Introduction
Endometrial cancer is one of the most common
gynecologic malignancies globally (1). Based on the current pathological
classification, endometrial cancer is primarily categorized into
two types: Type I, uterine endometroid carcinoma (UEC); and type
II, uterine serous carcinoma (USC). UECs, the majority of which are
moderately or well-differentiated, generally have a favorable
prognosis (2). However, according
to some reports in 2011 and 2012, USCs, which account for 5–10% of
all endometrial cancer types globally, are highly aggressive and
are associated with a poor outcome (2,3).
Compared with its counterpart UEC, USC is
hormone-receptor negative; thus, patients with USC rarely benefit
from hormonal treatment (2).
Additionally, the currently limited targeted therapies are
principally designed and indicated for UEC (3). Therefore, there is an urgent
requirement to identify potential targets and develop effective
treatment regimens for USCs (3).
Cyclin-dependent kinase inhibitor 2A (P16INK4A),
encoded by the P16INK4A gene (also known as CDKN2A),
has been reported to be a tumor suppressor, as it can inhibit
cyclin-dependent kinase 4/6 (CDK4/6) and cause cell cycle arrest
(4,5). Thus, deficiency of P16INK4A serves an
important role in the development of several types of cancer,
including breast and colorectal cancer (4,6).
However, a number of other studies indicate that P16INK4A is
frequently overexpressed in USC tissues and may serve as a protein
marker for the differential diagnosis of USC and UEC (7–9).
Previous studies demonstrated that P16INK4A is
required for cell proliferation and migration in cervical and
hepatic cancer (10–12). Additionally, P16INK4A overexpression
has been detected in certain types of cancer, including cervical
cancer (13). Although its
biological function in these tissue samples has yet to be fully
elucidated, researchers have reported that P16INK4A may serve a
crucial role in the survival of cervical cancer cells (11,12).
Furthermore, in another study, it has been reported that P16INK4A
was determined to promote the migration of liver cancer cells
(10). These aforementioned
observations indicate that P16INK4A may exert distinct biological
effects, as a tumor suppressor or an oncogenic protein, in a
tissue-specific manner.
It has been reported that P16INK4A expression is
regulated by the methylation level of histone 3 lysine 27 (H3K27)
(14). Targeting histone lysine
demethylase (KDM) 6B by GSK-J4, a KDM6B inhibitor, increases the
level of H3K27 trimethylation, which is an effective approach to
curtailing P16INK4A expression and reducing cell proliferation
(11). Thus, P16INK4A may be worthy
of further consideration as a therapeutic target for
P16INK4A-positive cancer cases.
The aim of the present study was to examine the
biological functions of P16INK4A in USC and to determine whether
targeting KDM6B is a viable therapeutic option for the treatment of
endometrial cancer.
Materials and methods
HEK293T and endometrial cancer cell
lines, cell culture and reagents
The human endometrial adenocarcinoma cell lines
AN3CA, EFE-184, ETN-1, Hec1A, Hec108 and Nou-1 were obtained from
the Dana-Farber/Harvard Cancer Center (Boston, MA, USA). AN3CA,
EFE-184, ETN-1, Hec108 and Nou-1 cells were maintained at 37°C in a
humidified normoxic atmosphere in the presence of 5% CO2
in RPMI-1640 (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA), and Hec1A cells were maintained in McCoy's 5A (Gibco; Thermo
Fisher Scientific, Inc.). HEK293T cells were maintained in
Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc.). All the media were supplemented with 10% fetal
bovine serum (FBS; HyClone; GE Healthcare, Logan, UT, USA). Among
the 6 cell lines, EFE-184 and ETN-1 cells were screened and used
the two for the subsequent experiments. Palbociclib was purchased
from Selleck Chemicals (Houston, TX, USA; cat. no. S1579), and
GSK-J4 was obtained from MedChemExpress (Shanghai, China; cat. no.
HY-15648B).
Proliferation assays
Cells were plated into 96-well plates at a density
of 5×103 cells/well, and incubated at 37°C with or
without increasing concentrations of palbociclib (0, 0.25, 0.5, 1,
2.5, 5 and 10 µM). Cells were harvested at 72 h after palbociclib
or vehicle [dimethyl sulfoxide (DMSO)] treatment, and evaluated
with a Cell Counting Kit-8 assay (Dojindo Molecular Technologies,
Inc., Kumamoto, Japan), according to the manufacturer's guidelines,
as previously described (15).
GraphPad Prism software 5.0 (GraphPad Software, Inc., San Diego,
CA, USA) was used to calculate the half-maximal inhibitory
concentration (IC50) values.
Short hairpin RNA (shRNA)
construction
The construction of shRNA P16INK4A was performed as
previously described (16).
Briefly, a doxycycline-inducible shRNA system was constructed and
the plasmids (~150 ng/µl) were transfected into cells with a
lentivirus. The following primers were designed and synthesized by
Sangon Biotech Co., Ltd. (Shanghai, China), and used for P16INK4A
knockdown: shP16INK4A-1, forward,
5′-CCGGCGCACATTCATGTGGGCATTTCTCGAGAAATGCCCACATGAATGTGCGTTTTTG-3′,
and reverse,
5′-AATTCAAAAACGCACATTCATGTGGGCATTTCTCGAGAAATGCCCACATGAATGTGCG-3′;
and shP16INK4A-2, forward,
5′-CCGGATCAGTCACCGAAGGTCCTACCTCGAGGTAGGACCTTCGGTGACTGATTTTTTG-3′,
and reverse
5′-AATTCAAAAAATCAGTCACCGAAGGTCCTACCTCGAGGTAGGACCTTCGGTGACTGAT-3′.
To generate pLKO-tet-on-shRNAs targeting human P16INK4A,
oligonucleotides were designed and synthesized (Sangon Biotech Co.,
Ltd.). Following annealination, double-stranded oligonucleotides
were directly ligated with pLKO-Tet-on vector that was digested
with Agel (New England Biolabs, Inc., Ipswich, MA, USA) and EcoRI
(Takara Biotechnology Co., Ltd., Dalian, China). Retroviruses were
generated by transfecting HEK293T cells with pWzl plasmids and
packaging DNA, A total of 1.6 µg pWzl DNA, 1.2 µg pCG-VSVG, 1.2 µg
pCG-gap/pol and 12 µl Lipofectamine® 3000 (Thermo Fisher
Scientific, Inc.) were used. DNA and lipid were diluted in 300 µl
DMEM and mixed, and following 15 min of incubation at room
temperature, they were added to one 6-cm dish that was seeded with
3×106 HEK293T cells 1 day earlier at room temperature.
Viral supernatant was collected 48 and 72 h after transfection.
After the supernatant was filtered through 0.45-µm membrane, it was
added to target cells in the presence of 8 µg/ml polybrene (EMD
Millipore, Billerica, MA, USA). Lentiviruses were generated with a
similar approach with the exception of HEK293T cells that were
transfected with 2 µg pLKO DNA, 1.5 µg pCMV-dR8.91 (Biovector,
Beijing, China) and 0.5 µg pMD2-VSVG (Biovector). Cells were
selected with antibiotics at 72 h after initial infection.
Puromycin (Thermo Fisher Scientific, Inc.) and blasticidine (Thermo
Fisher Scientific, Inc.) were used at the final concentrations of
1.5 and 4 µg/ml, respectively. The knockdown of P16INK4A was
confirmed by reverse transcription-quantitative polymerase chain
reaction (RT-qPCR) analysis and western blotting for at least 3
times each. The subsequent experiments were performed immediately
following the transfection.
Clonogenic assay
Following transfection with shRNA plasmids, cells
were seeded into 12/24-well plates at a density of
3×103/6×103 cells/well, respectively, with or
without doxycycline (1 µg/µl), and cultured at 37°C for 10 days.
ETN-1 and EFE-184 cells were also plated in 6-well plates at a
density of 1×104 cells/well with GSK-J4 (30 µM) or DMSO,
and were also cultured at 37°C for 10 days. The culture medium
RPMI-1640, containing doxycycline (1 µg/µl), GSK-J4 (30 µM) or
DMSO, was changed every 3 days. After 10 days of treatment, the
cells were fixed and stained with methanol and crystal violet
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) mixture (0.05%
concentration at room temperature for 30 min), and then extracted
with 10% glacial acetic acid. The optical density (OD) was measured
at 570 nm by EnSpire® Multimode Plate Readers
(PerkinElmer, Inc., Waltham, MA, USA).
