Introduction
Colorectal cancer (CRC) is one of the most common
causes of cancer-associated mortality worldwide (1,2).
Multiple clinical studies have reported that patients with CRC with
distant metastases (>50% of patients with HCC have metastasis in
the liver), have a poor prognosis, but the underlying molecular
mechanisms have not been fully elucidated (3–5). In
addition, phosphatase of regenerating liver-3 (PRL-3), a member of
the family of protein tyrosine phosphatases, is highly expressed in
~90% of liver metastases of CRC, whereas it is moderately expressed
in primary lesions (4). In
addition, primary solid tumors with high PRL-3 expression are prone
to metastasize to distant tissues (6–9). Many
studies have described PRL-3 as a biomarker of a poor prognosis and
shorter survival durations.
Recent studies have suggested that inflammatory
cells and cytokines present in tumors have a critical role in tumor
progression (10). This
inflammatory microenvironment is now considered to strongly
contribute to tumor metastasis by providing a better environment
for tumor cells to grow. Furthermore, evidence had demonstrated
that inflammation promotes processes by which cells switch from
epithelial phenotypes to mesenchymal phenotypes
(epithelial-mesenchymal transition; EMT), which increases
invasiveness and the chance of metastasis (2,11,12).
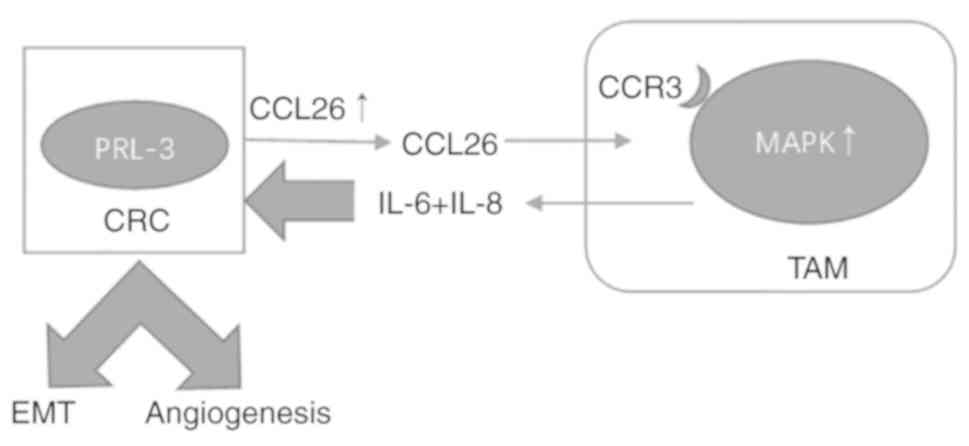
In addition, our previous research demonstrated that PRL-3
increases the expression of the chemokine ligand 26 (CCL26), which
induces infiltration of tumor-associated macrophages (TAMs), thus
enhancing invasion by increasing the expression of interleukin
(IL)-6 and IL-8 (4). Elevated
levels of serum IL-6 are associated with an increase in tumor size,
the occurrence of liver metastases and a poor prognosis. It has
also been reported that IL-8 is involved in EMT in breast cancer
(13). However, the underlying
mechanisms leading to the increased expression of IL-6 and IL-8,
and how these cytokines affect invasion and metastasis in CRC
remain unknown.
Angiogenesis is another key element in cancer
progression and metastasis (5). EMT
provides cancer cells with the ability to survive in the
circulatory system and then become established in a distant
location (2). However, blood
vessels are essential for supplying nutrients to new metastases, to
maintain the cells and enable further growth. It was previous
reported that PRL-3 participates in invasion, migration, metastasis
and angiogenesis (4,8). However, the potential role of TAMs in
the process of angiogenesis is not yet clear.
In the present study, it was aimed to determine how
the interaction of CRC cells and TAMs functions during the
processes of metastasis and angiogenesis in CRC.
Materials and methods
Reagents and antibodies
Fetal bovine serum (FBS) was purchased from
Biological Industries (Kibbutz Beit Haemek, Israel); phorbol ester
(PMA) was obtained from Sigma-Aldrich (Merck KGaA, Darmstadt,
Germany). Radioimmunoprecipitation assay (RIPA) buffer was
purchased from Thermo Fisher Scientific, Inc. (Waltham, MA, USA),
and RPMI and Dulbecco's modified Eagle's medium (DMEM) media were
purchased from Gibco (Thermo Fisher Scientific, Inc.). TRIzol and
Prime Script reverse transcription (RT) reagents were purchased
from Takara Bio, Inc. (Otsu, Japan), and recombinant CCL26 (rCCL26)
was obtained from PeproTech, Inc. (Rocky Hill, NJ, USA). Matrigel
matrix was purchased from BD Biosciences (Becton, Dickinson and
Company, Franklin, Lakes, NJ, USA). Anti-extracellular
signal-regulated 1/2 [ERK(1/2); cat. no. AF0155],
anti-phosphorylated (p)-ERK(1/2) (cat. no. AF1015), anti-c-Jun
N-terminal kinase (JNK; cat. no. AF6318), anti-IL-8 (cat. no.
DF6998), anti-Lamin B (cat. no. BF1002), horseradish peroxidase
(HRP)-labeled goat anti-mouse IgG (cat. no. S0002), HRP-labeled
goat anti-rabbit IgG (cat. no. S0001) and anti-phosphorylated
(p)-JNK antibodies (cat. no. AF3318) were purchased from Affinity
Biosciences (Cincinnati, OH, USA). Anti-IL-6 (cat. no. 12153),
anti-E-cadherin (cat. no. 3195), anti-Snail (cat. no. 3879),
anti-Vimentin (cat. no. 5741), anti-CD68 (cat. no. 79594),
anti-NF-κB inhibitor α (IκBα; cat. no. 4812), anti-inhibitor of
nuclear factor-κB kinase subunit α (IKKα; cat. no. 61294), anti-P50
(cat. no. 3035) and anti-GAPDH antibodies (cat. no. 5174) were
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Anti-CD206 (cat. no. AF2534-SP) was purchase from R&D Systems,
Inc. (Minneapolis, MN, USA). Inhibitors of p-JNK (SP600125) and
p-ERK (SCH772984) were purchased from MedChemExpress USA (Monmouth
Junction, NJ, USA), and IL-6 and IL-8 were purchased from
ProteinTech Group, Inc. (Chicago, IL, USA).
Cell cultures and treatments
LoVo, HT29 and THP-1 cells were purchased from the
Shanghai Cell Bank of the Chinese Academy of Sciences (Shanghai,
China). The cell lines were authenticated in July 2017 by Guangzhou
Cellcook Biotech Co., Ltd. (Guangzhou, China), with their STR
profiles compared to those in the American type Culture Collection
and DSMZ (Braunschweig, Germany) databases. LoVo and THP-1 lines
were cultured in RPMI-1640 medium, while HT29 cells were cultured
in in DMEM; all cells were supplemented with 10% FBS, 100 mg/ml
streptomycin and 100 mg/ml penicillin. Western blot analysis was
performed to identify PRL-3 expression in these CRC cell lines, and
the results showed that HT29 cells expressed high levels of PRL-3,
while LoVo cells expressed low levels. Therefore, LoVo cells were
selected for constructing PRL-3-overexpression lines, and HT29
cells were chosen for the knockdown groups. Stable transfection of
pAcGFP negative control (LoVo-NC) and pAcGFP-PRL-3 (LoVo-P) vectors
from Shanghai GeneChem (Shanghai, China) was performed, resulting
in control vector-transfected and PRL-3-overexpression cell lines,
respectively. To knock down PRL-3, HT29 cells were stably
transfected with PRL-3 short hairpin RNA (shRNA; HT29-P), and an
shRNA control was used in negative control groups (Fig. S1). The sequence of shRNA-PRL-3 was
as follows: 5′-ACAGAGGCTGCGGTTCAAA-3′. The sequence of negative
control was: 5′-TTCTCCGAACGTGTCACGT-3′. THP-1 cells were
transformed into M1 and M2 macrophages as previously described
(4,14). THP-1 cells differentiated into M2
macrophages after treatment with PMA for 6 h and IL-4 for 18 h (a
total of 24 h). The cells mentioned above were incubated at 37°C in
a humidified 5% CO2 atmosphere. Subsequent experiments
were performed in >24 h after the transfection.
