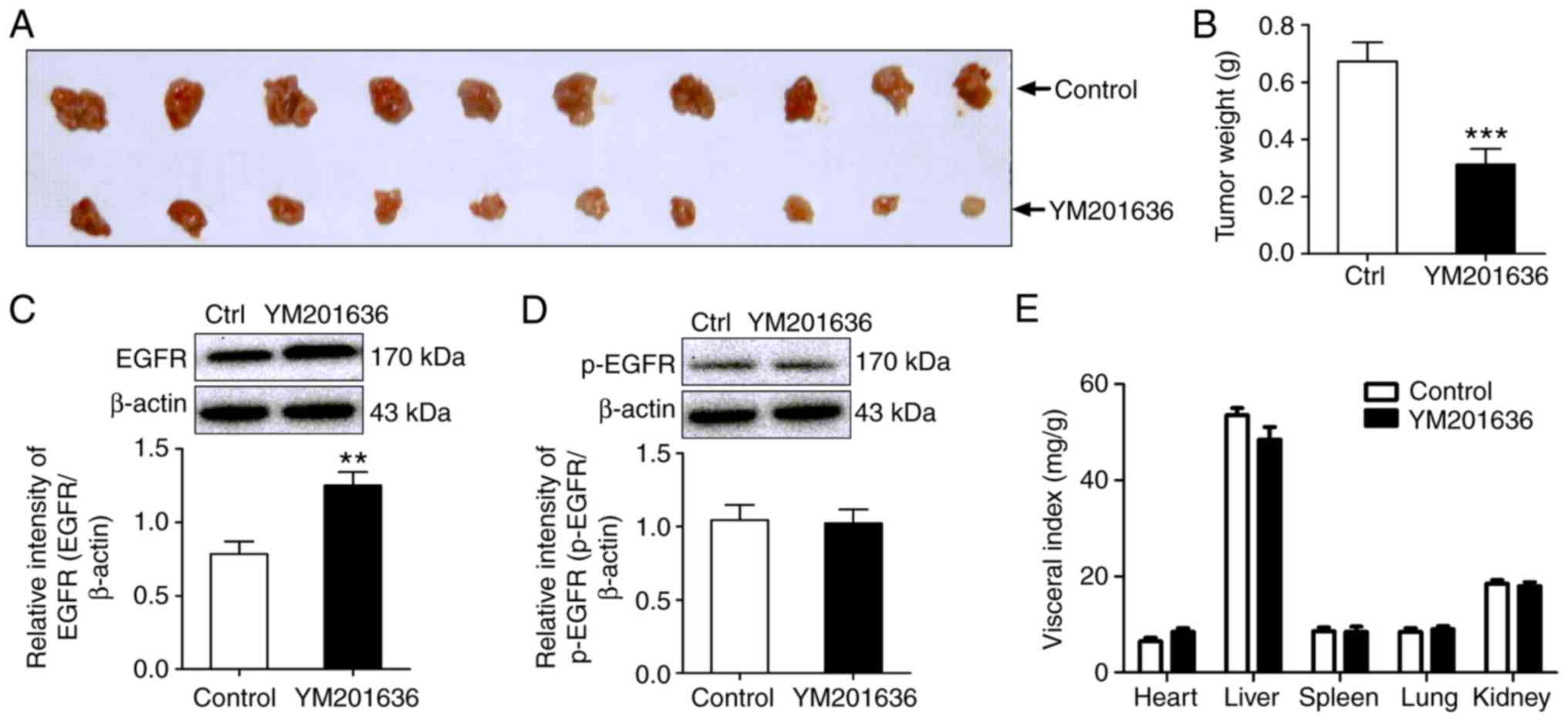
To further evaluate the antitumor activity of the
PIKfyve inhibitor, YM201636, in vivo, an allograft model of
mouse liver cancer was established by subcutaneously injecting H22
cells (mouse hepatoma cell line) into BALB/c mice. Transplantation
of H22 cells into BALB/c mice induced the in situ formation
of tumors. Following treatment with 2 mg/kg YM201636 for 7
consecutive days, the tumor weight of the YM201636 group was lower
than that of the negative control group. For example, the mean
weight of tumors in the YM201636-treated group was 0.31±0.05 g,
compared with 0.67±0.07 g in the control group (Fig. 10A and B). Additionally, YM201636
promoted the expression of EGFR total protein in allograft tumors;
however, the phosphorylation levels of EGFR at Tyr1068
were not notably altered (Fig. 10C and
D). In addition, visceral organ indices were also examined to
evaluate adverse effects of YM201636. No significant differences in
the visceral organ indexes (heart, liver, spleen, lung and kidney)
were observed in the YM201636 group compared with the control group
(Fig. 10E). These results
suggested that YM201636 may inhibit tumor growth without notable
systemic toxicity at the dose applied in the present study.
