Introduction
Metabolic reprogramming is a hallmark of cancer
cells, supporting the acquisition and maintenance of malignant
properties (1,2). The high rate of proliferation
characteristic of cancer cells requires an increase in nutrient
uptake and anabolism in order to produce energy and precursors
necessary for macromolecular biosynthesis. In this regard, glucose
and glutamine metabolism has attracted particular interest, as
these molecules are the main nutrients consumed by cancer cells
(3). Despite the presence of
oxygen, cancer cells mainly convert glucose into lactate through
glycolysis, as first described by Otto Warburg (4), while glutamine is the major source of
anaplerotic intermediates for the tricarboxylic acid (TCA) cycle,
thus maintaining its functionality for the production of ATP
(5). Through glucose and glutamine
catabolism, cancer cells maintain a high pool of diverse
intermediates that serve as building blocks for the biosynthesis of
macromolecules. Moreover, oxidation of the carbon skeletons of
glucose and glutamine allows cells to produce NADPH, which plays a
crucial role in reductive biosynthetic reactions and helps to
maintain cellular redox capacity (6). Analogously, glutamine is important for
the production of glutathione, another antioxidant molecule
(7).
In line with the multiple roles of glucose and
glutamine in cancer cell physiology, deprivation of these nutrients
impairs cellular viability, indicating that glucose and glutamine
metabolism may be targeted for cancer therapy (8). Given that different cancer cell types
can activate different responses to nutrient deprivation, such as
apoptosis, autophagy or cell growth arrest, depending on genetic,
epigenetic and environmental clues (9–12), it
is important to improve our current understanding of cell behavior
under conditions of starvation.
In the present study, well-characterized in
vitro transformed cells were used to investigate the cellular
and molecular response to glucose and/or glutamine starvation.
Transformed cen3tel cells were derived from human fibroblasts
immortalized by telomerase expression, which spontaneously became
tumorigenic during propagation in culture (13–16).
Following the transition from normal to transformed cells,
genome-wide gene and miRNA expression profiles of cells
representing different phases of transformation were examined by
microarray analysis. Transformed cells exhibited changes in gene
expression suggestive of a high usage of glutamine, such as
overexpression of the c-MYC oncogene, which is a regulator
of several genes involved in glutamine metabolism (14). High levels of the c-Myc protein were
paralleled by a reduced expression of miR-23a and miR-23b (14,16),
which negatively regulate glutaminase, the enzyme catalyzing the
first step of glutamine metabolism with the conversion of glutamine
to glutamate (17). Moreover,
microarray analysis revealed that tumorigenic cells overexpressed
the genes encoding the enzymes asparagine synthetase and aspartate
aminotransferase, which are involved in non-essential amino acid
biosynthesis from glutamine, as well as the enzymes pyruvate
dehydrogenase kinase 1 and 3, which inactivate pyruvate
dehydrogenase, thereby preventing pyruvate from entering into the
TCA cycle and downregulating aerobic respiration (National Center
for Biotechnology Information database Gene Expression Omnibus;
accession no. GSE15742).
The aim of the present study was to analyze the
effect of glutamine and/or glucose deprivation on cellular
viability in cen3tel cells at the latest stage of transformation,
as well as the expression of markers associated with different cell
death pathways and DNA double-strand break (DSB) induction.
Materials and methods
Cells and cell culture
The cen3tel cells used in this study belong to the
cen3tel cellular system, which was developed from
telomerase-immortalized fibroblasts that gradually underwent
transformation during propagation in culture (13–16,18–20).
Cen3tel cells underwent approximately 1,000 population doublings
after telomerase immortalization. These cells are tumorigenic and
metastatic in nude mice, overexpress c-MYC and carry a
mutation in the TP53 codon 161, which has been used to
confirm the identity of the cen3tel cells used in the present study
(14,15).
Cells were propagated in high-glucose Dulbecco's
modified Eagle's medium (DMEM; Euroclone S.p.A, Pero, MI, Italy)
supplemented with 10% newborn calf serum (EuroClone), 2 mM
glutamine (EuroClone) and non-essential amino acids (0.1 mM each
L-alanine, L-asparagine, L-aspartic acid, L-glycine, L-serine,
L-proline and L-glutamic acid, EuroClone). To determine the
cellular response to nutrient deprivation, cen3tel cells were
seeded in 3-cm Petri dishes in complete medium at a concentration
of 2–2.5×105 cells per dish and incubated at 37°C. When
the desired confluence was reached (~2-3×105
cells/cm2 in ~72 h), the medium was removed and cells
were accurately washed with phosphate-buffered saline, followed by
incubation for the desired time (12–144 h) in the appropriate
medium. Three dishes were set up for each experimental point. When
cells were grown in the absence of glutamine, non-essential amino
acids were not added. At each time point, for each dish, the
culture medium was collected and the cells were trypsinized and
resuspended with the same culture medium, to recover all the cells
of the culture. An aliquot of the cell suspension was counted using
Trypan blue staining to determine the total number of viable and
dead cells. Experiments were repeated at least three times. Cell
samples were then used for protein analysis.
The MTS assay was performed using the CellTiter
96® AQueous One Solution Cell Proliferation Assay
(Promega Corporation, Madison, WI, USA) according to the
manufacturer's instructions. Briefly, 7,500 cells/well were seeded
in a 96 multi-well dish in complete medium, and after 72 h the
medium was changed and cells were incubated with the appropriate
medium (triplicates were set up for each experimental point). To
test viability, the CellTiter solution was added to glucose-starved
cells 45 h after starvation, and to glucose- and glutamine-starved
cells 13 h after starvation. In both cases, the solution was added
in parallel to cells fed with complete medium. Cell samples were
then incubated for a further 3 h at 37°C before measuring
absorbance at 492 nm with the microplate reader (EZ Read 400;
Biochrom Ltd., Cambridge, UK). Absorbance values were considered as
a measure of viable cells in each sample.
The numbers of viable and dead cells in starved
samples and after recovery in complete medium were compared by
ANOVA performed using R version 3.5.2 (https://www.r-project.org/).
Olaparib treatment
To inhibit PARP-1 activity, cells were seeded as
described above, and incubated for 24 h in complete DMEM or in
glutamine-free medium, with or without addition of 10 or 20 µM
olaparib (Selleck Chemicals, Houston, TX, USA). At the end of the
incubation, cells were collected, counted and analyzed for PARP-1
expression and protein poly(ADP-ribo)sylation (PARylation).
Immunofluorescence
To detect S-phase cells, 5×104 cells/well
were seeded on coverslips in 12-well plates (Corning Inc., Corning,
NY, USA) in complete medium. After 72 h, the medium was replaced
with either complete medium or medium without glutamine, and the
cells were incubated for 24, 48 or 72 h. During the last 30 min of
incubation, 10 mM bromodeoxyuridine (BrdU) was added to each dish.
The cells were then fixed and permeabilized on ice with methanol
for 4 min, treated with 2 N HCl for 20 min and then incubated with
0.1 M sodium tetraborate (pH 8.5) for 5 min. Following incubation
with the primary antibody against BrdU (cat. no. 347580; Becton,
Dickinson and Company, Franklin Lakes, NJ, USA) diluted at 1:250,
the cells were incubated with a TRITC-labeled secondary antibody
(cat. no. 115-025-146; Jackson ImmunoResearch Laboratories, Inc.,
West Grove, PA, USA) diluted at 1:100. The slides were examined
with the Olympus microscope IX71 (Olympus Corp., Tokyo, Japan).