Protein isolation and western
blotting
The cells not treated were collected at 80%
confluence, while if treated with GSK-J4, the cells were collected
at the 24 h after the treatment, and if treated with Palbociclib,
they were collected at 12 h after treatment. The cells were seeded
at 1×104 cells/plate and maintained in 60-cm plates and
were lysed on ice in radioimmunoprecipitation assay buffer
supplemented with protease and phosphatase inhibitors (KGP2100;
Nanjing KeyGen Biotech Co., Ltd., Nanjing, China), as previously
described (17). Immunoblots were
conducted using a Bio-Rad system (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). A total of 25 µg protein was loaded for each
sample, the total protein was separated by 12% SDS-PAGE and
transferred to polyvinylidene difluoride membrane (EMD Millipore).
The membranes were blocked with 5% milk at room temperature for 1 h
and the target proteins were probed by the following antibodies for
12 h at 4°C: P16INK4A (OriGene Technologies, Inc., Rockville, MD,
USA; cat. no. ZS-0033; 1:500), KDM6B (ProteinTech Groups, Inc.,
Chicago, IL, USA; cat. no. 55354-1-AP; 1:1,000), H3K27-M3
(Immunoway Biotechnology Company, Plano, TX, USA; cat. no. YM3338;
1:1,000) and H3K27-M1 (Immunoway Biotechnology Company; cat. no.
YM3336; 1:1,000). Vinculin (Cell Signaling Technology, Inc.,
Danvers, MA, USA; cat. no. #4650; 1:1,000) was used as loading
control. The membrane was lastly stained with horseradish
peroxidase (HRP)-conjugated rabbit anti-sheep IgG H&L (1:5,000;
ab6747; Abcam, Shanghai, China) for 2 h at 4°C. The protein bands
were then detected using a radioactive detection system (SuperLumia
Enhanced Chemiluminescence kit; cat. no. K22020; Abbkine Scientific
Co., Ltd., Redlands, CA, USA, www.abbkine.com). The absolute density of P16INK4A
bands was later determined by ImageJ software 4.0 (National
Institutes of Health, Bethesda, MD, USA). The relative density of
P16INK4A was normalized to the corresponding vinculin bands. In
vehicle-treated cells, the relative density of P16INK4A was
assigned as 1.
RT-qPCR analysis
Total RNA was isolated from the cells with an
Illustra RNAspin kit (GE Healthcare) and reverse-transcribed using
a iScript cDNA synthesis kit (Bio-Rad Laboratories, Inc.),
according to the manufacturer's protocols. An universal
SYBR®-Green MasterMix and RT-qPCR was used to measure
gene expression. Briefly, the samples were incubated for 60 min at
42°C, 15 min at 72°C, and stored at −20°C. For qPCR, 1 µl of
diluted RT products were mixed with 10 µl of SYBR®-Green
MasterMix (Bio-Rad Laboratories, Inc.), 0.5 µl of forward and
reverse primers, and 4 µl of nuclease-free water for a final volume
of 20 µl, according to the manufacturer's protocols. The reactions
were run on the ABI-7500 Real-Time PCR System (Applied Biosystems;
Thermo Fisher Scientific, Inc.) using the conditions of 40 cycles
at 95°C for 20 sec and 60°C for 45 sec. Actb was used as the
loading control. The primers for P16INK4A were as follows:
Forward, 5′-ATATGCCTTCCCCCACTACC-3′, and reverse,
5′-CCCCTGAGCTTCCCTAGTTC-3′. The primers for Actb were: forward,
5′-CCTAGAAGCATTTGCGGTGG-3′, and reverse,
5′-GAGCTACGAGCTGCCTGACG-3′. Cq values were generated using the
default analysis settings. ΔCq was defined as Cq gene of interest -
Cq β-actin. ΔΔCqT was defined as ΔCq treated sample - Cq control
sample. Relative quantification was calculated as
2−ΔΔCq, as described previously (18).
3D Sphere-forming cultures
As previously described (19), the cells (2,000/well) were seeded on
96-well plates coated with Matrigel (BD Biosciences; Beckon,
Dickinson and Company, Franklin Lakes, NJ, USA). The cells were
grown in RPMI-1640 medium supplemented with 2% FBS and 2% Matrigel,
and allowed to grow for 96 h at 37°C. The original medium was
replaced with the fresh RPMI-1640 medium containing 2% FBS and 2%
Matrigel additional with GSK-J4 (30 µM) or vehicle (DMSO) at this
time point. For shRNA P16INK4A ETN-1 and EFE-184 cells, doxycycline
was added when seeding. Over 100 colonies were scored for each
condition. Quantitation of tumor spheres for structural integrity
was performed after a 96-h culture.
Wound healing assay
A wound healing assay was used to evaluate the
migration ability of ETN-1 and EFE-184 cells, as previously
described (20). Cells were plated
in 24-well plates at the density of 20,000/well and grown at 37°C
in RPMI-1640 medium supplemented with 10% FBS until confluence. A
scratch was created using sterile 200 µl pipette tips. PBS was used
twice to remove cell debris and fresh RPMI-1640 medium supplemented
with 2% FBS was added, with or without doxycycline. The mean width
of each scratch was measured using Image-Pro Plus software 4.0
(Media Cybernetics, Inc., Rockville, MD, USA).
Hematoxylin and eosin (H&E) and
immunohistochemistry (IHC)
For H&E, tissues were first fixed in 4%
paraformaldehyde solution at room temperature for 24 h. After
gradient tissue dehydration (75% for 24 h; 85% for 3 h; 95% for 1
h; 100% for 1 h; and 100% for 1 h; ethanol solution at room
temperature), followed by 100% xylene to remove alcohol, the
tissues were embedded in paraffin. Subsequently, paraffin-embedded
tissue sections (4-µm) were dewaxed with 100% xylene at room
temperature for 30 min and gradient ethanol solution (100% for 10
min; 100% for 10 min; 95% for 10 min; 80% and 10 min).
Subsequently, sections were immersed in 0.5% hematoxylin (cat. no.
H8070; Beijing Solarbio Science & Technology Co., Ltd.,
Beijing, China) for 10 min followed by 5 quick dips in 0.3% acid
alcohol at room temperature. The sections were then washed with
running water for 60 min. Following this, 1% of eosin (cat. no.
G1100; Beijing Solarbio Science & Technology Co., Ltd.) was
used for 1 min at room temperature to stain the cytoplasm. IHC was
performed using P16INK4A (OriGene Technologies, Inc.; cat. no.
ZS-0033; 1:200), H3K27-M3 (Immunoway Biotechnology Company; cat.
no. YM3338; 1:500) and H3K27-M1 (Immunoway Biotechnology Company;
cat. no. YM3336; 1:500), as previously described (21). Briefly, the IHC stainning of
paraffin-embedded samples was performed using a standard
Biotin-Streptavidin HRP Detection method, as previously described
(https://www.cellsignal.com/contents/resources-protocols/immunohistochemistry-protocol-(paraffin)/ihc-paraffin).
The 4-µm sections were deparaffinized as aforementioned and
antigens in the tissue were retrieved for 5 min. Following blocking
endogenous peroxidase activity with 30% hydrogen peroxide
formaldehyde solution at room temperature for 30 min, the sections
were incubated with 20% normal goat serum to block non-specific
binding sites for 30 min at room temperature. The primary antibody
used was a polyclonal antibody against human P16INK4A protein. The
samples were incubated overnight at 4°C in a moist chamber.
Following 3 washes with PBS, the sections were incubated for 30 min
at room temperature with goat anti-mouse secondary IgG (dilution
1:400; cat. no. SP-9001; OriGene Technologies, Inc.). The
3,5-diaminobenzidine detection kit (OriGene Technologies, Inc.) was
used for staining. Negative controls with PBS (0.01 mol/l, pH 7.4)
replacing the primary antibody were also included. Finally, the
tissue sections were counterstained with 0.5% hematoxylin for 1 min
at room temperature, dehydrated and mounted in resinous mountant at
room temperature. Digital images were captured using a light
microscope (Leica Microsystems GmbH, Wetzlar, German) at a ×400
magnification. The intensity of P16INK4A, H3K27M1 and H3K27M3
staining was quantified as the percentage of positive cells per
high-power field. The formalin fixed paraffin-embedded tissues
including malignant and normal endometrium were all obtained from
First Affiliated Hospital of Dalian Medical University (Dalian,
China). It was approved by the ethics committee of the hospital and
written informed consent were also obtained from the patients.