Discussion
PIKfyve is an enzyme that is crucial for the
synthesis of PtdIns(3,5)P2 and has been associated with various
membrane transport events (20,21).
Perturbations in the functions of PIKfyve lead to the formation of
swollen endosomal/lysosomal structures in a variety of mammalian
cell lines (22,23). Additionally, PIKfyve has been
proposed to be involved in oncogenesis and cancer cell migration.
For example, PIKfyve promotes cell migration and invasion through
the activation of Ras-related protein Rac1in lung carcinoma,
osteosarcoma and rhabdomyosarcoma cells (8,24);
knockdown of PIKfyve resulted in significant decreases in cell
migration velocity and persistence (10). Consistent with these observations,
in the present study, it was demonstrated that pharmacological
inhibition of PIKfyve using YM201636 resulted in an inhibitory
effect on tumor cell growth via the induction of autophagy in
hepatoma cell lines.
The present study demonstrated that EGFR
upregulation required for autophagy induced by the inhibition of
PIKfyve. In support of the results, Er et al (18) reported that suppression of PIKfyve
activity reduces the rate of EGFR degradation in MCF-10A cells.
EGFR is part of the ErbB family of receptor tyrosine kinases and is
overexpressed in many human cancers (25). Abnormal hyperactivation of EGFR is
associated with unregulated proliferation, malignant transformation
and metastasis in cancer cells (26,27).
It has been demonstrated that EGFR activation inhibits autophagy,
dependent upon its kinase domain, by maintaining high activation
levels of the PI3K/protein kinase B/mammalian target of rapamycin
signaling pathway; knockdown or inhibition of EGFR signaling
induces autophagy in various epithelial cancers (28,29).
However, in the present study, overexpression of EGFR induced
autophagy in liver cancer cells. The results suggested that the
increased EGFR may be inactive, as Tan et al (19) reported that inactive EGFR complexes
collaborate with lysosomal-associated transmembrane protein 4B and
exocyst complex component 2 at endosomes to disassociate Rubicon
from Beclin 1, which in turn initiates autophagy. Autophagy
exhibits a dual role that can induce a cell survival or cell death
response (30,31). In the present study, EGFR-mediated
autophagy was proposed to underlie cancer cell death.
Numerous studies have suggested EGFR overexpression
promotes tumor cell proliferation (26,32);
however, the findings of the present study indicated that PIKfyve
inhibitor YM201636-induced autophagy was dependent on EGFR
overexpression. As mentioned above, the increased EGFR total
protein was inactive EGFR, and inactive EGFR complexes could
initiate autophagy (19). In
support of the present results, Lanaya et al (33) demonstrated that mice lacking EGFR in
hepatocytes develop more hepatocellular carcinoma (33). However, there are limitations in
this study. Although the results suggested the increased EGFR total
protein was inactive EGFR, the expression of proteins downstream of
EGFR, such as Akt, p-Akt, ERK, p-ERK, was not analyzed. The
phosphorylation of Akt and ERK are indicators of EGFR activation
(34,35), therefore, the expression of these
downstream proteins will be examined in future studies.
In the present study, it was speculated that
YM201636 induced total EGFR expression through reducing the rate of
EGFR degradation in HepG2 and Huh-7 cells. Since EGF stimulation
causes EGFR degradation by delivery to the lysosomes (18), therefore, it was assumed that the
unchanged phosphorylation levels of EGFR at Tyr1068, was
due to lack of EGF stimulation.
In conclusion, the results of the present study
suggested that the PIKfyve inhibitor YM201636 may possess
therapeutic potential for the treatment of liver cancer. In
addition, EGFR overexpression induced by a PIKfyve inhibitor
contributes to autophagy, thus leading to an inhibitory effect on
cancer cell growth.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Science
Foundation of China (grant nos. 81573465, 81772832 and 81701110),
the Natural Science Foundation of Henan (grant no. 162300410039);
and the Program for Science and Technology of the Department of
Education of Henan Province (grant no. 16A350013).
Availability of data and materials
All data generated or analysed during this study are
available from the corresponding author on reasonable request.