Western blotting
Whole-cell lysates for western blot analysis were
prepared using RIPA lysis buffer [1% Nonidet P40, 50 mM Tris-HCl pH
8.0, 150 mM NaCl, 0.1% SDS, 0.1% DOC, 1X protease inhibitor
cocktail (Thermo Fisher Scientific, Inc., Waltham, MA, USA)], and
1X phosphatase inhibitor cocktail (Roche Diagnostics, Basel,
Switzerland)]. The following antibodies were used: anti-PARP-1
(cat. no. ab191217; Abcam, Cambridge, UK) diluted at 1:1,000;
anti-poly(ADP-ribose) chains (cat. no. sc-56198; Santa Cruz
Biotechnology Inc., Dallas, TX, USA) diluted at 1:1,000; anti-H2AX
(cat. no. ab11175; Abcam) diluted at 1:5,000; anti-γ-H2AX (cat. no.
JBW301; EMD Millipore, Billerica, MA, USA) diluted at 1:5,000;
anti-caspase 9 (cat. no. ALX-210-816; Enzo Life Sciences Inc.)
diluted at 1:500, it recognizes the caspase 9 cleaved active form
of 37 kDa; anti-caspase 3 (cat. no. ALX-210-807; Enzo Life Sciences
Inc.) diluted at 1:500, it recognizes the caspase 3 cleaved active
form of 19 kDa; anti-LC3 (cat. no. 2775; Cell Signaling Technology)
diluted at 1:1,000; anti-p62 (cat. no. BML-PW9860; Enzo Life
Sciences) diluted at 1:1,000; anti-beclin (cat. no. 3738; Cell
Signaling Technology, Inc.) diluted at 1:1,000; anti-γ-tubulin
(cat. no. T6557; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany),
diluted at 1:10,000. All primary antibodies were probed by a
secondary horseradish peroxidase-conjugated antibody (anti-mouse,
cat. no. 115-035-146; anti-rabbit, cat. no. 111-035-144; Jackson
ImmunoResearch Laboratories, Inc.). Chemiluminescent assay was used
for detection (Clarity™; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). To determined H2AX phosphorylation levels, the
intensity of the band in each sample was quantified using the
Quantity One 4.6.6 software (Bio-Rad Laboratories Inc.), corrected
for the intensity of the corresponding band obtained with the
anti-H2AX antibody and then normalized relatively to the
appropriate control sample.
Results
Cellular response to glutamine and/or
glucose deprivation
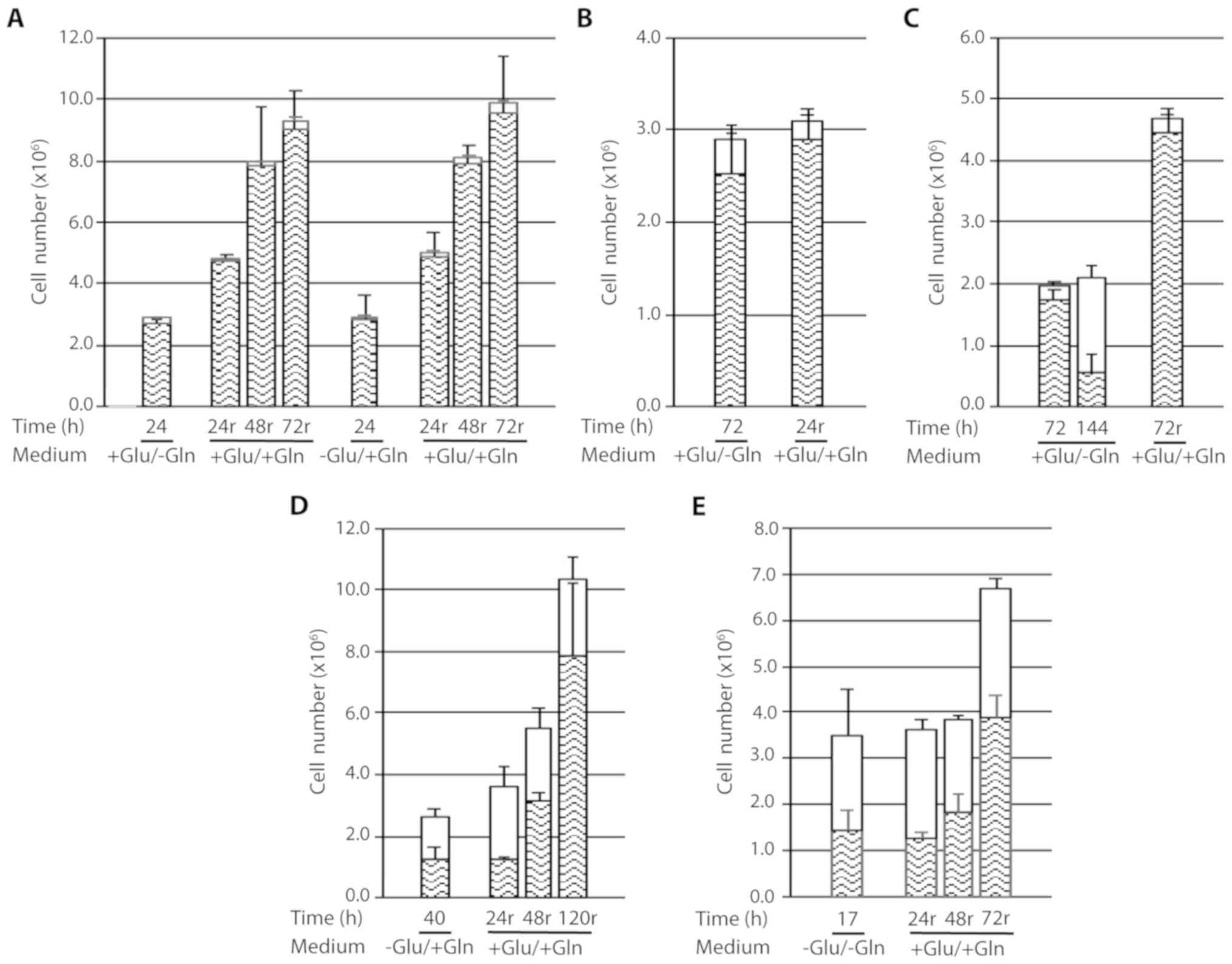
The results obtained from growing cen3tel cells in
the absence of glutamine and/or glucose for different time
intervals (12–144 h) are presented in Fig. 1A-C. Between 12 and 20 h of culture,
cells deprived of glutamine or glucose showed reduced growth
compared with the control cells. Cells starved for both glucose and
glutamine had become detached from the bottom of the dish at 16 h
after deprivation, floating as large sheets in the medium (Fig. 1D). However, despite all the cells
had lost adhesion to the bottom of the dish, only 36% of them were
positive to Trypan blue, suggesting that cell death followed cell
detachment; in fact, after an additional 4 h of deprivation, the
percentage of dead cells increased to 60% (Fig. 1A). To confirm the presence of viable
cells among the detached cells, the MTS assay was performed in
cells starved for glucose for 16 h, demonstrating the actual
reduction of MTS to formazan in these samples (data not shown).