Patient information, tissue
preparation, patient-derived xenograft (PDX) model establishment
and ex vivo culture of PDX tumor tissue
Fresh tumor tissue and 121 FFPE tumor samples from
other patients (age range, 38–77 years; mean age, 56 years) with
endometrial cancer were collected from the Biobank of First
Affiliated Hospital of Dalian Medical University. All were
processed according to the protocol approved by the Institutional
Review Board of the First Affiliated Hospital of Dalian Medical
University. Written informed consent was obtained from the
patients. A primary tumor tissue was obtained from a 57-year-old
female during hysterectomy who had been diagnosed with endometrial
serous carcinoma in dilatation and curettage, a pre-operative
endometrium biopsy. The fresh tumor tissue was maintained in normal
saline and transported to the laboratory in 4°C on ice. Prior to
transplanting the tumor into the mice, the tumor tissue was cut
into 5×5×5 mm tissue blocks. A total of 2 non obese-severe combined
immunodeficiency (NOD-SCID) female mice maintained in a
pathogen-free environment were used to establish the first
generation of PDX mouse models. The mice, each weighing 25–30 g
were provided by the Laboratory Animal Center of Dalian Medical
University. The 6-week old NOD-SCID mice were raised in large
plastic cages with a maximum of 6 mice/cage at 40–60% humidity and
19–23°C. The mice were maintained on a 12/12 h light/dark period
with 12–15 air exchanges/h and were fed with food and water ad
libitum. The mice bearing xenografts were housed under standard
conditions and monitored closely. During the whole experiments, a
total of 8 mice were used to establish models of different
generations. The PDX mouse model was constructed as previously
described (22). In brief, after
the mice were anesthetized with intraperitoneal injection of
Tribromoethanol (avertin 1.25%), 300 mg/kg, a 1-cm scalp incision
was produced on the skin near each leg. Subsequently, tissue blocks
were placed into the subcutaneous space and the incisions were
closed. Tumor volume, body weight, living activity and food intake
were monitored every 3 days. The timing of sacrifice depended on
the following two conditions: If multiple tumors were present, the
monitoring was terminated when the largest tumor volume reached 1
cm3 (with the combination of the two largest tumor size
<2 cm); but if only a single tumor was observed, monitoring was
terminated when the largest diameter of the tumor was near 2 cm.
Therefore, sufficient tissue for the experiments could be ensured.
After the mice were sacrificed with cervical dislocation and the
tumor tissue was transplanted into other NOD-SCID mice, as a next
generation model, the 2 mice for this experiment were the third
generation. In total, 5 tumors from 8 transplanting sites (4 for
each mouse) were obtained and the maximum tumor size was 1.8 cm
(the single tumor appeared in that mouse). The mice were then
sacrificed by decapitation and tumor specimens were used for ex
vivo culture, as previously described (23). Briefly, the fresh tumor specimen was
dissected into ~1-mm3 tissue blocks. The blocks were
cultured on an absorbable gelatin sponge in the presence of vehicle
control (DMSO) or GSK-J4 (50 µM) for 24 h. All animal procedures
were conducted under the approval of the Institutional Animal Care
and Use Committee of Dalian Medical University (Dalian, China) and
strictly followed the guideline for tumor induction in mice and
rats (http://igeam.dmu.edu.cn/jgsz1/SPFdwsyzx1/zcfg.htm).
Statistical analysis
Data are expressed as mean ± standard deviation or
percentage. In analyzing quantitative results, Student's
t-test and χ2 test were used for two group
comparison, analysis of variance and a post hoc test
(Student-Newman-Keuls) were used for multiple comparisons.
P<0.05 was considered to indicate a statistically significant
difference. All statistical analyses were performed by GraphPad
Prism 5.0 software.
Results
P16INK4A is overexpressed in the
majority of USC cases
To evaluate the levels of P16INK4A expression in
endometrial cancer, IHC staining was performed on 121 tumor samples
from patients (age range, 38–77 years, with the mean age of 56
years) with endometrial cancer at First Affiliated Hospital of
Dalian Medical University. In line with the observations of
previous studies (8,9,24–26),
it was observed that P16INK4A was overexpressed in the majority of
USC cases and was negative in the majority of UEC cases, with 75%
(25/33) of USCs and 12% (11/88) of UECs being P16INK4A-positive
(Fig. 1A and B, Table I). In normal endometrium from 8
individuals receiving hysterectomy due to of uterine leiomyoma, the
expression of P16INK4A was negative. The representative image was
depicted (Fig. 1A). Therefore,
these results demonstrated that P16INK4A is overexpressed in the
majority of USC samples.
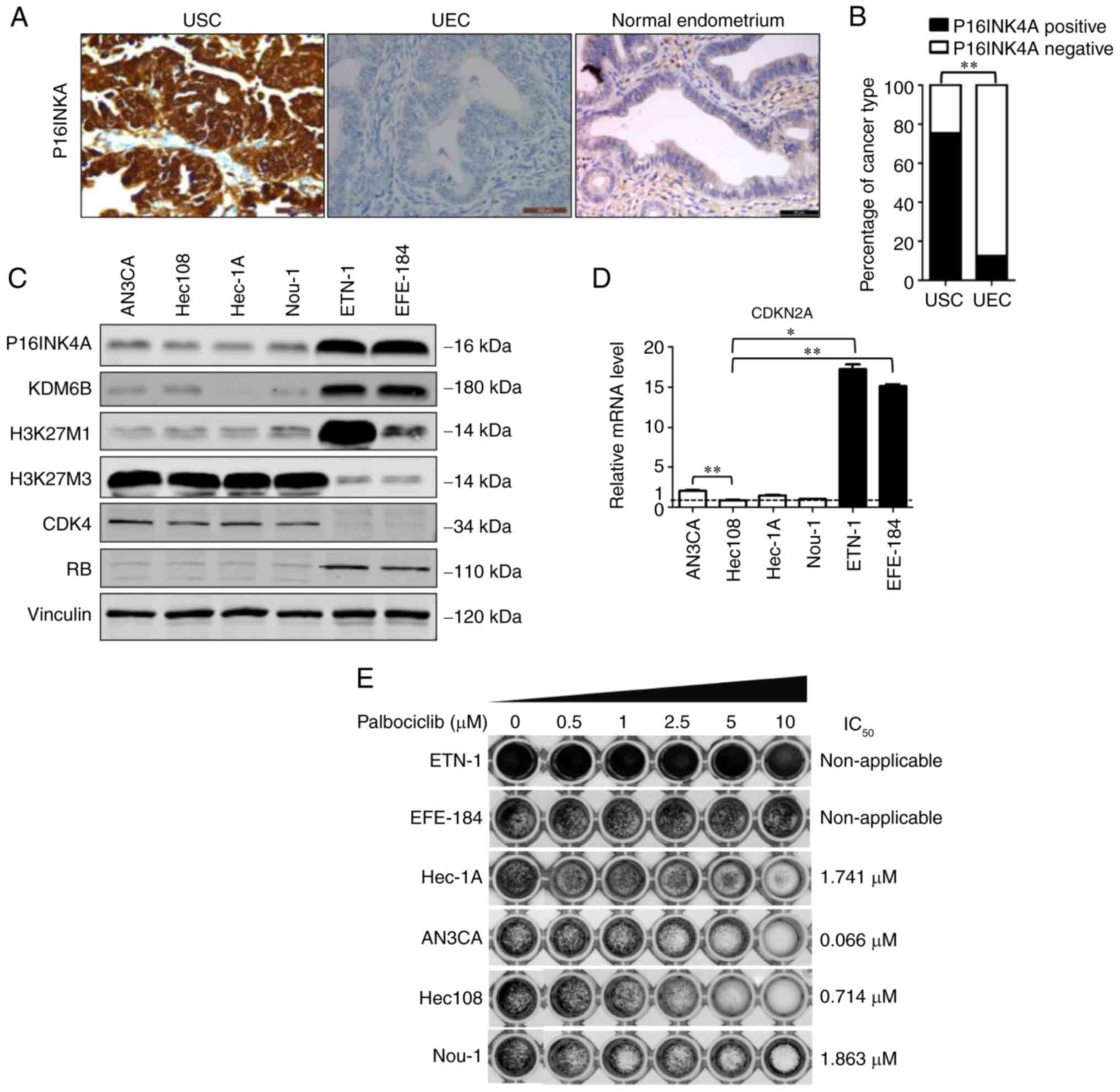 | Figure 1.Differential expression of P16INK4A
in tumor samples. (A) Representative images of P16INK4A positivity
in USC, P16INK4A negativity in UEC and P16INK4A negativity in
normal endometrium. The normal tissue was obtained from 8 patients
with leiomyoma who receiving hysterectomy procedures. Scale bar, 50
µm. (B) Percentage of P16INK4A-positive cases among 33 USC and 88
UEC samples. **P<0.01 (χ2 test). (C) Western blot
analysis of 6 endometrial cell lines (AN3CA, Hec1A, Hec108, Nou-1,
EFE-184 and ETN-1). Vinculin was used as a loading control. (D) The
reverse transcription-quantitative polymerase chain reaction
results of the 6 endometrial cell lines (AN3CA, Hec1A, Hec108,
Nou-1, EFE-184 and ETN-1) were analyzed and shown as relative
P16INK4A mRNA level of the Hec1A values, which were then
converted as fold change. *P<0.05 and **P<0.01 [analysis of
variance and a post hoc test (Student-Newman-Keuls)]. (E) Response
of different endometrial cell lines to the CDK4/6 inhibitor
palbociclib. The viability of ETN-1, EFE-184, Hec-1A, AN3CA, Hec108
and Nou-1 cells was measured with a Cell Counting Kit-8 assay
following treatment with vehicle (dimethyl sulfoxide) and
palbociclib (0.5, 1, 2.5, 5 and 10 µM) for 72 h. The
IC50 are depicted. P16INK4A, cyclin-dependent kinase
inhibitor 2A; CDK4, cyclin-dependent kinase 4; UEC, uterine
endometroid carcinoma; USC, uterine serous carcinoma; H3K27,
histone 3 lysine 27; KDM6B, histone lysine demethylase 6B;
IC50, half-maximal inhibitory concentration; RB,
retinoblastoma. |
 | Table I.Different expression of P16INK4A
between tumor samples of USCs and UECs. |
Table I.