Western blot assay
Cells were washed with phosphate-buffered saline
(PBS) and then lysed on ice with RIPA buffer [50 mM Tris, 150 mM
NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 2 mM EDTA (pH
7.5)] containing 1% cocktail and 1% phosphatase inhibitor. Protein
concentration was determined by the Bradford protein assay using
bovine serum albumin (BSA) as a standard. Equal amounts of proteins
(30 µg/lane) were separated by SDS-PAGE with 10 or 12%
polyacrylamide gels. Proteins separated in the gels were
transferred to PVDF membranes and then blocked in 5% BSA at room
temperature for 90 min. After blocking, the membranes were washed
three times with Tris-buffered saline + 0.1% Tween-20, incubated
overnight at 4°C with the relevant primary antibodies, washed again
and incubated with goat anti-rabbit IgG (1:8,000) or goat
anti-mouse IgG conjugated with HRP (1:8,000) for 1 h at room
temperature. The dilution of each primary antibody was as follows:
E-cadherin, Snail, Vimentin, VEGF-A, IL-6 and IL-8 were 1:600;
GAPDH and Lamin B were 1:1,000; ERK(1/2), p-ERK(1/2), JNK and p-JNK
are 1:500; IκBα, p-IκBα, IKKα and p-IKKα were 1:1000; P50 was
1:800. Labeled proteins were visualized and measured using
chemiluminescence (eECL Western Blot Kit, Beijing ComWin Biotech
Co., Ltd., Beijing, China).
Co-culture of TAMs (M2 macrophages)
with CRC cells
M2 macrophages (a total of 5×105) were
transformed and seeded onto 6-well plates, with HT29-NC, HT29-P,
LoVo-P and LoVo-NC cells (2×106 each) co-cultured in
upper Transwell inserts for 48 h. The ratio of M2 cells to tumor
cells was 1:4. After co-culturing for 24 h, the cells were washed,
and the cell culture supernatant was collected for further
experiments. Flow cytometry was used to confirm M2 cells by
identifying CD68 and CD206 as markers.
Cell invasion, migration and
wound-healing assays
Transwell inserts were applied to detect cell
invasion. LoVo-P, LoVo-NC, HT29-NC or HT29-P cells
(8×104 each) were diluted in 0.2 ml of serum-free
RPMI-1640 medium and added to the upper chamber that had 10%
Matrigel on the surface, with M2 macrophages (5×105)
seeded in the lower chamber. Before combining the chambers, JNK/ERK
inhibitors, anti-IL-6 or anti-IL-8 were added to the M2 macrophage
culture for 2 h; then, the medium was replaced with RPMI-1640
medium. The cells were incubated at 37°C in a humidified atmosphere
containing 5% CO2. After 24 h, invasive CRC cells
located on the lower side of the chamber were fixed with 4%
paraformaldehyde for 25 min at room temperature, stained with 0.1%
crystal violet in methanol for 15 min at room temperature, air
dried, screened and counted under a microscope. Similarly, LoVo-P
and LoVo-NC (1×106) were seeded in 6-well plates and
cultured overnight. The cells were divided into three groups. Two
of the groups were exposed to inhibitors of p-JNK (SP600125; 2.5
and 5 nM) and p-ERK (SCH772984; 4 and 8 nM) for 2 h, while the
third served as the negative control. Cell migration was measured
by determining the movement of cells into a scraped area created by
the tip of a 200-µl pipette. The degree of ‘wound closure’ was
examined after 24 and 48 h. After cell adherence, the remaining gap
was then assessed using light microscopy and quantified using
ImageJ (National Institutes of Health, Bethesda, MA, USA; ImageJ
bundled with 64-bit Java 1.8.0_112).
ELISA
IL-6, IL-8 and vascular endothelial growth factor-A
(VEGF-A) were assayed in the culture supernatants from the tested
cells using a Quantikine Kit (cat. no. CSB-E04638h for IL-6; cat.
no. CSB-E04641h for IL-8; CUSABIO Technology LLC, Houston, TX, USA)
according to the manufacturer's protocol. The specific cells and
the various conditions applied to these cells are listed in the
Results.
Immunohistochemistry (IHC) assay
IHC was performed to detect p-JNK, and p-ERK using
an Ultra-Sensitive SP IHC Kit (cat. no. KIT-9710; Fuzhou Maixin
Biotech Co., Ltd., Fuzhou, China) according to the manufacturer's
instructions. Normal IgG was used as a negative control. Images
were captured with a Zeiss laser-scanning microscope (Carl Zeiss
AG, Oberkochen, Germany) with a core data acquisition system (NIS
elements 4.0; Nikon Corporation, Tokyo, Japan).
Human angiogenesis array analysis
The RayBio Human Angiogenesis Array G1000
(RayBiotech Life, Norcross, GA, USA), which consists of 43
different human angiogenesis-associated antibodies spotted on a
membrane, was used in the present study according to the
manufacturer's instructions. The membranes were blocked in blocking
buffer at 37°C for 30 min and incubated with protein samples at
37°C for 1 h. After washing, the membranes were incubated with
diluted biotin-conjugated antibodies at 37°C for 2 h. After the
membranes were washed again, HRP-conjugated streptavidin (1:1,000
dilution) was added and incubated for 2 h. Membranes were then
washed thoroughly and exposed to detection buffer in the dark. By
comparing the signal intensities, relative expression levels of
proteins were determined. Signal intensities were quantified by
densitometry (ImageJ; National Institutes of Health; ImageJ bundled
with 64-bit Java 1.8.0_112).
Mouse tumor model
Athymic nude mice (BALB/c nude, 6-week-old males)
were purchased from the Laboratory Animal Science Center of
Guangdong Province (Guangzhou, China) and maintained under defined
conditions (12-h light cycle in a room maintained at 21±1°C and
50±10% humidity) at the Animal Experiment Center of Sun Yat-Sen
University. Food was provided at the same time each day with ad
libitum access to water in the cage. All animal protocols were
approved by the Institutional Animal Care and Use Committee and
Welfare Committee of Sun Yat-Sen University (Guangzhou, China).
These mice were divided into four groups, with six mice randomly
chosen for each group. Mice in each group were injected with
LoVo-NC, LoVo-P, HT29-NC or HT29-P cells at 5×106 cells
each into the subcutaneous tissue of the left flank. After
injection, the mice were maintained in pathogen-free environments.
All mice were sacrificed on day 30, and the xenografted tumors were
excised from the animals for further study, including IsHC assays.
The formula we used to calculate the volume of xenograft was as
follows: Volume = [major axis × (minor axis)2]/2.
Statistical analysis
Statistical analysis was performed using SPSS 22.0
(IBM Corp., Armonk, NY, USA). All data obtained from each
experiment are presented as the mean ± standard deviation of three
separate experiments. A post hoc test (Bonferroni) was used
following one-way analysis of variance (ANOVA) for statistical
analysis. The differences between two groups and among three or
more groups were determined using Student t-tests and one-way
ANOVAs, respectively. All experiments were performed independently.
P<0.05 was considered to indicate a statistically significant
difference.