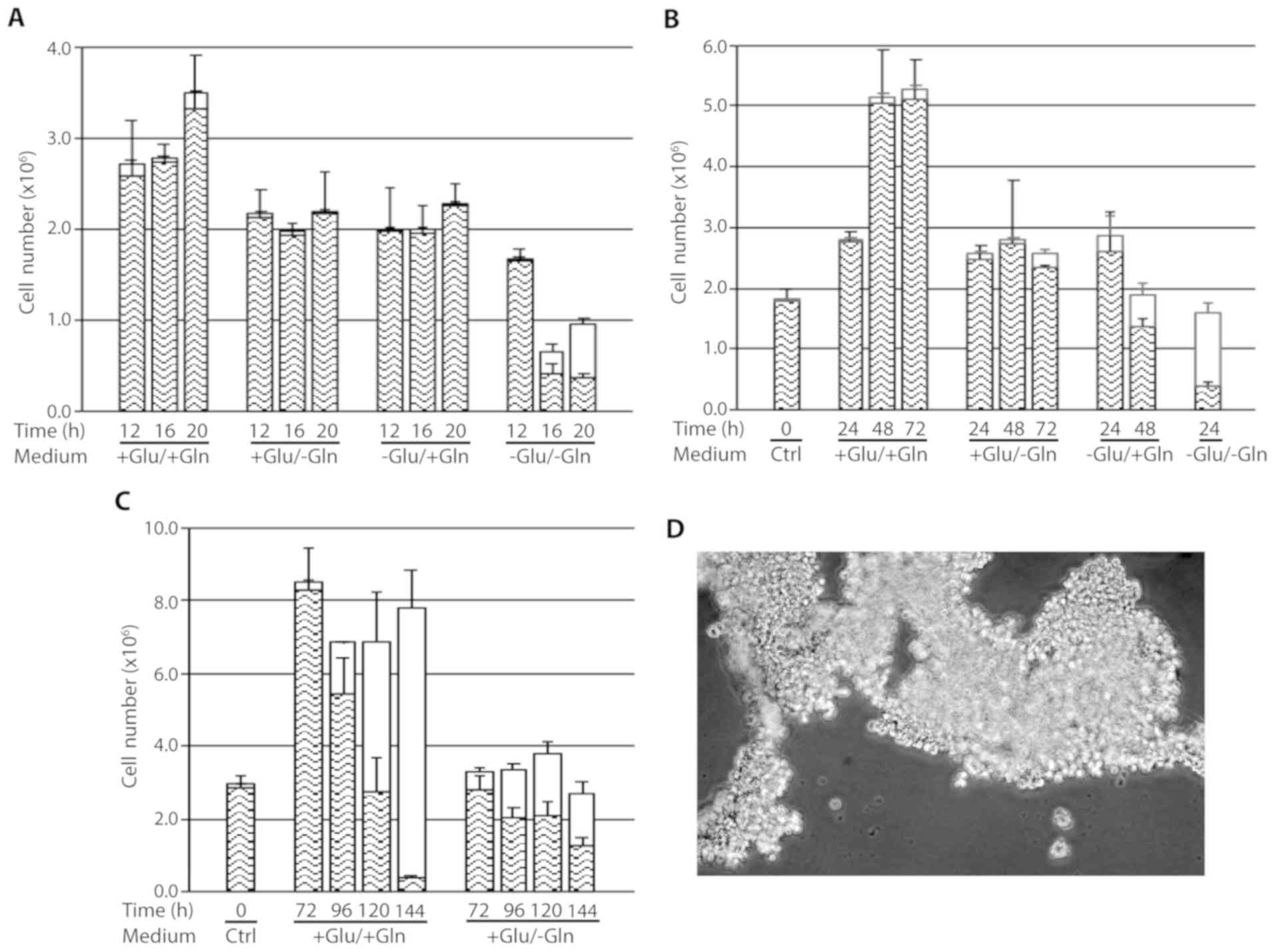 | Figure 1.Response of cen3tel cells to
glutamine and/or glucose starvation. (A-C) Results obtained in
representative experiments are shown. In each plot, the mean number
of viable cells (lower part of each column) and dead cells [upper
part of each column (white box)] obtained in three replicates is
reported for each sample, together with the standard deviations
(bars). (D) Cell sheets detached from the bottom of the dish after
16 h of glutamine and glucose starvation. The image was captured
with a ×4 objective with the Olympus microscope IX71 equipped with
the CoolSnap CCD camera (Teledyne Photometrics, Tucson, AZ, USA),
using MetaMorph 7.7.5 software (Molecular Devices, San Jose, CA,
USA). Glu, glucose; Gln, glutamine. |
These results indicate that short term glutamine or
glucose starvation reduces cell growth, while the absence of both
nutrients leads to cell detachment from the dish and, subsequently,
to cell death.
Upon prolonging cell culture up to 144 h (Fig. 1B and C), we observed that cells
propagated in complete medium grew actively for up to 48 h of
incubation, when reached a very high density (~8×105
cells/cm2), then remained stable in number and started
to die after 96 h, likely because of medium exhaustion due to their
high number. The percentage of dead cells markedly increased at
subsequent time points, being >90% at 144 h (Fig. 1C). Of note, at that time point, many
cells remained attached to the dish, despite they were mostly
positive to Trypan blue staining, whereas detached cells floated as
single cells (data not shown).
Cells grown in the absence of glutamine remained
viable and stable in number up to 72 h, indicating that glutamine
starvation led to an arrest in cell growth. This was confirmed by
immunofluorescence on cells incubated with BrdU, which demonstrated
that the percentage of cells in the S-phase at 24 h after
starvation was lower compared with that in control cells (~14 vs.
~50%, respectively) and decreased to 1.7 and <1% at 48 and 72 h
after starvation, respectively. From 96 h of incubation in the
absence of glutamine, the total number of cells remained almost
stable, but the number of dead cells increased to a percentage of
~50% (Fig. 1C). As observed in
control cells, at these time points, the majority of cells remained
adherent to the dish despite the high percentage of dead cells.
At ~48 h after glucose deprivation, all the cells
had become detached from the bottom of the dish; however, the
majority remained alive, with a percentage of dead cells ~30%
(Fig. 1B). The presence of viable
cells among the detached cells was again confirmed using the MTS
assay (data not shown). This finding was similar to that observed
in glucose- and glutamine-starved cells at shorter times of
deprivation. At 24 h of deprivation, cells starved for both
nutrients were completely detached from the dish, as expected, and
the percentage of dead cells had increased to ~70% (Fig. 1B).
Based on these results, it may be concluded that
glutamine deprivation is associated with a prolonged proliferation
arrest in cen3tel cells. Deprivation of both glucose and glutamine
rapidly led to cell detachment and cell death; the cen3tel response
to glucose starvation was similar to that observed in the absence
of both glucose and glutamine, albeit with a slower kinetics.
Starved cell response after recovery
in complete medium
Whether the cellular response observed after
nutrient deprivation was reversible upon supply of complete medium
was next investigated. As shown in Fig.
2A, cells starved for either glutamine or glucose for 24 h were
able to restart proliferating once nutrients were supplied. In both
conditions, cells performed ~0.8 PDs every 24 h up to 48 h of
recovery, after which time proliferation slowed down (~0.2 PDs
between 48 and 72 h), likely because of the high cell density in
the dish.
Given that cells starved for glutamine remained
viable and stable in number for up to 72 h, we tested whether they
were also able to resume growing after a 72-h starvation period
(Fig. 2B). At 24 h after recovery,
there was a small, but not statistically significant, increase in
the number of viable cells and a mild but statistically significant
decline in the number of dead cells (dead cells in 24-h recovery
sample vs. dead cells in 72-h starved samples P=0.0305); when
starved cells were recovered in complete medium for 72 h, the
number of viable cells clearly increased (cells performed ~1.3 PDs)
and the number of dead cells was negligible. This behavior was
completely different compared with cells that were continuously
starved for glutamine for 144 h, which exhibited a net increase in
dead cells, as expected (Fig. 2C,
middle column).
Thus, cells grown for 72 h in the absence of
glutamine are able to restart growing when nutrients are
replenished, although with slower kinetics compared with cells
deprived of glutamine for 24 h.
