Introduction
Glioma is the most common and aggressive form of
primary malignant brain tumor, which is characterized by a high
proliferation rate (1,2). Despite the continuous development of
novel clinical therapies, the prognosis and survival rates of
patients with grade IV glioma [glioblastoma multiforme (GBM), the
most malignant form of glioma] remain poor. One of the most
important reasons for this may be that the proliferation of glioma
cells is a complex process regulated by a network of various
regulatory molecules (3,4). Elucidation of the interaction between
various molecules in this network is critical for understanding the
mechanisms underlying glioma development. Emerging evidence has
revealed that numerous types of protein or RNA either positively
(5–7) or negatively (8,9)
regulate glioma cell proliferation, whereas the molecular pathways
underlying this proliferative behavior remain largely unknown or
disputed.
Although there are few reports of the molecular
pathways involved in the proliferation of glioma cells, it has been
reported that the PI3K-AKT pathway serves an important role in the
process (7,10,11).
Signal transducer and activator of transcription 3 (STAT3)-cyclin
D1 is another well-known pathway in cancer cells, which has been
implicated in the control of cellular responses to diverse
cytokines and growth factors, including cell proliferation
(12). Currently, whether the
STAT3-cyclin D1 pathway has a role in glioma is disputed.
Overexpression of cyclin D1 has been observed in glioma cells, and
is closely related to the oncogenesis and progression of glioma
(13). Similarly, another study
demonstrated that the levels of JAK2 and STAT3 are significantly
upregulated and exhibit pairwise correlation in human glioma
tissues (14). The overexpression
of constitutively active STAT3 has also been observed to be
accompanied by the restoration of cyclin D1 expression (15); however, there is evidence that
increased STAT3 and decreased cyclin D1 protein levels may
contribute to the recurrence of astrocytic tumors (16). Therefore, the exact regulation of
and relationship between STAT3 and cyclin D1 in glioma remains
unclear. In addition, upstream regulatory molecules of STAT3
signaling in glioma cells are worthy of further study. In breast
cancer cells, RNA interference (RNAi)-mediated silencing of Annexin
A2 (ANXA2) inhibits proliferation by downregulating cyclin D1 in
the STAT3-dependent pathway (17).
Additionally, ANXA2 reduction has been reported to inhibit
epidermal growth factor (EGF)-induced epithelial-mesenchymal
transition (EMT) in a STAT3-dependent manner (18). Therefore, ANXA2 may be considered an
attractive putative upstream regulator of STAT3 signaling in glioma
cells. Therefore, the present study aimed to identify the role of
ANXA2 and the phosphorylated (p)STAT3-cyclin D1 pathway in the
proliferation of glioma cells.
ANXA2 is a multifunctional phospholipid-binding
protein that is expressed in various cell types (19). High ANXA2 expression is a common
feature of numerous types of tumor cells, suggesting that it is a
crucial regulator of these cells (20–22).
In GBM, ANXA2 is overexpressed and is positively correlated with
tumor aggressiveness and low patient survival (23), whereas ANXA2-dependent gene
expression profiles are strictly correlated with the regulation of
fundamental cellular features, including migration, invasion,
cytoskeletal remodeling and the cell cycle, which have all been
examined in vitro and in vivo in primary human GBM
cells (24,25). Accumulating evidence indicates that
ANXA2 is involved in the proliferation of cancer cells.
Transfection of HeLa or 293T cells with an antisense ANXA2 vector
results in the inhibition of cell division and proliferation with a
concomitant reduction in ANXA2 signaling and protein levels
(26,27). The proliferation of MHCC97-H cells
is strongly suppressed by short hairpin RNA (shRNA)-mediated ANXA2
silencing in vitro (28).
Similarly, silencing of ANXA2 in breast cancer tissue leads to the
accumulation of G0/G1 phase cells and a
reduction in S/G2+M phase cells (17,18,29).
However, there are also opposing examples of the proliferative
effects of ANXA2. A previous study demonstrated that pancreatic
cancer cells exhibit high ANXA2 expression, but this is inversely
related to cell proliferation in vitro (30), indicating that the molecular
mechanisms of ANXA2 vary among different types of tumor cells. In
human GBM cells, numerous reports regarding ANXA2 focus on
pathological specimens and the mechanisms of tumor invasion and
metastasis. It has been reported that ANXA2 knockdown decreases
tumor size and slows tumor progression, as evidenced by decreased
invasion, angiogenesis, migration and proliferation, as well as
increased apoptosis in the tumor tissue of the ANXA2 knockdown
group (31,32). Nevertheless, details of the
molecular mechanisms underlying tumor proliferation in glioma
remain unclear.
The present study aimed to investigate the effect of
ANXA2 knockdown on glioma cell proliferation. The results
demonstrated that ANXA2 depletion in glioma cells significantly
inhibited proliferation by decelerating cell cycle progression. The
arrested G1-to-S phase transition observed in
ANXA2-silenced cells was attributed to reduced activity of the
STAT3-cyclin D1 pathway, which is a classic proliferative factor in
breast cancer cells (17). Rescue
experiments indicated that ANXA2 may specifically and directly
regulate STAT3, leading to decreased cyclin D1 levels in
ANXA2-silenced glioma cells. In addition, in glioma cells, ANXA2 is
usually overexpressed and in a redundant state; however, in the
present study, a positive synergistic effect between ANXA2 and EGF
was detected on the pSTAT3 pathway, which may be related to cell
proliferation. The present results revealed a novel mechanism by
which ANXA2 may regulate glioma proliferation.
Materials and methods
Cell lines and cell culture
Human U251, U87 and 293T cells were obtained from
the Cell Bank of the Chinese Academy of Sciences, Shanghai
Institute for Biological Science. The U87 cell line used in the
present study is the American Type Culture Collection version
(glioblastoma of unknown origin). Cells were cultured in DMEM
(HyClone; GE Healthcare Life Sciences) supplemented with 10% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc.) at 37°C in a
humidified incubator containing 5% CO2. For serum-free,
low-density culture, cells were routinely cultured to a cell
density of 1×104 cells/ml and were then cultured in
serum-free OPTI-MEM medium (Gibco; Thermo Fisher Scientific, Inc.)
for another 24 h. For EGF induction, cells were starved of serum
for 12 h and were then stimulated with 500 ng/ml EGF (cat. no.
P5552; Beyotime Institute of Biotechnology) for 0, 5, 15 and 30
min. All cells used in this study were passaged for <3
months.
Plasmid construction, lentivirus
packaging, and stable cell line generation
A lentiviral vector, pLVX-shRNA2 (Takara Bio, Inc.),
was constructed to express human ANXA2-specific shRNAs
(5′-CGGGATGCTTTGAACATTGAA-3′; cat. no. TRCN0000056145;
Sigma-Aldrich; Merck KGaA) or human STAT3-specific shRNAs
(5′-GCACAATCTACGAAGAATCAA-3′; cat. no. TRCN0000329887;
Sigma-Aldrich; Merck KGaA). A scrambled sequence (ANXA2,
5′-CCGGGACATCACGGATCATAT-3′; STAT3, 5′-TGGCCAGTTTGCTTTCCACAT-3′)
that does not target any known human coding sequence was used as a
negative control. The lentiviral vector pLVX–IRES-ZsGreen1 (Takara
Bio, Inc.) was employed to overexpress ANXA2 or FLAG-HA-ANXA2-His
fusion protein in glioma cells. The ANXA2 or FLAG-HA-ANXA2-His
coding sequence was synthesized by Takara Bio, Inc. and then
subcloned into the EcoRI and BamHI sites of the
vector. For lentivirus packing, 1×106 293T cells were
seeded into a 100-mm dish and cultured to 70–80 % confluence. The
cells were then cotransfected at 37°C for 10 h with 10 µg
lentiviral vectors and 5 µg packaging plasmids (pMD2.G and psPAX2;
Addgene, Inc.) using Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.). Virus-containing media were
collected 48 and 72 h post-transfection and were then centrifuged
at 2,000 × g for 5 min at 4°C to remove cell debris. The
supernatant was ultrafiltrated via centrifugation in
ultrafiltration columns (Merck KGaA) at 1,500 × g for 1 h at 4°C to
concentrate lentiviral particles. U251 and U87 cells were seeded at
a density of 1×105 cells/ml in 6-well plates and
cultured to 80–90 % confluence. The cells were infected with
lentivirus (MOI=10) in the presence of 8 mg/ml polybrene (cat. no.
107689; Sigma-Aldrich; Merck KGaA), and stable expressing cells
were enriched and purified according to their green fluorescent
protein fluorescence.
Western blotting
The cells were lysed in RIPA buffer (Beyotime
Institute of Biotechnology) for total protein extraction and
protein concentrations were determined using the bicinchoninic acid
protein assay reagent (Beyotime Institute of Biotechnology). Equal
amounts of protein (40 µg/lane) were separated by 12% SDS-PAGE and
transferred onto a PVDF membrane (Merck KGaA). The membrane was
blocked at room temperature with 5% nonfat milk for 1 h and
incubated with anti-ANXA2 (cat. no. sc-47696; 1:2,000; Santa Cruz
Biotechnology, Inc.), anti-cyclin D1 (cat. no. sc-8396; 1:500;
Santa Cruz Biotechnology, Inc.), anti-STAT3 (cat. no. 9139s;
1:1,000; Cell Signaling Technology, Inc.),
anti-pSTAT3(Y705) (cat. no. 9145s; 1:1,000; Cell
Signaling Technology, Inc.), anti-FLAG/HA/6X HIS (cat. no.
AF0036/AF0039/AH367; 1:1,000; Beyotime Institute of Biotechnology)
and anti-GAPDH (cat. no. sc-293335; 1:5,000; Santa Cruz
Biotechnology, Inc.) primary antibodies overnight at 4°C.