Results
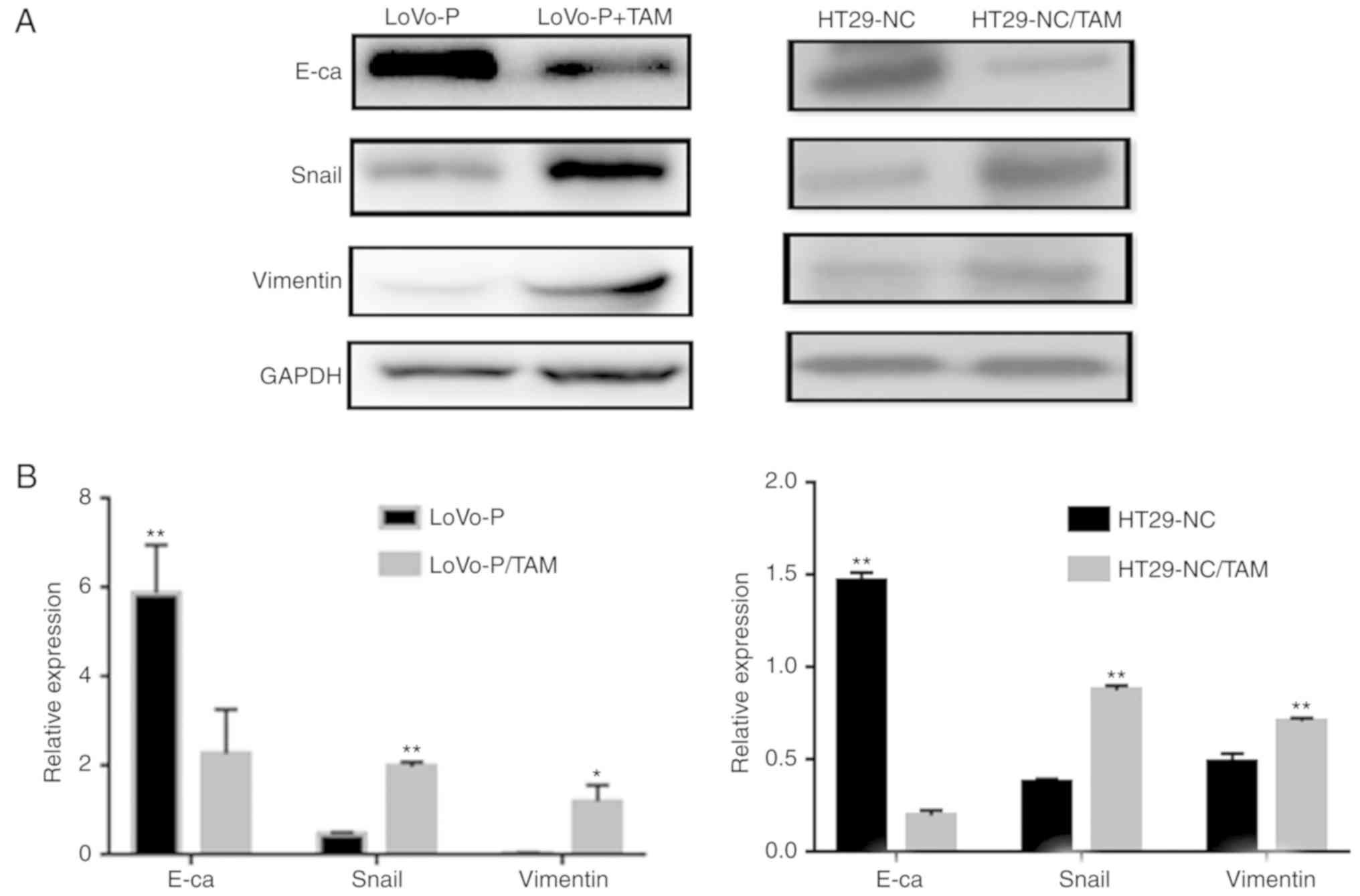
Co-culture of TAMs and CRC cells with
high PRL-3 expression promotes EMT
EMT is believed to have a critical role in cancer
metastasis, during which cancer cells tend to become a more
invasive and develop a metastatic phenotype. In addition, the
extent of EMT can be characterized by detecting several proteins,
including E-cadherin, Snail and Vimentin, via western blot
analysis. To explore the effect of co-culturing, LoVo-P or HT29
cells, both with high PRL-3 expression levels, and TAMs were used
in a co-culture system. After 24 h of co-culture, EMT markers in
CRC cells, including E-cadherin, Snail and Vimentin expression in
LoVo-P and HT29 cells, were significantly altered (Fig. 1). Co-culturing CRC cells and TAMs
downregulated the expression of E-cadherin, and upregulated the
expression of Snail and Vimentin, which suggested that CRC cells
acquired a mesenchymal phenotype when co-cultured with TAMs.
PRL-3-induced activation of IL-6 and
IL-8 is based on the MAPK pathway in TAMs
Our previous study suggested that PRL-3 promoted CRC
cell invasion by initiating signaling pathways in TAMs (4). To explore the molecular mechanism
underlying PRL-3-induced IL-6 and IL-8 production, western blot
assays were performed to elucidate the phosphorylation status of
proteins that may be involved after the co-culture of CRC cells
(LoVo-P, LoVo-NC, HT29-NC and HT29-P) and TAMs, such as the
phosphorylated forms of JNK and ERK. PRL-3 induced the
phosphorylation of JNK and ERK in TAMs. MAPK pathway activation was
suppressed after downregulating PRL-3 (Fig. 2A). Additionally, IL-6 and IL-8
levels were changed by silencing/overexpression of PRL-3
levels.
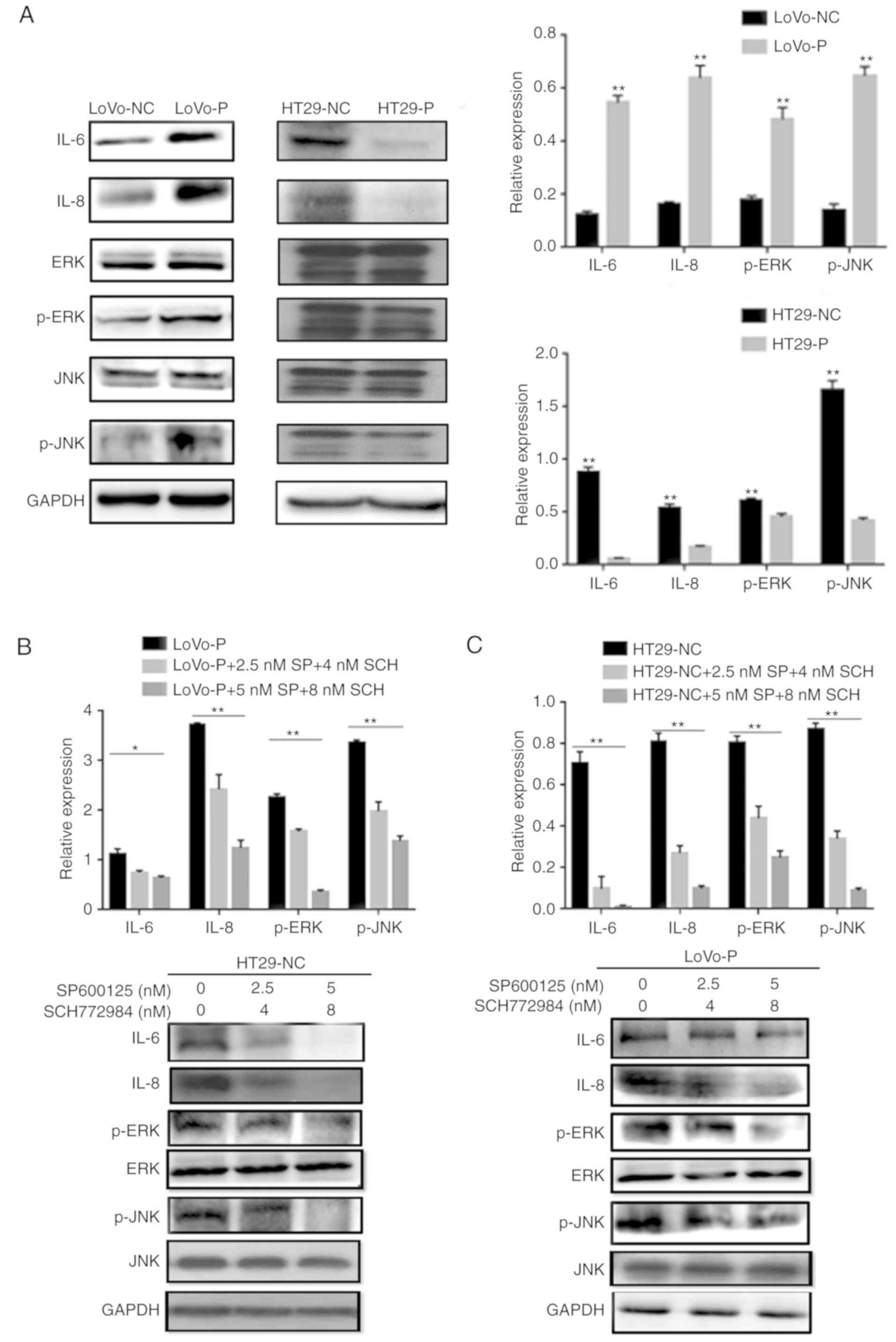 | Figure 2.PRL-3-induced activation of IL-6 and
IL-8 by initiating JNK and ERK pathways in TAMs. (A) After
co-culture with colorectal cancer cells, IL-6, IL-8, p-JNK, and
p-ERK levels in TAMs were examined using western blot assays. In
(B) LoVo-P and (C) HT29-NC cells, IL-6 and IL-8 expression was
detected after the addition of an inhibitor to p-JNK (SP600125; 2.5
and 5 nM) or p-ERK (SCH772984; 4 and 8 nM) into the co-culture
system with LoVo-P cells. *P<0.05, **P<0.01. PRL-3,
phosphatase of regenerating liver-3; NC, negative control; LoVo-P,
LoVo PRL-3 overexpression; HT29-P, HT29 PRL-3 knockdown; IL,
interleukin; ERK, extracellular signal-regulated kinase; p,
phosphor; JNK, c-Jun N-terminal kinase; SP, SP600125; SCH,
SCH772984. |
To examine whether the MAPK/JNK pathway or the
MAPK/ERK pathway regulated IL-6 and IL-8 production, inhibitors of
p-JNK (SP600125, 2.5 and 5 nM) and p-ERK (SCH772984, 4 and 8 nM)
were added to the TAM cultures prior to co-culture with CRC cells.
The western blot analysis illustrated that both p-JNK and p-ERK
were suppressed as the inhibitor concentration increased (Fig. 2B and C). Similarly, IL-6 and IL-8
levels declined significantly when the MAPK signaling pathways were
suppressed. Additionally, ELISAs were performed to identify the
extracellular levels of IL-6 and IL-8, which confirmed the results
of the western blot assays. The results showed that the
PRL-3-induced IL-6 and IL-8 expression was abolished by the
addition of JNK and ERK inhibitors, resulting in the downregulation
of IL-6 and IL-8 (Fig. 3A and B).