We also investigated whether cells starved for
glucose, which were detached from the bottom of the dish but were
mostly negative for Trypan blue staining, were committed to die or
were able to restart proliferating when recovered in complete
medium (Fig. 2D). Cells starved for
glucose for 40 h were collected by simply washing the dish with the
culture medium and, after centrifugation (400 × g), were replated
in complete medium. A 24-h recovery in complete medium had little
effect on starved cells; in fact, the number of viable cells was
approximately the same as in the starved samples, whereas there was
a small, albeit not statistically significant, increase in the
number of dead cells. By contrast, at 48 h after recovery, the
number of viable cells increased (~1.4 PDs), and increased even
further after 120 h of recovery (~2.7 PDs). This suggests that the
population of cells detaching from the dish upon glucose
deprivation includes cells which can proliferate when the
appropriate culture conditions are re-established. However, at each
recovery time point, the proportion of dead cells was high (~40% at
72 h of recovery vs. ~5% in glutamine-starved cells recovering in
complete medium; Fig. 2A and B),
suggesting that cells starved for glucose are prone to cell
death.
Similar results and conclusions were reached testing
the recovery capacity of cells after deprivation of both glucose
and glutamine. At 17 h after starvation, the detached cells were
collected and reseeded in complete medium (Fig. 2E), and were found to be able to
restart proliferating (~1.4 PDs in 72 h), but were also found to be
prone to cell death (the percentage of dead cells was ~40% after 72
h of recovery).
Analysis of autophagic and apoptotic
markers in glutamine and/or glucose-deprived cells
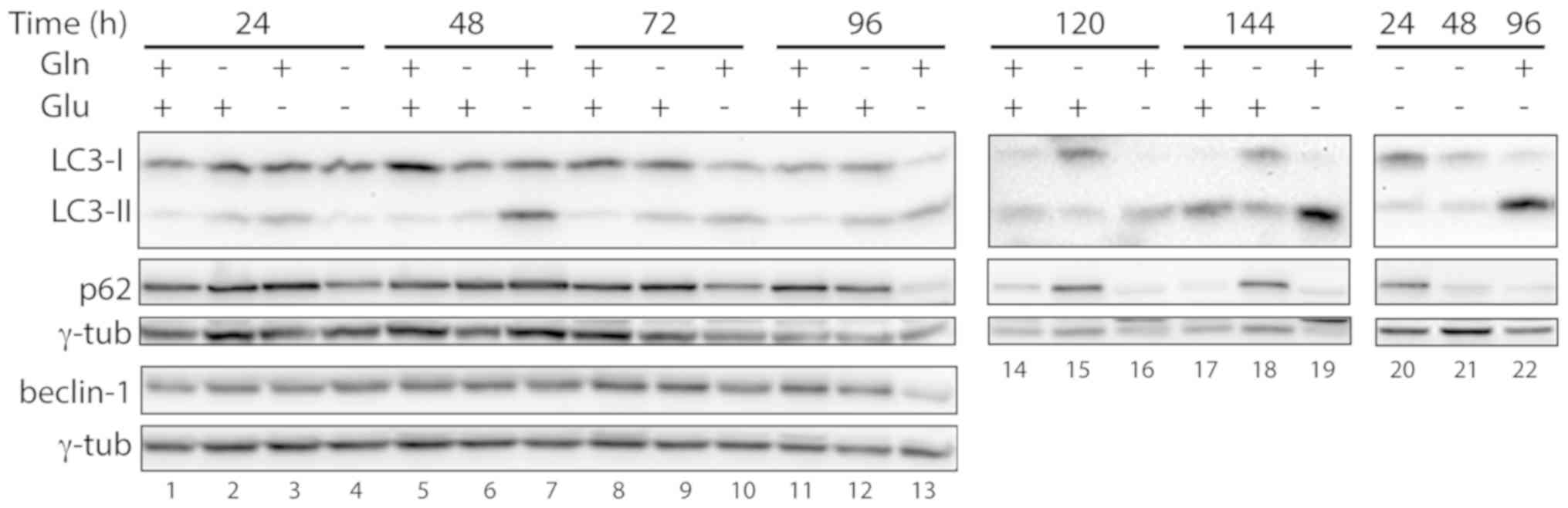
To investigate whether autophagy was activated in
starved cells, the expression of the autophagic markers LC3-II and
p62 was analyzed (21). In control
cells, autophagy activation was observed at 120 and 144 h of
incubation, with a relative increase in LC3-II vs. LC3-I and a
decrease in p62 levels (Fig. 3,
lanes 14 and 17), likely due to medium exhaustion. In cells grown
in the absence of glutamine and in the presence of glucose, there
were no indications of autophagy; in fact, the levels of p62
remained constant, and there was no increase in LC3-II levels
relative to LC3-I at any given time point (Fig. 3, lanes 2, 6, 9, 12, 15 and 18). In
the cell samples grown in the absence of glucose but in the
presence of glutamine, an increase in the ratio between LC3-II and
LC3-I was observed, starting from 48 h of starvation (Fig. 3, lanes 7, 10, 13, 16 and 19); at the
latest stages of deprivation (120 and 144 h) the LC3-I band was
barely detectable (Fig. 3, lanes 16
and 19). A clear decrease in p62 levels was observed 72 h after
starvation and at the subsequent time points, indicating the
occurrence of autophagy (Fig. 3,
lanes 10, 13, 16 and 19). In glucose-starved cells, the expression
of the autophagic protein beclin-1 was not altered up to 48 h after
starvation (Fig. 3, lanes 3 and 7)
and was found to be decreased at later time points (Fig. 3, lanes 10 and 13), suggesting that a
beclin-independent autophagic mechanism (22) was activated in these cells. As was
mentioned above, at 48 h after glucose deprivation the cells were
completely detached from the bottom of the dish. However, in cells
deprived of both glutamine and glucose for 24 or 48 h, a decrease
in p62 levels was observed (Fig. 3,
lanes 4, 20 and 21), but it was not paralleled by an increase in
the levels of LC3-II (Fig. 3, lanes
4, 20 and 21), suggesting that autophagy was not activated in these
cells.
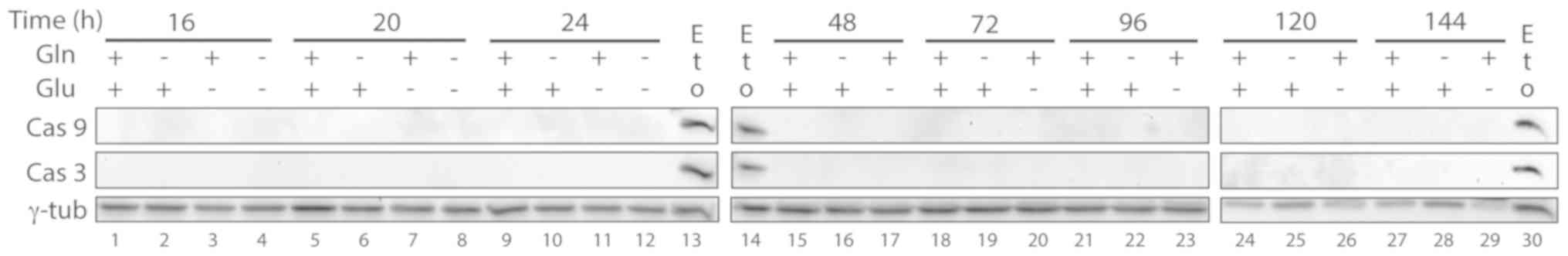
Apoptotic markers, such as the proteolytic fragments
of caspase 3, caspase 9 and PARP-1 (23) were then analyzed in starved cells.