Immunodetection was subsequently performed at room temperature for
1 h with horseradish peroxidase-linked goat anti-rabbit or goat
anti-mouse IgG (cat. nos. A0208 and A0216; 1:5,000; Beyotime
Institute of Biotechnology) and enhanced chemiluminescence reagents
(PerkinElmer, Inc.), according to the manufacturers' protocols. The
bands were detected using an ImageQuant LAS 4000 mini (GE
Healthcare) and band intensities were semi-quantified using
ImageJ2× software (National Institutes of Health); the relative
intensities to the internal GAPDH control were calculated. Western
blotting was repeated at least three times to confirm the
results.
MTT assay
The cells were seeded into 96-well plates at a
density of 2×103 cells/well. At each time-point, 20 µl
MTT solution (5 mg/ml; Beyotime Institute of Biotechnology) in PBS
was added to each well and the cells were stained for 4 h at 37°C.
The supernatant was then aspirated carefully. The formazan in the
plate was dissolved by adding 200 µl DMSO. Absorbance was
determined at 490 nm on a micro-ELISA reader. The assays were
performed using five replicates at each time-point and were
repeated three times. In addition, transfected cells were treated
with the STAT3 inhibitor NSC 74859 (cat. no. SD4794; Beyotime
Institute of Biotechnology) at a final concentration of 50 µM and
then incubated at 37°C for 48 h. Subsequently, the MTT assay was
performed as aforementioned.
Colony formation assay
The cells were seeded in 6-cm dishes at a density of
1,000 cells/dish and were then cultured for 14 days. Subsequently,
the medium was removed, and the cells were washed three times with
PBS, fixed with 100% methanol at 4°C for 10 min and stained with
0.5% crystal violet solution (Beyotime Institute of Biotechnology)
at room temperature for 2 h. The number of colonies containing
>50 cells was then counted under an inverted light microscope
(Primo Vert; Carl Zeiss AG). The assay was performed in triplicate
and repeated three times.
Cell cycle assay
After culturing to 70–80 % confluence, the cells
were collected, washed three times with PBS, and fixed with
ice-cold 70% ethanol at 4°C overnight. The cells were then washed
three times with ice-cold PBS and stained with 500 µl propidium
iodide (PI; BD Biosciences) containing 1 µg/ml RNase (Beyotime
Institute of Biotechnology) at 37°C for 30 min. Flow cytometric
analysis was performed on a Beckman Coulter EPICS analyzer (Beckman
Coulter, Inc.) and cell cycle phase distribution was analyzed with
FlowJo 10 (FlowJo, LLC) and revealed in the three major phases
(G0/G1 vs. S vs. G2/M). The assay
was performed in triplicate and repeated three times.
Subcutaneous tumorigenesis in nude
mice
BALB/c nude mice (age, 6 weeks; weight, 20±3 g;
Laboratory Animal Center, Fujian Medical University) were housed
and bred at 18–22 °C and 50–60 % relative humidity under a 12-h
light/dark cycle with ad libitum access to food and water.
For injection, mice were randomly divided into four groups
(n=5/group) and were injected subcutaneously using U251 glioma
cells with different ANXA2 expression levels. Cells were
trypsinized, collected and adjusted to a concentration of
5.0×107 cells/ml. Subsequently, ~200 µl cells were
injected subcutaneously into one side of the axillary region of
nude mice. After 2 weeks of normal feeding, the mice were
euthanized by intraperitoneal injection of an overdose of
pentobarbital sodium. Tumor tissues were dissected and tumor weight
was statistically analyzed.
Electrophoretic mobility shift assay
(EMSA)
Nuclear extracts of U251 cells were prepared using
the Nuclear and Cytoplasmic Protein Extraction kit (Beyotime
Institute of Biotechnology). EMSA was conducted using the
EMSA/Gel-Shift kit (Beyotime Institute of Biotechnology), according
to the manufacturer's instructions. Briefly, the STAT3 consensus
oligonucleotide probe (5′-GATCCTTCTGGGAATTCCTAGATC-3′) was
end-labeled with biotin. For the assay, 30 µg nuclear protein and
0.02 µM biotin-labeled probe (final concentration) were used in the
20-µl reaction system. The STAT3 probe binding activity was
determined using a chemiluminescent EMSA kit (cat. no. GS002;
Beyotime Institute of Biotechnology), according to the
manufacturer's protocol. The specificity of the DNA-protein complex
was confirmed with biotin-labeled, unlabeled and mutated STAT3
probes (cat. nos. GS083B, GS083 and GS083M; Beyotime Institute of
Biotechnology) added to the mixture.
Immunofluorescence and confocal
microscopy
Cells (1 ml/well) were plated at a density of
1×105/ml on slides in 12-well plates. After 24 h, the
cells were fixed in 4% paraformaldehyde for 30 min at room
temperature. After permeabilization in PBS containing 0.2% Triton
X-100 at room temperature for 10 min, the slides were blocked in 3%
BSA (cat. no. ST023; Beyotime Institute of Biotechnology) for 1 h
at room temperature, and then incubated with anti-ANXA2 (cat. no.
sc-47696; 1:200; Santa Cruz Biotechnology, Inc.) and anti-STAT3
(cat. no. 9139s; 1:200; Cell Signaling Technology, Inc.) antibodies
in a humidified chamber at 4°C overnight. After washing with PBS,
the slides were incubated with rhodamine- or FITC-conjugated
secondary antibodies (cat. nos. AP124R and AP124F; 1:200; Merck
KGaA) at 4°C for 2 h. DNA was stained with a solution of PBS
containing 10 µg/ml DAPI for 5 min. The slides were mounted with
Mowiol-based anti-fading medium and visualized under a
laser-scanning confocal microscope (Leica TCS SP5; Leica
Microsystems, Inc.).
Co-immunoprecipitation (IP) assay
Vectors expressing 3×FLAG-ANXA2 fusion protein were
constructed using p3×FLAG-CMV™-10 system (cat. no. E7658;
Sigma-Aldrich; Merck KGaA). Briefly, 1×106 U251 or U87
cells were seeded into 100-mm dishes, allowed to grow to 80–90 %
confluence, and transfected at 37°C for 10 h with 10 µg fusion
protein-expressing vector p3×FLAG-ANXA2-CMV™-10 using the
Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). The blank plasmid p3×FLAG-CMV™-10 was used as
the control. At 48 h after transfection, the cells were lysed for
30 min at 4°C in 1 ml IP lysis buffer containing 10 mM Tris-HCl (pH
7.4), 1 mM EDTA, 250 mM NaCl, 1% NP-40 (Beyotime Institute of
Biotechnology), and protease inhibitor cocktail (Sangon Biotech
Co., Ltd.). Supernatants were incubated with 1 µg normal mouse IgG
(cat. no. sc-2025; Santa Cruz Biotechnology, Inc.) and 40 µl
protein A&G sepharose beads (Beyotime Institute of
Biotechnology) with gentle agitation on a rotator at 4°C for 2 h.
After centrifugation at 12,000 × g for 10 min at 4°C, the
precleared supernatants were incubated at 4°C overnight on a
rotator with anti-FLAG antibodies (Beyotime Institute of Biotech).
Subsequently, 40 µl protein A&G sepharose beads were added for
an additional 3 h with gentle agitation at 4°C. After
centrifugation at 12,000 × g for 10 min at 4°C, the beads were
washed with IP lysis buffer three times and then boiled in SDS
loading buffer. The final samples were assessed by western blotting
with anti-ANXA2, anti-STAT3 and anti-FLAG antibodies. Whole cell
lysate was used as a control.