Additionally, cell invasion assays were performed to examine
whether invasiveness was regulated by MAPK pathways. The invasive
and metastatic capacities of HT29-NC and LoVo-P cells were reduced
by blocking JNK and ERK signaling. As the concentrations of
inhibitors increased, the number of cells invading through the
membrane down from the upper chamber gradually declined (Fig. 3C and D).
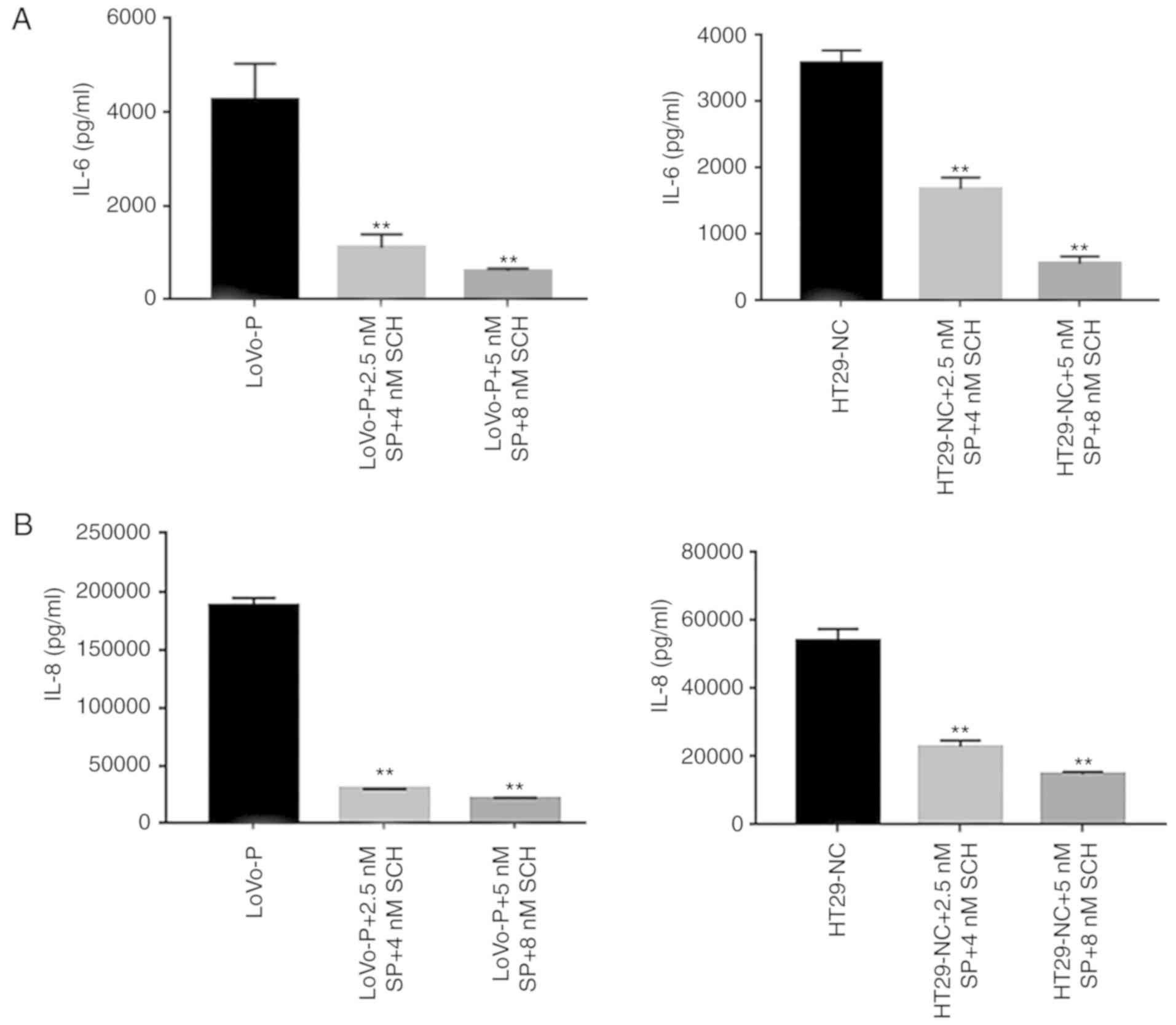 | Figure 3.MAPK pathway regulates invasion of
CRCs. (A) IL-6 and (B) IL-8 expression in supernatants from
co-culture systems measured by ELISA. (C) Blocking ERK and JNK
decreases the invasiveness of colorectal cancer cells. (D) A
wound-healing assay showed the same trend. *P<0.05, **P<0.01
vs. respective control. PRL-3, phosphatase of regenerating liver-3;
ERK, extracellular signal-regulated kinase; p, phosphor; JNK, c-Jun
N-terminal kinase; LoVo-P, LoVo PRL-3 overexpression; NC, negative
control; SP, SP600125; SCH, SCH772984; IL, interleukin. |
PRL-3 and activated MAPK pathways in
TAMs
Our previous study demonstrated that PRL-3 in CRC
cells upregulated the expression of CCL26, which induced TAM
infiltration, enhancing the invasiveness of CRC cells (4). As it was found that PRL-3 was involved
in the initiation of MAPK pathways, the potential association
between CCL26 and MAPK signaling was examined. CCL26 was added to
the lower chamber of the co-culture system with CRC cells (HT29-P,
PRL-3 shRNA knockdown) and TAMs to detect the phosphorylation of
JNK and ERK, and the expression of IL-6 and IL-8 in TAMs via
western blot analysis. ELISAs were used to identify the
extracellular levels of IL-6 and IL-8 after collecting the cell
culture supernatant. In addition, cell invasion assays were
performed to clarify whether CCL26 increased the invasiveness of
HT29-P cells. The analysis showed that JNK and ERK signaling in
TAMs were both triggered by the addition of CCL26 (10 and 100
ng/ml) even when PRL-3 expression in HT29 cells was suppressed
(Fig. 4A and B). Furthermore,
analysis of the lower Transwell chamber containing CCL26 (100
ng/ml) showed that the migratory activity of HT29-P cells was more
pronounced than that of those without CCL26 after 24 h in
co-culture (Fig. 4C and D).
Meanwhile, the levels of IL-6 and IL-8 in co-cultured TAMs, both
intracellular and extracellular, were upregulated compared with
those in cells not treated with CCL26 (Fig. 4E and F).
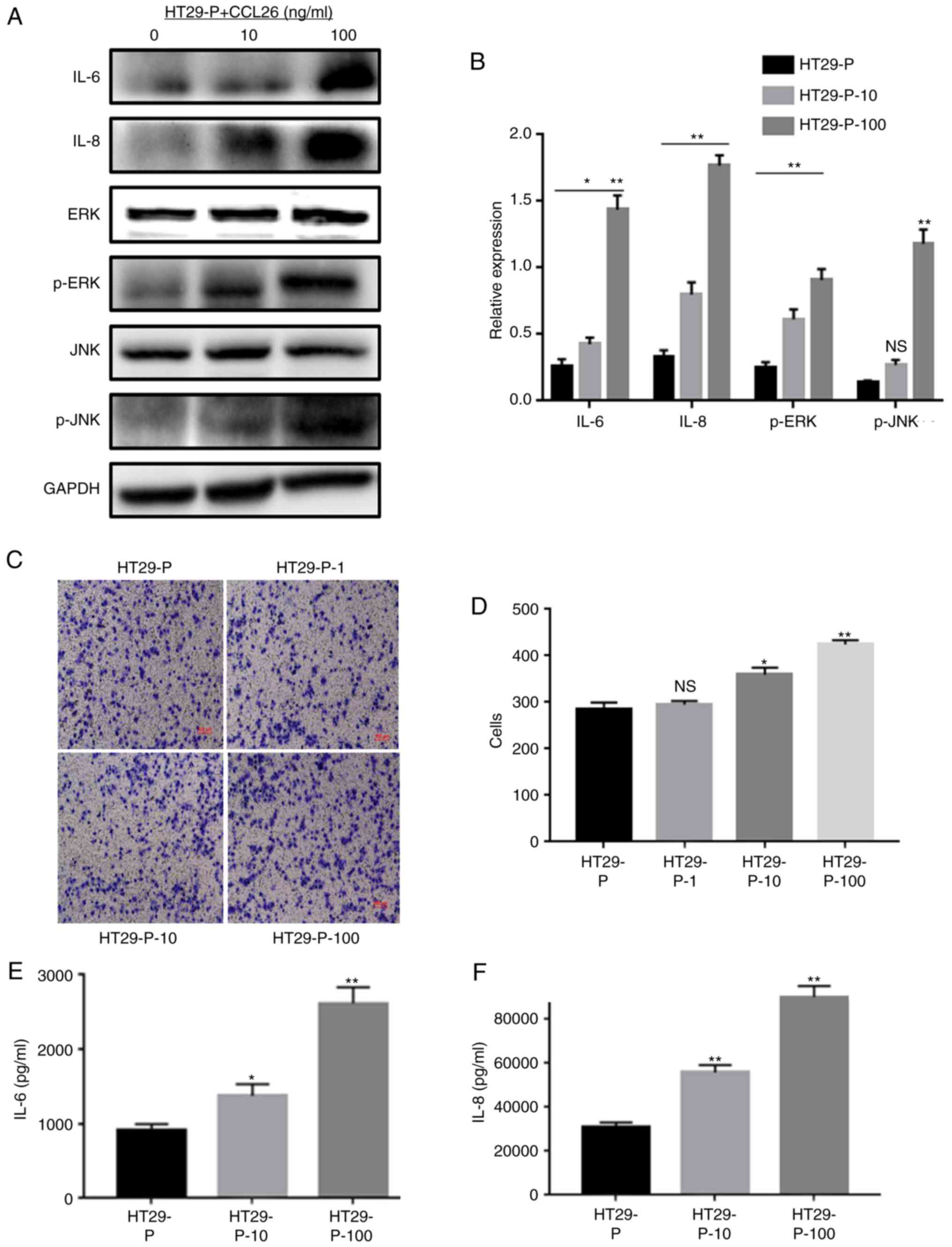 | Figure 4.CCL26 increases the expression of IL-6
and IL-8 and the activation of p-JNK and p-ERK in HT29-P cells,
resulting in an increase in the invasiveness of CRC cells. (A)
Western blot assays and (B) densitometry analysis were performed to
measure the expression of IL-6, IL-8, p-JNK, and p-ERK. (C and D)
Invasion and migration were analyzed in Matrigel invasion assays.