Cleaved caspase 3, caspase 9 or the proteolytic band of PARP-1 were
not found in any of the cell samples (Figs. 4 and 5), indicating that massive apoptosis does
not occur in cen3tel cells starved for glutamine and/or
glucose.
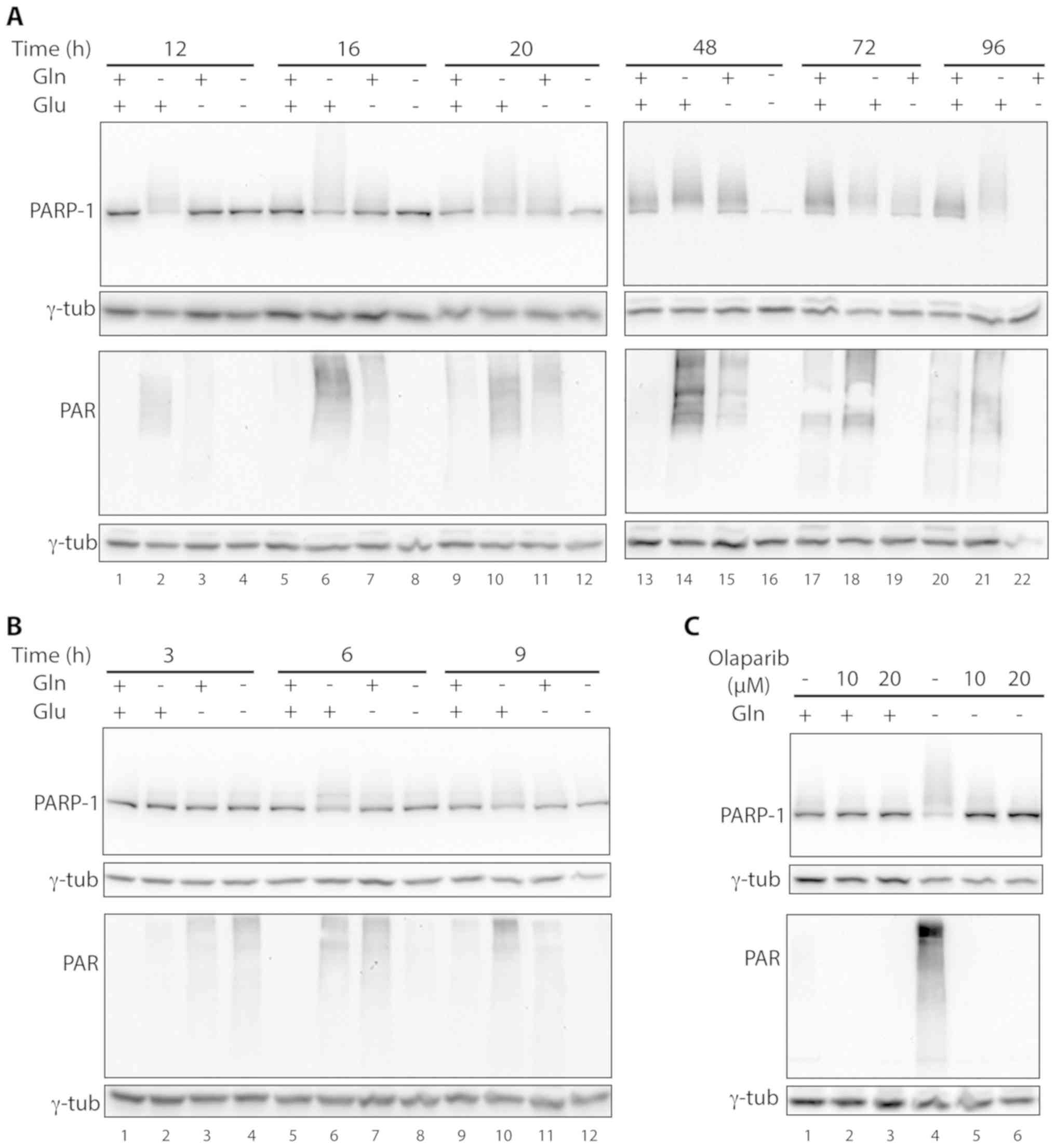
However, in several samples a series of bands with a
molecular weight higher than PARP-1 were detected by the
anti-PARP-1 antibody, suggesting that PARP-1 was modified in those
samples. Modified PARP-1 was first observed in cells starved for
glutamine. In fact, in the samples grown without glutamine, a
smeared PARP-1 signal was already visible at 12 h after deprivation
(Fig. 5A, lane 2). Upon increasing
glutamine starvation times, the PARP-1 canonical band became almost
undetectable, suggesting that the vast majority of PARP-1 was
modified (Fig. 5A, lanes 6, 10, 14,
18 and 21). In the samples deprived of glucose, the smeared signal
above the PARP-1 band was barely detectable at 12 h after
starvation and became progressively more intense at longer
deprivation time, but the canonical band was visible up to 72 h of
deprivation (Fig. 5A lanes 3, 7,
11, 15 and 19). In these samples the overall intensity of the
PARP-1 signals started decreasing 72 h after deprivation and was
not detectable at 96 h (Fig. 5A,
lane 22). In cells deprived of both glucose and glutamine,
additional PARP-1 bands were not detected at any time point
(Fig. 5A, lanes 4, 8, 12 and 16).
PARP-1 modifications were also gradually detected in control cells
starting from 16 h and a subsequent time points (Fig. 5A lanes 5, 9, 13, 17 and 20). In
control samples, canonical PARP-1 exhibited a lower degree of
modification compared with the corresponding glutamine- or
glucose-starved specimens.
PARP-1 modification was paralleled by the activation
of protein PARylation as shown in western blotting with an antibody
against PAR chains (Fig. 5A).
Again, protein PARylation occurred at higher levels in
glutamine-starved cells (Fig. 5A,
lanes 2, 6, 10, 14, 18 and 21) and became undetectable in glucose
starved cells at late time points (72 and 96 h) (Fig. 5A, lanes 19 and 22). In cells starved
for both glucose and glutamine protein PARylation was not
observable already at the first time point of analysis (12 h after
starvation, Fig. 5A, lane 4). We
thus analyzed this protein modification in cells starved for
shorter time periods (3, 6 and 9 h), finding that glucose and
glutamine starvation led to protein PARylation. Under these drastic
growth conditions, protein PARylation peaked at 3 h after
starvation, when it was higher than in the other starved samples
(Fig. 5B, lane 4 vs. lanes 2 and
3), and then rapidly decreased, becoming lower than in glutamine-
or glucose-starved cells at 6 h after starvation (Fig. 5B, lane 8 vs. lanes 6 and 7) and
undetectable at 9 h (Fig. 5B, lane
12). The same trend was observed for PARP-1 modification in cells
grown in the absence of both nutrients (Fig. 5B, lanes 4, 8 and 12). Cells grown
without glutamine exhibited a clear PARP-1 modification already at
6 and 9 h after starvation, and to a greater extent compared with
the other starved samples (Fig. 5B,
lanes 6 and 10).
Given that PARP-1 activation is linked to its
auto-PARylation, we tested whether the multiple PARP-1 isoforms
detected by western blotting corresponded to PARylated PARP-1 by
exposing glutamine-starved cells to the PARP inhibitor olaparib
(24,25). As shown in Fig. 5C, olaparib led to the loss of
protein PARylation, confirming its activity as a PARP inhibitor
(Fig. 5C lanes 5 and 6 vs. lane 4).
