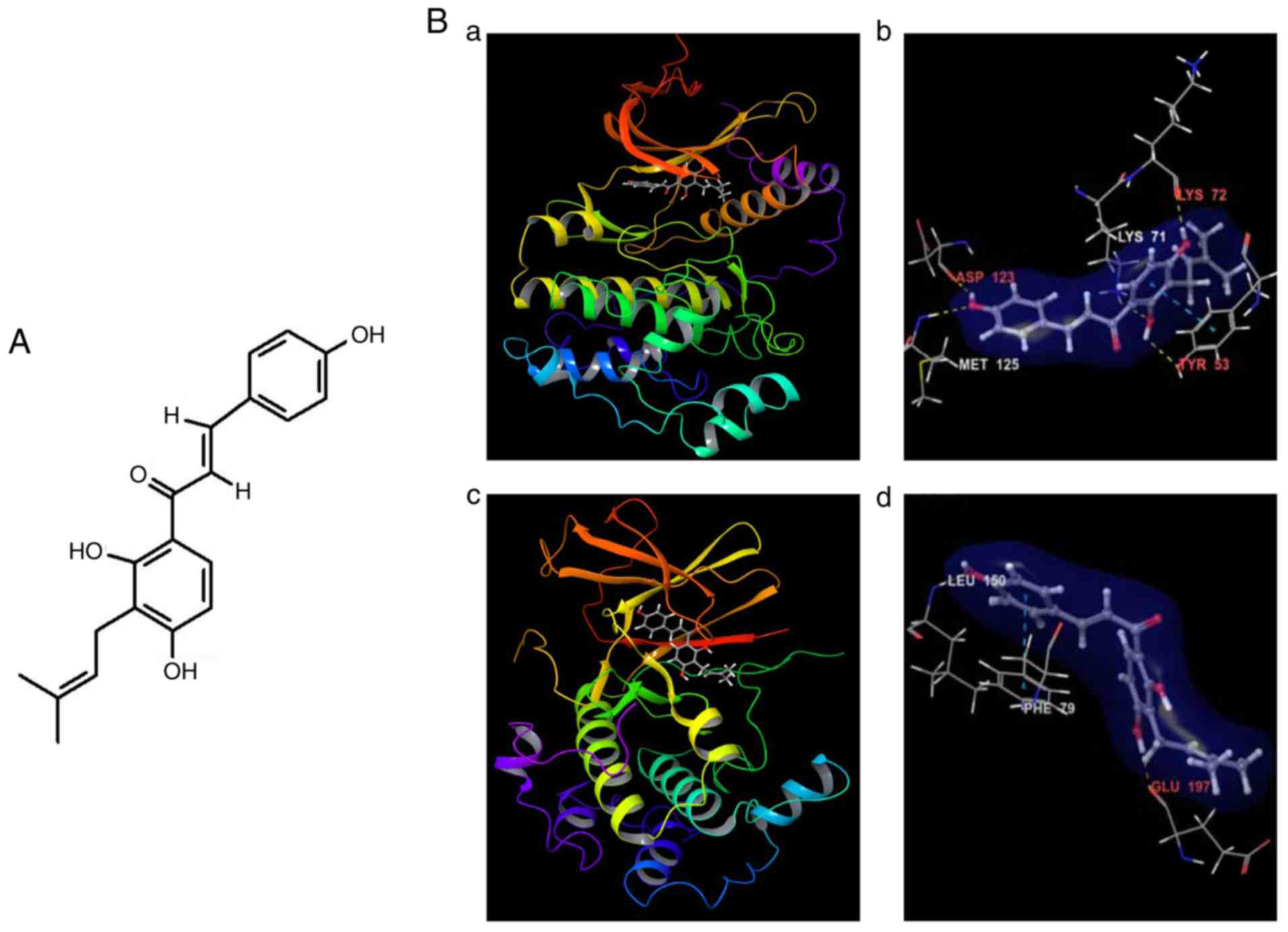
Introduction
Primary liver cancer is predicted to be the sixth
most common neoplasm and the fourth leading cause of
cancer-associated mortality globally in 2018, responsible for
~782,000 mortalities annually (1).
Additionally, 75–85% of primary liver cancer cases occurring
globally are hepatocellular carcinoma (1). Etiologically, the carcinogenesis of
liver cancer is a complex, multistep and multifactorial process
involving environmental risk factors, genetic derangement and
aberrant signal transduction (2).
The mitogen-activated protein kinase (MAPK) pathways are implicated
in various cellular processes, including cell survival,
proliferation, differentiation and apoptosis. The components of
MAPK signaling have been indicated to be promising targets in
developing novel chemoprevention and targeted therapies for cancer
(3). It was notable that the
aberrant activation of the Raf/mitogen-activated protein kinase
kinase (MEK)/extracellular signal-regulated kinases (ERKs) pathway
serves a pivotal role in the development, progression and
invasiveness of liver cancer and is associated with poor survival
rate and multidrug resistance (4,5).
Sorafenib and BAY86-9766, inhibitors targeting the Raf/MEK/ERKs
pathway, have been evaluated in clinical trials and have
demonstrated promising results in the treatment of unresectable
liver cancer (6,7). However, as this protein kinase pathway
is central to numerous signaling networks, including
phosphoinositide 3-kinase (PI3K)/Akt, Janus kinase/signal
transducer and activator of transcription and protein kinase
C/nuclear factor-κB, blocking Raf/MEK/ERKs signaling may result in
a number of adverse events and drug resistance (3,8).
Sorafenib suppresses the MAPK signaling pathways, but partially
induces the activation of PI3K/Akt signaling, which is responsible
for sorafenib-promoted invasion and metastasis in liver cancer
(9). Therefore, it is proposed that
blocking a subset of downstream functions of the MAPK pathways may
be a more effective strategy with fewer side effects.
Ribosomal S6 kinase 2 (RSK2), a member of the p90RSK
protein family, is a direct substrate kinase of ERK1/2 and is
activated in response to oncogenic signals and/or growth factor
stimuli (10). Alterations of the
ERKs pathway have been well documented in human liver cancer.
Increased expression and functional activity of ERK1/2 were
observed in primary liver cancer, compared with the adjacent
non-cancerous tissues (11,12). Constitutively activated ERK1/2 was
demonstrated to be required for the proliferation and invasion of
live cancer cells (13,14). Hepatitis B and C viruses, the two
most common causes of liver cancer, express viral proteins that
activate the ERKs signaling cascade (15,16).
The ERKs/RSK pathway was demonstrated to mediate ethanol-induced
proliferation of HepG2 cells (17).
Collectively, these observations implied that aberrant activation
of the ERKs/RSK2 pathway may be an important molecular event in
hepatocarcinogenesis, and it is desirable that the ERKs/RSK2
signaling axis becomes the subject of target-based therapies for
liver cancer.
In the last few decades, natural products extracted
from plants have attracted notable attention as potential
anticancer agents possessing efficacy and safety (18). Considering the increasing drug
resistance, the broad inhibition of multiple signaling pathways or
targets, rather than a single specific target, may represent a more
promising strategy for cancer treatment (19). Recently, computational biology has
been frequently applied for virtual screening of multiple-target
inhibitors, with the advantage of selecting a smaller number of
lead compounds in a large database for biological testing, while
avoiding expensive and time-consuming experiments (20). In the present study, using
structure-based virtual screening and molecular docking, >500
traditional Chinese medicine compounds available from TianJin
ShiLan Technology Company were screened for identification of
potential inhibitors targeting the ERKs/RSK2 pathway. The results
demonstrated that isobavachalcone (IBC), a natural chalcone
compound, can be bound inside the ATP binding pocket of ERK1/2 and
RSK2 with high affinity, implicating that IBC may be an
ATP-competitive inhibitor targeting ERK1/2 and RSK2. The antitumor
activity of IBC against liver cancer cells and its inhibitory
effect on the ERKs/RSK2 signaling pathway were further evaluated,
in the hope of providing novel insight into the potential
application of IBC as a chemotherapeutic agent for liver
cancer.
Materials and methods
Virtual screening and molecular
modeling
The three-dimensional structures of RSK2 and ERKs
were obtained from the PDB databank (http://www.rcsb.org/) [RSK2 N-terminal kinase domain,
PDB ID 3UBD (21); RSK2 C-terminal
kinase domain, PDB ID 4D9U (22);
ERK1 kinase domain, PDB ID 4QTB; and ERK2 kinase domain, PDB ID
4QTA (23)]. Prior to virtual
screening, the raw PDB structures were converted into an all-atom,
fully prepared receptor model structure with the Protein
Preparation Wizard module in Schrödinger (24). Subsequently, the docking receptor
grids were created by Glide's Receptor Grid Generation (25). The grid boxes and centers were set
to default with the co-crystal ligands in their ATP binding
sites.
The structures of >500 traditional Chinese
medicine compounds, which were downloaded from the PubChem Compound
database (http://www.ncbi.nlm.nih.gov/pccompound/), were
downloaded and a small compound database referred to as the ShiLan
database was built using the LigPrep module (26). These compounds were available from
TianJin ShiLan Technology Company (Tianjin, China). Glide was used
to run the extra precision (XP)-docking screening of this ShiLan
database targeted to two protein receptor structures (3UBD and
4D9U) (27,28). The screening processes outputted two
rank lists of nearly 50 compounds each. The compound IBC
(CID5281255) appeared in each list, and was manually selected for
further experimental tests.
Flexible ligand-protein docking was performed using
the Induced Fit Docking (IFD) Module (29) in Schrödinger to assess the possible
binding modes between ERKs/RSK2 and IBC. The induced fit docking
can capture the possible conformational changes in receptor active
site upon ligand binding. During IFD, the grid box and center for
each receptor structure were set to default with the co-crystal
ligand in its active binding site. Docking of IBC using its
LigPrep's minimized structure into each receptor structure was also
performed with Glide in XP mode (28). The binding pose with the lowest
docking score was considered as the correct binding structure. The
docking structures for each of the ERKs/RSK2-IBC complexes were
generated using Maestra (30) in
Schrödinger.
