Introduction
The liver is the metabolic repository and the
largest internal organ in the human body. Metabolic or nutritional
disorders can often lead to the tumorigenesis of hepatocellular
carcinoma (HCC) (1,2). HCC remains one of the most deadly
cancers in the world, particularly in China (3). MicroRNAs (miRNAs) regulate
post-transcriptional gene expression and participate in the
biological processes of a variety of diseases, including HCC
(4). Recently, long non-coding RNAs
(lncRNAs) are attracting widespread attention (5). These RNAs contain more than 200
nucleotides and do not encode any protein. With the rapid
development of high-throughput sequencing technology, lncRNAs have
been found to be involved in a large number of biological processes
(6). In addition, recent studies have
found that lncRNAs can form competitive endogenous RNAs (ceRNAs)
through sponge adsorption of miRNAs to regulate mRNA expression.
This plays an important role in the pathological development of
tumors (7–12). For example, lncRNA HULC plays an
important regulatory role in lung cancer through ceRNA (13). Currently, some related ceRNA databases
have been developed (11,14,15), but
research on ceRNAs is still relatively scarce in regards to their
implications in HCC.
In the ceRNA network, there are multiple miRNA
binding sites on each ceRNA, and the number of the same miRNA
binding sites also differs; thus, there are multiple targets for
each miRNA (16). This makes the
ceRNA network a complex large-scale post-transcriptional regulatory
network. Changing one node or edge in the network can affect the
entire network (17,18). These promote ceRNA interactions among
different signaling pathways (19).
ceRNA network members are varied and complicated: mRNA, lncRNA and
circRNA transcripts could interact with miRNAs involved in network
regulation (20). Further exploration
of the ceRNA network will facilitate a better understanding of
post-transcriptional regulation. In addition, the imbalance of the
ceRNA network may lead to the occurrence of various diseases
(21). Further study concerning the
mechanisms of ceRNAs may expand our understanding of the
pathogenesis of diseases.
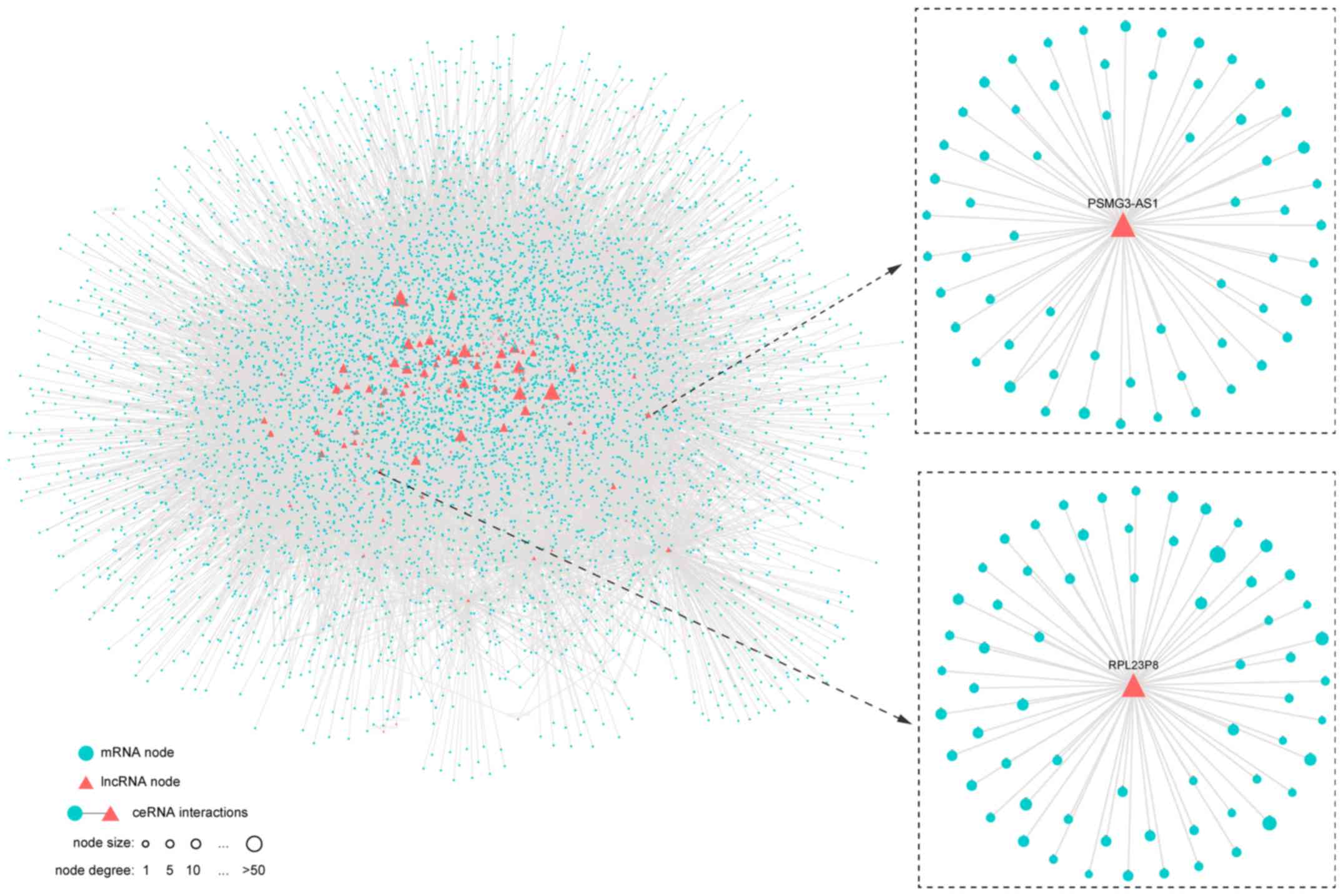
In the present study, we constructed a ceRNA network
and identified a number of the significant lncRNAs in HCC in a