Different expression of P16INK4A
between tumor samples of USCs and UECs.
| Investigator | Year | P16INK4A positive
in USCs (%) | P16INK4A positive
in UECs (%) | Method | P-value | Refs no. |
|---|
| Yemelyanova et
al | 2009 | 95 (47/49) | 38 (38/101) | IHC | <0.01 | (8) |
| Netzer et
al | 2011 | 78 (24/31) | 35 (11/31) | IHC | <0.001 | (24) |
| Levine DA et
al | 2013 | 31 (31/101) | 2.9 (13/447) | mRNA, RPPA | <0.01 | (26) |
| Han et
al | 2013 | 80 (12/15) | 11 (5/43) | IHC | <0.001 | (25) |
| Chen et
al | 2017 | 92 (48/52) | 26 (17/65) | IHC | <0.01 | (9) |
| Xiao et
al | 2017 | 75 (25/33) | 12 (11/88) | IHC | <0.01 | Present study |
Overexpression of P16INK4A acts as an
oncogenic event for a number of endometrial cancer cell lines
As P16INK4A overexpression in tumor tissue
contradicts its known role as a tumor suppressor, the aim was to
investigate its function in endometrial cancer cells. Firstly, the
expression of P16INK4A was tested in 6 endometrial cancer cell
lines (AN3CA, EFE-184, ETN-1, Hec108, Hec-1A and Nou-1) and the
expression level of P16INK4A was determined to be highest in ETN-1
and EFE-184 cells, whereas it was relatively low in AN3CA, Hec108,
Hec-1A and Nou-1 cells (Fig. 1C and
D).
Tumor suppressor retinoblastoma (RB) acts downstream
of CDK4/6 and serves an important role in the cell cycle (4,5).
RB-deficient tumors frequently express high levels of P16INK4A
(4–6). A research group reported that, when
P16INK4A is overexpressed, CDK4/6 expression is suppressed to
prevent cancer cells from responding to CDK4/6 inhibitors (27). Thus, to investigate the function of
CDK4/6 in the aforementioned endometrial cancer cell lines, their
treatment response to the CDK4/6 inhibitor palbociclib were
evaluated. Highly P16INK4A-expressing ETN-1 and EFE-184 cells were
determined to be resistant to palbociclib. The remaining cell lines
(AN3CA, Hec108, Hec-1A and Nou-1), in which P16INK4A expression was
low or negative, were sensitive to the treatment, with an
IC50 of 0.066, 0.714, 1.741 and 1.863 µM, respectively
(Fig. 1E). Therefore, ETN-1 and
EFE-184 cells were selected for further experiments.
To determine whether P16INK4A acts as a driving
factor in the proliferation of ETN-1 and EFE-184 cells, P16INK4A