Statistical analysis
All data are presented as the means ± standard
deviation of at least three independent experiments. Statistical
analyses were performed using GraphPad Prism version 6.01 software
(GraphPad Software, Inc.). Differences between groups were analyzed
by one-way analysis of variance and Holm-Sidak's multiple
comparisons test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Knockdown of ANXA2 expression inhibits
proliferation of glioma cells
It has previously been reported that ANXA2 serves an
important role in the proliferation of tumor cells (19–22);
however, its role in the proliferation of glioma cells remains to
be clarified. To obtain evidence regarding the function of ANXA2 on
glioma cells, this study investigated the effects of ANXA2 with
RNAi. When ANXA2 knockdown was performed using the lentiviral
vector pLVX-shRNA2-ZsGreen, the rate of ANXA2 knockdown was
72.2±3.2 and 80.2±4.3% (P<0.01) in glioma U251/U87 cell lines,
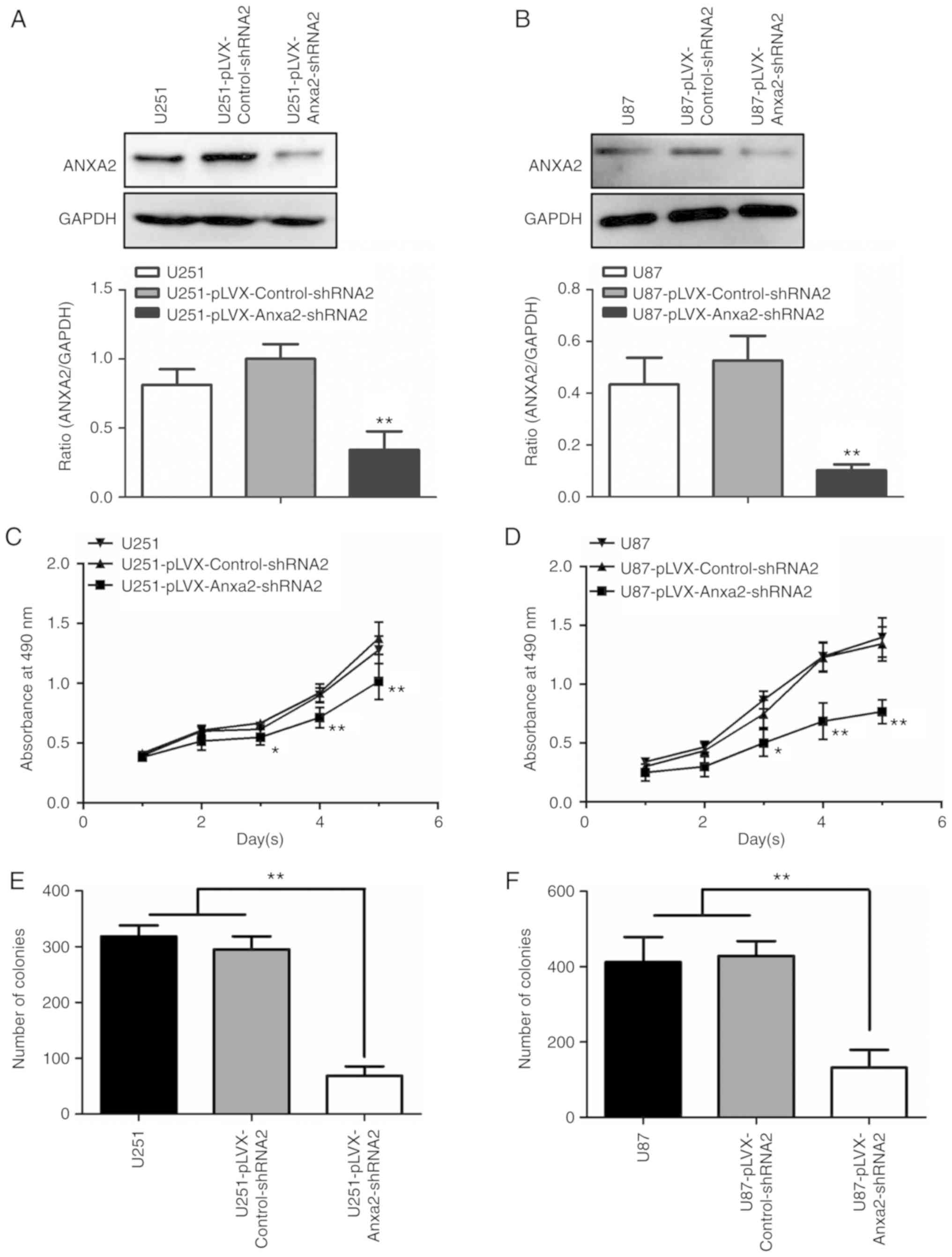
respectively (data not shown). As shown in Fig. 1A and B, the protein expression
levels of ANXA2 in U251/U87 cells were significantly reduced
compared with in control cells post-infection with a lentivirus
expressing specific shRNAs targeting ANXA2. An MTT assay was used
to detect the proliferation of each cell group from day 1 to 5
post-inoculation; significant differences between test and control
cells were observed on day 3 (P<0.05) and days 4 and 5
(P<0.01), indicating that ANXA2 knockdown may inhibit the
proliferation of U251 and U87 cells (Fig. 1C and D). Consistent with the results
of the MTT assay, cell colony formation was reduced in
ANXA2-knockdown U251 and U87 cells (Fig. 1E and F). These results suggested
that ANXA2 may serve an essential role in the proliferation of
glioma cells in vitro.
Knockdown of ANXA2 expression affects
cell cycle progression
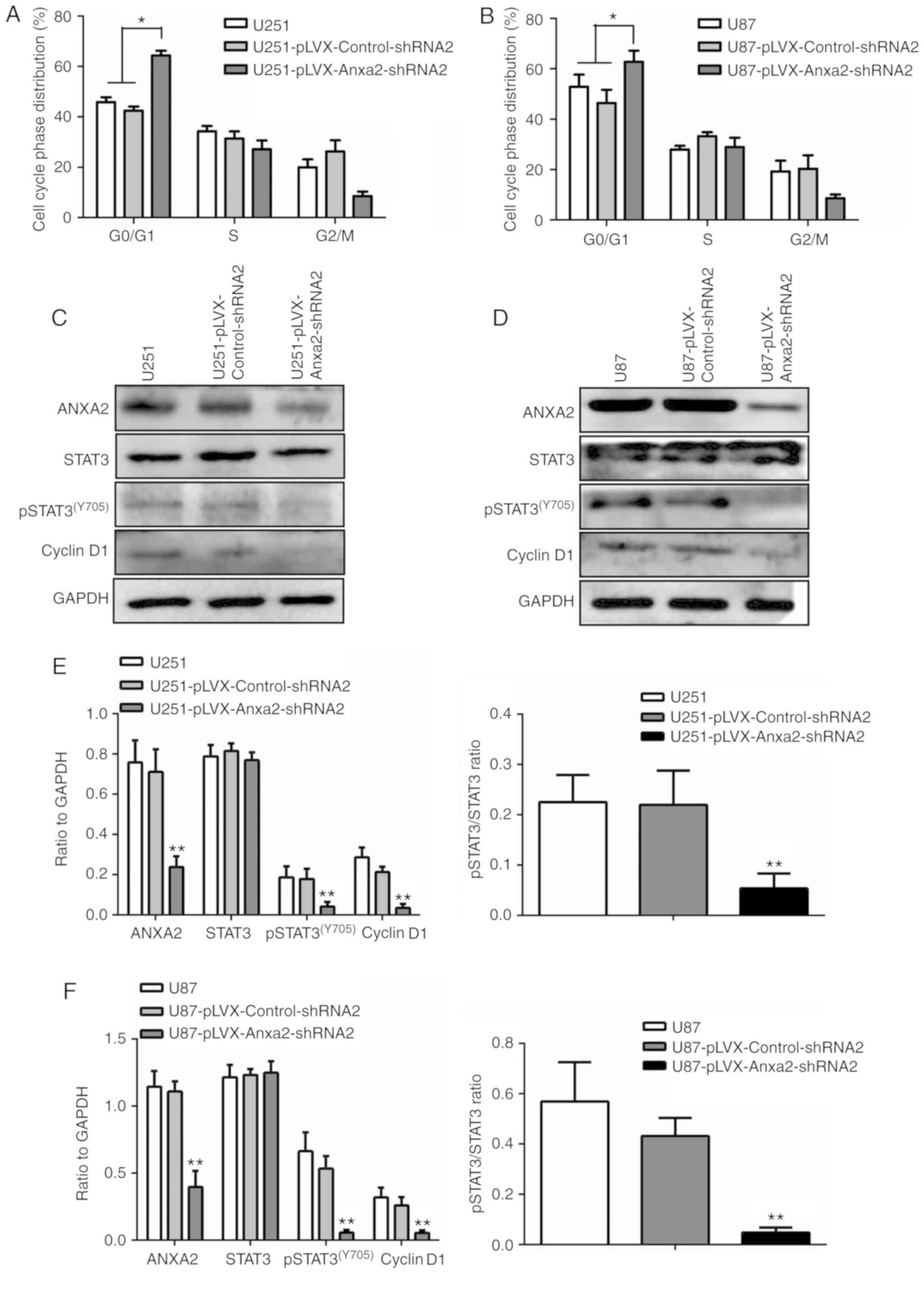
It has been hypothesized that ANXA2 may affect cell
proliferation through regulation of the cell cycle. To investigate
this, the present study measured the proportion of cells at
different stages of the cell cycle in two ANXA2-knockdown glioma
cell lines by flow cytometry. The results indicated that inhibition
of ANXA2 significantly increased the proportion of
G0/G1 phase cells (Fig. 2A and B), leading to a decrease in
the cell proliferation index and a potential G1/S block.
Therefore, ANXA2 may serve a critical role in the
G1-to-S phase transition in glioma cells.
This study also aimed to determine whether ANXA2
affects cell proliferation through the pSTAT3-cyclin D1 signaling
pathway. Western blotting revealed that the downregulation of ANXA2
had little effect on total STAT3 levels; however, it did reduce the
expression of pSTAT3(Y705) and cyclin D1 (Fig. 2C-F). These findings suggested that
ANXA2 may be involved in the phosphorylation of STAT3 and that
knockdown of ANXA2 may inhibit cell proliferation by reducing the
levels of pSTAT3(Y705) and cyclin D1.