(E) IL-6 and (F) IL-8 were determined by ELISA. *P<0.05,
**P<0.01, NS, not significant. PRL-3, phosphatase of
regenerating liver-3; HT29-P, HT29 PRL-3 knockdown; CCL26,
chemokine ligand 26; IL, interleukin; ERK, extracellular
signal-regulated kinase; p, phosphor; JNK, c-Jun N-terminal kinase;
10, 10 ng/ml CCL26; 100, 100 ng/ml CCL26; 1, 1 ng/ml CCL26. |
IL-6 and IL-8 induced EMT in CRC
cells
To further determine the role of IL-6 and IL-8 in
EMT in CRC, the protein levels of E-cadherin, Snail and Vimentin
were analyzed in LoVo-NC, LoVo-P, HT29-NC and HT29-P cells after 24
h of co-culture with TAMs. LoVo-P and HT29-NC cells expressed
higher levels of IL-6 and IL-8 than LoVo-NC and HT29-P cells,
respectively (Fig. 2A). And after
adding inhibitors of JNK (SP600125, 2.5 and 5 nM) and ERK
(SCH772984, 4 and 8 nM) into co-culture system, E-cadherin was
increased, and Snail and Vimentin were reduced as the concentration
of inhibitors increased (Fig.
5A).
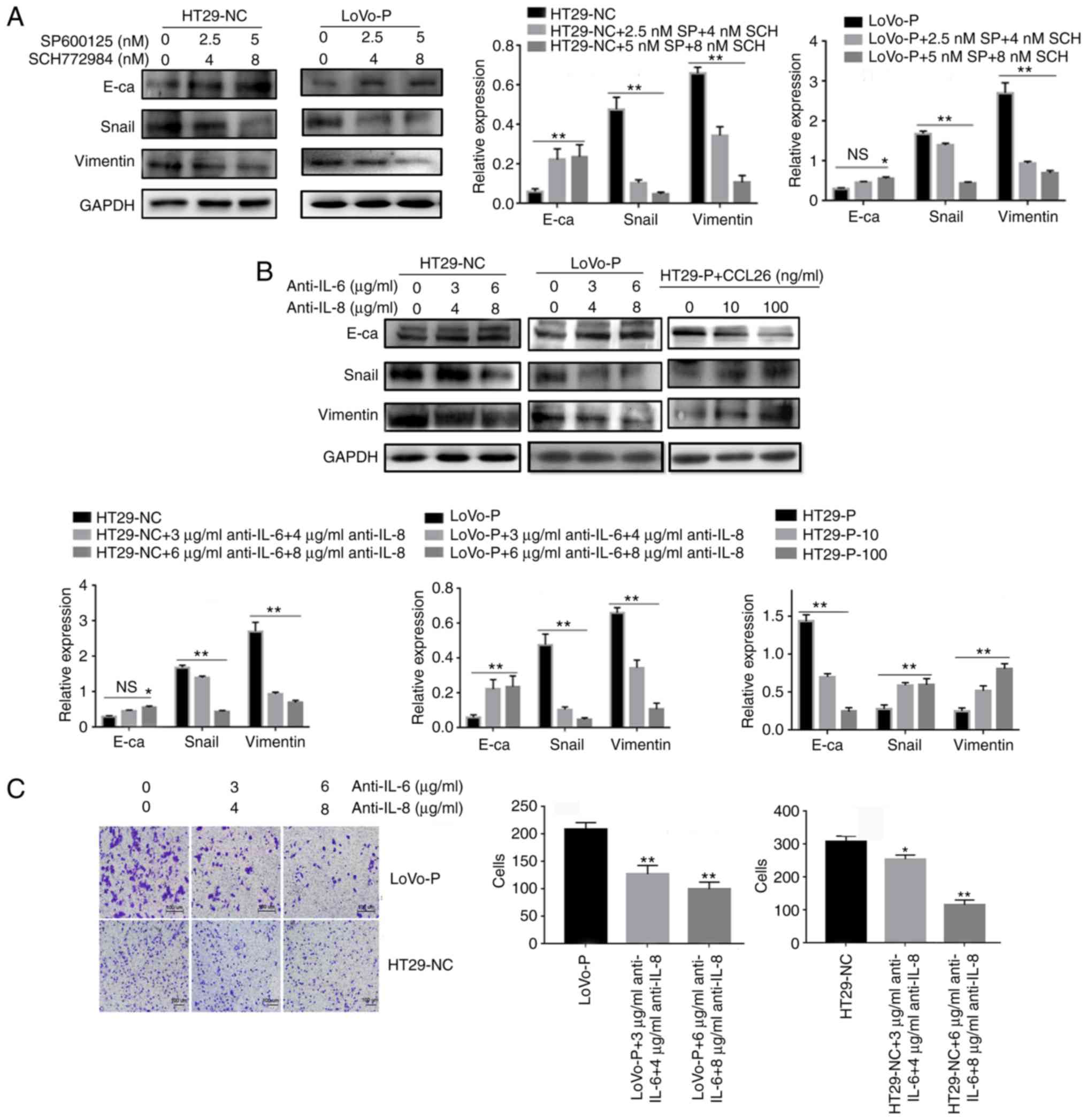 | Figure 5.IL-6 and IL-8 induced
epithelial-mesenchymal transition in CRC cells, which was regulated
by MAPK and PRL-3. (A) After neutralizing the MAPK pathways in
TAMs, the levels of E-cadherin, Snail and Vimentin in colorectal
cancer cells were examined using western blot assays. (B) IL-6 (3
and 6 µg/ml) and IL-8 (4 and 8 µg/ml) antibodies, and the addition
of CCL26, were used and the levels of E-cadherin, Snail and
Vimentin in CRC cells were examined using western blot assays. (C)
Invasion was evaluated by Matrigel invasion assays after blocking
IL-6 (3 and 6 µg/ml) and IL-8 (4 and 8 µg/ml) *P<0.05,
**P<0.01 vs. appropriate controls, NS, not significant. PRL-3,
phosphatase of regenerating liver-3; MAPK, mitogen-activated
protein kinase; NC, negative control; LoVo-P, LoVo PRL-3
overexpression; HT29-P, HT29 PRL-3 knockdown; E-ca, E-cadherin; SP,
SP600125; SCH, SCH772984; IL, interleukin; CCL26, chemokine ligand
26. |
Additionally, antibodies against IL-6 and IL-8 were
added to the co-culture system, and protein levels in the HT29-NC
and LoVo-P cells, in the upper chamber, were detected by western
blot analysis. The neutralizing antibodies increased the expression
of E-cadherin and reduced the expression of Snail and Vimentin in
CRC cells (Fig. 5B). Transwell
analysis also indicated that CRC cells treated with neutralizing
antibodies targeting IL-6 and IL-8 tended to have fewer cells that
migrated successfully (Fig.
5C).
PRL-3 promotes angiogenesis by
activating the nuclear factor-κB (NF-κB) pathway
After co-culture with TAMs, angiogenesis-associated
proteins in the supernatants of LoVo-P and LoVo-NC cells were
evaluated using a Human Angiogenesis Array to identify the
potential effects. Protein levels of neurotrophin-4, VEGF, VEGF
receptor 2 and VEGF-D in LoVo-P cells were at least 2-fold higher
than in LoVo-NC cells (Fig. 6A).