Reagents and antibodies
IBC (98% purity) was purchased from Tianjin ShiLan
Technology Company, and then it was dissolved in dimethyl sulfoxide
(DMSO; 100 mM stock solution). CNBr-activated Sepharose 4B and
epidermal growth factor (EGF) were purchased from Sigma-Aldrich
(Merck KGaA, Darmstadt, Germany). The primary antibodies were
purchased from the following companies: Mouse monoclonal antibodies
against p53 (cat. no. 2524; 1:1,000) and caspase-9 (cat. no. 9508;
1:1,000), rabbit monoclonal antibodies against cleaved caspase-9
(cat. no. 7237; 1:1,000), caspase-7 (cat. no. 12827; 1:1,000),
cleaved caspase-7 (cat. no. 8438; 1:1,000), caspase-3 (cat. no.
9665; 1:1,000), cleaved caspase-3 (cat. no. 9664; 1:1,000), poly
ADP-ribose polymerase (PARP; cat. no. 9542; 1:1,000), ERK1/2 (cat.
no. 5695; 1:2,000), phospho-ERK1/2 (Thr202/Tyr204; cat. no. 4370;
1:1,000), phospho-RSK2 (Ser227; cat. no. 3556; 1:1,000) and Flag
tag (cat. no. 14793; 1:1,000), and rabbit polyclonal antibodies
against RSK2 (cat. no. 9340; 1:1,000), stress-activated protein
kinase/c-Jun NH2-terminal kinase (SAPK/JNK; cat. no. 9525;
1:1,000), histone H3 (cat. no. 9715; 1:1,000) and phospho-histone
H3 (Ser10) (cat. no. 9701; 1:1,000) purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA); mouse monoclonal antibodies
against mouse double minute 2 homolog (MDM2; cat. no. sc-13161;
1:500), RSK2 (cat. no. sc-9968; 10 µl) and β-actin (cat. no.
sc-8432; 1:1,000) purchased from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA); rabbit polyclonal antibodies against cAMP
response element-binding protein (CREB; cat. no. ab31387; 1:1,000)
and activating transcription factor 1 (ATF1; cat. no. ab225880;
1:1,000), and rabbit monoclonal antibodies against phospho-CREB
(Ser133; cat. no. ab32096; 1:1,000) and phospho-ATF1 (Ser63; cat.
no. ab76085; 1:1,000) purchased from Abcam (Cambridge, MA, USA);
mouse monoclonal antibodies against B-cell lymphoma 2 (Bcl-2; cat.
no. 610538; 1:1,000) and Bcl-2 associated X protein (Bax; cat. no.
610982; 1:1,000) purchased from BD Transduction Laboratory (BD
Biosciences; Becton, Dickinson and Company, Franklin Lakes, NJ,
USA). Dylight 680-conjugated anti-mouse IgG (cat. no. 610-144-002;
1:10,000) and Dylight 800-conjugated anti-rabbit IgG (cat. no.
611-145-002; 1:10,000) secondary antibodies were purchased from
Rockland Immunochemicals, Inc. (Limerick, PA, USA).
Cell culture and transfection
The human liver cancer cell lines HepG2 and Hep3B
and the normal immortalized liver cell line L02 were obtained from
American Type Culture Collection (Manassas, VA, USA). HepG2 and
Hep3B cells were maintained in Dulbecco's modified Eagle's medium
(DMEM; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (FBS), 100 mg/ml
streptomycin, 100 IU/ml penicillin at 37°C in an incubator
containing 5% CO2, and L02 cells were cultured in DMEM
with 20% FBS. Cells freshly revived from cryopreservation were
maintained and cultured for ≤8 weeks, as aforementioned. Activating
protein-1 (AP-1) luciferase reporter vector was provided by Dr
ArndKieser (Helmholtz Zentrum München, Munich, Germany). The
pCMV3-C-Flag-RSK2 (pCMV3-RSK2) vector and pCMV3-C-Flag (pCMV3)
control vector were obtained from Sino Biological Inc. (Beijing,
China). To construct the short hairpin RNA (shRNA) vector targeting
RSK2 (shRSK2) and the scramble shRNA control vector (shCtrl), the
mU6pro vector was digested with XbaI and BbsI. The
annealed synthetic primers (shRSK2, sense,
5′-TTTGAAGGCAGATCCTTCCCAGTTTCAAGAGAACTGGGAAGGATCTGCCTTTTTTT-3′, and
antisense,
5′-CTAGAAAAAAAGGCAGATCCTTCCCAGTTCTCTTGAAACTGGGAAGGATCTGCCTT-3′; and
shCtrl, sense,
5′-TTTGACTACCGTTGTTATAGGTGTTCAAGAGACACCTATAACAACGGTAGTTTTTT-3′, and
antisense,
5′-CTAGAAAAAAACTACCGTTGTTATAGGTGTCTCTTGAACACCTATAACAACGGTAGT-3′)
were then introduced into the mU6pro vector (31). The recombinant plasmids were
confirmed by DNA sequencing. L02 cells (5×105 cells/well) or HepG2
cells (4×105 cells/well) were seeded in 6-well plates and cultured
to 60–70% confluence. Then, pCMV3-RSK2 (3 µg), shRSK2 (3 µg) and
their corresponding controls were transiently transfected into
cells with jetPEI™ DNA transfection reagent (Polyplus-Transfection
SA, Illkirch, France), according to the manufacturer's protocols.
After being transfected for 24 h, cells were used for subsequent
experimentation.
Cell viability assay
HepG2, Hep3B or L02 cells were seeded into 96-well
plates at a density of 3×103 cells/well, and then
incubated with 5, 10, 20 or 40 µM IBC for 24 or 48 h at 37°C. Cell
viability was determined with a Cell Counting Kit-8 (CCK-8; Dojindo
Molecular Laboratories, Inc., Kumamoto, Japan) assay. A total of 10
µl CCK-8 solution was added to each well and cells were incubated
for 2 h at 37°C. The absorbance was measured at a wavelength of 450
nm using a Synergy 2 Multi-Mode Microplate Reader (BioTek
Instruments, Inc., Winooski, VT, USA). The half-maximal inhibitory
concentration (IC50) of IBC was calculated by Probit
analysis using SPSS 16.0 software (SPSS, Inc., Chicago, IL,
USA).
EdU incorporation assay
Cell proliferation was assessed using a Cell-Light
EdU DNA Cell Proliferation kit (Guangzhou RiboBio Co., Ltd.,
Guangzhou, China). HepG2 or Hep3B cells were seeded into 96-well
plates at a density of 3×103 cells/well, and then
incubated with 10 or 20 µM IBC for 48 h at 37°C. Following
treatment with IBC, cells were exposed to 50 µM EdU for 2 h at
37°C, and then fixed with 4% formaldehyde for 15 min and
permeabilized with 0.5% Triton X-100 for 15 min at room
temperature. Subsequently, cells were incubated with 100 µl 1X
Apollo® reaction cocktail for 30 min at room
temperature, followed by Hoechst 33342 (5 µg/ml) for 30 min at room
temperature in the dark. The stained cells were imaged under a
fluorescence microscope (Olympus Corporation, Tokyo, Japan) at a
magnification of ×200, and the EdU positive ratio was calculated as
(EdU-labeled cells/Hoechst-stained cells) × 100%.
Colony formation assay
HepG2 or Hep3B cells were seeded in 6-well plates at
a density of 500 cells/well and cultured with DMEM complete medium
containing 5 or 10 µM IBC for 2 weeks at 37°C. The cell colonies
were fixed with methanol for 15 min at room temperature and then
stained with 0.5% crystal violet for 15 min at room temperature.
The number of colonies containing ≥50 cells was counted under an
inverted optical microscope at a magnification of ×40.
Analysis of cell cycle and
apoptosis
HepG2 or Hep3B cells were starved in serum-free DMEM
medium for 24 h at 37°C and treated with 10 or 20 µM IBC for
another 48 h at 37°C. Subsequently, cells were harvested with
trypsin and fixed with 70% ice-cold ethanol at 4°C overnight. The
fixed cells were stained via incubation with 10 µg/ml propidium
iodide (PI) and 100 µg/ml RNase for 30 min at room temperature in
the dark. For the apoptosis analysis, cells were treated with 10 or
20 µM IBC for 48 h at 37°C and stained with Annexin V-fluorescein
isothiocyanate and PI (Nanjing KeyGen Biotech Co., Ltd., Nanjing,
China) for 15 min at room temperature in the dark, according to the
manufacturer's instructions. All of the samples were analyzed using
a FACS Calibur flow cytometer with Cell Quest software version 3.0
(BD Biosciences; Becton, Dickinson and Company).
Protein extraction and western
blotting
HepG2 or Hep3B cells were harvested and total
protein was extracted using Radioimmunoprecipitation Assay lysis
buffer (Beyotime Institute of Biotechnology, Haimen, China) with
phenylmethane sulfonyl fluoride (PMSF). The isolation of histone
protein was performed as described previously (32). Protein concentration was determined
using a bicinchoninic acid kit (Pierce; Thermo Fisher Scientific,
Inc.). Equal amounts of total protein (50 µg) or histone protein
(20 µg) were resolved by 10 or 15% SDS-PAGE, respectively, and
transferred to polyvinylidene fluoride membranes (EMD Millipore,
Billerica, MA, USA). The membranes were incubated with blocking
buffer (PBS with 5% non-fat milk and 0.1% Tween-20) for 1 h at room
temperature, and then probed with specific primary antibodies
overnight at 4°C. Subsequently, the membranes were incubated with
the species-appropriate infrared-dye-conjugated secondary
antibodies for 2 h at 4°C, and then protein bands were visualized
using an Odyssey Infrared Imaging System (LI-COR Biosciences,
Lincoln, NE, USA).