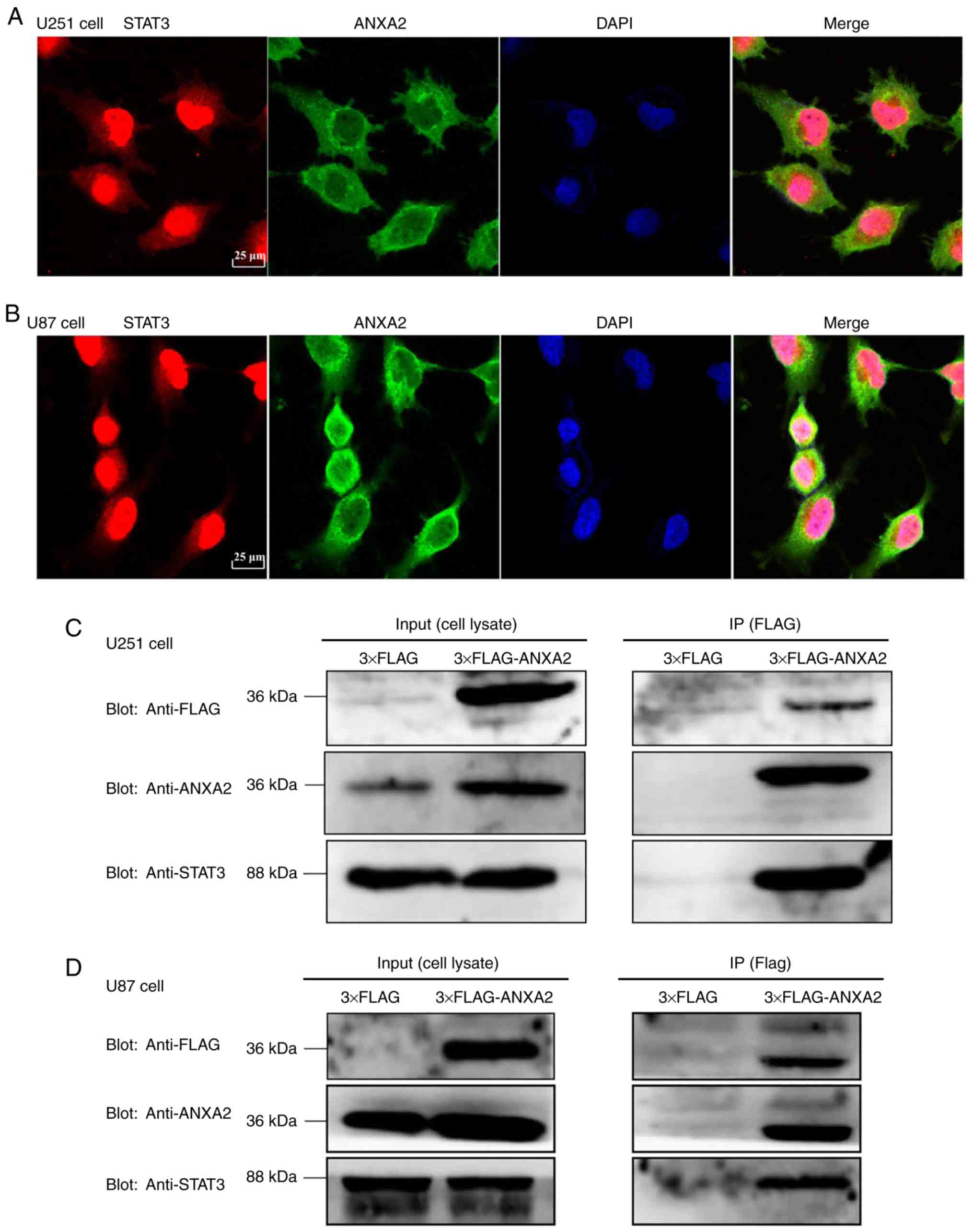
ANXA2 affects cell proliferation
through direct binding with STAT3
ANXA2 is mainly located in the cytoplasm and cell
membrane, suggesting that it may function via direct or indirect
interaction with other proteins. Immunofluorescence was performed
to detect protein interactions between ANXA2 and STAT3 in glioma
cells. Confocal images revealed that ANXA2 was mainly located in
the cytoplasm and that there was an overlap in the cytoplasmic
distribution of ANXA2 and STAT3, particularly in the peripheral
margin of the nucleus, suggesting colocalization of, and possible
interactions between, ANXA2 and STAT3 (Fig. 3A and B). IP and western blotting
further confirmed direct binding between ANXA2 and STAT3. ANXA2 and
STAT3 were both detected using FLAG IP (Fig. 3C and D), thus indicating that ANXA2
may interact directly with STAT3 in glioma cells.
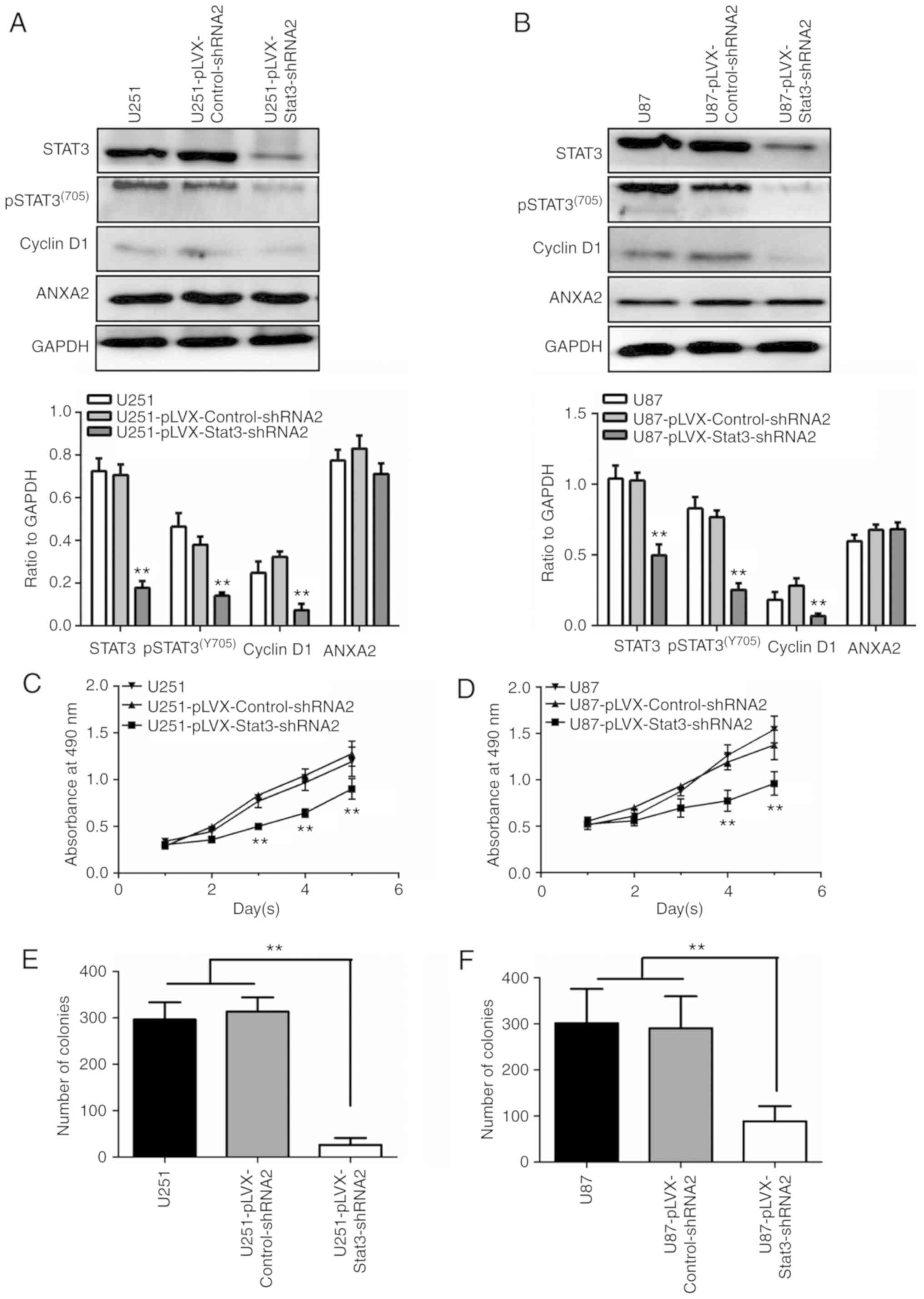
STAT3 knockdown inhibits proliferation
by downregulating cyclin D1
It has been reported that STAT3 can regulate the
transcriptional expression of cyclin D1 and further regulate cell
proliferation in various types of tumor cells (12). To date, however, there have been no
reports on the effect of the pSTAT3-cyclin D1 pathway on glioma
cell proliferation. In the present study, the shRNA sequence of the
STAT3 gene was cloned into pLVX-shRNA2 and infected into U251 or
U87 cells to obtain stable cell lines. MTT and colony formation
assays, and western blotting, were used to assess cell
proliferation. The results revealed that knockdown of STAT3 did not
reduce the expression of ANXA2, but did reduce the expression of
total STAT3 and pSTAT3(Y705), which in turn further
downregulated the expression levels of cyclin D1 (Fig. 4A and B). The MTT assay revealed that
the proliferation rate in the STAT3 RNAi group from day 3 to 5 was
significantly reduced compared with in the nonsense or empty
control groups (P<0.01; Fig. 4C and
D). The colony formation assay also demonstrated that
downregulation of pSTAT3(Y705) significantly inhibited
the colony-forming ability of cells (Fig. 4E and F). These data suggested that
the pSTAT3-cyclin D1 pathway may affect glioma cell
proliferation.