computational way. According to the edgeR algorithm, 7,334 mRNAs
and 138 lncRNAs were detected as being differentially expressed
between HCC and normal liver tissues. Based on these mRNAs and
lncRNAs, we constructed a ceRNA network with 35,637 edges
connecting 113 lncRNAs and 6,136 mRNAs by integrating both the
miRNA interaction-based and expression-based mRNA-lncRNA
correlations. Various connections in the network were also
validated by another independent dataset. Further network and
functional enrichment analyses may identify various hub lncRNAs in
the network that may play key roles in HCC.
Materials and methods
Gene expression profiles and clinical
information
RNASeq based gene expression data which included the
expression of 20,531 genes in 372 HCC tissues and 50 normal liver
tissues, were downloaded from TCGA (https://www.cancer.gov/tcga.). The edgeR algorithm was
used to identify differentially expressed genes between HCC and
normal liver samples. The lncRNA expression profiles and lncRNA
transcript sequences were separated from the lncRNA annotation,
which was downloaded from the GENCODE database (v19) (22). In addition to the TCGA data, another
independent dataset to verify the accuracy of the constructed ceRNA
network was used. A public data set GSE62232 (23) was downloaded from the GEO database. It
contained expression profile data from 81 HCC patients detected by
the Affymetrix Human Genome U133 Plus 2.0 Array platform (Thermo
Fisher Scientific, Inc., Waltham, MA, USA).
Probe annotation
The probe annotation sequences supported by
Affymetrix (http://www.affymetrix.com/) were compared with human
long non-coding transcript sequences and human coding transcript
sequences from the GENCODE database (http://www.gencodegenes.org/) using the BLASTn tool
separately. The sequence comparison results are filtered as
follows: i) Removal of non-coding transcripts and
transcript-encoding probes that were compared simultaneously; and
ii) removal of probes that were compared with multiple
transcripts.
miRNA target information
A total of 410,384 and 423,975 miRNA-mRNA
interactions were downloaded from the miTarBase (http://mirtarbase.mbc.nctu.edu.tw/php/index.php) and
StarBase (http://starbase.sysu.edu.cn/) databases, respectively.
A total of 713,391 interactions remained after removing the
duplicate interactions.