was transfected into ETN-1 and EFE-184 cells by transfecting two
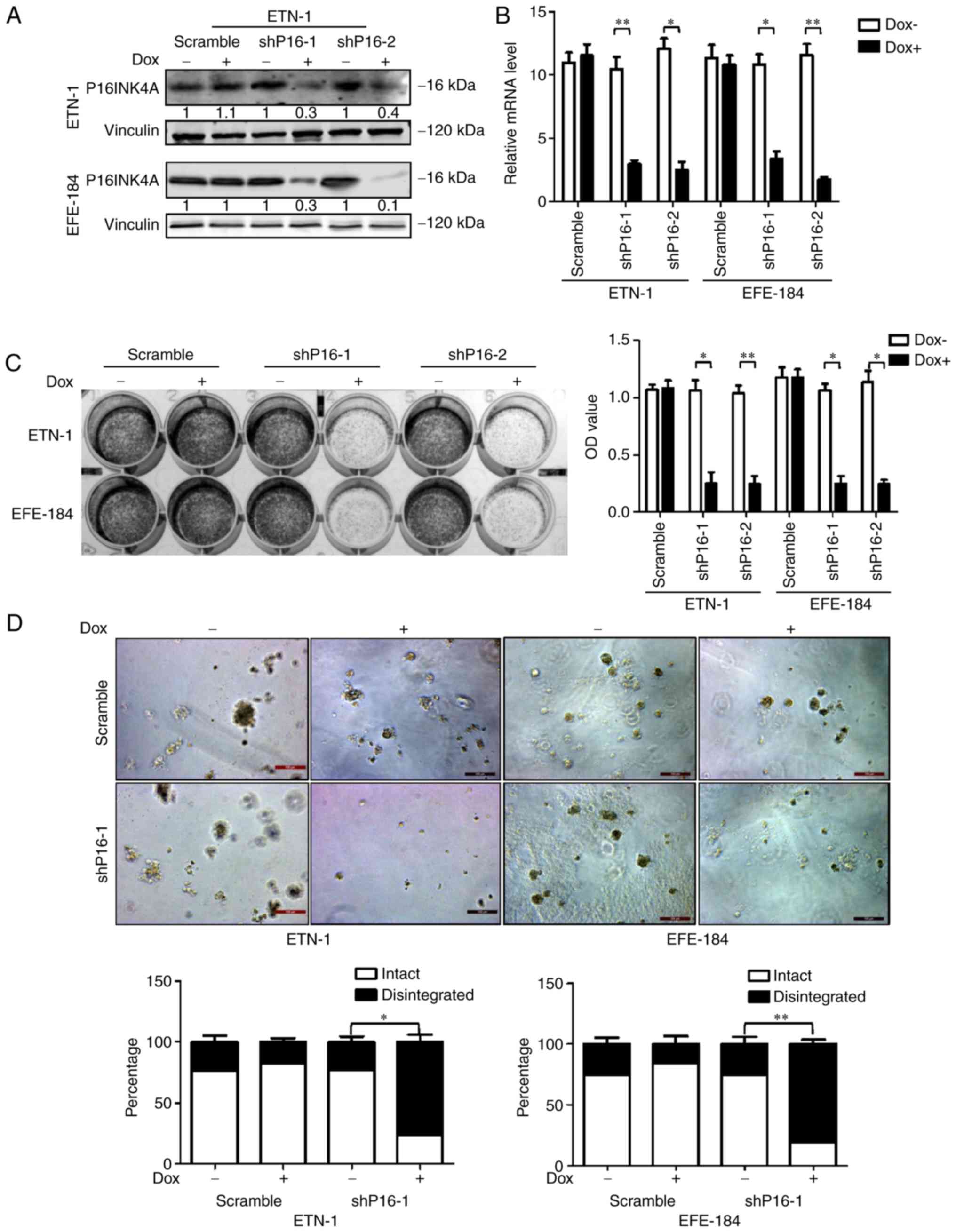
P16INK4A-specific shRNAs. The downregulation of P16INK4A was
verified by RT-qPCR and western blot analyses (Fig. 2A and B). Following
doxycycline-induced P16INK4A depletion, the growth potential of
ETN-1 and EFE-184 cells was significantly decreased, as depicted in
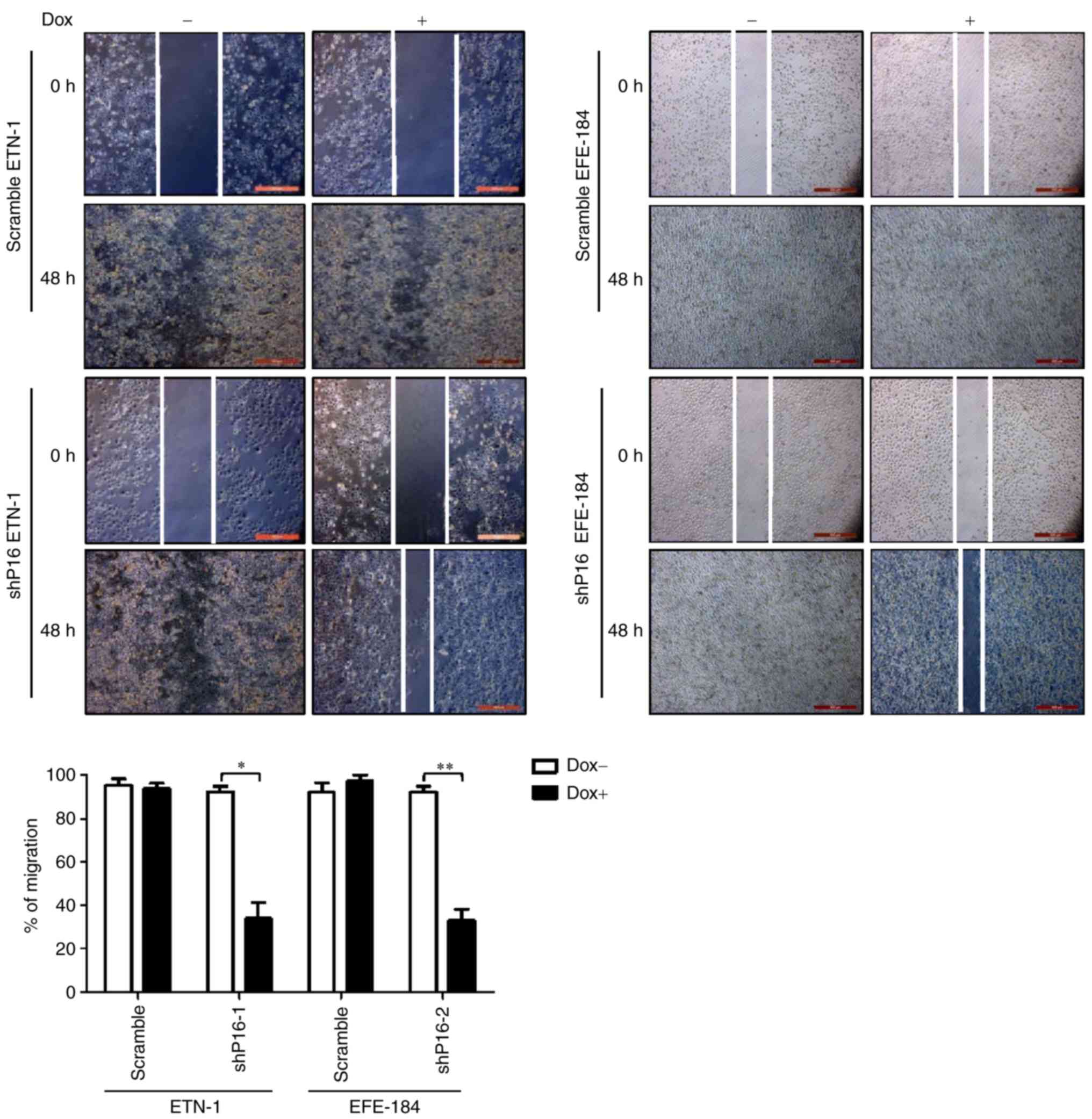
2D colony-forming and 3D culture assays (Fig. 2C and D). A wound healing assay also
demonstrated that cells with P16INK4A knocked down exhibit
significantly reduced migration ability, compared with those with
intact P16INK4A expression (Fig.
3). These results indicated that P16INK4A, in addition to its
canonical role as a tumor suppressor, may also serve an oncogenic
role in P16INK4A-positive cells, although the underlying mechanism
remains elusive.
P16INK4A overexpression in USCs
coexists with the demethylation of H3K27
Previous studies demonstrated that the transcription
of P16INK4A is primarily regulated by the methylation level
of H3K27 in cervical cancer (14,28,29).
To determine whether a similar mechanism is involved in the
abundant expression of P16INK4A in USC, 20 cases of
P16INK4A-positive USC tissue samples was investigated. Using IHC,
70% (14/20) samples in this cohort were identified as
H3K27M3-negative and H3K27M1-positive (Fig. 4A and B). A similar result was also
observed in the ETN-1 and EFE-184 cell lines. Western blot analysis
revealed weak H3K27M3 and robust H3K27M1 expression in ETN-1 and
EFE-184 cancer cells (Fig. 4C).
These observations indicate that the upregulation of P16INK4A in
USC may share the same mechanism as reported in cervical cancer
aforementioned.
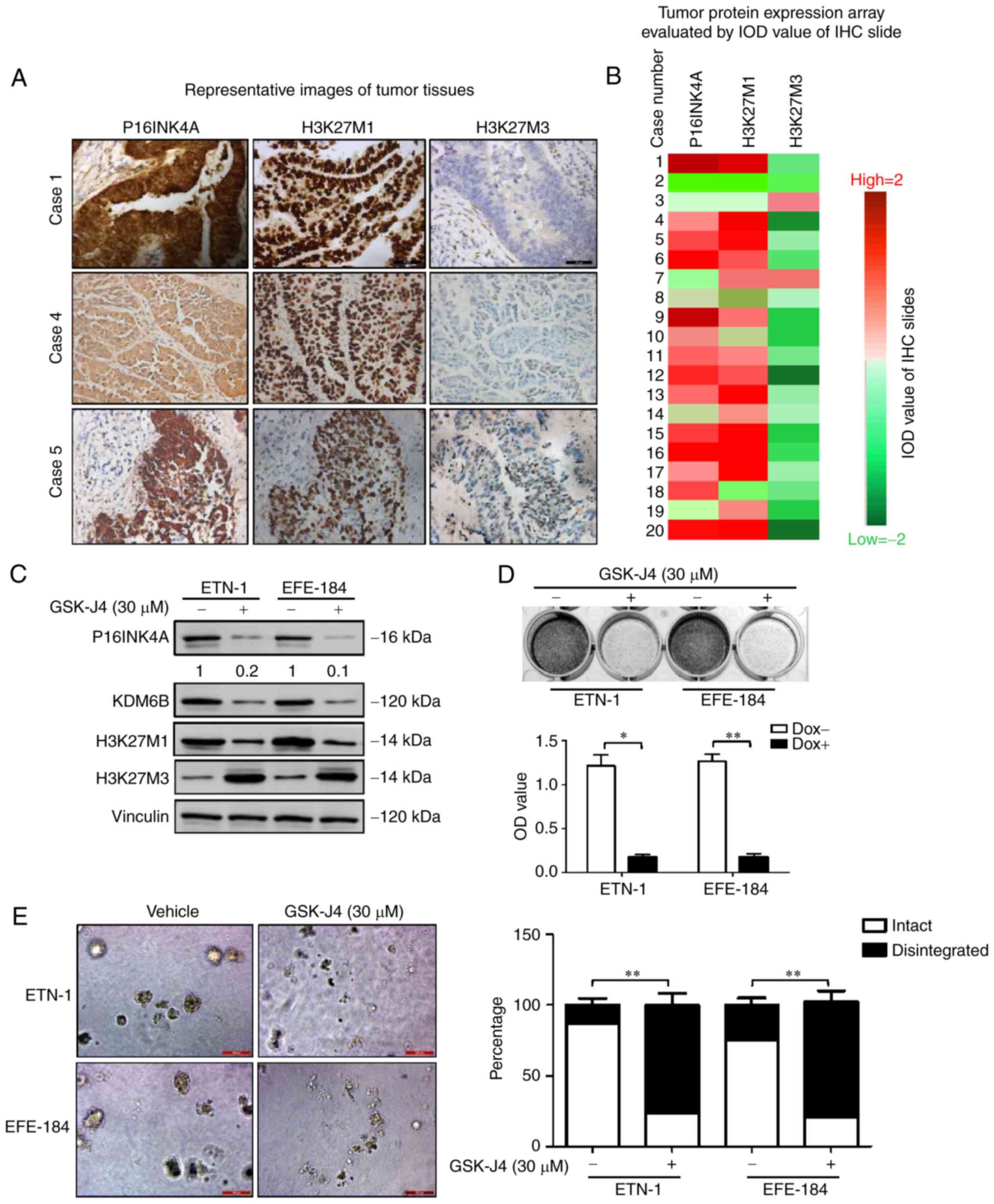 | Figure 4.P16INK4A overexpression coexists with
demethylation of H3K27 in endometrial cancer, and the effects of
target inhibition of H3K27 demethylation on cellular P16INK4A and
growth potential of ETN-1 and EFE-184 cells. (A) Representative
images for IHC analyses in 3 independent cases. H3K27M1, H3K27M3
and P16INK4A were evaluated in these tumor samples. Scale bar, 50
µm. (B) A total of 70% (14/20) of uterine serous carcinoma samples
in the study cohort were H3K27M3-negative and H3K27M1-positive.