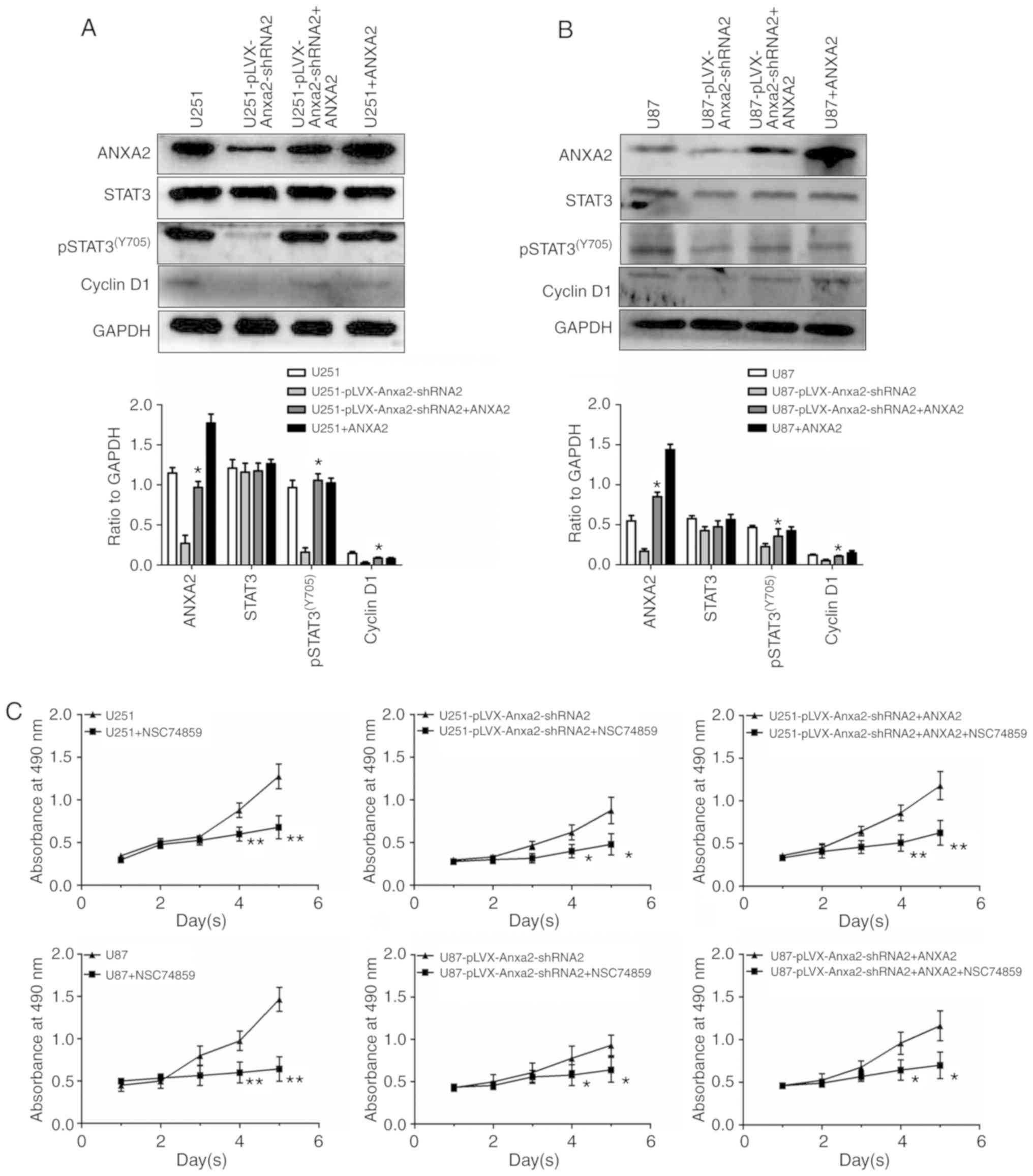
Restoration of pSTAT3-cyclin D1 by
ANXA2 re-expression in ANXA2-knockdown glioma cells
To further confirm that ANXA2 affects the
proliferation of glioma cells through the pSTAT3-cyclin D1 pathway,
ANXA2-knockdown cell lines were transiently transfected with a
pLVX-ANXA2-ZsGreen expression plasmid to determine whether
re-expression of ANXA2 could restore the expression of
proliferation- associated molecules that were inhibited by ANXA2
knockdown. The results revealed that ANXA2 re-expression could
partially compensate for the decrease of proliferation- associated
pSTAT3(Y705)-cyclin D1 signaling caused by ANXA2
knockdown (Fig. 5A and B); however,
in the presence of a STAT3 inhibitor, NSC 74859, the recovery of
proliferation did not occur (Fig.
5C), thus suggesting that ANXA2 may have a specific role in
pSTAT3-cyclin D1-mediated proliferation of glioma cells.
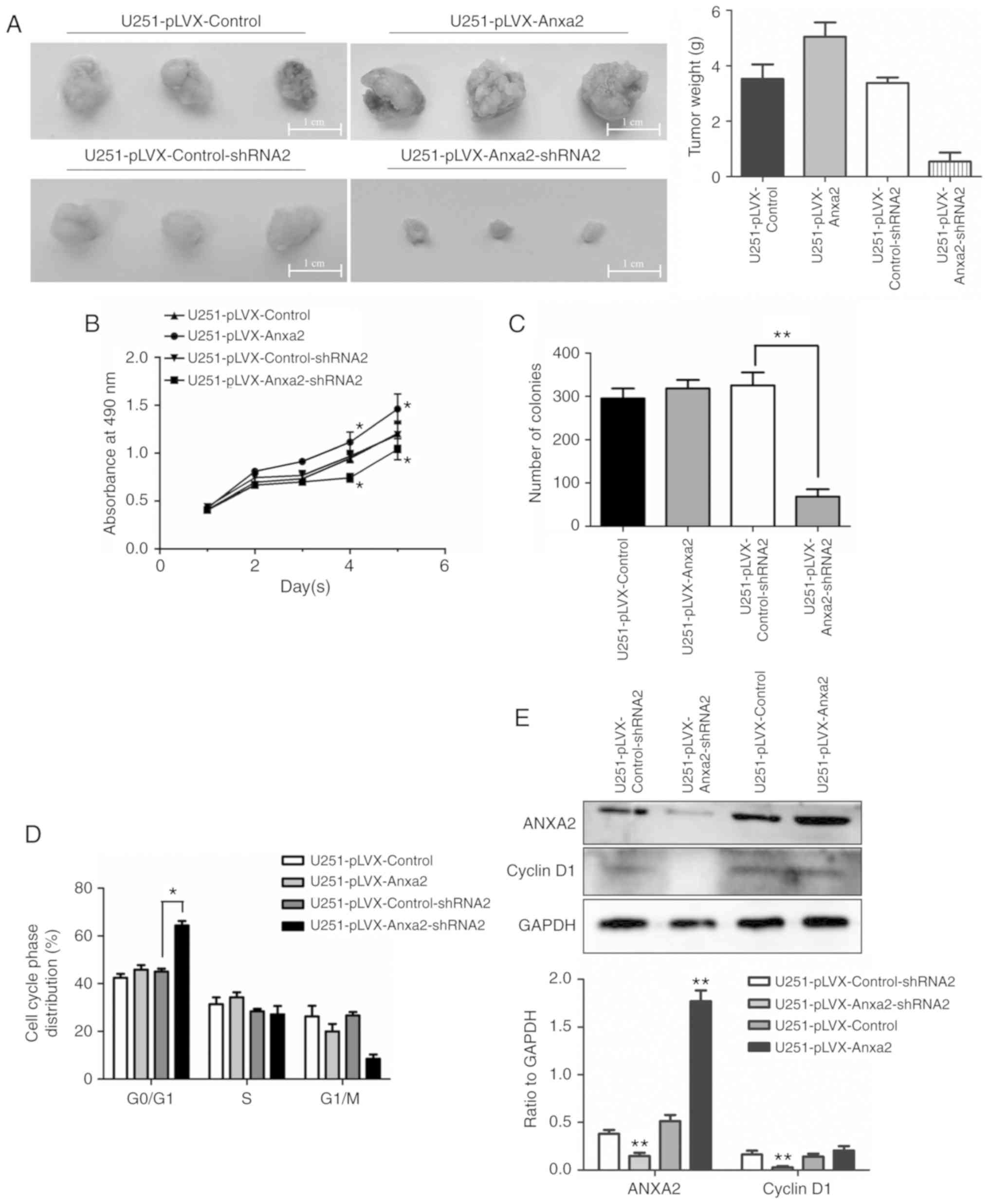
Effects of ANXA2 overexpression on
proliferation appear to be ambiguous
This study demonstrated that ANXA2 knockdown
significantly inhibited proliferation through pSTAT3-cyclin D1
signaling in glioma cells. Subsequently, this study aimed to reveal
the effects of ANXA2 overexpression on proliferation. In a
subcutaneous tumorigenesis model in nude mice, the volume or weight
of transplanted tumors formed by ANXA2-knockdown U251 cells was
smaller than those formed by control cells; however, the tumors
were much larger in the ANXA2 overexpression group compared with in
the control group (Fig. 6A). The
results of the MTT assay revealed that the proliferation rate of
U251 cells with ANXA2 overexpression was significantly increased on
days 4 and 5, whereas it was significantly decreased in U251 cells
with ANXA2 knockdown, compared with in the control group
(P<0.05; Fig. 6B). Surprisingly,
overexpression of ANXA2 had no significant effect on the colony
formation rate of U251 cells and did not change the expression
level of cycle-related proteins cyclin D1 and the proportion of
G0/G1 phase cells (Fig. 6C-E); this is in contrast to the
results obtained using ANXA2-knockdown cells. These ambiguous
results may be due to complex environmental conditions in
vitro and in vivo.