Preparation of Sepharose 4B beads and
in vitro pull-down assay
CNBr-activated Sepharose 4B beads (0.1 g) were
washed five times with 1 mM HCl and collected by centrifugation at
500 × g for 3 min at 4°C. Subsequently, the beads were incubated
with 3 mg IBC or DMSO in coupling buffer [0.1 M NaHCO3
and 0.5 M NaCl (pH 8.3)] with gentle rocking overnight at 4°C. The
beads were then washed five times with coupling buffer to remove
excess ligand and incubated in blocking buffer [0.1 M Tris-HCl (pH
8.0)] overnight at 4°C. Afterwards, the beads were alternatively
washed in three cycles with 0.1 M acetic acid buffer (pH 4.0)
containing 0.5 M NaCl and 0.1 M Tris-HCl (pH 8.0) containing 0.5 M
NaCl, and then resuspended in 500 µl PBS. IBC-Sepharose 4B beads
(or only Sepharose 4B as a control) were then incubated with
cellular supernatant fractions of HepG2 or Hep3B cells (500 µg) in
reaction buffer [50 mM Tris (pH 7.5), 5 mM EDTA, 150 mM NaCl, 1 mM
DTT, 0.01% NP40 and 0.02 mM PMSF] containing 2 µg/ml BSA and
protease inhibitor cocktail (Roche Applied Science, Penzberg,
Germany) with gentle rocking overnight at 4°C. Finally, the beads
were washed five times with reaction buffer, and the bound proteins
were visualized by western blotting according to the aforementioned
protocol.
Immunoprecipitation (IP) and in vitro
kinase assay
The pCMV3-RSK2 expression vector was transiently
introduced into HepG2 cells, and total protein was extracted using
IP lysis buffer (Beyotime Institute of Biotechnology). Cell
extracts (300 µg) were incubated with 2 µg anti-Flag tag antibody
overnight at 4°C, followed by incubation with 20 µl of protein A/G
agarose beads (Santa Cruz Biotechnology, Inc.) for 2 h at 4°C. The
precipitated beads were washed three times with lysis buffer and
twice with 1X kinase buffer (Cell Signaling Technology, Inc.). The
kinase reactions were performed in a total of 40 µl 1X kinase
buffer supplemented with 200 µM ATP (Cell Signaling Technology,
Inc.), 1 µg recombinant human histone H3 (New England BioLabs,
Inc., Ipswich, MA, USA), and 10 or 20 µM IBC at 30°C for 30 min.
Reactions were terminated with 6X SDS sample buffer (Beyotime
Institute of Biotechnology), and then the supernatants were boiled
for 5 min at 95°C and resolved by 15% SDS-PAGE. The phosphorylation
levels of histone H3 were detected by western blotting according to
the aforementioned protocol.
The effect of IBC on the endogenous RSK2 activity
was further analyzed. HepG2 cells were starved in serum-free DMEM
medium for 24 h at 37°C and treated with 10 or 20 µM IBC for 2 h
prior to exposure to EGF (10 ng/ml) for 15 min at 37°C. Cell
extracts (500 µg) were used for IP with RSK2 antibody according to
the aforementioned protocol, and then an in vitro kinase
assay was performed with histone H3 peptide as a substrate.
Reporter gene assay
HepG2 or Hep3B cells were seeded into 24-well plates
at a density of 1×105 cells/well, and then were
co-transfected with AP-1 luciferase reporter vector (1.0 µg) and
pRL-TK Renilla luciferase vector (0.02 µg) (Promega
Corporation, Madison, WI, USA) using jetPEI DNA transfection
reagent. At 24 h after transfection, cells were serum-starved for
another 12 h and pretreated with 10 or 20 µM IBC for 4 h at 37°C,
followed by stimulation with EGF (10 ng/ml) for 12 h at 37°C. Cells
were lysed with passive lysis buffer (Promega Corporation) for 20
min with gentle shaking, and then the firefly luciferase and
Renilla luciferase activities were measured using a
Dual-Luciferase assay system (Promega Corporation) in a FB12
Luminometer from Titertek-Berthold (Berthold Detection Systems
GmbH, Pforzheim, Germany). The firefly luciferase activity was
normalized to Renilla luciferase activity to equalize the
transfection efficiency.
Statistical analysis
All data were analyzed using SPSS 16.0 and GraphPad
Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA, USA).
Data from at least three independent experiments are expressed as
the mean ± standard deviation. An unpaired Student's t-test was
employed to evaluate the difference between two groups. The
differences were determined using one-way analysis of variance with
Dunnett's post hoc corrections for multiple comparisons. P<0.05
was considered to indicate a statistically significant
difference.
Results
Computational modeling between IBC and
ERKs/RSK2
IBC is a prenylated chalcone of the class flavonoid
(Fig. 1A). The predicted docking
structures of IBC into the binding pockets of ERKs and RSK2 kinases
are depicted in Fig. 1B.
Additionally, the ligand-protein interactions of IBC-ERKs/RSK2
complexes in two-dimensional diagrams are further depicted in
Fig. S1. IBC docked inside the ATP
binding pocket of ERK1 (Fig. 1B-a)
and formed five hydrogen-bonds with ERK1, with three involved in
the backbone atoms of the residues of Asp123, Met125 and Lys72,
while the other two engaged with the side-chain atoms of the
residues of Lys71 and Tyr53 in close proximity to the ATP binding
pocket (Fig. 1B-b and S1A). Among these five residues, Asp123
and Met125 were located in the hinge loop and Tyr53 was the gate
residue in the glycine-rich loop, which also developed
π-interactions with IBC between their benzene rings (Fig. 1B-b and S1A). Additionally, the 3-methyl-2-butene
tail of the ligand located inside a hydrophobic pocked formed by
the residues of Tyr53, Ile73, Ile89 and Ile120, while the other
carbon atoms of IBC also created the hydrophobic interaction with
the side-chains of the residues Ile48, Tyr53, Met55, Val56, Ala69,
Ile101, Leu124, Met125 and Leu173 around the ATP binding site
(Fig. S1A). The docking of this
compound to ERK2 provided a similar binding pose as that between
IBC and ERK1.
However, the docking of IBC to the N-terminal kinase
domain (NTKD) of RSK2 provided a binding pose (Fig. 1B-c) similar to the
SL0101/afzelin-bound form (21),
which demonstrated notable structural rearrangements of the N-lobe,
compared with the AMP-PNP-bound form (33) as a typical molecular architecture of
AGC kinases (34). The distinct
features of the conformational changes of RSK2NTKD from
the AMP-PNP-bound form (33) were
described previously (21). In the
docking model of IBC-RSK2NTKD, IBC formed two hydrogen
bonds with RSK2, with one involved in the backbone amide group of
the hinge loop residue Leu150, and the other engaged with the
backbone carbonyl oxygen of the residue Glu197 (Fig. 1B-d). IBC also developed
π-interactions between the benzene rings with the gate residue
Phe79 in the glycine-rich loop (Fig.
1B-d) and Phe212 in the DFG motif of the activation loop
(Fig. S1B). Additionally, the
3-methyl-2-butene tail of the ligand located inside a hydrophobic
pocked formed by the residues of Ile50, Leu155, Phe212 and Leu214,
while the other carbon atoms of IBC also created hydrophobic
interactions with the side-chains of the residues Phe79, Val82,
Ala98, Leu102, Val131, Leu145, Leu147, Phe149, Leu150, Leu155,
Leu200 and Phe212 around the ATP binding site (Fig. S1B). The docking energy between this
compound and RSK2 C-terminal kinase domain (CTKD) was demonstrated
to be >2.0 kcal/mol higher, compared with between IBC and
RSK2NTKD from virtual screening and induced fit docking
(Table SI). Thus, it was
considered that this ligand could be primarily bound inside the ATP
binding pocket in the N-terminal kinase domain of RSK2. Finally,
these computational results indicate that IBC possibly exhibits
ATP-competitive inhibitory effects on ERK1/2 and RSK2 kinases.
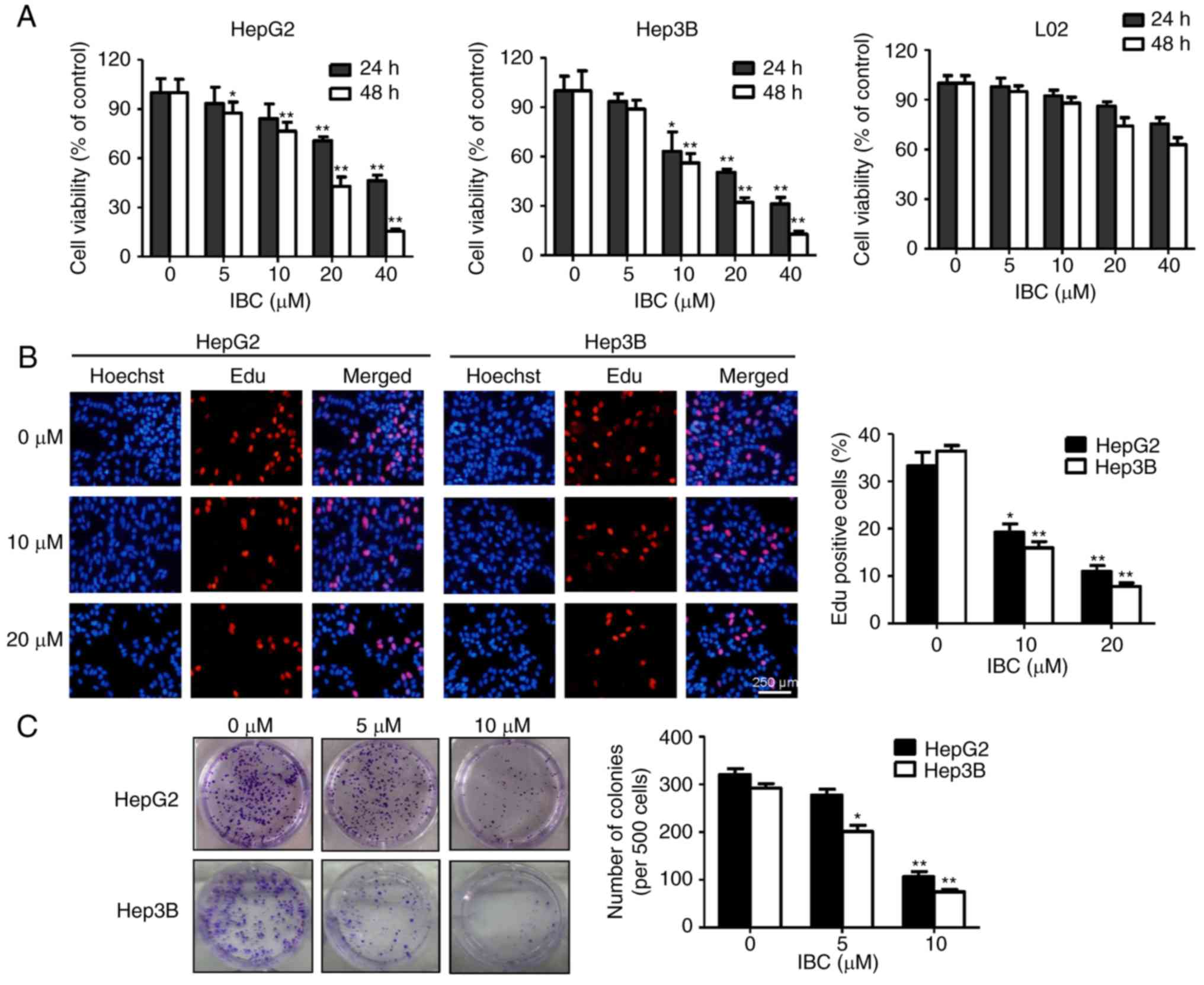
IBC inhibits the proliferation of
liver cancer cells
In view of the computer predication that IBC may be
a potential inhibitor of ERKs/RSK2 signaling, the cytotoxic effects
of IBC on liver cancer cells and immortalized hepatocytes were
investigated. As depicted in Fig.