Hotmap was depicted according to the IOD value of each slides
[ranging from −2 (green) to +2 (red)], red indicates positive
staining and green represents negative staining. (C) Western blot
analyses of P16INK4A, H3K27M1 and H3K27M3 in ETN-1 and EFE-184
cells when left untreated or treated with vehicle (DMSO) and GSK-J4
(30 µM). Vinculin was used as a loading control. (D) ETN-1 cells
were treated with DMSO and GSK-J4 (30 µM) for 5 days. Means ±
standard deviation for 3 independent experiments are depicted.
*P<0.05; **P<0.01 (Student's t-test). (E) The ETN-1 and
EFE-184 cells treated with DMSO and GSK-J4 (30 µM) were maintained
in 3D culture medium for 4 days. Representative images of scored
structures (intact vs. disintegrated) are depicted. Scale bar, 100
µm. **P<0.01 (Student's t-test). DMSO, dimethyl sulfoxide;
P16INK4A, cyclin-dependent kinase inhibitor 2A; H3K27, histone 3
lysine 27; KDM6B, histone lysine demethylase 6B; IHC,
immunohistochemistry; IoD, integrated option density; OD, optical
density. |
KDM6B inhibition reduces P16INK4A
expression and suppresses the proliferation of cancer cells in cell
lines and USC explants
As histone KDM6B mediates H3K27 demethylation and
increases the expression of P16INK4A (11,14),
it was hypothesized that treatment with the KDM6B inhibitor GSK-J4
may be effectively used to target P16INK4A-positive cancer cells.
As expected, GSK-J4 treatment significantly suppressed cell growth,
compared with the control vehicle (Fig.
4D), which was also verified in 3D culture assays (Fig. 4E). Furthermore, following GSK-J4
treatment, P16INK4A expression was markedly inhibited with the
increase of H3K27M3 and the decrease of KDM6B and H3K27M1 (Fig. 4C), supporting our hypothesis that
target inhibition of H3K27 demethylation is a potential treatment
for P16INK4A-positive endometrial cancer.
To further confirm the treatment effects ex
vivo, a PDX mouse model of USC was then established (Fig. 5A). The model maintained a number of
histopathological characteristics of the human primary tumor,
including P16INK4A overexpression (Fig.
5B). In the ex vivo experiment, following GSK-J4
treatment, samples were subjected to histological examination. It
was observed that, following treatment with GSK-J4, cellular
integrity was markedly disrupted, without distinct USC-specific
structures. However, explants treated with DMSO retained the
architecture and cellularity of non-treated tumors. IHC analysis
also demonstrated that the expression of P16INK4A and H3K27M1 was
significantly reduced, while H3K27M3 staining was significantly
increased in GSK-J4-treated explants (Fig. 5C). The results from the tumor
explants were consistent with those obtained from cell lines,
indicating that targeting KDM6B by GSK-J4 may be a viable treatment
strategy for P16INK4A-positive endometrial cancer cases.
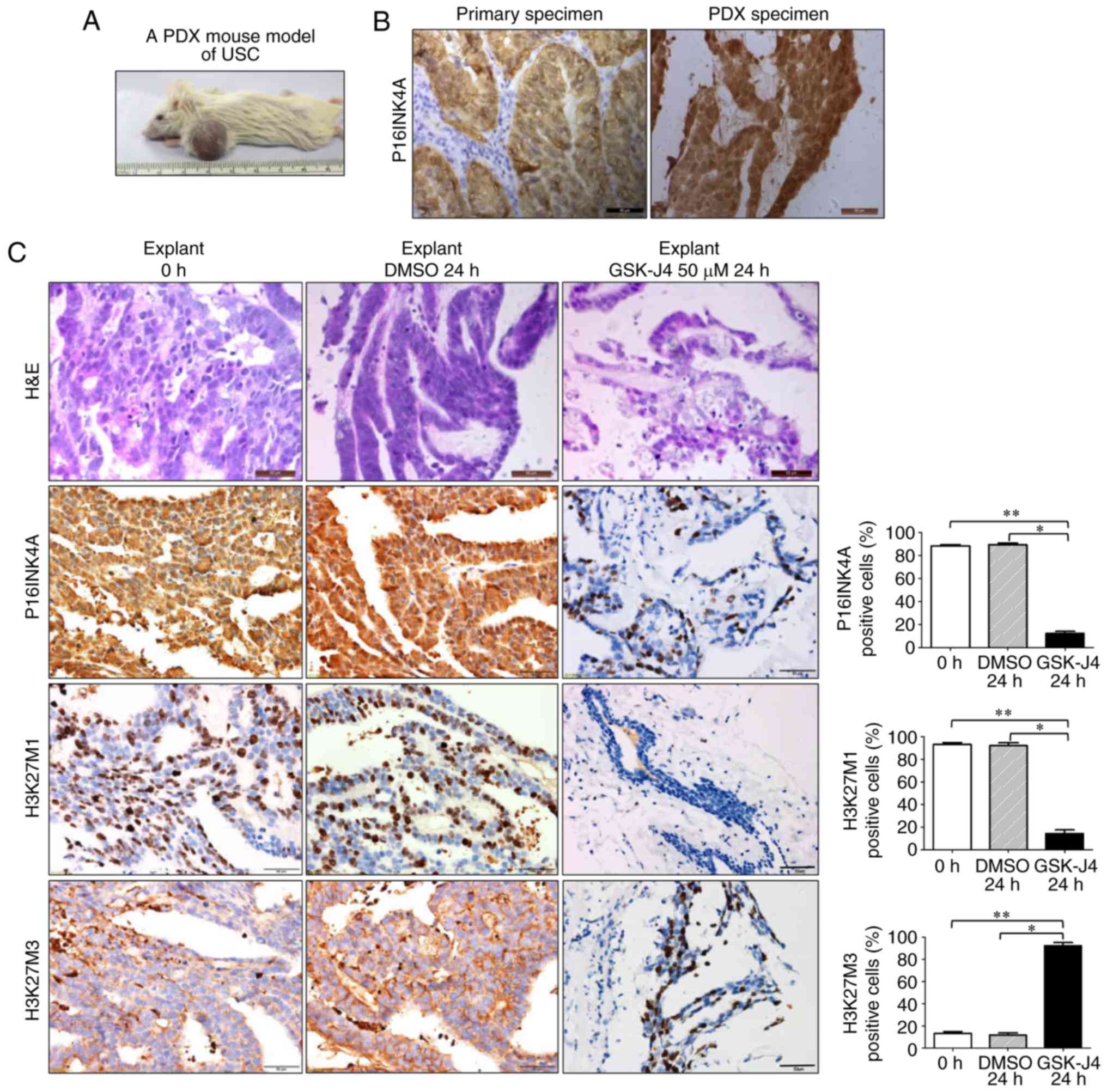 | Figure 5.Target inhibition of H3K27
demethylation by GSK-J4 exerts therapeutic effects on USC PDX tumor
explants. (A) Establishment of a PDX mouse model with a USC tumor.
(B) Representative P16INK4A staining images of human primary
specimen and tumor from the PDX mouse model. Scale bar, 50 µm. (C)
Representative H&E and immunohistochemistry staining images of
P16INK4A, H3K27M1 and H3K27M3 on tumor explants when left untreated
or treated with DMSO or GSK-J4 (50 µM). The expression of P16INK4A
and H3K27M1 was significantly reduced, while H3K27M3 staining was
significantly increased in GSK-J4-treated explants, compared with
the tissues treated by vehicle or untreated. Scale bar, 50 µm.
*P<0.05 and **P<0.01 [analysis of variance and a post hoc
test (Student-Newman-Keuls)]. H&E, hematoxylin and eosin;
P16INK4A, cyclin-dependent kinase inhibitor 2A; H3K27, histone 3
lysine 27; DMSO, dimethyl sulfoxide; PDX, patient-derived
xenograft; USC, uterine serous carcinoma. |
Discussion
To the best of our knowledge, the present study is
the first to investigate the oncogenic role of P16INK4A in
endometrial cancer. The present data demonstrated that P16INK4A is
able to promote proliferation of endometrial cancer cells, and
targeting P16INK4A by altering H3K27 methylation may be a
beneficial strategy for the treatment of endometrial cancer.
Contrary to the known role of P16INK4A as a tumor
suppressor, the present study revealed its oncogenic effects. In a
previous review, the mechanism underlying P16INK4A overexpression
in cancer includes P16INK4A-CDK4/6-Rb pathway alterations, such as
the E7 of human papillomavirus (HPV) targeting RB (13); however, in endometrial cancer, this
may not be the case. Firstly, there is no evidence that endometrial
cancer is associated with HPV infection, and secondly, there are
few mutations or mRNA changes in downstream molecules, such as
CDK4/6 and RB, in endometrial cancer, as detailed in The Cancer
Genome Atlas database (26).