Authors' contributions
DF and SQX participated in the design of the study,
data analysis and manuscript writing. JZH, ZQX, JN conducted MTT
assays and flow cytometry, participated in the design of the study
and data analysis. WL, XW, CL and HS performed the assays and
analysis. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All experiments were approved by the Ethics
Committee of University of Henan University (Kaifeng, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lozano R, Naghavi M, Foreman K, Lim S,
Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, et
al: Global and regional mortality from 235 causes of death for 20
age groups in 1990 and 2010: A systematic analysis for the global
burden of disease study 2010. Lancet. 380:2095–2128. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yang Y, Nagano H, Ota H, Morimoto O,
Nakamura M, Wada H, Noda T, Damdinsuren B, Marubashi S, Miyamoto A,
et al: Patterns and clinicopathologic features of extrahepatic
recurrence of hepatocellular carcinoma after curative resection.
Surgery. 141:196–202. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sbrissa D, Ikonomov OC and Shisheva A:
PIKfyve, a mammalian ortholog of yeast Fab1p lipid kinase,
synthesizes 5-phosphoinositides. Effect of insulin. J Biol Chem.
274:21589–21597. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shisheva A: PIKfyve: The road to PtdIns
5-P and PtdIns 3,5-P2. Cell Biol Int. 25:1201–1206.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Takasuga S and Sasaki T:
Phosphatidylinositol-3,5-bisphosphate: Metabolism and physiological
functions. J Biochem. 154:211–218. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
McCartney AJ, Zolov SN, Kauffman EJ, Zhang
Y, Strunk BS, Weisman LS and Sutton MA: Activity-dependent
PI(3,5)P2 synthesis controls AMPA receptor trafficking
during synaptic depression. Proc Natl Acad Sci USA.
111:E4896–E4905. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim J, Jahng WJ, Di Vizio D, Lee JS,
Jhaveri R, Rubin MA, Shisheva A and Freeman MR: The
phosphoinositide kinase PIKfyve mediates epidermal growth factor
receptor trafficking to the nucleus. Cancer Res. 67:9229–9237.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Oppelt A, Haugsten EM, Zech T, Danielsen
HE, Sveen A, Lobert VH, Skotheim RI and Wesche J: PIKfyve, MTMR3
and their product PtdIns5P regulate cancer cell migration and
invasion through activation of Rac1. Biochem J. 461:383–390. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ikonomov OC, Filios C, Sbrissa D, Chen X
and Shisheva A: The PIKfyve-ArPIKfyve-Sac3 triad in human breast
cancer: Functional link between elevated Sac3 phosphatase and
enhanced proliferation of triple negative cell lines. Biochem
Biophys Res Commun. 440:342–347. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Oppelt A, Lobert VH, Haglund K, Mackey AM,
Rameh LE, Liestol K, Schink KO, Pedersen NM, Wenzel EM, Haugsten
EM, et al: Production of phosphatidylinositol 5-phosphate via
PIKfyve and MTMR3 regulates cell migration. EMBO Rep. 14:57–64.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dupuis-Coronas S, Lagarrigue F, Ramel D,
Chicanne G, Saland E, Gaits-Iacovoni F, Payrastre B and Tronchere
H: The nucleophosmin-anaplastic lymphoma kinase oncogene interacts,
activates, and uses the kinase PIKfyve to increase invasiveness. J
Biol Chem. 286:32105–32114. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sano O, Kazetani K, Funata M, Fukuda Y,
Matsui J and Iwata H: Vacuolin-1 inhibits autophagy by impairing
lysosomal maturation via PIKfyve inhibition. FEBS Lett.
590:1576–1585. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hessvik NP, Overbye A, Brech A, Torgersen
ML, Jakobsen IS, Sandvig K and Llorente A: PIKfyve inhibition
increases exosome release and induces secretory autophagy. Cell Mol
Life Sci. 73:4717–4737. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Galluzzi L, Baehrecke EH, Ballabio A, Boya
P, Bravo-San Pedro JM, Cecconi F, Choi AM, Chu CT, Codogno P,
Colombo MI, et al: Molecular definitions of autophagy and related
processes. EMBO J. 36:1811–1836. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee YK and Lee JA: Role of the mammalian
ATG8/LC3 family in autophagy: Differential and compensatory roles
in the spatiotemporal regulation of autophagy. BMB Rep. 49:424–430.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bhat P, Kriel J, Shubha Priya B, Basappa,
Shivananju NS and Loos B: Modulating autophagy in cancer therapy:
Advancements and challenges for cancer cell death sensitization.