2A, IBC significantly decreased the viability of liver cancer
HepG2 and Hep3B cells in a concentration- and time-dependent
manner. The IC50 values of IBC on HepG2 and Hep3B cells
at 48 h were 16.45 and 13.22 µM, respectively. In contrast, a less
significant cytotoxic effect was observed in immortalized normal
liver L02 cells. The EdU incorporation assay was performed to
further determine the effects of IBC on the proliferation of HepG2
and Hep3B cells. As depicted in Fig.
2B, following exposure to various concentrations of IBC for 48
h, the ratio of EdU-positive cells was gradually reduced with the
increasing concentrations of IBC. Furthermore, the colony-forming
ability of HepG2 and Hep3B cells was attenuated by IBC in a
concentration-dependent manner (Fig.
2C). Collectively, these observations indicated that IBC
effectively suppresses the viability and proliferation of liver
cancer cells rather than normal hepatocytes.
IBC induces cell apoptosis in liver
cancer cells
The effects of IBC on cell cycle distribution and
apoptosis were further analyzed using flow cytometry. Notably, it
was determined that IBC exerted no notable influence on cell cycle
distributions of HepG2 and Hep3B cells, even up to the
concentration of 20 µM (Fig. S2).
However, treatment with IBC could induce a notable
concentration-dependent increase of cell apoptosis, particularly
early apoptosis, in HepG2 and Hep3B cells (Fig. 3A). Subsequently, the effect of IBC
on apoptosis-associated molecules was investigated. As depicted in
Fig. 3B, exposure of HepG2 cells to
IBC caused the downregulation of MDM2 and Bcl-2, along with the
increase of p53 and Bax, in a concentration-dependent manner.
Additionally, procaspase-9 exhibited a significant decrease
response to IBC treatment, concomitant with the increasing levels
of caspase-3 and −7 activities and cleavage of PARP (Fig. 3C), indicating a hierarchical
activation of the apoptotic caspase cascade. Collectively, these
results indicated that the induction of caspase-mediated apoptosis
was involved in the antitumor activity of IBC on liver cancer
cells.
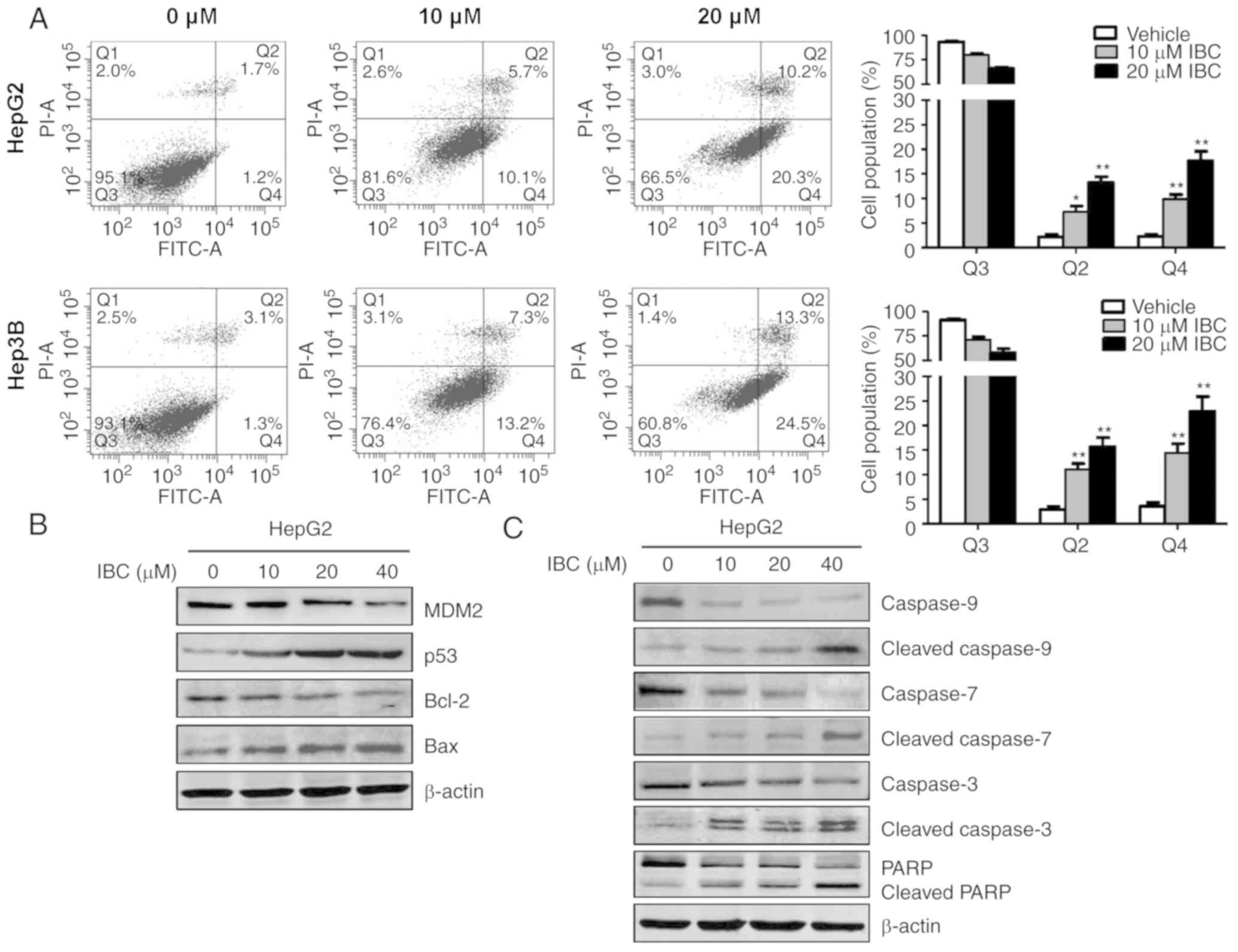 | Figure 3.IBC induces cell apoptosis in liver
cancer cells. (A) HepG2 or Hep3B cells were treated with different
concentrations of IBC for 48 h, and apoptosis was analyzed using
flow cytometry with Annexin V/PI double staining assay. Cells were
categorized into viable cells (Q3), early apoptotic cells (Q4),
late apoptotic cells (Q2) and dead cells (Q1). (B) HepG2 cells were
treated with different concentrations of IBC for 48 h. Cell lysates
were harvested and immunoblotted with anti-MDM2, p53, Bcl-2 and Bax
antibodies. (C) Cells were treated with different concentrations of
IBC for 48 h, and then were subjected to immunoblotting analysis
with caspase-9, caspase-7, caspase-3 and PARP. β-actin was used as
the loading control. Data are presented as the mean ± standard
deviation (n=3). *P<0.05 and **P<0.005 vs. vehicle-treated
control. IBC, isobavachalcone; PI, propidium iodide; FITC,
fluorescein isothiocyanate; MDM2, mouse double minute 2 homolog;
Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X; PARP, poly
ADP-ribose polymerase. |
IBC directly binds with ERKs/RSK2 and
inhibits RSK2 kinase activity
It has been determined from computer-aided virtual
screening and molecular modeling that ERK1/2 and RSK2 may represent
potential targets of IBC. To further determine whether IBC directly
binds with ERK1/2 and RSK2, an in vitro pull-down assay
using HepG2 or Hep3B cell lysates was conducted. As depicted in
Fig. 4A, ERK1/2 and RSK2 could bind
to the IBC-Sepharose 4B beads, but not to DMSO-Sepharose 4B beads.
Additionally, IBC could not selectively bind to JNK/SAPK (Fig. 4A).