As aforementioned, a number of previous independent
reports support the present conclusions. The studies on cervical
and hepatic cancer demonstrate that P16INK4A is necessary for cell
survival and migration (10,11).
These studies further indicated that the oncogenic role of P16INK4A
is dependent on its inhibition of CDK4/6 (10,11).
Additionally, researchers also identified that different cellular
P16INK4A localization may be associated with different biological
functions. It has been demonstrated that cytoplasmic P16INK4A
expression is frequently associated with poor survival in head and
neck (30), and breast cancer
(31), and cytoplasmic P16INK4A can
promote cell proliferation and migration via a cell cycle-unrelated
pathway (32). In the present
study, P16INK4A was overexpressed in the nucleus and cytoplasm of
USC cells, and following GSK-J4-induced P16INK4A depletion, cancer
cell proliferation and migration were markedly reduced. Thus, the
oncogenic role P16INK4A in endometrial cancer may be mediated
through its classic CDK4/6 inhibitory effect on the nucleus and an
uncertain cell cycle-unrelated pathway in the cytoplasm.
Another observation of the present study was how
P16INK4A is overexpressed in endometrial cancer. Consistently with
a previous report (14), this
upregulation of P16INK4A also results from demethylation of H3K27.
Based on this observation, it appeared reasonable to develop a
strategy for USC treatment by inhibiting P16INK4A through targeting
KDM6B. The present experiments, consistent with previous studies
(14,33–35),
revealed strong treatment effects of GSK-J4 on ETN-1 cells and USC
explants. Thus, the present study not only revealed the role of
demethylation of H3K27 in the progression of endometrial cancer,
but also indicated a novel targeting strategy for the treatment of
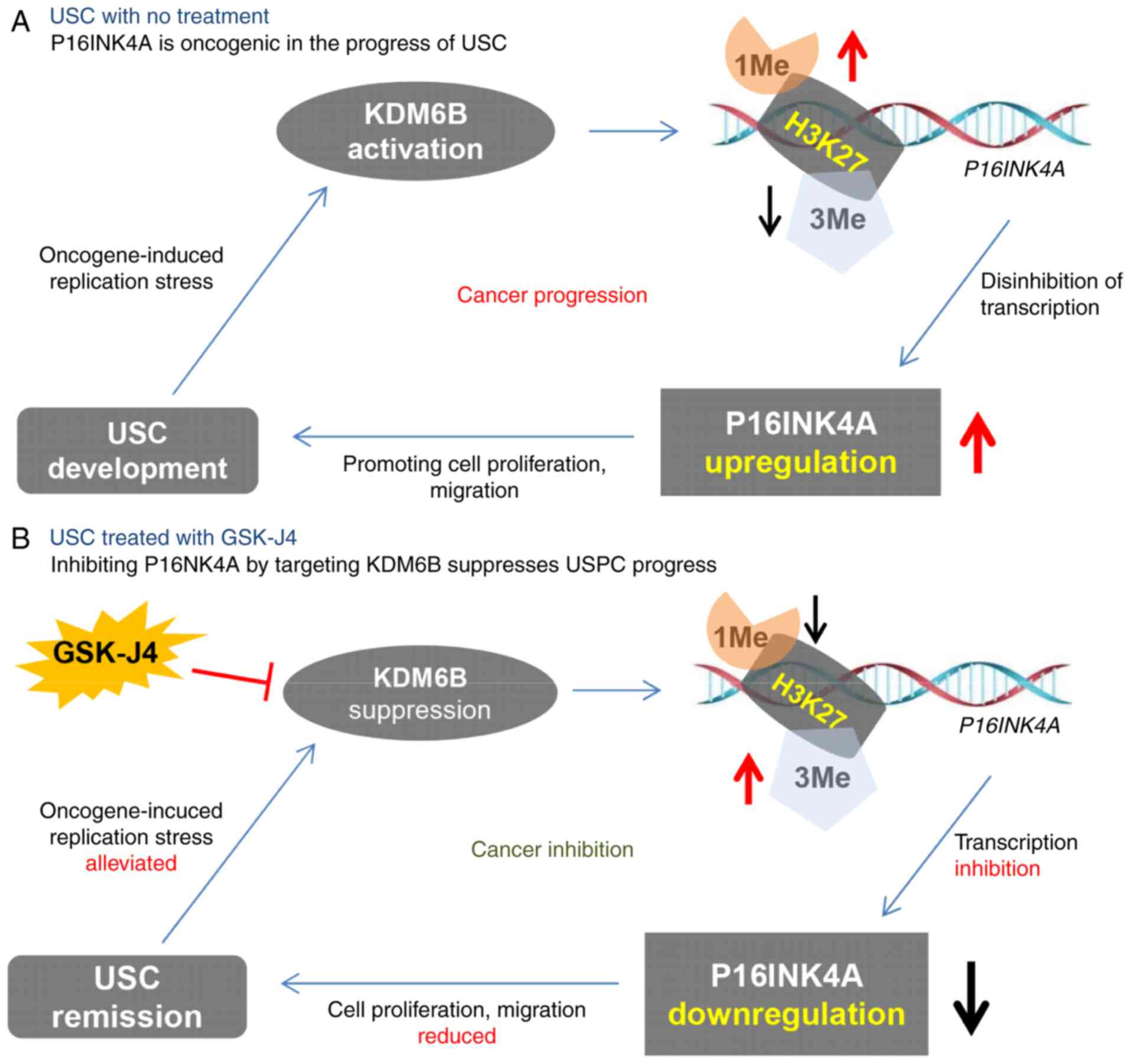
USC. The content of the present study is graphically summarized in
Fig. 6.
There were certain limitations to the present study.
Firstly, as relatively few endometrial cell lines were
P16INK4A-positive, only ETN-1 and EFE-184 cells were included in
the experiments aforementioned. Further research should be
performed on more P16INK4A-positive cell lines in order to verify
these conclusions. Another weakness is that the effects of GSK-J4
was only tested on cell lines and ex vivo tissues. Animal
studies should be performed in the future to further confirm the
validity of this treatment strategy. The third controversial issue
is about the concentration of GSK-J4 (30 µM), which was a slightly
increased, compared with other common small molecular inhibitors.
However, this concentration had been frequently used in a number of
studies on cervical cancer (11),
brain glioma (34) and immunologic
diseases (36,37), demonstrating that this concentration
of GSK-J4 exerted prominent in vitro and in vivo
anti-proliferative effects on cancerous and non-cancerous cells and
tissues.
In conclusion, despite certain limitations, the
present results revealed an oncogenic role of the traditional tumor
suppressor P16INK4A in endometrial cancer, indicating a possible
novel approach to developing promising therapeutic paradigms for
P16INK4A-positive USC.
Acknowledgements
The authors would like to thank Dr Yuan Zhang, Dr
Jinglei Ding and Dr Penglong Cao (Dalian Medical Univercity,
Dalian, China), for their help and support in our experiments and
manuscript preparation.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81372853 and
81572586 to P.L.), Liaoning Provincial Climbing Scholars Supporting
Program of China (grant no. 2012086524 to P.L.), Provincial Natural
Science Foundation of Liaoning (grant no. 2014023002 to P.L.) and
Youth Natural Science Foundation of First Affiliated Hospital of
Dalian Medical University (grant no. 2014QN003/2017FH004 to
Z.X.).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZX, YH, HS and PL are responsible for the conception
and design of the study. ZX, YH, CL, PL, LX, JY and MW performed
the majority of the cell experiments. ZX, YH, PL and HS performed
the acquisition of data, and provided animals, facilities and
selected the patients. ZX, YH, CL, TS, HS and PL performed the
writing, review and revision of the manuscript. ZX, YH, CL, LX, JY,
MW, YX, TS, PL and LS performed the animal-model establishments and
ex vivo experiments. YH, CL, ZX, LS and YX performed the
H&E and IHC staining, and examinations. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The study and the use of human samples were approved
by the ethics committee of First Affiliated Hospital of Dalian
Medical University (Dalian, China). The use of experimental animals
was approved by the ethics committee of Dalian Medical University
and strictly followed the guideline for tumor induction in mice and
rats. Written informed consent was obtained from the patient.
Patient consent for publication
The authors declared that the patients provided
written informed consent for the publication of the associated data
in this manuscript.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wright JD, Barrena Medel NI, Sehouli J,
Fujiwara K and Herzog TJ: Contemporary management of endometrial
cancer. Lancet. 379:1352–1360. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Salvesen HB, Haldorsen IS and Trovik J:
Markers for individualised therapy in endometrial carcinoma. Lancet
Oncol. 13:e353–e361. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Asghar U, Witkiewicz AK, Turner NC and
Knudsen ES: The history and future of targeting cyclin-dependent
kinases in cancer therapy. Nat Rev Drug Discov. 14:130–146. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sherr CJ, Beach D and Shapiro GI:
Targeting CDK4 and CDK6: From discovery to therapy. Cancer Discov.