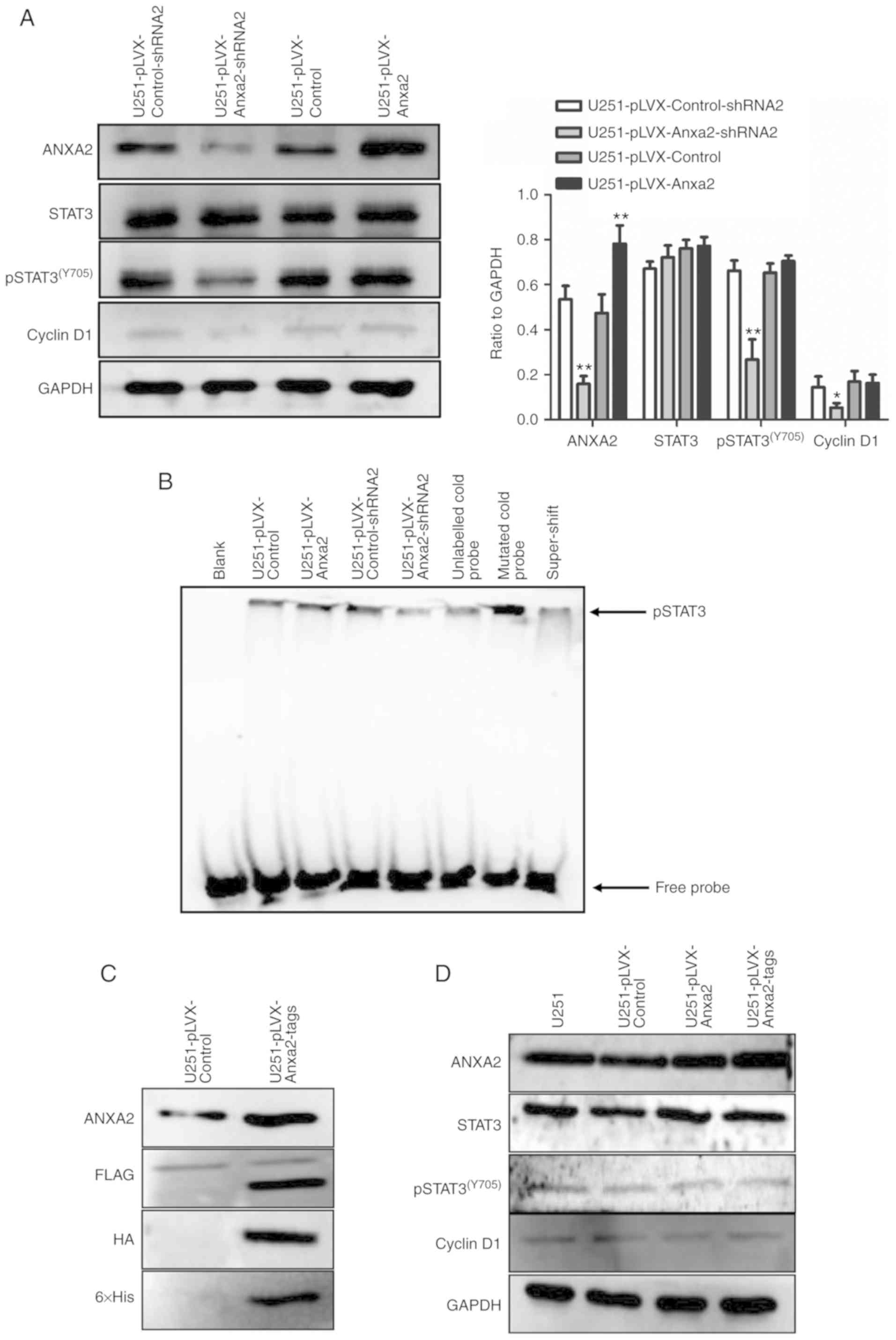
Overexpression of ANXA2 does not
promote pSTAT3-cyclin D1 in glioma U251 cells in serum-free
medium
It was hypothesized that different experimental
conditions, particularly differences in extracellular paracrine
factors and serum, may have led to these mixed results. Therefore,
this study aimed to further determine the effects of ANXA2
overexpression on the pSTAT3-cyclin D1 pathway in U251 cells
cultured in serum-free, low-density conditions. The results of
western blotting indicated that under serum-free, low-density
culture, knockdown of ANXA2 significantly reduced the expression of
pSTAT3(Y705) and cyclin D1, whereas ANXA2 overexpression
did not induce elevation of pSTAT3(Y705) or cyclin D1
(Fig. 7A).
The DNA binding ability of the transcriptional
factor pSTAT3(Y705) was detected using an EMSA assay in
U251 cell lines differentially expressing ANXA2. As shown in
Fig. 7B, the binding of the
STAT3-specific probe in the ANXA2-knockdown nucleoprotein was
decreased compared with the control. However, there was no
significant increase in the amount of probe binding in cells
overexpressing ANXA2. To obtain more convincing evidence,
FLAG-HA-ANXA2-His fusion protein was expressed in U251 cells under
serum-free, low-density conditions and the three tags were used to
detect the fusion protein, in order to ensure the integrity of
ANXA2. The results confirmed that FLAG, HA and His tags were all
detected at 36 kDa, which was exactly the position of ANXA2,
indicating that intact ANXA2 was overexpressed in U251 cells
(Fig. 7C), however, this still did
not enhance the expression of pSTAT3(Y705) or cyclin D1
(Fig. 7D). This further confirmed
that overexpression of ANXA2 in U251 cells may have no significant
effect on the pSTAT3-cyclin D1 pathway in low-density, serum-free
conditions, thus suggesting that ANXA2 cannot activate the STAT3
pathway alone. Therefore, some paracrine factors may be necessary
for ANXA2 to function in cell proliferation.
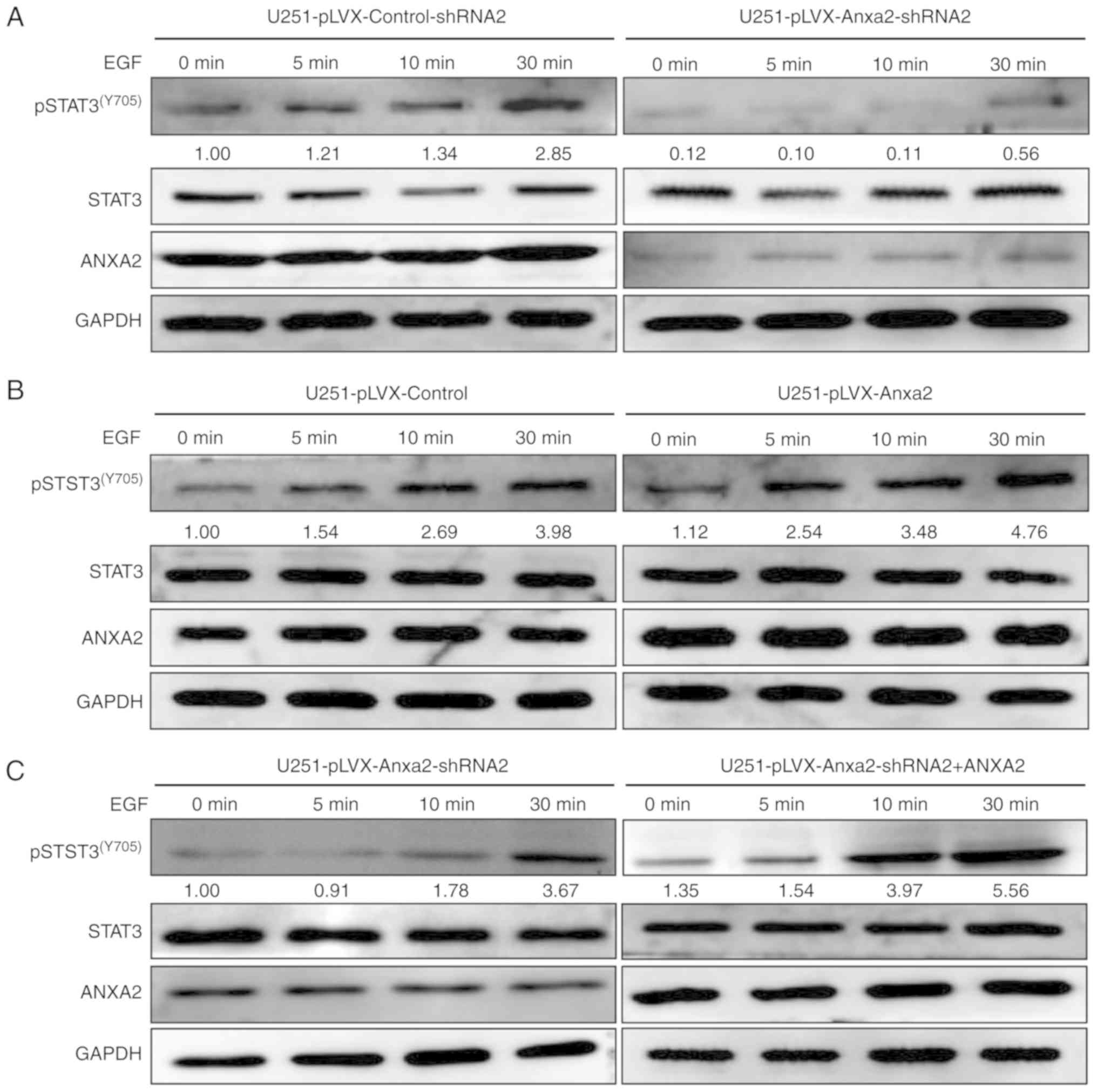
ANXA2 and EGF synergistically promote
pSTAT3-cyclin D1 signaling in serum-free culture
It was hypothesized that overexpression of ANXA2 may
indirectly promote cell proliferation by synergizing with a
paracrine factor. In order to investigate this,
ANXA2-overexpressing or -knockdown U251 cells were treated with
EGF, which has been reported to effectively activate pSTAT3-cyclin
D1 signaling (17). After treatment
with EGF for 0, 5, 15 and 30 min, total cellular protein was
extracted and subjected to western blotting. In ANXA2-knockdown
cells, the initial level of pSTAT3(Y705) was lower than
in control cells and was not significantly increased during EGF
treatment (Fig. 8A). Meanwhile, in
ANXA2-overexpressing cells, the expression levels of
pSTAT3(Y705) were initially not significantly different
to those in control cells; however, they were significantly
increased during EGF induction in a time-dependent manner (Fig. 8B). To further validate the specific
synergy between ANXA2 and EGF, ANXA2 was re-expressed in
ANXA2-knockdown U251 cells. Under the same conditions of EGF
induction, there was a significant recovery of
pSTAT3(Y705) in ANXA2-re- exepressing cells compared
with the knockdown control (Fig.
8C). These findings suggested that EGF-induced activation of
pSTAT3(Y705) may depend on ANXA2 expression in glioma
U251 cells. Additionally, ANXA2 overexpression alone did not affect
pSTAT3-cyclin D1; however, in the presence of EGF, redundant ANXA2
can significantly activate the pathway. Therefore, ANXA2 and EGF
may act synergistically in the activation of
pSTAT3(Y705), but neither can activate
pSTAT3(Y705) activity alone.