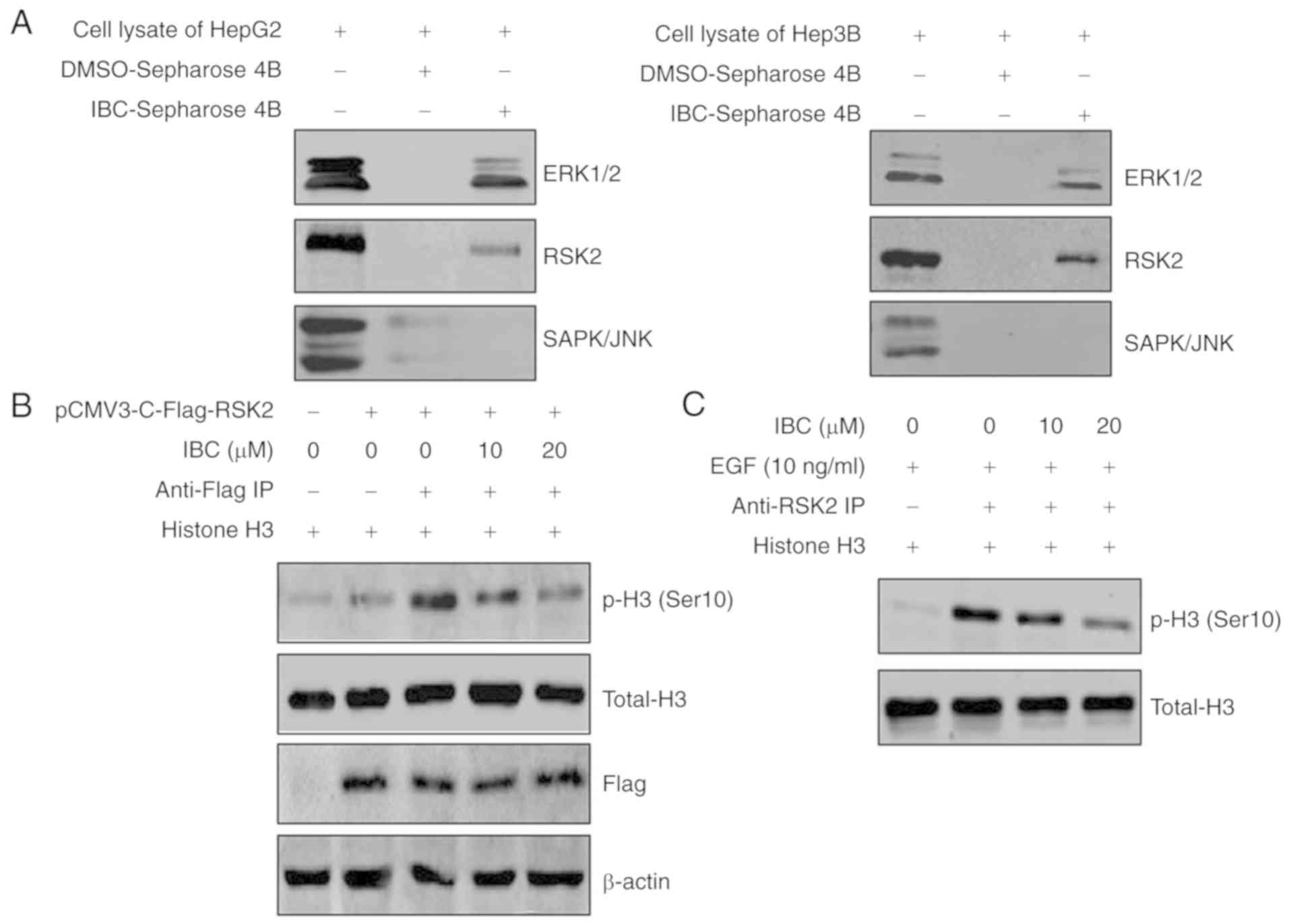 | Figure 4.IBC directly binds with ERK1/2 and
RSK2, and inhibits RSK2 kinase activity. (A) For the in
vitro IBC pull-down assay, lane 1 depicts cell lysates from
HepG2 or Hep3B cells used as input controls, lane 2 depicts cell
lysates incubated with DMSO-Sepharose4B beads used as the negative
controls and lane 3 depicts cell lysates incubated with
IBC-Sepharose4B beads, and then the precipitated proteins were
visualized by western blotting using antibodies against ERK1/2,
RSK2 and SAPK/JNK. (B) HepG2 cells were transfected with pCMV3-RSK2
or control vector, and then cell lysates were immunoprecipitated
with Flag-tagged antibody. The kinase assay was performed using
different concentrations of IBC and histone H3 peptide as a
substrate. The expression levels of p-histone H3 (Ser10), histone
H3 and Flag-tag were detected by western blotting. (C) HepG2 cells
were serum-starved for 24 h, treated with different concentrations
of IBC for 2 h and then stimulated with EGF (10 ng/ml) for 15 min.
Cell lysates were immunoprecipitated with RSK2 antibody, and then
RSK2 kinase activity was determined using an in vitro kinase
assay with histone H3 peptide as a substrate. IBC, isobavachalcone;
ERK1/2, extracellular signal-regulated kinase 1/2; RSK2, ribosomal
S6 kinase 2; DMSO, dimethyl sulfoxide; SAPK/JNK, stress-activated
protein kinase/c-Jun NH2-terminal kinase; IP, immunoprecipitation;
EGF, epidermal growth factor; p-, phospho-. |
Subsequently, an IP kinase assay was conducted to
investigate the effect of IBC on RSK2 kinase activity. Histone H3
is a well-known phosphorylation substrate of RSK2 (35). The full-length RSK2 was transfected
into HepG2 cells and RSK2 protein was immunoprecipitated by
Flag-tagged antibody. The precipitates were subjected to an in
vitro kinase assay with various concentrations of IBC and
histone H3 peptide as a substrate. As depicted in Fig. 4B, treatment with IBC inhibited the
levels of histone H3 phosphorylation at Ser10 in a
concentration-dependent manner, compared with untreated control,
indicating that IBC could effectively block RSK2 kinase activity
in vitro. Additionally, whether IBC interferes with the
activity of endogenous RSK2 in HepG2 cells was investigated. Cells
were treated with various concentrations of IBC for 2 h, followed
by stimulation with EGF. The results from RSK2 IP kinase assay
revealed that IBC also significantly inhibited EGF-induced
endogenous RSK2 kinase activity (Fig.
4C). Overall, these observations indicated that IBC could
effectively suppress the RSK2 activity by directly binding to
ERK1/2 and RSK2 in liver cancer cells.
IBC suppresses EGF-induced activation
of the ERKs/RSK2 signaling pathway
Based on the aforementioned experimental results, it
was concluded that IBC exerted antitumor activity on liver cancer
cells through regulating the ERKs/RSK2 signaling pathway. The
effects of IBC on EGF-induced phosphorylation of ERK1/2 and RSK2
were examined by western blotting. As expected, EGF, a tumor
promoter, induced the phosphorylation of ERK1/2 and RSK2 in HepG2
and Hep3B cells (Fig. 5A).
Treatment with IBC significantly inhibited EGF-induced
phosphorylation of RSK2 in a concentration-dependent manner
(Fig. 5A), consistent with the
decrease of endogenous RSK2 activity by IBC. However, the
EGF-induced phosphorylation of ERK1/2 does not appear to be
affected by IBC (Fig. 5A). To
demonstrate the inhibitory role of IBC on the ERKs/RSK2 signaling
pathway, the status of its downstream target proteins, including
CREB, ATF1 and histone H3, whose phosphorylation is increased in
response to EGF-induced ERKs/RSK2 activation, were evaluated. As
depicted in Fig. 5B, the
phosphorylation levels of CREB, ATF1 and histone H3 induced by EGF
were notably abrogated by IBC in a concentration-dependent manner.
AP-1 is a dimeric transcription factor composed of Jun, Fos or ATF
protein family members (36). It is
well known that RSK2 is involved in the regulation of AP-1
transcriptional activity through its phosphorylation of histone H3
at Ser10 (31). The present results
indicated that EGF promoted AP-1 transcriptional activity and that
treatment with IBC resulted in a concentration-dependent inhibition
of AP-1 transactivation in liver cancer cells (Fig. 5C). Collectively, these results
provided evidence that IBC could effectively block the ERKs/RSK2
downstream signaling pathway, which may be responsible for the
anti-proliferation activity of IBC to some extent.
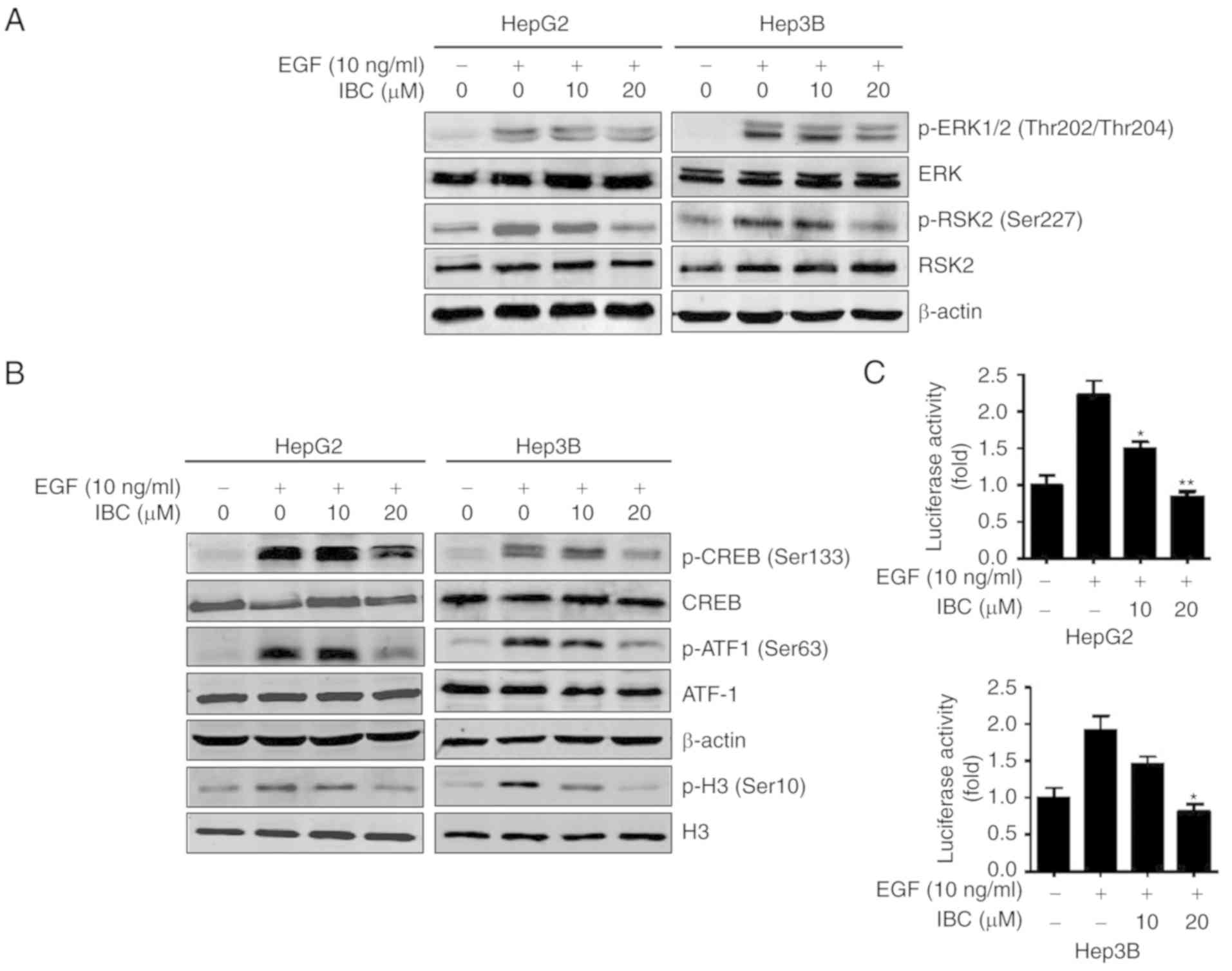 | Figure 5.IBC suppresses EGF-induced activation
of the ERKs/RSK2 signaling pathway in liver cancer cells. (A) HepG2
or Hep3B cells were serum-starved for 24 h, and then treated with
various concentrations of IBC for 2 h followed by exposure to EGF
(10 ng/ml) for 15 min. Cell lysates were harvested for
immunoblotting with anti-ERK1/2, p-ERK1/2 (Thr202/Thr204), RSK2 and
p-RSK2 (Ser227) antibodies. (B) HepG2 or Hep3B cells were serum
starved for 24 h, and then treated with various concentrations of
IBC for 2 h followed by stimulation with EGF. The expression levels
of CREB, p-CREB (Ser133), ATF1, p-ATF1 (Ser63) and p-histone H3
(Ser10) were detected by western blotting. β-actin and histone H3
were used as loading controls. (C) HepG2 or Hep3B cells were
co-transfected with AP-1 luciferase reporter gene and pRL-TK
Renilla luciferase vector. At 24 h after transfection, cells
were serum-starved for 12 h, and then treated with various
concentrations of IBC for 4 h followed by exposure to EGF for 12 h.