Then, VEGF-A, which is the major factor in blood vessel formation,
was chosen for further examination. RT-qPCR results indicated that
VEGF-A expression in LoVo-P cells was upregulated significantly in
3 days of co-culture, while VEGF-A expression in LoVo-NC cells
showed no significant differences over the whole experimental
period. In addition, supernatants were examined via ELISA assays
and found that VEGF-A expression in LoVo-P cells increased to
~2,000 ng/ml after 3 days of co-culture, while in LoVo-NC cells and
TAMs, the levels remained steady (Fig.
6B).
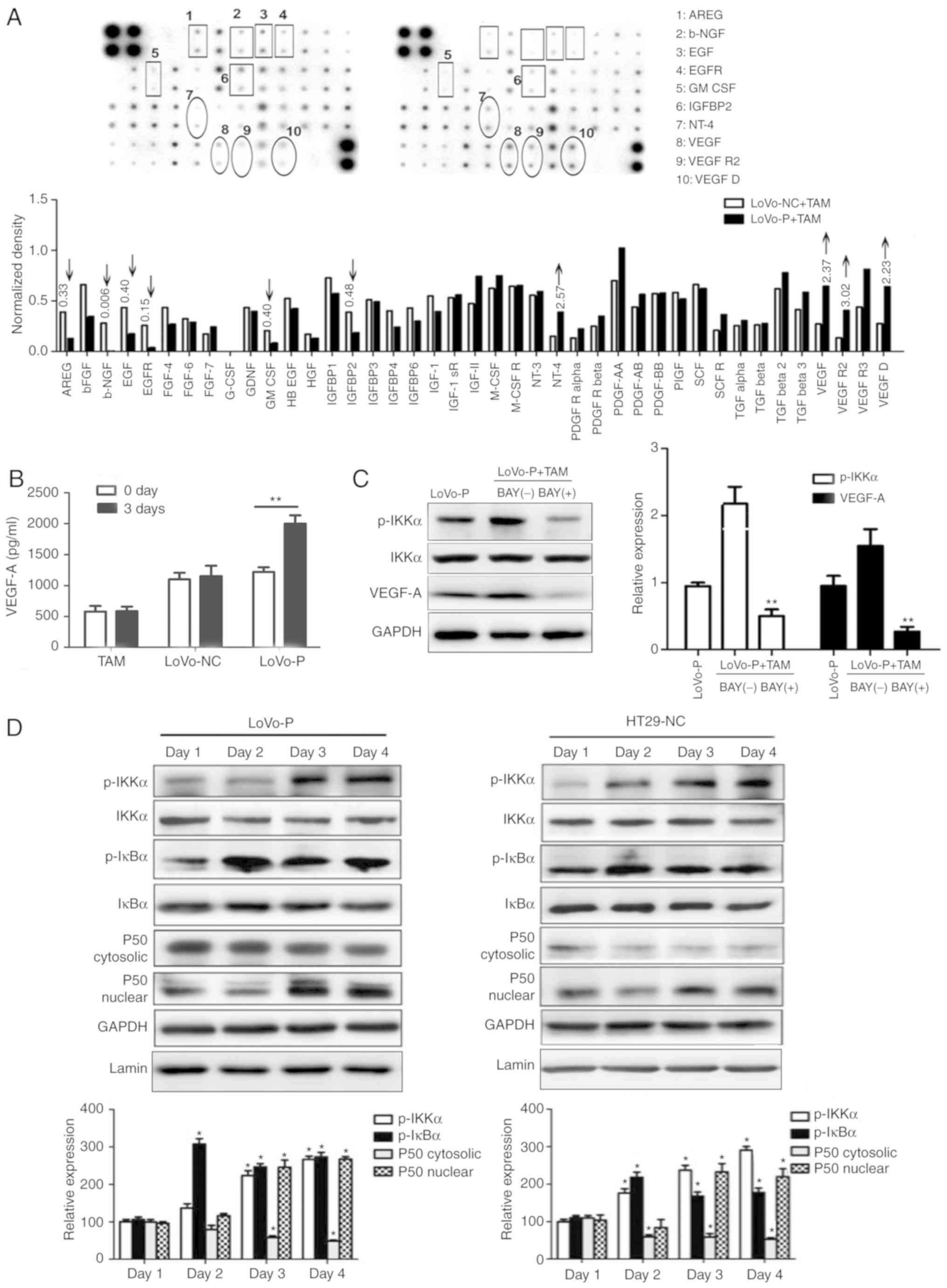 | Figure 6.PRL-3 promotes angiogenesis by
activating the NF-κB pathway. (A) Angiogenesis-associated proteins
in supernatants from LoVo-P and LoVo-NC cells. (B) ELISAs were used
to detect VEGF-A in LoVo-P and LoVo-NC cells after co-culture with
TAMs. **P<0.01. (C) After suppressing the NF-κB pathway, VEGF-A
expression in LoVo-P cells was determined. **P<0.01 vs. BAY(−).
(D) NF-κB pathway activation was examined by western blot analysis.
*P<0.05. PRL-3, phosphatase of regenerating liver-3; VEGF,
vascular endothelial growth factor; TAM, tumor-associated
macrophage; NC, negative control; LoVo-P, LoVo PRL-3
overexpression; p, phosphor; IKKα, nuclear factor κ-B kinase
subunit α; AREG, amphiregulin; b-NGF, β nerve growth factor; EGF,
epidermal growth factor; EGFR, epidermal growth factor receptor;
GM-CSF, granulocyte-macrophage colony-stimulating factor; IGFBP2,
insulin-like growth factor-binding protein 2; NT-4, neurotrophin-4;
VEGFR2, vascular endothelial growth factor receptor 2; IκBα, NF-κB
inhibitor α. |
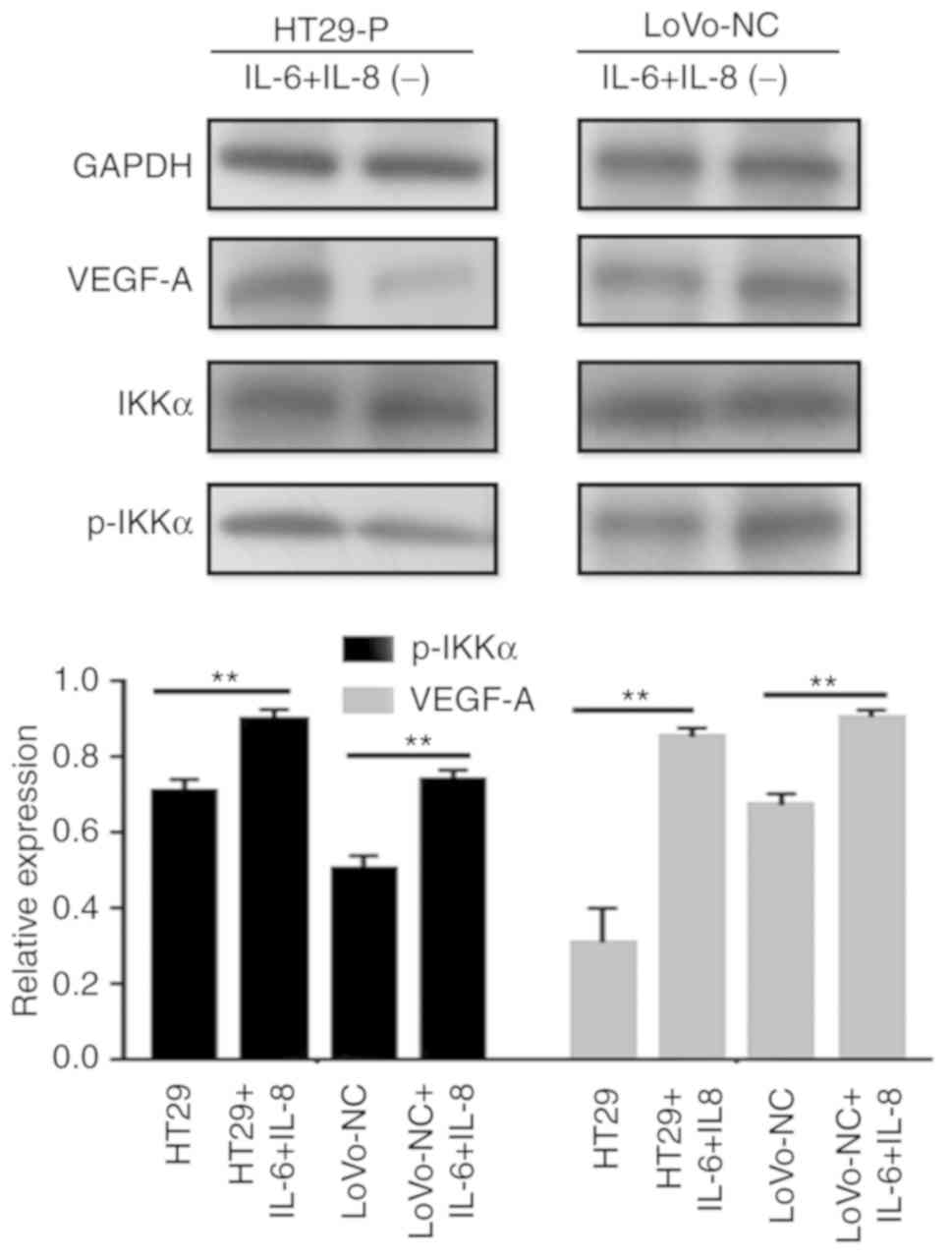
NF-κB is a key regulator of innate
immunity and inflammation
Jedinak et al (15) suggested that NF-κB might be
associated with the expression of VEGF. Therefore, it whether the
NF-κB pathway was activated in CRC cells was explored by performing
western blot analysis (Fig. 6C and
D). The levels of the phosphorylated forms of IκBα and IKKα
increased, and nuclear P50 showed a similar trend during to
co-culture. To determine the association between NF-κB and VEGF-A,
an inhibitor of NF-κB (BAY 11–7082) was added to the co-culture
system. Consequently, p-IKKα levels were reduced by BAY 11–7082, as
was VEGF-A. Additionally, to explore the potential mechanism
between the MAPK and NF-κB pathways, IL-6 (200 ng/ml) and IL-8 (10
ng/ml) was added to LoVo-NC and HT29-P co-culture with TAMs to
detect NF-κB pathway activation. The result showed that IL-6 and
IL-8 activated the NF-κB pathway (Fig.