6:353–367. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Witkiewicz AK, Knudsen KE, Dicker AP and
Knudsen ES: The meaning of p16ink4a expression in
tumors: Functional significance, clinical associations and future
developments. Cell Cycle. 10:2497–2503. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Halperin R, Zehavi S, Habler L, Hadas E,
Bukovsky I and Schneider D: Comparative immunohistochemical study
of endometrioid and serous papillary carcinoma of endometrium. Eur
J Gynaecol Oncol. 22:122–126. 2001.PubMed/NCBI
|
|
8
|
Yemelyanova A, Ji H, Shih IeM, Wang TL, Wu
LS and Ronnett BM: Utility of p16 expression for distinction of
uterine serous carcinomas from endometrial endometrioid and
endocervical adenocarcinomas: Immunohistochemical analysis of 201
cases. Am J Surg Pathol. 33:1504–1514. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen W, Husain A, Nelson GS, Rambau PF,
Liu S, Lee CH, Lee S, Duggan MA and Köbel M: Immunohistochemical
profiling of endometrial serous carcinoma. Int J Gynecol Pathol.
36:128–139. 2017.PubMed/NCBI
|
|
10
|
Chen YW, Chu HC, Ze-Shiang Lin, Shiah WJ,
Chou CP, Klimstra DS and Lewis BC: p16 Stimulates CDC42-dependent
migration of hepatocellular carcinoma cells. PLoS One.
8:e693892013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
McLaughlin-Drubin ME, Park D and Munger K:
Tumor suppressor p16INK4A is necessary for survival of
cervical carcinoma cell lines. Proc Natl Acad Sci USA.
110:16175–16180. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pauck A, Lener B, Hoell M, Kaiser A,
Kaufmann AM, Zwerschke W and Jansen-Dürr P: Depletion of the cdk
inhibitor p16INK4a differentially affects proliferation
of established cervical carcinoma cells. J Virol. 88:5256–5262.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Romagosa C, Simonetti S, Lopez-Vicente L,
Mazo A, Lleonart ME, Castellvi J and Ramon y Cajal S:
p16Ink4a overexpression in cancer: A tumor suppressor
gene associated with senescence and high-grade tumors. Oncogene.
30:2087–2097. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
McLaughlin-Drubin ME, Crum CP and Munger
K: Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B
histone demethylase expression and causes epigenetic reprogramming.
Proc Natl Acad Sci USA. 108:2130–2135. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Junttila TT, Akita RW, Parsons K, Fields
C, Lewis Phillips GD, Friedman LS, Sampath D and Sliwkowski MX:
Ligand-independent HER2/HER3/PI3K complex is disrupted by
trastuzumab and is effectively inhibited by the PI3K inhibitor
GDC-0941. Cancer Cell. 15:429–440. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu P, Cheng H, Santiago S, Raeder M,
Zhang F, Isabella A, Yang J, Semaan DJ, Chen C, Fox EA, et al:
Oncogenic PIK3CA-driven mammary tumors frequently recur via PI3K
pathway-dependent and PI3K pathway-independent mechanisms. Nat Med.
17:1116–1120. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Juvekar A, Burga LN, Hu H, Lunsford EP,
Ibrahim YH, Balmana J, Rajendran A, Papa A, Spencer K, Lyssiotis
CA, et al: Combining a PI3K inhibitor with a PARP inhibitor
provides an effective therapy for BRCA1-related breast cancer.
Cancer Discov. 2:1048–1063. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee GY, Kenny PA, Lee EH and Bissell MJ:
Three-dimensional culture models of normal and malignant breast
epithelial cells. Nat Methods. 4:359–365. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li L, Chang W, Yang G, Ren C, Park S,
Karantanos T, Karanika S, Wang J, Yin J, Shah PK, et al: Targeting
poly(ADP-ribose) polymerase and the c-Myb-regulated DNA damage
response pathway in castration-resistant prostate cancer. Sci
Signal. 7:ra472014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xiao Y, Wang J, Qin Y, Xuan Y, Jia Y, Hu
W, Yu W, Dai M, Li Z, Yi C, et al: Ku80 cooperates with CBP to
promote COX-2 expression and tumor growth. Oncotarget. 6:8046–8061.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Morton CL and Houghton PJ: Establishment
of human tumor xenografts in immunodeficient mice. Nat Protoc.
2:247–250. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang D, Li C, Zhang Y, Wang M, Jiang N,
Xiang L, Li T, Roberts TM, Zhao JJ, Cheng H, et al: Combined
inhibition of PI3K and PARP is effective in the treatment of
ovarian cancer cells with wild-type PIK3CA genes. Gynecol
Oncol. 142:548–556. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Netzer IM, Kerner H, Litwin L, Lowenstein
L and Amit A: Diagnostic implications of p16 expression in serous
papillary endometrial cancer. Int J Gynecol Cancer. 21:1441–1445.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Han G, Sidhu D, Duggan MA, Arseneau J,
Cesari M, Clement PB, Ewanowich CA, Kalloger SE and Köbel M:
Reproducibility of histological cell type in high-grade endometrial
carcinoma. Modern Pathol. 26:1594–1604. 2013. View Article : Google Scholar
|
|
26
|
Cancer Genome Atlas Research Network, ;
Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H,
Robertson AG, Pashtan I, Shen R, Benz CC, et al: Integrated genomic
characterization of endometrial carcinoma. Nature. 497:67–73. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Konecny GE, Winterhoff B, Kolarova T, Qi
J, Manivong K, Dering J, Yang G, Chalukya M, Wang HJ, Anderson L,
et al: Expression of p16 and retinoblastoma determines response to
CDK4/6 inhibition in ovarian cancer. Clin Cancer Res. 17:1591–1602.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Groves IJ, Knight EL, Ang QY, Scarpini CG
and Coleman N: HPV16 oncogene expression levels during early
cervical carcinogenesis are determined by the balance of epigenetic
chromatin modifications at the integrated virus genome. Oncogene.
35:4773–4786. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Soto DR, Barton C, Munger K and
McLaughlin-Drubin ME: KDM6A addiction of cervical carcinoma cell
lines is triggered by E7 and mediated by p21CIP1
suppression of replication stress. PLoS Pathog. 13:e10066612017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhao N, Ang MK, Yin XY, Patel MR, Fritchie
K, Thorne L, Muldrew KL, Hayward MC, Sun W, Wilkerson MD, et al:
Different cellular p16INK4a localisation may signal
different survival outcomes in head and neck cancer. Br J Cancer.
107:482–490. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Di Vinci A, Perdelli L, Banelli B, Salvi
S, Casciano I, Gelvi I, Allemanni G, Margallo E, Gatteschi B and
Romani M: p16INK4a promoter methylation and protein
expression in breast fibroadenoma and carcinoma. Int J Cancer.
114:414–421. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gray-Schopfer VC, Cheong SC, Chong H, Chow
J, Moss T, Abdel-Malek ZA, Marais R, Wynford-Thomas D and Bennett
DC: Cellular senescence in naevi and immortalisation in melanoma: A
role for p16? Br J Cancer. 95:496–505. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hashizume R, Andor N, Ihara Y, Lerner R,
Gan H, Chen X, Fang D, Huang X, Tom MW, Ngo V, et al: Pharmacologic
inhibition of histone demethylation as a therapy for pediatric
brainstem glioma. Nat Med. 20:1394–1396. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cregan S, Breslin M, Roche G, Wennstedt S,
MacDonagh L, Albadri C, Gao Y, O'Byrne KJ, Cuffe S, Finn SP, et al:
Kdm6a and Kdm6b: Altered expression in malignant pleural
mesothelioma. Int J Oncol. 50:1044–1052. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Morozov VM, Li Y, Clowers MM and Ishov AM:
Inhibitor of H3K27 demethylase JMJD3/UTX GSK-J4 is a potential
therapeutic option for castration resistant prostate cancer.
Oncotarget. 8:62131–62142. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kruidenier L, Chung CW, Cheng Z, Liddle J,
Che K, Joberty G, Bantscheff M, Bountra C, Bridges A, Diallo H, et
al: A selective jumonji H3K27 demethylase inhibitor modulates the
proinflammatory macrophage response. Nature. 488:404–408. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wu W, Qin M, Jia W, Huang Z, Li Z, Yang D,
Huang M, Xiao C, Long F, Mao J, et al: Cystathionine-γ-lyase
ameliorates the histone demethylase JMJD3-mediated autoimmune
response in rheumatoid arthritis. Cell Mol Immunol. May
29–2018.(Epub ahead of print). doi: 10.1038/s41423-018-0037-8.
View Article : Google Scholar
|