The firefly luciferase activity was determined in cell lysates and
normalized against Renilla luciferase activity. Data are
presented as the mean ± standard deviation (n=3). *P<0.05 and
**P<0.01 vs. vehicle-treated control stimulated by EGF. IBC,
isobavachalcone; EGF, epidermal growth factor; ERKs, extracellular
signal-regulated kinases; RSK2, ribosomal S6 kinase 2; CREB, cAMP
response element-binding protein; ATF1, activating transcription
factor 1; AP-1, activator protein 1; p-, phospho-. |
RSK2-mediated signaling is involved in
IBC-induced suppression of liver cancer cells
To further determine the role of RSK2 in IBC-induced
suppression of liver cancer cells, the expression levels of RSK2 in
HepG2, Hep3B and L02 cells were detected. As depicted in Fig. 6A, compared with L02 cells, elevated
total- and phospho-RSK2 protein levels were observed in HepG2 and
Hep3B cells. On this basis, a vector encoding RSK2 was transfected
into L02 cells, and then cell viability was evaluated with a CCK-8
assay under the conditions with or without IBC. Expectedly,
overexpression of RSK2 significantly promoted the proliferation of
L02 cells (Fig. S3). Furthermore,
enforced RSK2 expression in L02 cells notably enhanced the
inhibitory effect of IBC on cell viability (Fig. 6B). Additionally, it was observed
that RSK2 overexpression notably increased colony formation
capacity in L02 cells, whereas IBC effectively counteracted the
effect of RSK2 on inducing colony formation (Fig. 6C). Conversely, RSK2 knockdown in
HepG2 cells reduced the effect of IBC on suppressing cell
proliferation in a concentration-dependent manner, compared with
the control cells (Fig. 6D).
Overall, these results supported the assertion that the
RSK2-mediated signaling pathway serves a critical role in the
inhibitory effect of IBC on liver cancer cells.
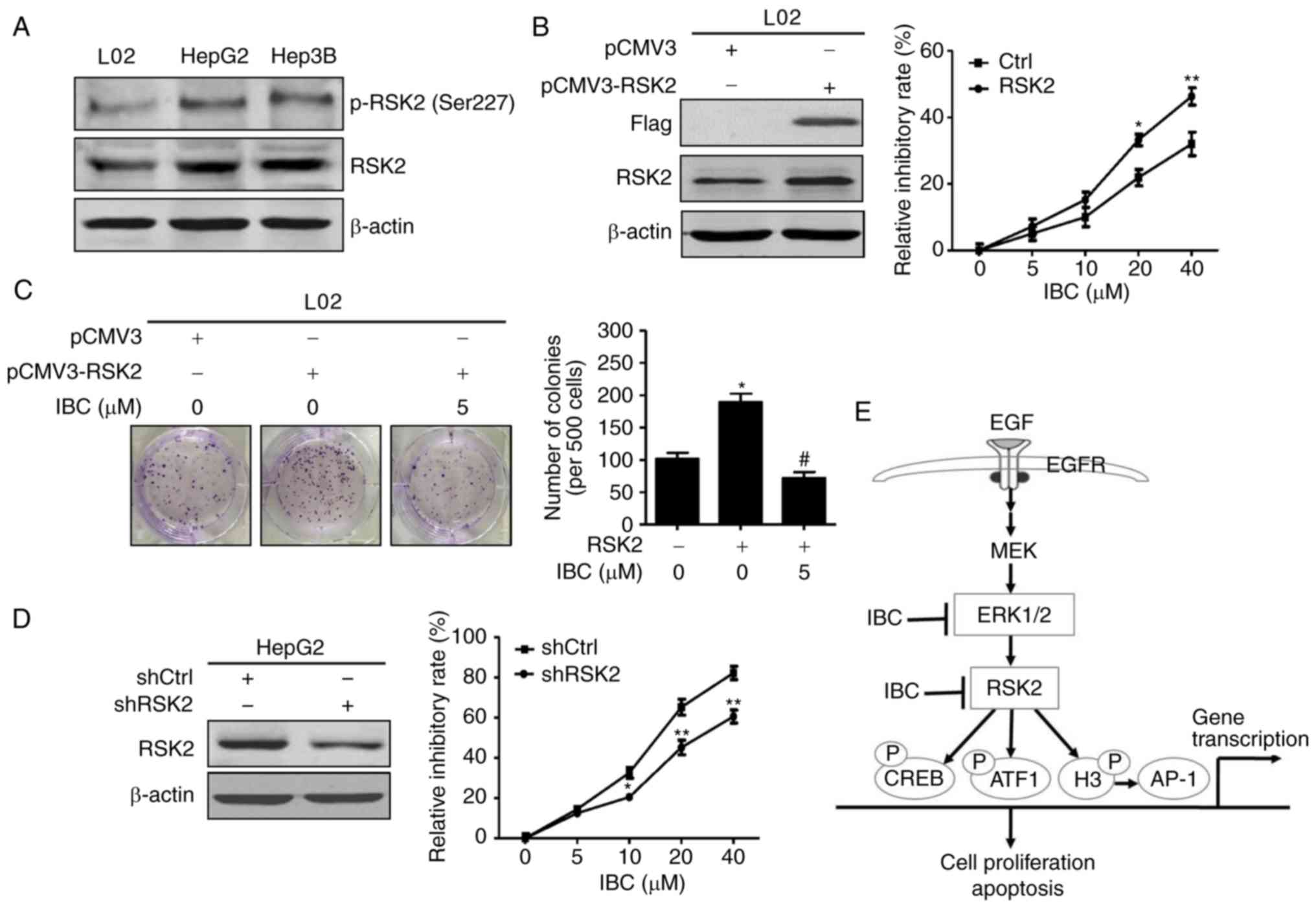 | Figure 6.IBC blocks RSK2-mediated cell
proliferation in liver cancer cells. (A) Cell lysates from L02,
HepG2 or Hep3B cells were harvested and analyzed by immunoblotting
with anti-RSK2 or p-RSK2 (Ser227). β-actin was used as the loading
control. (B) L02 cells were transfected with pCMV3-RSK2 or pCMV3
control vector. After being transfected for 24 h, cells were
aliquoted into 96-well plates and incubated with different
concentrations of IBC for another 48 h. Cell viability was assessed
with a CCK-8 assay. (C) L02 cells transfected with pCMV3-RSK2 or
control vector were treated with indicated concentrations of IBC
for 2 weeks, and then cell growth was detected with a colony
formation assay. (D) HepG2 cells were transfected with shRSK2 or
shCtrl control vector. After being transfected for 24 h, cells were
aliquoted into 96-well plates and incubated with different
concentrations of IBC for another 48 h. Cell viability was assessed
with a CCK-8 assay. Data are expressed as the mean ± standard
deviation (n=3). *P<0.05 and **P<0.01 vs. cells transfected
with control vector, #P<0.005 vs. vehicle-treated
cells transfected with RSK2. (E) Proposed signal transduction
pathways modulated by IBC in liver cancer cells. IBC suppressed
cell proliferation and induced cell apoptosis by directly targeting
ERK1/2 and RSK2, thereby blocking the activation of its downstream
transcription factors, including CREB, ATF1, histone H3 and AP-1.
IBC, isobavachalcone; RSK2, ribosomal S6 kinase 2; CCK-8, Cell
Counting Kit-8; Ctrl, control; sh, short hairpin; EGF, epidermal
growth factor; EGFR, epidermal growth factor receptor; MEK,
mitogen-activated protein kinase kinase; ERK1/2, extracellular
signal-regulated kinase 1/2; CREB, cAMP response element-binding
protein; ATF1, activating transcription factor 1; AP-1, activator
protein 1; P, phosphorylation. |
Discussion
Deregulation of Raf/MEK/ERKs signaling is implicated
in cell proliferation, survival, metastasis and tumorigenesis,
along with the overwhelming frequency in which this pathway is
aberrantly activated in human cancer types, including melanomas,
colorectal cancer, non-small cell lung cancer and liver cancer
(8). RSK2 is a downstream kinase of
ERKs and, functionally speaking, is located between ERKs and its
own target transcription factors. RSK2 was reportedly involved in
the proliferation of various cancer cells and neoplastic cell
transformation induced by tumor promoters, including EGF and
12-O-tetradecanoylphorbol-13-acetate (31,37).
Mice lacking RSK2 exhibited the reduced c-Fos-dependent
osteosarcoma formation (38).