In glutamine-deprived cells, PARP-1 was highly modified and
olaparib treatment led to a concentration-dependent decrease in the
smeared signal, with a parallel increase in the intensity of the
canonical PARP-1 band (Fig. 5C,
lanes 5 and 6 vs. lane 4), confirming the hypothesis that
post-translationally modified PARP-1 corresponds to PARylated
PARP-1.
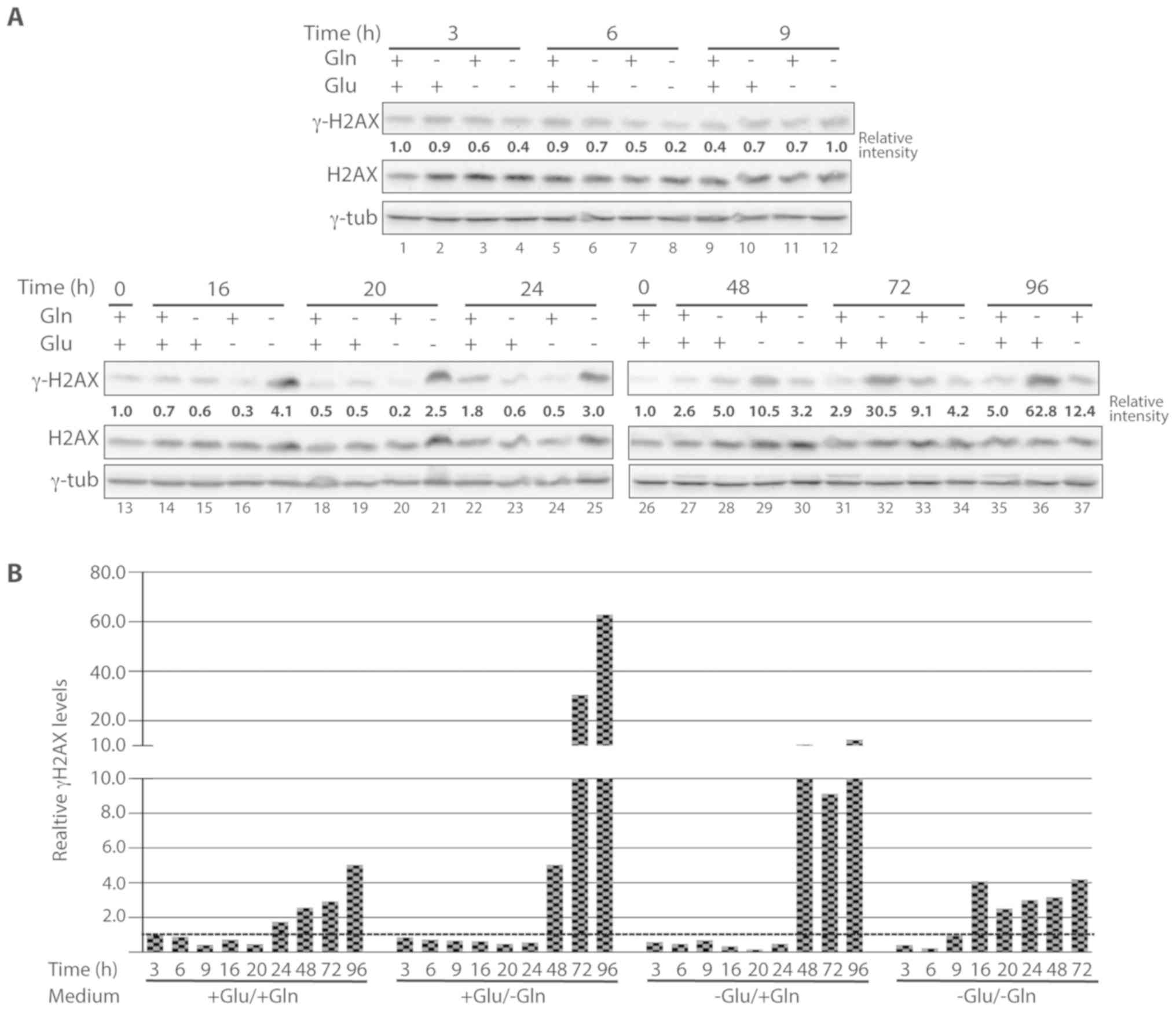
Nutrient starvation and DNA DSBs
The results thus far indicated that nutrient
starvation induces PARP-1 activation. As PARP-1 is activated by
different types of DNA damage, including DNA DSBs, we tested
whether cell growth in nutrient-deficient media induced the
formation of DSBs. We thus analyzed the DSB marker γ-H2A histone
family member X (γ-H2AX) (26) by
western blotting in samples starved for different time intervals.
Representative western blotting results are shown in Fig. 6A and the results of the
quantification of the signal intensities are plotted in Fig. 6B.
Of the cells grown in complete medium, γ-H2AX levels
gradually increased from 48 h and reached the highest levels at 96
h (~x3 compared with control cells; Fig. 6A, lanes 27, 31 and 35). In the
absence of glutamine, H2AX phosphorylation started to increase ~48
h after deprivation and dramatically increased at 72 and 96 h
(Fig. 6A, lanes 28, 32 and 36; ×5,
×30.5 and ×62.8, respectively, compared with control cells). In
glucose-deprived cells, H2AX phosphorylation was always lower than
in control cells up to 24 h (Fig.
6A, lanes 3, 6, 9, 16, 20 and 24; between ×0.2 and ×0.7,
compared with control cells). Between 48 and 96 h of starvation,
phosphorylation levels increased up to approximately ×10 compared
to control cells (Fig. 6A, lane 29,
33 and 37). Finally, starvation for glutamine and glucose led to an
early decrease in γ-H2AX levels (3 and 6 h after starvation,
Fig. 6A, lanes 4 and 8; ×0.4 and
×0.2, respectively, compared with control cells) followed by an
increase from 16 h after deprivation (Fig. 6A, lane 17; ×4.1 compared with
control cells) and similar levels of phosphorylation at the
subsequent time points (20, 24, 48 and 72 h) (Fig. 6A, lanes 21, 25, 30 and 34; ×2.5,
×3.0, ×3.2 and ×4.2, respectively, compared with control
cells).
Taken together, these results indicate that
long-term glutamine deprivation is associated with high levels of
γ-H2AX, while glucose deprivation alone and glucose-glutamine
deprivation led to lower levels of γ-H2AX compared with glutamine
deficiency. Moreover, this analysis revealed that there was no
correlation between PARP-1 activation and γ-H2AX levels. In fact,
PARP-1 activation occurred earlier than H2AX phosphorylation after
nutrient deprivation, indicating that PARP-1 activation was not
triggered by DNA DSBs.
Discussion
Given the importance of glucose and glutamine
metabolism for tumor cell survival, nutrient withdrawal may
contribute to the suppression of tumor cell proliferation and,
eventually, cancer progression. Thus, it is important to explore
how different tumor cells respond to nutrient deprivation. In the
present study, a well-characterized in vitro cellular system
of human transformed fibroblasts was used to compare the cellular
and molecular response to deprivation of either glucose or
glutamine, or both.