7).
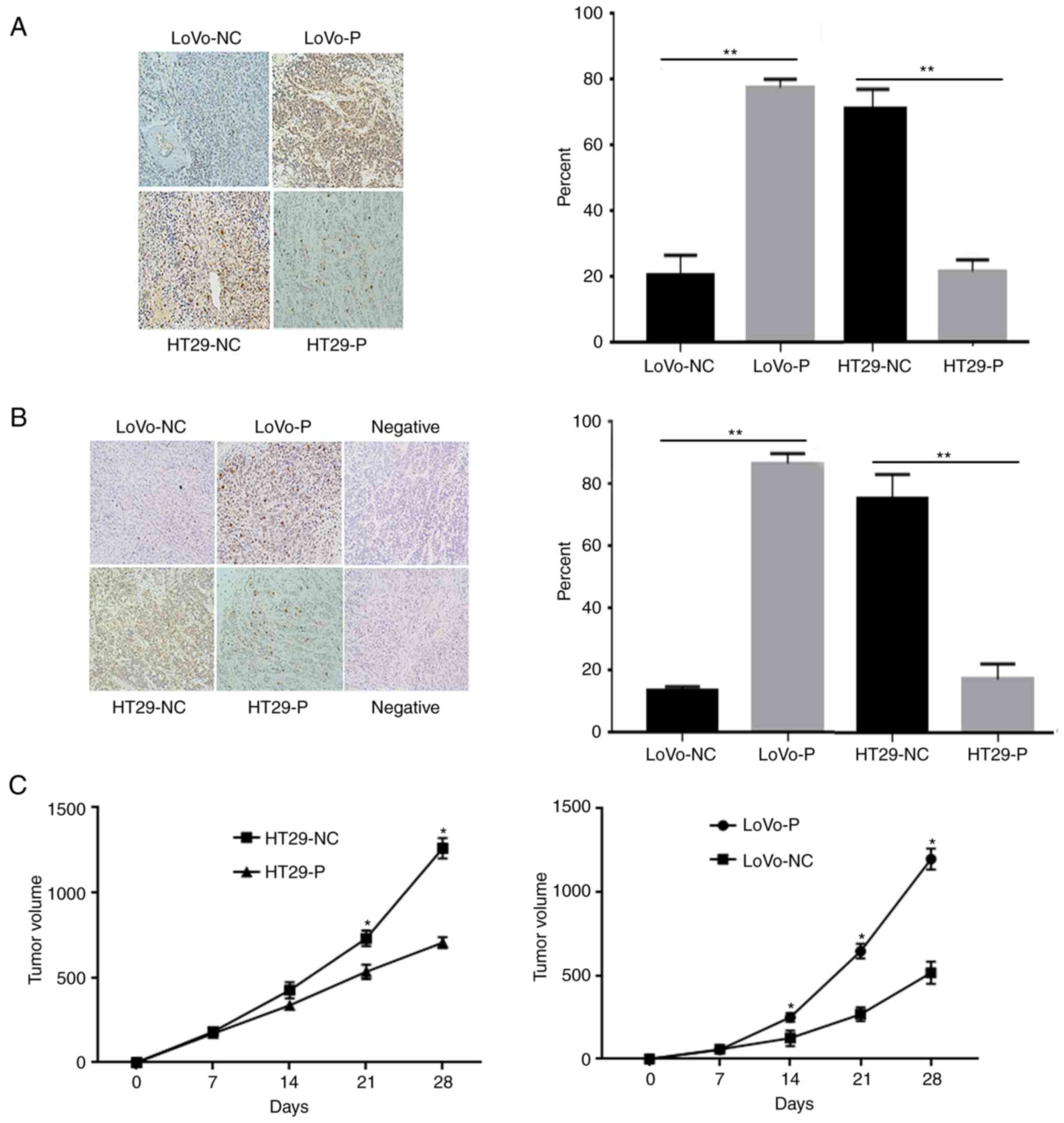
JNK and ERK activation in CRC
xenografts
To investigate the effect of TAM infiltration
induced by PRL-3 on tumor growth and angiogenesis in vivo,
mice were inoculated with equal numbers of LoVo-P, LoVo-NC, HT29-NC
and HT29-P cells into the subcutaneous tissue of the left flank of
nude mice (Fig. 8). After 30 days,
the animals were sacrificed, and the tumor xenografts were excised
for further experiments. IHC analyses of were performed using these
xenografts to compare the level of p-JNK and p-ERK (Fig. 8A and B). The results showed that the
proteins mentioned above were obviously higher in xenografts from
LoVo-P and HT29-NC cells than those from LoVo-NC and HT29-P cells,
respectively, which suggested that MAPK activation induced by PRL-3
was associated with tumor growth (Fig.
8C). Tumors were larger in groups with high levels in PRL-3
expression from cells (LoVo-P, HT29-NC) than in cells with low
PRL-3 (LoVo-NC, HT29-P; Fig. 8C).
TAMs were present in the tumor xenografts and showed no obvious
difference between groups (Fig.
S2).
Discussion
Increasing numbers of studies have emphasized the
interaction of the tumor microenvironment and tumor cells in the
promotion of tumor progression, angiogenesis and invasion (11,14–17).
In addition, TAMs, one of the most investigated cancer-associated
factors, have been demonstrated to be associated with cancer
prognosis (15–17). Various inflammatory cells and
cytokines associated with this process have been investigated
(10). Our previous studies found
that TAM infiltration plays a critical role in CRC progression, and
IL-6 and IL-8 secretion is upregulated when PRL-3 is overexpressed
in cancer cells (4,14). Additionally, previous studies have
suggested that PRL-3 might be a crucial effector in the MAPK
cascade (18). In the present
study, the phosphorylation of JNK and ERK in TAMs was increased
after co-culture with HT29-NC and LoVo-P cells, both of which
overexpressed PRL-3. Additionally, the activation of JNK and ERK
was associated with high levels of IL-6 and IL-8. Notably, the two
MAPK pathways were blocked with specific inhibitors, IL-6 and IL-8
expression was notably reduced. These results illustrated that
PRL-3 activated JNK and ERK signaling pathways in TAMs, resulting
in the secretion of IL-6 and IL-8.
In CRC, EMT is associated with an invasive and
metastatic phenotype (19). Rokavec
et al (12) demonstrated
that the activation of the IL-6 receptor/STAT3/microRNA-34a loop by
IL-6 induces EMT, which promotes invasion and a metastatic cascade.
In our experiments, the upregulation of IL-6 and IL-8 was
consistent with the downregulation of E-cadherin and the
upregulation of Snail and Vimentin. In addition, invasion assays
demonstrated that IL-6 and IL-8 are indicators of the number of
migrating cells. An opposite trend was produced when JNK and ERK
were inhibited, according to the western blot analysis and invasion
assay. Furthermore, the migratory ability of CRC cells declined
when IL-6 and IL-8 were neutralized by antibodies. All of these
results suggested that PRL-3 promoted the invasion of CRC cells by
initiating MAPK pathways in TAMs and activating EMT. Our previous
experiments found that PRL-3 promoted CCL26 expression in CRC
cells, which induced TAM infiltration to establish the tumor
microenvironment (4). CCL26 was
added to HT29-P cell cultures to determine the potential effects.