Inhibition of RSK2 significantly blocked the proliferation and
invasion of human glioblastoma cells and enhanced the effectiveness
of temozolomide and irradiation therapy in temozolomide-resistant
glioblastoma cells (39). Notably,
increased total and activated RSK2 protein levels are exhibited in
various cancer cell lines and solid cancer types, including
melanoma, glioblastoma, and multiple myeloma, and are correlated
with advanced tumor stage and poor survival prognosis of patients
(39–41). The mRNA expression of RSK2 was
significantly increased in HCC tissues, compared with adjacent
non-tumor liver tissues according to the analysis of microarray
gene expression data (42). It was
also observed that the levels of total and phosphorylated RSK2 were
elevated in HepG2 and Hep3B cells, compared with normal liver L02
cells. Enforced expression of RSK2 in L02 cells substantially
promoted cell proliferation and colony formation. These
observations indicated that RSK2 serves a key role in cell
proliferation, transformation and cancer development and that
targeting RSK2 may represent a potential therapeutic strategy for
numerous human cancer types, including liver cancer.
RSK2 is composed of two functional kinase domains
that are activated by a series of phosphorylations in a sequential
manner (10). The NTKD of RSK2
serves a key role in the transducing RSK2 activation signal to its
substrates (10). However, the
activation of CTKD by upstream ERK1/2 is required for the
initiation of the activation process, resulting in activation of
RSK2 NTKD (10). Therefore,
high-throughout virtual screening was performed to identify the
potential inhibitors targeting ERKs/RSK2 from >500 traditional
Chinese medicine compounds. The results demonstrated that IBC, a
chalcone constituent from Psoralea corylifolia and
Angelica keiskei (43),
could bind to the ATP binding pocket of ERK1/2, as well as NTKD of
RSK2, indicating that IBC may be an ATP-competitive inhibitor
targeting ERK1/2 and RSK2. An in vitro pull-down assay
further demonstrated that IBC directly bound with ERK1/2 and RSK2.
Furthermore, IBC was demonstrated not only to suppress RSK2 kinase
activity, but also to abate EGF-induced phosphorylation of RSK2.
The decrease in ERK1/2 activity caused by IBC may account for the
abrogation of RSK2 phosphorylation. Notably, it was observed that
IBC did not modulate the phosphorylation of ERK1/2. Binding with
IBC may abrogate the catalytic activity of ERK1/2 on its downstream
substrates or directly interfere the interaction between ERK1/2 and
its substrates, but does not appear to affect the phosphorylation
of ERK1/2 mediated by its upstream kinases. Collectively, these
results demonstrated that ERK1/2 and RSK2 are potential targets of
IBC.
Among numerous biologically active chalcones, IBC is
one of the most resourceful compounds and is present in a variety
of plant families and species, particularly Fabaceae and Moraceae
(43). It is well-known that IBC
possesses a wide range of pharmacological effects, including
anti-inflammatory, antifungal, antimicrobial, antioxidant and other
activities (43). Previously, IBC
has been demonstrated to exert antitumor activity against a number
of cancer types. IBC exhibited notable inhibitory effects on mouse
skin tumor promotion in in vivo two-stage skin
carcinogenesis (44). IBC could
induce apoptosis in neuroblastoma cells with no significant
cytotoxicity against normal cerebellar granule cells (45). IBC was demonstrated to induce more
growth limitations and apoptosis for cancer cells with elevated Akt
activation rather than umbilical vein endothelial cells (HUVEC) and
normal hepatocyte L02 cells (46).
In the present study, it was observed that IBC could notably
suppress the proliferation of HepG2 and Hep3B cells, while it has
minimal effect on L02 cells, further indicating that IBC may be
applicable as an efficacious and safe anticancer agent candidate.
Flow cytometric analysis further revealed that IBC dose-dependently
promoted the apoptosis of liver cancer cells, whereas no notable
alteration was observed in cell cycle distributions following
treatment with the same dose of IBC. Furthermore, the expression of
apoptotic markers, including active caspase-3, −7 and −9, and
cleaved PARP, was also detected in the IBC-treated liver cancer
cells. These observations revealed that the antitumor activity of
IBC may be associated with caspase-mediated cell apoptosis in liver
cancer cells.
RSK2 inhibition was indicated to be involved in
IBC-induced apoptosis. Exposure of cells to IBC caused the
downregulation of MDM2, concomitant with an increase of p53, which
may be associated with the inhibition of the RSK2/Akt pathway by
IBC, resulting in a decrease of MDM2 phosphorylation and stability
(47,48). The p53 protein is also an important
substrate of RSK2, and the RSK2-p53-histone H3 complex may
contribute to chromatin remodeling and cell cycle regulation, but
is not required for cell apoptosis (49). Additionally, RSK2-mediated
phosphorylation and inactivation of Bcl-2-associated agonist of
cell death and death-associated protein kinase exerted an
anti-apoptosis effect via the modulation of Bcl-2/Bax (50,51).
The downregulation of Bcl-2 by IBC is expected to reduce the drug
resistance in liver cancer cells (52,53).
The possibility will be further investigated in the future.
Overall, these data implied the association between RSK2 inhibition
and the pro-apoptotic effect of IBC.
Various physiological and pathological functions of
RSK2 are attributed to its extensive substrate specificity
(10,37). When activated, RSK2 is translocated
to the nucleus and phosphorylates a number of diverse substrates
that regulate cell proliferation, transformation, cell cycle and
apoptosis, depending on the specific situation (10,37).
The CREB/ATF family of transcription factors are notable substrates
of RSK2, and the RSK2-CREB/ATF pathway modulates transcriptional
activation of numerous target genes, including the proto-oncogenes
c-Fos and c-Jun, the cell cycle genes Cyclin D
and Cyclin A, and the anti-apoptotic gene Bcl-2
(10,37). RSK2 mediates EGF-induced
phosphorylation of histone H3 at Ser10 and promotes the expression
of c-Fos and c-Jun, as well as AP-1 transactivation
(31). In the present study, IBC
was demonstrated to effectively inhibit EGF-induced phosphorylation
of CREB, ATF1 and histone H3, along with AP-1 transactivation
activity, which was responsible for the antitumor effect of IBC on
liver cancer cells. The functional association between IBC and RSK2
was further demonstrated by in vitro functional experiments.
Enforced RSK2 expression in L02 cells could significantly increase
the selectivity of IBC to suppress cell viability of normal
hepatocytes. Furthermore, RSK2 overexpression promoted cell
proliferation and colony formation, which could be notably
counteracted by IBC. On the contrary, silencing of RSK2 expression
in HepG2 cells could decrease the inhibitory efficacy of IBC on
cell proliferation. Collectively, these results demonstrated that
IBC exerted antitumor activity against liver cancer cells through
regulating the ERKs/RSK2 signaling pathway.
In conclusion, to the best of our knowledge, the
present study demonstrated for the first time that IBC is a
potential inhibitor targeting ERK1/2 and RSK2 and that inhibition
of the ERKs/RSK2 signaling pathway serves a pivotal role in the
anti-proliferative and pro-apoptotic effects of IBC on liver cancer
cells (Fig. 6E). Therefore, IBC may
represent a promising therapeutic candidate for human cancer cases
with elevated ERKs/RSK2 activity.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81502411, 31770774
and 81372137) and the National Undergraduate Training Program for
Innovation and Entrepreneurship (grant no. 201810571007).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
BL, ZHu and ZHe contributed to the conception and
design of the study. BL, NX, ZW, HL, WC and XC performed the
experiments and analyzed data. ZHu and LM performed bioinformatic
analysis. BL, ZHu and ZHe contributed to drafting and revising the
manuscript. All authors read and approved the manuscript and agree
to be accountable for all aspects of the research in ensuring that
the accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
IBC
|
isobavachalcone
|
|
ERK1/2
|
extracellular signal-regulated kinase
1/2
|
|
RSK2
|
ribosomal S6 kinase 2
|
|
MAPK
|
mitogen-activated protein kinase
|
|
EGF
|
epidermal growth factor
|
|
CREB
|
cAMP response element-binding
protein
|
|
ATF1
|
activating transcription factor 1
|
|
AP-1
|
activating protein-1
|
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kumar M, Zhao X and Wang XW: Molecular
carcinogenesis of hepatocellular carcinoma and intrahepatic
cholangiocarcinoma: One step closer to personalized medicine? Cell
Biosci. 1:52011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Santarpia L, Lippman SM and El-Naggar AK:
Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy.
Expert Opin Ther Targets. 16:103–119. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yip-Schneider MT, Klein PJ, Wentz SC, Zeni
A, Menze A and Schmidt CM: Resistance to mitogen-activated protein
kinase kinase (MEK) inhibitors correlates with up-regulation of the
MEK/extracellular signal-regulated kinase pathway in hepatocellular
carcinoma cells. J Pharmacol Exp Ther. 329:1063–1070. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang S and Liu G: Targeting the
Ras/Raf/MEK/ERK pathway in hepatocellular carcinoma. Oncol Lett.
13:1041–1047. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hsu CH, Shen YC, Shao YY, Hsu C and Cheng
AL: Sorafenib in advanced hepatocellular carcinoma: Current status
and future perspectives. J Hepatocell Carcinoma. 1:85–99.
2014.PubMed/NCBI
|
|
7
|
Lim HY, Heo J, Choi HJ, Lin CY, Yoon JH,
Hsu C, Rau KM, Poon RT, Yeo W, Park JW, et al: A phase II study of
the efficacy and safety of the combination therapy of the MEK
inhibitor refametinib (BAY 86-9766) plus sorafenib for Asian
patients with unresectable hepatocellular carcinoma. Clin Cancer
Res. 20:5976–5985. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen-activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang H, Xu L, Zhu X, Wang P, Chi H and
Meng Z: Activation of phosphatidylinositol 3-kinase/Akt signaling
mediates sorafenib-induced invasion and metastasis in
hepatocellular carcinoma. Oncol Rep. 32:1465–1472. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Romeo Y, Zhang X and Roux PP: Regulation
and function of the RSK family of protein kinases. Biochem J.
441:553–569. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ito Y, Sasaki Y, Horimoto M, Wada S,
Tanaka Y, Kasahara A, Ueki T, Hirano T, Yamamoto H, Fujimoto J, et
al: Activation of mitogen-activated protein kinases/extracellular
signal-regulated kinases in human hepatocellular carcinoma.