The first effect of starvation for each nutrient was
a slowdown in cellular proliferation. This was protracted for
>72 h after glutamine deprivation, while cells grown in the
absence of glucose detached from the bottom of the dish as floating
sheets within 48 h after starvation. Withdrawal of both glucose and
glutamine caused cell detachment more quickly compared with glucose
starvation alone (within 16 h). Under both conditions, a high
percentage of floating cells was negative to Trypan blue staining,
suggesting that cell detachment occurred prior to cell death. This
was confirmed by the fact that some of the detached cells were able
to resume proliferation when cultured in complete medium. However,
a high percentage of dead cells was also found during the recovery
period, possibly because the damage induced by nutrient starvation
made cells prone to death. The observation that, in the absence of
glucose, cells first detached from the growth substrate and then
died, suggests that glucose absence reduces the cells' capacity to
adhere to the extracellular matrix and, once in suspension, cells
cannot survive due to the metabolic impairment. It is worth
mentioning that cen3tel cells are actually able to grow in
suspension under non-adherent culture conditions and in the
presence of nutrients (13,27). An old study by McGuire (28) reported a similar result, showing
that liver cells exposed to glucose metabolism inhibitors lost
their adhesion capacity. Under those conditions, the cells remained
viable and viability could be rescued by eliminating the inhibitor.
The mechanism through which glucose deprivation impairs cell
adhesion remains to be fully elucidated. There is evidence that
glutamine deprivation can decrease melanoma cell attachment to the
extracellular matrix (29); this
was not the case in our cellular system. In fact, upon glutamine
deprivation only, cells mostly died prior to detaching from the
dish or immediately after detachment. However, removal of glucose
and glutamine accelerated the detachment process observed in the
absence of glucose, suggesting that the metabolic impairment due to
glutamine withdrawal acts synergistically with glucose in reducing
cell adhesion.
In the literature, the activation of different cell
death pathways, mainly apoptosis and autophagy, has been reported
in cells under glucose or glutamine deprivation, indicating that
different cell types can respond in different ways to nutrient
withdrawal, possibly depending on their genetic background
(10–12). In glutamine-starved cen3tel cells,
no detectable levels of activated caspase 3 and caspase 9 or the
proteolytic PARP-1 fragment were observed, indicating that
apoptosis is not a process massively induced in these cells by
glutamine withdrawal. In contrast to what has been reported in the
literature (30), glutamine
withdrawal did not even activate autophagy, either as a rescue
mechanism at the earliest stages of deprivation or as a death
pathway at the latest stages. Under our experimental conditions,
the main response to glutamine starvation appeared to be growth
arrest, followed by caspase-independent cell death only after
prolonged starvation (120–144 h). In glucose-starved cells,
autophagy activation was detected, starting at 48 h of deprivation,
when all the cells had become detached from the bottom of the dish.
Autophagy activation has been described under glucose deprivation
conditions and is mainly due to ATP depletion (30). Furthermore, autophagy activation was
found in control cells, occurring at the latest time points of
analysis (120 and 144 h), when the percentage of dead cells was
very high (>90% at 144 h); thus, in this context, autophagy acts
as a death mechanism and is not activated to prolong cell survival
during medium exhaustion. When cells were grown in the absence of
both glucose and glutamine, and became extensively detached from
the dish, we did not identify markers of either apoptosis or
autophagy. Under these conditions, cells may activate a
non-canonical mechanism of death, due to the highly impaired
metabolism.
PARP-1 analysis in the different cell samples
revealed, in addition to the expected PARP-1 band, the presence of
a series of bands suggestive of proteins of molecular weight higher
than PARP-1 that were able to react with the antibody. It is well
known that, in response to several stimuli, including DNA damage,
PARP-1 is activated and becomes able to catalyze the PARylation of
itself and several other proteins using NAD+ as a
substrate (31). As much of the
PARP-1 activity is self-directed (31), the additional PARP-1 bands detected
by western blotting may correspond to PARylated PARP-1. Indeed, we
found that PARP-1 modifications disappeared upon treatment of
glutamine-starved cells with the PARP-1 inhibitor olaparib,
demonstrating that they were due to auto-PARylation. At equal
deprivation times, glutamine-starved cells exhibited more prominent
PARP-1 auto-modification, as well as higher protein PARylation
levels, compared with glucose-starved cells. By contrast, in cells
grown in the absence of both glucose and glutamine, PARP-1 and
cellular proteins were PARylated only at very early times after
starvation (3 and 6 h). The lack of PARylation at later times may
be attributed to the dramatic decrease in the levels of the
substrate NAD+, due to a deep impairment in the energy
metabolism under highly stressful conditions.
Evidence indicates that nutrient deprivation
stimulates the formation of reactivate oxygen species (ROS), which
can in turn induce DNA double-strand breaks (DSBs) (32–34).
Analyzing DNA DSBs through H2AX phosphorylation in different cancer
cells lines, Tran et al (34) reported high levels of γ-H2AX already
24 h after glutamine deprivation and prior to cell death. We herein
demonstrated that H2AX phosphorylation is not rapidly induced by
glutamine and/or glucose deprivation, but occurs when cellular
viability is highly compromised. γ-H2AX reached its highest levels
in glutamine-starved cells compared with cells under different
growth conditions. However, it has to be pointed out that H2AX
phosphorylation was detected in glucose- or double-deprived cells
after they had already become detached from the bottom of the dish,
and it may be less efficient in carrying out this modification
after prolonged detachment. In glucose- and double-starved cells,
γ-H2AX levels were actually reduced compared with those in the
control cells at the earlier stages of starvation. As hypothesized
by Hruda et al (35), this
may be due to the high glycolytic and mitochondrial metabolic
activities that produce high ROS levels in cells grown in the
presence of high glucose concentration.
Under all nutrient deprivation conditions, H2AX
phosphorylation was not found to correlate with either protein
PARylation or PARP-1 PARylation, PARylation always preceded the
increase in γ-H2AX levels. In particular, in cells starved for both
glucose and glutamine, protein PARylation was detected only early
after starvation, when the γ-H2AX levels were lower compared with
those in control cells. Thus, other types of DNA damage rather than
DSBs are likely responsible for PARP-1 activation after glucose
and/or glutamine starvation.
Recently, Aguilera-Gomez et al (36) reported that protein
mono(ADP)-ribosylation in Drosophila cells is crucial for
Sec body formation and cell survival. Sec bodies are pro-survival
cytoplasmic assemblies produced in Drosophila cells upon
amino acid starvation. Despite the counterpart of Sec bodies not
being present in mammalian cells, it cannot be excluded, as
Aguilera-Gomez et al (36)
hypothesized, that mammalian PARPs activated upon glutamine
deprivation may act in concert to PARylate different target
proteins to promote the formation of assemblies similar to Sec
bodies, which may improve cell survival under metabolic stress.
In conclusion, the results of the present study may
widen our current knowledge concerning the consequences of
glutamine and or glucose starvation in transformed cells. Glucose
deprivation was found to be a harsher condition compared with
glutamine starvation. In transformed cells, the absence of glucose
rapidly induced a decrease in cell attachment to the substrate and
autophagic death. By contrast, glutamine starvation first led to an
arrest in cell growth, and cells died only after prolonged absence
of glutamine, without activating autophagy or massive apoptosis.
The absence of glutamine and/or glucose caused extensive PARP-1
PARylation, which was not associated with H2AX phosphorylation,
suggesting that PARylation is not due to DSBs, at least at the
early stages after nutrient deprivation. Moreover, the consequences
of glucose or glutamine deprivation were shown to be reversible,
indicating that starved tumor cells can recover if nutrient
availability in the microenvironment is restored.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
IC, GP and CMa performed the experiments and
analyzed the data. IC and CMo conceived the experiments and wrote
the manuscript. All authors read and approved the manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interest
The authors declare that they have no competing
interests.
References
|
1
|
Hanahan D and Weinberg RA: Hallmarks of
Cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Robey RB, Weisz J, Kuemmerle NB, Salzberg
AC, Berg A, Brown DG, Kubik L, Palorini R, Al-Mulla F, Al-Temaimi
R, et al: Metabolic reprogramming and dysregulated metabolism:
Cause, consequence and/or enabler of environmental carcinogenesis?