Biochem Pharmacol. 147:170–182. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Heckmann BL, Yang X, Zhang X and Liu J:
The autophagic inhibitor3-methyladenine potently stimulates
PKA-dependent lipolysis in adipocytes. Br J Pharmacol. 168:163–171.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Er EE, Mendoza MC, Mackey AM, Rameh LE and
Blenis J: AKT facilitates EGFR trafficking and degradation by
phosphorylating and activating PIKfyve. Sci Signal. 6:ra452013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tan X, Thapa N, Sun Y and Anderson RA: A
kinase-independent role for EGF receptor in autophagy initiation.
Cell. 160:145–160. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hirano T, Munnik T and Sato MH: Inhibition
of phosphatidylinositol 3,5-bisphosphate production has pleiotropic
effects on various membrane trafficking routes in Arabidopsis.
Plant cell Physiol. 58:120–129. 2017.PubMed/NCBI
|
|
21
|
Ikonomov OC, Fligger J, Sbrissa D,
Dondapati R, Mlak K, Deeb R and Shisheva A: Kinesin adapter JLP
links PIKfyve to microtubule-based endosome-to-trans-Golgi network
traffic of furin. J Biol Chem. 284:3750–3761. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jin N, Jin Y and Weisman LS: Early
protection to stress mediated by CDK-dependent PI3,5P2
signaling from the vacuole/lysosome. J Cell Biol. 216:2075–2090.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bissig C, Hurbain I, Raposo G and van Niel
G: PIKfyve activity regulates reformation of terminal storage
lysosomes from endolysosomes. Traffic. 18:747–757. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dayam RM, Sun CX, Choy CH, Mancuso G,
Glogauer M and Botelho RJ: The lipid kinase PIKfyve coordinates the
neutrophil immune response through the activation of the Rac
GTPase. J Immunol. 199:2096–2105. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Du Z and Lovly CM: Mechanisms of receptor
tyrosine kinase activation in cancer. Mol Cancer. 17:582018.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ye QH, Zhu WW, Zhang JB, Qin Y, Lu M, Lin
GL, Guo L, Zhang B, Lin ZH, Roessler S, et al: GOLM1 modulates
EGFR/RTK cell-surface recycling to drive hepatocellular carcinoma
metastasis. Cancer Cell. 30:444–458. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sainsbury JR, Farndon JR, Needham GK,
Malcolm AJ and Harris AL: Epidermal-growth-factor receptor status
as predictor of early recurrence of and death from breast cancer.
Lancet. 1:1398–1402. 1987.PubMed/NCBI
|
|
28
|
Li X and Fan Z: The epidermal growth
factor receptor antibody cetuximab induces autophagy in cancer
cells by downregulating HIF-1alpha and Bcl-2 and activating the
beclin 1/hVps34 complex. Cancer Res. 70:5942–5952. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li H, You L, Xie J, Pan H and Han W: The
roles of subcellularly located EGFR in autophagy. Cell Signal.
35:223–230. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen Y, Henson ES, Xiao W, Huang D,
McMillan-Ward EM, Israels SJ and Gibson SB: Tyrosine kinase
receptor EGFR regulates the switch in cancer cells between cell
survival and cell death induced by autophagy in hypoxia. Autophagy.
12:1029–1046. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jacob JA, Salmani JMM, Jiang Z, Feng L,
Song J, Jia X and Chen B: Autophagy: An overview and its roles in
cancer and obesity. Clin Chim Acta. 468:85–89. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sato H, Yamamoto H, Sakaguchi M, Shien K,
Tomida S, Shien T, Ikeda H, Hatono M, Torigoe H, Namba K, et al:
Combined inhibition of MEK and PI3K pathways overcomes acquired
resistance to EGFR-TKIs in non-small cell lung cancer. Cancer Sci.
109:3183–3196. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lanaya H, Natarajan A, Komposch K, Li L,
Amberg N, Chen L, Wculek SK, Hammer M, Zenz R, Peck-Radosavljevic
M, et al: EGFR has a tumour-promoting role in liver macrophages
during hepatocellular carcinoma formation. Nature Cell Biol.
16:972–977. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang Y, Wang L, Zhang M, Jin M, Bai C and
Wang X: Potential mechanism of interleukin-8 production from lung
cancer cells: An involvement of EGF-EGFR-PI3K-Akt-Erk pathway. J
Cell Physiol. 227:35–43. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang L, Li Y, Lan L, Liu R, Wu Y, Qu Q
and Wen K: Tamoxifen has a proliferative effect in endometrial
carcinoma mediated via the GPER/EGFR/ERK/cyclin D1 pathway: A
retrospective study and an in vitro study. Mol Cell Endocrinol.
437:51–61. 2016. View Article : Google Scholar : PubMed/NCBI
|