Hepatology. 27:951–958. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tsuboi Y, Ichida T, Sugitani S, Genda T,
Inayoshi J, Takamura M, Matsuda Y, Nomoto M and Aoyagi Y:
Overexpression of extracellular signal-regulated protein kinase and
its correlation with proliferation in human hepatocellular
carcinoma. Liver Int. 24:432–436. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bessard A, Frémin C, Ezan F, Fautrel A,
Gailhouste L and Baffet G: RNAi-mediated ERK2 knockdown inhibits
growth of tumor cells in vitro and in vivo. Oncogene. 27:5315–5325.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xie YX, Liao R, Pan L and Du CY: ERK
pathway activation contributes to the tumor-promoting effects of
hepatic stellate cells in hepatocellular carcinoma. Immunol Lett.
188:116–123. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liao B, Zhou H, Liang H and Li C:
Regulation of ERK and AKT pathways by hepatitis B virus X protein
via the Notch1 pathway in hepatocellular carcinoma. Int J Oncol.
51:1449–1459. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schmitz KJ, Wohlschlaeger J, Lang H,
Sotiropoulos GC, Malago M, Steveling K, Reis H, Cicinnati VR,
Schmid KW and Baba HA: Activation of the ERK and AKT signalling
pathway predicts poor prognosis in hepatocellular carcinoma and ERK
activation in cancer tissue is associated with hepatitis C virus
infection. J Hepatol. 48:83–90. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim HS, Kim SJ, Bae J, Wang Y, Park SY,
Min YS, Je HD and Sohn UD: The p90rsk-mediated signaling of
ethanol-induced cell proliferation in HepG2 cell line. Korean J
Physiol Pharmacol. 20:595–603. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yuan R, Hou Y, Sun W, Yu J, Liu X, Niu Y,
Lu JJ and Chen X: Natural products to prevent drug resistance in
cancer chemotherapy: A review. Ann N Y Acad Sci. 1401:19–27. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Raghavendra NM, Pingili D, Kadasi S, Mettu
A and Prasad S: Dual or multi-targeting inhibitors: The next
generation anticancer agents. Eur J Med Chem. 143:1277–1300. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Leelananda SP and Lindert S: Computational
methods in drug discovery. Beilstein J Org Chem. 12:2694–2718.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Utepbergenov D, Derewenda U, Olekhnovich
N, Szukalska G, Banerjee B, Hilinski MK, Lannigan DA, Stukenberg PT
and Derewenda ZS: Insights into the inhibition of the p90 ribosomal
S6 kinase (RSK) by the flavonol glycoside SL0101 from the 1.5 Å
crystal structure of the N-terminal domain of RSK2 with bound
inhibitor. Biochemistry. 51:6499–6510. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Serafimova IM, Pufall MA, Krishnan S, Duda
K, Cohen MS, Maglathlin RL, McFarland JM, Miller RM, Frödin M and
Taunton J: Reversible targeting of noncatalytic cysteines with
chemically tuned electrophiles. Nat Chem Biol. 8:471–476. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chaikuad A, Tacconi EM, Zimmer J, Liang Y,
Gray NS, Tarsounas M and Knapp S: A unique inhibitor binding site
in ERK1/2 is associated with slow binding kinetics. Nat Chem Biol.
10:853–860. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schrödinger Release 2015-2; Schrödinger
Suite 2015-2 Protein Preparation Wizard; Epik version 3.2,
Schrödinger, LLC, New York, NY, 2015; Impact version 6.7,
Schrödinger, LLC, New York, NY, 2015; Prime version 4.0, .
Schrödinger, LLC; New York, NY: 2015
|
|
25
|
Small-Molecule Drug Discovery Suite
2015-2; Glide, version 6.7, . Schrödinger, LLC; New York, NY:
2015
|
|
26
|
Schrödinger Release 2015-2; LigPrep,
version 3.4, . Schrödinger, LLC; New York, NY: 2015
|
|
27
|
Small-Molecule Drug Discovery Suite
2015-2; Glide, version 6.7, . Schrrödinger, LLC; New York, NY:
2015
|
|
28
|
Friesner RA, Murphy RB, Repasky MP, Frye
LL, Greenwood JR, Halgren TA, Sanschagrin PC and Mainz DT: Extra
precision glide: Docking and scoring incorporating a model of
hydrophobic enclosure for protein-ligand complexes. J Med Chem.
49:6177–6196. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Small-Molecule Drug Discovery Suite
2015-2: Schrödinger Suite 2015-2 Induced Fit Docking protocol.
Glide version 6.7, Schrödinger, LLC, New York, NY, 2015; Prime
version 4.0, . Schrödinger, LLC; New York, NY: 2015
|
|
30
|
Schrödinger Release 2015-2; Maestro,
version 10.2, . Schrödinger, LLC; New York, NY: 2015
|
|
31
|
Cho YY, Yao K, Kim HG, Kang BS, Zheng D,
Bode AM and Dong Z: Ribosomal S6 kinase 2 is a key regulator in
tumor promoter induced cell transformation. Cancer Res.
67:8104–8112. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li B, Huang G, Zhang X, Li R, Wang J, Dong
Z and He Z: Increased phosphorylation of histone H3 at serine 10 is
involved in Epstein-Barr virus latent membrane protein-1-induced
carcinogenesis of nasopharyngeal carcinoma. BMC Cancer. 13:1242013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Malakhova M, Kurinov I, Liu K, Zheng D,
D'Angelo I, Shim JH, Steinman V, Bode AM and Dong Z: Structural
diversity of the active N-terminal kinase domain of p90 ribosomal
S6 kinase 2. PLoS One. 4:e80442009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pearce LR, Komander D and Alessi DR: The
nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol.
11:9–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sassone-Corsi P, Mizzen CA, Cheung P,
Crosio C, Monaco L, Jacquot S, Hanauer A and Allis CD: Requirement
of Rsk-2 for epidermal growth factor-activated phosphorylation of
histone H3. Science. 285:886–891. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Karin M, Liu Z and Zandi E: AP-1 function
and regulation. Curr Opin Cell Biol. 9:240–246. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cho YY: RSK2 and its binding partners in
cell proliferation, transformation and cancer development. Arch
Pharm Res. 40:291–303. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
David JP, Mehic D, Bakiri L, Schilling AF,
Mandic V, Priemel M, Idarraga MH, Reschke MO, Hoffmann O, Amling M,
et al: Essential role of RSK2 in c-Fos-dependent osteosarcoma
development. J Clin Invest. 115:664–672. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sulzmaier FJ, Young-Robbins S, Jiang P,
Geerts D, Prechtl AM, Matter ML, Kesari S and Ramos JW: RSK2
activity mediates glioblastoma invasiveness and is a potential
target for new therapeutics. Oncotarget. 7:79869–79884. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shimura Y, Kuroda J, Ri M, Nagoshi H,
Yamamoto-Sugitani M, Kobayashi T, Kiyota M, Nakayama R, Mizutani S,
Chinen Y, et al: RSK2Ser227 at N-terminal kinase domain
is a potential therapeutic target for multiple myeloma. Mol Cancer
Ther. 11:2600–2609. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cho YY, Lee MH, Lee CJ, Yao K, Lee HS,
Bode AM and Dong Z: RSK2 as a key regulator in human skin cancer.
Carcinogenesis. 33:2529–2537. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Roessler S, Jia HL, Budhu A, Forgues M, Ye
QH, Lee JS, Thorgeirsson SS, Sun Z, Tang ZY, Qin LX, et al: A
unique metastasis gene signature enables prediction of tumor
relapse in early-stage hepatocellular carcinoma patients. Cancer
Res. 70:10202–10212. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kuete V and Sandjo LP: Isobavachalcone: An
overview. Chin J Integr Med. 18:543–547. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Akihisa T, Tokuda H, Hasegawa D, Ukiya M,
Kimura Y, Enjo F, Suzuki T and Nishino H: Chalcones and other
compounds from the exudates of Angelica keiskei and their
cancer chemopreventive effects. J Nat Prod. 69:38–42. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Nishimura R, Tabata K, Arakawa M, Ito Y,
Kimura Y, Akihisa T, Nagai H, Sakuma A, Kohno H and Suzuki T:
Isobavachalcone, a chalcone constituent of Angelica keiskei,
induces apoptosis in neuroblastoma. Biol Pharm Bull. 30:1878–1883.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Jing H, Zhou X, Dong X, Cao J, Zhu H, Lou
J, Hu Y, He Q and Yang B: Abrogation of Akt signaling by
Isobavachalcone contributes to its anti-proliferative effects
towards human cancer cells. Cancer Lett. 294:167–177. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Feng J, Tamaskovic R, Yang Z, Brazil DP,
Merlo A, Hess D and Hemmings BA: Stabilization of Mdm2 via
decreased ubiquitination is mediated by protein kinase
B/Akt-dependent phosphorylation. J Biol Chem. 279:35510–35517.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Qiu Q, Jiang J, Lin L, Cheng S, Xin D,
Jiang W, Shen J and Hu Z: Downregulation of RSK2 influences the
biological activities of human osteosarcoma cells through
inactivating AKT/mTOR signaling pathways. Int J Oncol.
48:2508–2520. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cho YY, He Z, Zhang Y, Choi HS, Zhu F,
Choi BY, Kang BS, Ma WY, Bode AM and Dong Z: The p53 protein is a
novel substrate of ribosomal S6 kinase 2 and a critical
intermediary for ribosomal S6 kinase 2 and histone H3 interaction.
Cancer Res. 65:3596–3603. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
She QB, Ma WY, Zhong S and Dong Z:
Activation of JNK1, RSK2, and MSK1 is involved in serine 112
phosphorylation of bad by ultraviolet B radiation. J Biol Chem.
277:24039–24048. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Anjum R, Roux PP, Ballif BA, Gygi SP and
Blenis J: The tumor suppressor DAP kinase is a target of
RSK-mediated survival signaling. Curr Biol. 15:1762–1767. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Chen KF, Lin JP, Shiau CW, Tai WT, Liu CY,
Yu HC, Chen PJ and Cheng AL: Inhibition of Bcl-2 improves effect of
LCL161, a SMAC mimetic, in hepatocellular carcinoma cells. Biochem
Pharmacol. 84:268–277. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Jiang L, Zhang Q, Ren H, Ma S, Lu C, Liu
B, Liu J, Liang J, Li M and Zhu R: Dihydromyricetin enhances the
chemo-sensitivity of nedaplatin via regulation of the p53/Bcl-2
pathway in hepatocellular carcinoma Cells. PLoS One.
10:e01249942015. View Article : Google Scholar : PubMed/NCBI
|