Even though PRL-3 expression was reduced, the addition of CCL26
still activated JNK and ERK pathways, contributing to EMT.
Therefore, CCL26 might be the key factor connecting TAMs and the
events that trigger a series of processes associated with tumor
progression.
PRL-3 has been reported as a biomarker of an
increased risk of metastasis and poor prognosis (20). For example, Sahai and Marshall
(21) found that PRL-3 can
stimulate Rho family GTPase signaling pathways to promote cell
motility and invasion. Furthermore, PRL-3 has been suggested to
regulate several pathways, such as the PI3K/AKT (22), Src (7) and ERK (6) pathways. In the current experiments,
high levels of PRL-3 were with larger xenografts in nude mice.
Additionally, p-JNK, p-ERK and VEGF-A expression in cells
overexpressing PRL-3 was also upregulated. These results illustrate
that PRL-3 has an important role in tumor progression.
The growth of solid tumors is dependent on
angiogenesis, which provides the blood supply needed for
metastasizing cells to flourish. Various studies have explored the
relationship between PRL-3 and angiogenesis. For instance, Guo
et al (8) suggested that
PRL-3 may be involved in triggering angiogenesis and establishing
the tumor microvasculature. Zimmerman et al (9) found that PRL-3 is involved in VEGF
signaling and blood vessel formation in vitro and in
vivo. In our research, a Human Angiogenesis Array was used and
found that VEGF expression was significantly increased in LoVo-P
cells compared with that in LoVo-NC cells in co-culture with TAMs.
Additionally, a prolonged co-culture resulted in a marked
upregulation of VEGF-A expression in LoVo-P cells, while expression
in LoVo-NC cells showed no notable difference. Previous research
demonstrated that cytokines secreted by TAMs might induce NF-κB
activation in CRC cells and VEGF expression. (15). NF-κB is a key regulator of innate
immunity and inflammation; the results are consistent with this
function. Both in the LoVo and HT29 cell lines, NF-κB activation
was enhanced with an increase in co-culturing time, indicating the
importance of PRL-3 and the tumor microenvironment in the process
of angiogenesis. Furthermore, the addition of IL-6 and IL-8
activated the NF-κB pathway in CRC, which indicated that MAPK
signaling is involved in the activation of NF-κB (Fig. 9). There are still some experiments
required to fully illustrate the mechanisms of angiogenesis, such
as IHC to identify microvessels in the mice xenograft tissues,
which would confirm the promotion of angiogenesis in
vivo.
In summary, PRL-3 promotes CRC cell invasion and
metastasis by activating MAPK pathways in TAMs to initiate EMT, and
PRL-3 may promote angiogenesis by activating the NF-κB pathway in
CRC cells.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (no.81871981), the National
Natural Science Foundation of Guangdong Province (nos.
2016A030313353 and 2016A030310183), the National Natural Science
Foundation of China (nos. 81602539, 81702902 and 81602125) and the
Science and Technology Project of Guangdong Province (no.
2015A050502021).
Availability of data and materials
The datasets used and analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
TZ and LL acquired the data after carrying out the
experiments, performed the statistical analysis of all experimental
results, and created a draft of the manuscript; YZ, HX and PS
prepared the experimental materials; WL, QL and ZC revised and
approved the final version of the manuscript and were also involved
in the conception of the study. All authors read and approved the
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
The protocol of the present study was approved by
the Institutional Research Ethics Committee of the Sun Yat-Sen
Memorial Hospital of Sun Yat-Sen University. All animal protocols
were approved by the Institutional Animal Care and Use Committee
and Welfare Committee of Sun Yat-Sen University (Guangzhou,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sheng N, Yan L, Wu K, You W, Gong J, Hu L,
Tan G, Chen H and Wang Z: TRIP13 promotes tumor growth and is
associated with poor prognosis in colorectal cancer. Cell Death
Dis. 9:4022018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vu T and Datta PK: Regulation of EMT in
colorectal cancer: A culprit in metastasis. Cancers. 9:E1712017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lan Q, Lai W, Zeng Y, Liu L, Li S, Jin S,
Zhang Y, Luo X, Xu H, Lin X, et al: CCL26 participates in the
PRL-3-induced promotion of colorectal cancer invasion by
stimulating tumor-associated macrophage infiltration. Mol Cancer
Ther. 17:276–289. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xu H, Zhang Y, Peña MM, Pirisi L and Creek
KE: Six1 promotes colorectal cancer growth and metastasis by
stimulating angiogenesis and recruiting tumor-associated
macrophages. Carcinogenesis. 38:281–292. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ming J, Liu N, Gu Y, Qiu X and Wang EH:
PRL-3 facilitates angiogenesis and metastasis by increasing ERK
phosphorylation and up-regulating the levels and activities of
Rho-A/C in lung cancer. Pathology. 41:118–126. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liang F, Liang J, Wang WQ, Sun JP, Udho E
and Zhang ZY: PRL3 promotes cell invasion and proliferation by
down-regulation of Csk leading to Src activation. J Biol Chem.
282:5413–5419. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guo K, Li J, Wang H, Osato M, Tang JP,
Quah SY, Gan BQ and Zeng Q: PRL-3 initiates tumor angiogenesis by
recruiting endothelial cells in vitro and in vivo. Cancer Res.
66:9625–9635. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zimmerman MW, McQueeney KE, Isenberg JS,
Pitt BR, Wasserloos KA, Homanics GE and Lazo JS: Protein-tyrosine
phosphatase 4A3 (PTP4A3) promotes vascular endothelial growth
factor signaling and enables endothelial cell motility. J Biol
Chem. 289:5904–5913. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mager LF, Wasmer MH, Rau TT and Krebs P:
Cytokine-induced modulation of colorectal cancer. Front Oncol.
6:962016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lin X, Yi Z, Diao J, Shao M, Zhao L, Cai
H, Fan Q, Yao X and Sun X: ShaoYao decoction ameliorates
colitis-associated colorectal cancer by downregulating
proinflammatory cytokines and promoting epithelial-mesenchymal
transition. J Transl Med. 12:1052014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rokavec M, Öner MG, Li H, Jackstadt R,
Jiang L, Lodygin D, Kaller M, Horst D, Ziegler PK, Schwitalla S, et
al: IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated
colorectal cancer invasion and metastasis. J Clin Invest.
125:13622015. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang L, Tang C, Cao H, Li K, Pang X, Zhong
L, Dang W, Tang H, Huang Y, Wei L, et al: Activation of IL-8 via
PI3K/Akt-dependent pathway is involved in leptin-mediated
epithelial-mesenchymal transition in human breast cancer cells.
Cancer Biol Ther. 16:1220–1230. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu H, Lai W, Zhang Y, Liu L, Luo X, Zeng
Y, Wu H, Lan Q and Chu Z: Tumor-associated macrophage-derived IL-6
and IL-8 enhance invasive activity of LoVo cells induced by PRL-3
in a KCNN4 channel-dependent manner. BMC Cancer. 14:3302014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jedinak A, Dudhgaonkar S and Sliva D:
Activated macrophages induce metastatic behavior of colon cancer
cells. Immunobiology. 215:242–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Erreni M, Mantovani A and Allavena P:
Tumor-associated macrophages (TAM) and inflammation in colorectal
cancer. Cancer Microenviron. 4:141–154. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li S, Xu F, Zhang J, Wang L, Zheng Y, Wu
X, Wang J, Huang Q and Lai M: Tumor-associated macrophages
remodeling EMT and predicting survival in colorectal carcinoma.
Oncoimmunology. 7:e13807652017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Peng L, Jin G, Wang L, Guo J, Meng L and
Shou C: Identification of integrin alpha1 as an interacting protein
of protein tyrosine phosphatase PRL-3. Biochem Biophys Res Commun.
342:179–183. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shi G, Zheng X, Zhang S, Wu X, Yu F, Wang
Y and Xing F: Kanglaite inhibits EMT caused by TNF-α via NF-κΒ
inhibition in colorectal cancer cells. Oncotarget. 9:6771–6779.
2017.PubMed/NCBI
|
|
20
|
Zhao WB, Li Y, Liu X, Zhang LY and Wang X:
Evaluation of PRL-3 expression, and its correlation with
angiogenesis and invasion in hepatocellular carcinoma. Int J Mol
Med. 22:187–192. 2008.PubMed/NCBI
|
|
21
|
Sahai E and Marshall CJ: RHO-GTPases and
cancer. Nat Rev Cancer. 2:133–142. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang H, Quah SY, Dong JM, Manser E, Tang
JP and Zeng Q: PRL-3 down-regulates PTEN expression and signals
through PI3K to promote epithelial-mesenchymal transition. Cancer
Res. 67:2922–2926. 2007. View Article : Google Scholar : PubMed/NCBI
|