Discussion
Accumulating evidence has suggested that ANXA2 is
highly expressed in numerous types of tumor cells, and serves a
critical role in proliferation, migration, invasion, metastasis and
angiogenesis (19–22). However, ANXA2 overexpression has an
opposing role in various tumor cell types (23) and details of its function in glioma
remain elusive. In the present study, it was demonstrated that
ANXA2 knockdown inhibited cell proliferation by impeding the
G1-to-S phase transition via the pSTAT3-cyclin D1
pathway through a direct interaction with STAT3. The present
results indicated that EGF-induced activation of
pSTAT3(Y705) may depend on the presence of ANXA2, and
that ANXA2 and EGF may have a positive synergistic effect on
pSTAT3-cyclin D1 signaling, which is related to proliferation of
human glioma cells. Overall, the present study provided novel
insights into the functions of ANXA2 as a critical molecule in
glioma cells, and may improve understanding of the mechanism
underlying ANXA2-mediated proliferation in cancer cells.
In glioma cells, ANXA2 can participate in
invasion-associated processes (22); in a previous study, it was revealed
that ANXA2 knockdown decreases glioma cell migration, tumor size
and tumor progression, but does not affect proliferation (26). In addition, it has been suggested
that GBM cell migration and invasion are sustained by ANXA2, and
that ANXA2 impairment induces differentiation and inhibits
proliferation of GBM cells (27).
However, to the best of our knowledge, no further studies have been
performed regarding the underlying molecular mechanisms of these
processes. A previous study demonstrated that ANXA2 depletion in
breast cancer cells significantly inhibits cell proliferation by
decelerating progression of the cell cycle (17). In agreement with this observation,
the results of the present MTT and colony formation assays
demonstrated that knockdown of ANXA2 in U251 or U87 glioma cells
inhibited proliferation, thus indicating that ANXA2 may serve a
critical role in the proliferation of glioma cells and encouraging
us to further validate the potential underlying molecular
mechanisms. Subsequently, this study demonstrated that knockdown of
ANXA2 significantly decreased the expression levels of
pSTAT3(Y705) and cyclin D1, and increased the proportion
of G1 phase cells, thus reducing the cell proliferation
index and suggesting that ANXA2 knockdown may lead to a
G1/S block. Cyclin D1 is a protein required for
progression through the G1 phase of the cell cycle
(33), and it has been reported
that G1 phase cell cycle arrest is induced by a
reduction in STAT3, which is consistent with a decrease in cyclin
D1 protein expression (34). In
GBM, STAT3 mutations contribute to a concomitant suppression of
proliferation and survival of U251 cells (35). The present results revealed that
knockdown of ANXA2 significantly reduced the expression levels of
pSTAT3 and cyclin D1 in U251 and U87 cells. Furthermore, it has
been reported that impairment of ANXA2 is sufficient to partially
arrest GBM cells at the S-G2/M cell cycle checkpoint
(24), which differs from the
results of the present study; however, there may be a potential
difference between primary GBM cells and GBM cell lines.
The pSTAT3-cyclin D1 pathway is considered to serve
a crucial role in the proliferation of several tumor types
(12). Therefore, the arrested cell
cycle progression in ANXA2-knockdown cells may be attributed to
inhibition of pSTAT3(Y705) activity. Immunofluorescence
and IP suggested that a direct interaction existed between ANXA2
and STAT3, thus suggesting that ANXA2 may regulate STAT3
phosphorylation via direct binding in glioma cells, thus affecting
pSTAT3-cyclin D1-mediated cell proliferation. When STAT3 was
knocked down in glioma cells, the expression levels of
pSTAT3(Y705) and cyclin D1 were consistently
downregulated, and cell proliferation and colony formation were
also inhibited.
It has been reported that ANXA2 regulates the
pSTAT3-cyclin D1 pathway in breast cancer cells, and affects breast
cancer progression (17,18); however, these studies did not
perform rescue experiments. To further verify the specificity of
ANXA2 regulation on the pSTAT3 pathway, ANXA2 was re-expressed in
ANXA2-knockdown glioma cells; as a result, the reduction in
pSTAT3(Y705) and cyclin D1 expression caused by ANXA2
deletion was partially restored. To validate the specific binding
of ANXA2 and STAT3, the STAT3 inhibitor NSC 74859 was used in this
rescue experiment. Similar to the previous results, re-expression
of ANXA2 partially compensated for the decrease of
proliferation-associated pSTAT3(Y705)-cyclin D1
signaling caused by ANXA2 knockdown; however, in the presence of
STAT3 inhibitors, there was no recovery of proliferation. This
finding confirmed that ANXA2 may affect glioma proliferation by
regulating the pSTAT3-cyclin D1 pathway. In summary, the present
findings suggested that ANXA2 may directly bind to STAT3 and
enhance its transcriptional activity, thus regulating the
proliferation of glioma cells.
Few studies of gene function have investigated gene
overexpression. The main reason for this is that genes are often
redundant in tumor cells, and the effect of their overexpression is
not obvious. ANXA2 is highly expressed in glioma cells, and its
expression level is positively correlated with glioma grade
(24,25). However, to the best of our
knowledge, there have been no reports to date on whether or how the
overexpression of ANXA2 can accelerate glioma cell proliferation.
Therefore, the present study aimed to elucidate the effects of
ANXA2 overexpression on glioma cell proliferation. The results
revealed that the overexpression of ANXA2 had no significant effect
on colony formation rate, cell cycle progression or the
pSTAT3-cyclin D1 pathway in the U251 glioma cell line. However,
proliferation was markedly increased by ANXA2 overexpression in the
MTT and subcutaneous tumorigenesis experiments. Previous studies
have reported that the role of ANXA2 is associated with multiple
paracrine factors, including EGF (18) and interleukin-6 (36). As for experimental methods, MTT and
tumor transplant experiments may be affected by paracrine factors
due to cell culture density and complex tumor formation in
vivo. Therefore, it was hypothesized that the observed effect
of overexpression on cell proliferation may be due to synergy
between paracrine factors and ANXA2. To further assess whether
paracrine factors are involved in ANXA2-regulated cell
proliferation, serum-free medium and low-density cell culture were
used to eliminate possible interference from paracrine factors. The
results revealed that knockdown of ANXA2 significantly reduced the
expression levels of pSTAT3(Y705) and cyclin D1, whereas
overexpression of ANXA2 did not upregulate either
pSTAT3(Y705) or cyclin D1 in the U251 cell line in
low-density, serum-free conditions. An EMSA experiment further
demonstrated that knockdown of ANXA2 decreased the DNA binding
activity of pSTAT3(Y705); however, there was no obvious
increase in the amount of probe binding in ANXA2-overexpressing
cells. Intact expression of ANXA2 also did not enhance the
expression of pSTAT3(Y705) or cyclin D1. Furthermore,
rescue experiments indicated that re-expressed ANXA2 exhibits
biological activity by partially activating pSTAT3 signaling in
ANXA2-knockdown cells. Together, these results demonstrated that
ANXA2 may not activate the pSTAT3-cyclin D1 pathway in the absence
of essential paracrine and serum components.
EGF receptor (EGFR) is a common transmembrane
tyrosine kinase receptor. EGF and EGFR recognition can activate
tyrosine kinase Janus kinase activity, resulting in phosphorylation
of the transcription factor STAT3 at Tyr705, which may be involved
in the regulation of cell proliferation (37). ANXA2 alone may not be able to
activate STAT3 activity, but EGF, which is closely related to the
pSTAT3-cyclin D1 signaling pathway, may be its extracellular
cofactor. The latest reports in this field suggest that ANXA2 and
EGF signaling have a positive synergistic effect on EMT
transformation in CaSki human cervical cancer cells (38). In order to assess this possibility
in glioma cells, EGF induction was performed on each cell line and
on control cells. The results revealed that pSTAT3(Y705)
expression was not markedly increased when EGF was added in the
absence of ANXA2. However, in cells overexpressing ANXA2, the
addition of EGF markedly increased the expression levels of
pSTAT3(Y705). In addition, when ANXA2 was re-expressed
in ANXA2-knockdown U251 cells, there was a significantly recovery
of pSTAT3(Y705) in response to EGF. These results
suggested that EGF-induced activation of pSTAT3(Y705)
may depend on the presence of ANXA2. It was further speculated that
ANXA2 and EGF may be synergistic in the activation of
pSTAT3(Y705), as neither can effectively activate
pSTAT3(Y705) activity alone. In the present MTT assay
and nude mouse model, ANXA2 overexpression may have resulted in a
more significant proliferation-promoting effect due to the
cumulative and synergistic effect of EGF secretion.
In conclusion, the present study revealed that ANXA2
may affect the proliferation of human glioma cells via the
pSTAT3-cyclin D1 pathway by direct interaction with STAT3 in U251
and U87 glioma cells. Overexpresson of ANXA2 cannot activate the
pSTAT3 pathway alone, while positive synergy may exist between
ANXA2 and EGF. Activated EGF and elevated levels of ANXA2 are
frequently observed in a large number of human malignancies,
including glioma (24,25). Therefore, these findings may provide
novel insight into the functions of ANXA2 as a critical molecule in
glioma cells and may improve our understanding of the mechanism
underlying ANXA2-mediated proliferation in cancer cells.
Acknowledgements
Not applicable.
Funding
The present study was sponsored by the Project of
the Education Department of Fujian Province of China (grant no.
JA12148), the Natural Science Foundation of Fujian Province in
China (grant nos. 2008J0089 and 2013J01372), the Research Fund from
Fujian Medical University (grant no. 2013JY030) and the Nursery
Research Fund Project of Fujian Medical University (grant no.