Carcinogenesis. 36 (Suppl 1):S203–S231. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pavlova NN and Thompson CB: The emerging
hallmarks of cancer metabolism. Cell Metab. 23:27–47. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Daye D and Wellen KE: Metabolic
reprogramming in cancer: Unraveling the role of glutamine in
tumorigenesis. Semin Cell Dev Biol. 23:362–369. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jones DP and Sies H: The redox code.
Antioxid Redox Signal. 23:734–746. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Altman BJ, Stine ZE and Dang CV: From
Krebs to clinic: Glutamine metabolism to cancer therapy. Nat Rev
Cancer. 16:619–634. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wolpaw AJ and Dang CV: Exploiting
metabolic vulnerabilities of cancer with precision and accuracy.
Trends Cell Biol. 28:201–212. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fuchs BC and Bode BP: Stressing out over
survival: Glutamine as an apoptotic modulator. J Surg Res.
131:26–40. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yuneva M, Zamboni N, Oefner P,
Sachidanandam R and Lazebnik Y: Deficiency in glutamine but not
glucose induces MYC-dependent apoptosis in human cells. J Cell
Biol. 178:93–105. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shanware NP, Bray K, Eng CH, Wang F,
Follettie M, Myers J, Fantin VR and Abraham RT: Glutamine
deprivation stimulates mTOR-JNK-dependent chemokine secretion. Nat
Commun. 5:49002014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Choi SW, Song JK, Yim YS, Yun HG and Chun
KH: Glucose deprivation triggers Protein kinase C Dependent
β-catenin proteasomal degradation. J Biol Chem. 290:9863–9873.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mondello C, Chiesa M, Rebuzzini P, Zongaro
S, Verri A, Colombo T, Giulotto E, D'Incalci M, Franceschi C and
Nuzzo F: Karyotype instability and anchorage-independent growth in
telomerase-immortalized fibroblasts from two centenarian
individuals. Biochem Biophys Res Commun. 308:914–921. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zongaro S, de Stanchina E, Colombo T,
D'Incalci M, Giulotto E and Mondello C: Stepwise neoplastic
transformation of a telomerase immortalized fibroblast cell line.
Cancer Res. 65:11411–11418. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Belgiovine C, Frapolli R, Bonezzi K,
Chiodi I, Favero F, Mello-Grand M, Dei Tos AP, Giulotto E,
Taraboletti G, D'Incalci M and Mondello C: Reduced expression of
the rOCK inhibitor Rnd3 Is associated with increased invasiveness
and metastatic potential in mesenchymal tumor cells. PLoS One.
5:e141542010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ostano P, Bione S, Belgiovine C, Chiodi I,
Ghimenti C, Scovassi AI, Chiorino G and Mondello C: Cross-analysis
of gene and miRNA genome-wide expression profiles in human
fibroblasts at different stages of transformation. OMICS. 16:24–36.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gao P, Tchernyshyov I, Chang TC, Lee YS,
Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT and
Dang CV: c-Myc suppression of miR-23a/b enhances mitochondrial
glutaminase expression and glutamine metabolism. Nature.
458:762–765. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Belgiovine C, Chiodi I and Mondello C:
Relocalization of cell adhesion molecules during neoplastic
transformation of human fibroblasts. Int J Oncol. 39:1199–1204.
2011.PubMed/NCBI
|
|
19
|
Chiodi I, Belgiovine C, Zongaro S, Ricotti
R, Horard B, Lossani A, Focher F, Gilson E, Giulotto E and Mondello
C: Super-telomeres in transformed human fibroblasts. Biochim
Biophys Acta. 1833:1885–1893. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Belgiovine C, Chiesa G, Chiodi I, Frapolli
R, Bonezzi K, Taraboletti G, D'Incalci M and Mondello C: Snail
levels control the migration mechanism of mesenchymal tumor cells.
Oncol Lett. 12:767–771. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Codogno P, Mehrpour M and Proikas-Cezanne
T: Canonical and non-canonical autophagy: Variations on a common
theme of self-eating? Nat Rev Mol Cell Biol. 13:7–12. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ward TH, Cummings J, Dean E, Greystoke A,
Hou JM, Backen A, Ranson M and Dive C: Biomarkers of apoptosis. Br
J Cancer. 99:841–846. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bryant HE, Schultz N, Thomas HD, Parker
KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ and Helleday T:
Specific killing of BRCA2-deficient tumours with inhibitors of
poly(ADP-ribose) polymerase. Nature. 434:913–917. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Farmer H, McCabe N, Lord CJ, Tutt AN,
Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I,
Knights C, et al: Targeting the DNA repair defect in BRCA mutant
cells as a therapeutic strategy. Nature. 434:917–921. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kuo LJ and Yang LX: Gamma-H2AX-a novel
biomarker for DNA double-strand breaks. In Vivo. 22:305–309.
2008.PubMed/NCBI
|
|
27
|
Bono B, Ostano P, Peritore M, Gregnanin I,
Belgiovine C, Liguori M, Allavena P, Chiorino G, Chiodi I and
Mondello C: Cells with stemness features are generated from in
vitro transformed human fibroblasts. Sci Rep. 8:138382018.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
McGuire EJ: Intercellular adhesive
selectivity. II. Properties of embryonic chick liver cell-cell
adhesion. J Cell Biol. 68:90–100. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fu YM, Zhang H, Ding M, Li YQ, Fu X, Yu ZX
and Meadows GG: Specific amino acid restriction inhibits attachment
and spreading of human melanoma via modulation of the
integrin/focal adhesion kinase pathway and actin cytoskeleton
remodeling. Clin Exp Metastasis. 21:587–598. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Galluzzi L, Pietrocola F, Levine B and
Kroemer G: Metabolic control of autophagy. Cell. 159:1263–1276.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gupte R, Liu Z and Kraus WL: PARPs and
ADP-ribosylation: Recent advances linking molecular functions to
biological outcomes. Genes Dev. 31:101–126. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Scherz-Shouval R, Shvets E, Fass E, Shorer
H, Gil L and Elazar Z: Reactive oxygen species are essential for
autophagy and specifically regulate the activity of Atg4. EMBO J.
26:1749–1760. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rodríguez-Vargas JM, Ruiz-Magaña MJ,
Ruiz-Ruiz C, Majuelos-Melguizo J, Peralta-Leal A, Rodríguez MI,
Muñoz-Gámez JA, de Almodóvar MR, Siles E, Rivas AL, et al:
ROS-induced DNA damage and PARP-1 are required for optimal
induction of starvation-induced autophagy. Cell Res. 22:1181–1198.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tran TQ, Ishak Gabra MB, Lowman XH, Yang
Y, Reid MA, Pan M, O'Connor TR and Kong M: Glutamine deficiency
induces DNA alkylation damage and sensitizes cancer cells to
alkylating agents through inhibition of ALKBH enzymes. PLoS Biol.
15:e20028102017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hruda J, Sramek V and Leverve X: High
glucose increases susceptibility to oxidative-stress-induced
apoptosis and DNA damage in K-562 cells. Biomed Pap Med Fac Univ
Palacky Olomouc Czech Repub. 154:315–320. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Aguilera-Gomez A, van Oorschot MM,
Veenendaal T and Rabouille C: In vivo vizualisation of
mono-ADP-ribosylation by dPARP16 upon amino-acid starvation. Elife.
5:e214752016. View Article : Google Scholar : PubMed/NCBI
|