2010MP023).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZZ was involved in study concept, design and
supervision, and provided final approval of the version to be
published. LC was involved in drafting of the manuscript, data
analysis and interpretation, and performed experiments and obtained
funding. LL assisted with the experimental design, data
interpretation and acquisition of funding. NX performed
experiments, and was involved in analysis and interpretation of the
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
This study was conducted in accordance with ethical
standards, according to the Declaration of Helsinki and national
and international guidelines, and was approved by the Ethics
Committee of Fujian Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ohgaki H: Epidemiology of brain tumors.
Methods Mol Biol. 472:323–342. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ohgaki H and Kleihues P: Epidemiology and
etiology of gliomas. Acta Neuropathol. 109:93–108. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al: Radiotherapy plus concomitant and adjuvant temozolomide
for glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stylli SS and Kaye AH: Photodynamic
therapy of cerebral glioma-a review. Part II-clinical studies. J
Clin Neurosci. 13:709–717. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pan DS, Feng SZ, Cao P and Li JJ:
Endothelin B receptor promotes the proliferation and immune escape
of malignant gliomas. Artif Cells Nanomed Biotechnol. 46:1230–1235.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ruokun C, Yake X, Fengdong Y, Xinting W,
Laijun S and Xianzhi L: Lentivirus-mediated silencing of HSDL2
suppresses cell proliferation in human gliomas. Tumour Biol.
37:15065–15077. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jiang L, Wang C, Lei F, Zhang L, Zhang X,
Liu A, Wu G, Zhu J and Song L: miR-93 promotes cell proliferation
in gliomas through activation of PI3K/Akt signaling pathway.
Oncotarget. 6:8286–8299. 2015.PubMed/NCBI
|
|
8
|
Pan Y, Liang W, Zhao X, Liu L, Qing Y and
Li Y: miR-548b inhibits the proliferation and invasion of malignant
gliomas by targeting metastasis tumor-associated protein-2.
Neuroreport. 27:1266–1273. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liang ML, Hsieh TH, Ng KH, Tsai YN, Tsai
CF, Chao ME, Liu DJ, Chu SS, Chen W, Liu YR, et al: Downregulation
of miR-137 and miR-6500-3p promotes cell proliferation in pediatric
high-grade gliomas. Oncotarget. 7:19723–19737. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Catanzaro G, Besharat ZM, Miele E,
Chiacchiarini M, Po A, Carai A, Marras CE, Antonelli M, Badiali M,
Raso A, et al: The miR-139-5p regulates proliferation of
supratentorial paediatric low-grade gliomas by targeting the
PI3K/AKT/mTORC1 signalling. Neuropathol Appl Neurobiol. 44:687–706.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang G, Kang C and Pu P: Increased
expression of Akt2 and activity of PI3K and cell proliferation with
the ascending of tumor grade of human gliomas. Clin Neurol
Neurosurg. 112:324–327. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Leslie K, Lang C, Devgan G, Azare J,
Berishaj M, Gerald W, Kim YB, Paz K, Darnell JE, Albanese C, et al:
Cyclin D1 is transcriptionally regulated by and required for
transformation by activated signal transducer and activator of
transcription 3. Cancer Res. 66:2544–2552. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang X, Zhao M, Huang AY, Fei Z, Zhang W
and Wang XL: The effect of cyclin D expression on cell
proliferation in human gliomas. J Clin Neurosci. 12:166–168. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu S, Fu J, Dong Y, Yi Q, Lu D, Wang W, Qi
Y, Yu R and Zhou X: GOLPH3 promotes glioma progression via
facilitating JAK2-STAT3 pathway activation. J Neurooncol.
139:269–279. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ren Z, Zou W, Cui J, Liu L, Qing Y and Li
Y: Geraniin suppresses tumor cell growth and triggers apoptosis in
human glioma via inhibition of STAT3 signaling. Cytotechnology.
69:765–773. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang K, Pang B, Xin T, Hou X, Jia J, Feng
B, Meng L, Xu S and Pang Q: Increased signal transducer and
activator of transcription 3 (STAT3) and decreased cyclin D1 in
recurrent astrocytic tumours compared with paired primary
astrocytic tumours. J Int Med Res. 39:2103–2109. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang F, Wang Z, Yuan J, Wei X, Tian R and
Niu R: RNAi-mediated silencing of Anxa2 inhibits breast cancer cell
proliferation by downregulating cyclin D1 in STAT3-dependent
pathway. Breast Cancer Res Treat. 153:263–275. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang T, Yuan J, Zhang J, Tian R, Ji W,
Zhou Y, Yang Y, Song W, Zhang F and Niu R: Anxa2 binds to STAT3 and
promotes epithelial to mesenchymal transition in breast cancer
cells. Oncotarget. 6:30975–30992. 2015.PubMed/NCBI
|
|
19
|
Lokman NA, Ween MP, Oehler MK and
Ricciardelli C: The role of Annexin A2 in tumorigenesis and cancer
progression. Cancer Microenviron. 4:199–208. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sharma MR, Koltowski L, Ownbey RT,
Tuszynski GP and Sharma MC: Angiogenesis-associated protein Annexin
II in breast cancer: Selective expression in invasive breast cancer
and contribution to tumor invasion and progression. Exp Mol Pathol.
81:146–156. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Inokuchi J, Narula N, Yee DS, Skarecky DW,
Lau A, Ornstein DK and Tyson DR: Annexin A2 positively contributes
to the malignant phenotype and secretion of IL-6 in DU145 prostate
cancer cells. Int J Cancer. 124:68–74. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shiozawa Y, Havens AM, Jung Y, Ziegler AM,
Pedersen EA, Wang J, Wang J, Lu G, Roodman GD, Loberg RD, et al:
Annexin II/Annexin II receptor axis regulates adhesion, migration,
homing, and growth of prostate cancer. J Cell Biochem. 105:370–380.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Villano JL, Seery TE and Bressler LR:
Temozolomide in malignant gliomas: Current use and future targets.
Cancer Chemother Pharmacol. 64:647–655. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Maule F, Bresolin S, Rampazzo E, Boso D,
Della Puppa A, Esposito G, Porcù E, Mitola S, Lombardi G, Accordi
B, et al: Annexin 2A sustains glioblastoma cell dissemination and
proliferation. Oncotarget. 7:54632–54649. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Reeves SA, Chavez-Kappel C, Davis R,
Rosenblum M and Israel MA: Developmental regulation of Annexin II
(Lipocortin 2) in human brain and expression in high grade glioma.
Cancer Res. 52:6871–6876. 1992.PubMed/NCBI
|
|
26
|
Aukrust I, Hollas H, Strand E, Evensen L,
Travé G, Flatmark T and Vedeler A: The mRNA-binding site of Annexin
A2 resides in helices C-D of its domain IV. J Mol Biol.
368:1367–1378. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chiang Y, Rizzino A, Sibenaller ZA, Wold
MS and Vishwanatha JK: Specific down-regulation of Annexin II
expression in human cells interferes with cell proliferation. Mol
Cell Biochem. 199:139–147. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang HJ, Yao DF, Yao M, Huang H, Wang L,
Yan MJ, Yan XD, Gu X, Wu W and Lu SL: Annexin A2 silencing inhibits
invasion, migration, and tumorigenic potential of hepatoma cells.
World J Gastroenterol. 19:3792–3801. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang J, Guo B, Zhang Y, Cao J and Chen T:
Silencing of the Annexin II gene down-regulates the levels of
S100A10, c-Myc, and plasmin and inhibits breast cancer cell
proliferation and invasion. Saudi Med J. 31:374–381.
2010.PubMed/NCBI
|
|
30
|
Kumble KD, Hirota M, Pour PM and
Vishwanatha JK: Enhanced levels of annexins in pancreatic carcinoma
cells of Syrian hamsters and their intrapancreatic allografts.
Cancer Res. 52:163–167. 1992.PubMed/NCBI
|
|
31
|
Zhai H, Acharya S, Gravanis I, Mehmood S,
Seidman RJ, Shroyer KR, Hajjar KA and Tsirka SE: Annexin A2
promotes glioma cell invasion and tumor progression. J Neurosci.
31:14346–14360. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tatenhorst L, Rescher U, Gerke V and
Paulus W: Knockdown of annexin 2 decreases migration of human
glioma cells in vitro. Neuropathol Appl Neurobiol. 32:271–277.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Baldin V, Lukas J, Marcote MJ, Pagano M
and Draetta G: Cyclin D1 is a nuclear protein required for cell
cycle progression in G1. Genes Dev. 7:812–821. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhou C, Ma J, Su M, Shao D, Zhao J, Zhao
T, Song Z, Meng Y and Jiao P: Down-regulation of STAT3 induces the
apoptosis and G1 cell cycle arrest in esophageal carcinoma ECA109
cells. Cancer Cell Int. 18:532018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rahaman SO, Harbor PC, Chernova O, Barnett
GH, Vogelbaum MA and Haque SJ: Inhibition of constitutively active
Stat3 suppresses proliferation and induces apoptosis in
glioblastoma multiforme cells. Oncogene. 21:8404–8413. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yuan J, Yang Y, Gao Z, Wang Z, Ji W, Song
W, Zhang F and Niu R: Tyr23 phosphorylation of Anxa2 enhances STAT3
activation and promotes proliferation and invasion of breast cancer
cells. Breast Cancer Res Treat. 164:327–340. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sansone P and Bromberg J: Targeting the
interleukin-6/Jak/stat pathway in human malignancies. J Clin Oncol.
30:1005–1014. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cui L, Song J, Wu L, Cheng L, Chen A, Wang
Y, Huang Y and Huang L: Role of Annexin A2 in the EGF-induced
epithelial-mesenchymal transition in human CaSki cells. Oncol Lett.
13:377–383. 2017. View Article : Google Scholar : PubMed/NCBI
|