A total of 62,838 and 10,213 miRNA-lncRNA
interactions were downloaded from the lncBase (http://carolina.imis.athena-innovation.gr/diana_tools/web/index.php?r=lncbasev2%2Findex-experimental)
and StarBase databases, respectively. A total of 68,773
interactions remained after removing the duplicate data. Among
these, 18,972 interactions finally remained after GENCODE database
filtration.
ceRNA network construction
Acquisition of candidate ceRNA
relationship based on miRNA interactions
The potential ceRNA relationship for lncRNA-mRNA met
two basic requirements. Firstly, the number of shared target miRNAs
between lncRNA and mRNA was >3. Secondly, the hypergeometric
test false discover rate (FDR) on the significance of the shared
miRNAs between one mRNA and one lncRNA was <0.01. The
hypergeometric test formula is as follows:
P=1-∑i=0r-1(ti)(m-tn-i)(mn)
where, m, is the number of all miRNAs, t, represents
the number of miRNAs that interact with mRNA, n, stands for the
number of miRNAs that interact with lncRNA, and r, indicates the
number of miRNAs that are shared by mRNAs and lncRNAs.
Further filtration by expression-based
correlations
The spearman correlation coefficient between the
lncRNA and mRNA was calculated for each potential lncRNA-mRNA pair.
A correlation coefficient >0 and the corresponding FDR <0.01
were retained to construct the ceRNA network only.
Validation of the ceRNA network by
independent dataset
Using probe re-annotation, microarray profiling data
were obtained and the constructed ceRNA network was validated.
Network analysis
First, Cytoscape 3.1.1 (https://cytoscape.org/) was utilized to display the
constructed ceRNA network, where the built-in Network Analyzer tool
was used to analyze the topological properties of the network and
calculate the degree, betweenness and closeness of the nodes in the
network.
Second, the hub nodes in the ceRNA network were
identified based on the above 3 node centrality measurements, and
then the DAVID database (24) was
applied for Gene Ontology (GO) function enrichment and functional
annotations of these hub nodes. The R package 3.4.1 (https://www.r-project.org/) ‘go Profiles’ was used for
result visualization.
Results
ceRNA network construction
Gene expression profiling data of 372 HCC samples
and 50 normal liver tissues were downloaded from the TCGA database.
Based on the edgeR algorithm, 7,919 differentially expressed genes
were identified. Among them, 7,334 mRNAs and 138 lncRNAs were also
annotated with at least one miRNA partner based on the collected
miRNA-mRNA and miRNA-lncRNA interactions, and the basic nodes of
the ceRNA network were utilized.
Based on these basic nodes, 370,256 miRNA-mRNA
interactions and 5,504 miRNA-lncRNA interactions from the miRNA
interaction information were extracted, of which 133,300
mRNA-lncRNA interactions shared >3 miRNA targets. Based on
hypergeometric enrichment analysis, 10,2031 mRNA-lncRNA
interactions were with significantly shared (FDR <0.01) miRNAs,
and they were taken as potential pairs for ceRNA network
construction. Considering the competitive binding with miRNA, the
mRNA and lncRNA expression should show a positive correlation.
Using Spearman rank correlation, 35,637 mRNA-lncRNA pairs had
significantly positive correlations among the 10,2031 potential
interactions, including 6,136 mRNAs and 113 lncRNA; these nodes and
their interactions constituted the HCC ceRNA network (Fig. 1; Table
SI).
Verification analysis of the
constructed HCC ceRNA network
The annotation data for lncRNA and mRNA based on the
re-annotation of ‘BLASTn’ were obtained due to the lack of
annotation data for the lncRNA chip in GEO database. First, the
public data set GSE62232 which contained gene expression profiles
for 81 HCC patients was downloaded from the GEO database.
Furthermore, using the BLASTn algorithm, the platform probe
sequences were aligned to non-coding and protein-coding transcripts
respectively (see Materials and methods section). A total of 16,074
mRNA-probes and 6,273 lncRNA-probes were obtained by the previous
filter condition introduced in Materials and methods. For
mRNA/lncRNA which was matched by multiple probes, the average value
of the detection probe was taken as the expression value, and
finally the expression profile of 4,846 mRNA and 47 lncRNA were
obtained. By mapping these mRNAs and lncRNAs to the ceRNA network
we constructed, 13,419 mRNA-lncRNA pairs were obtained.
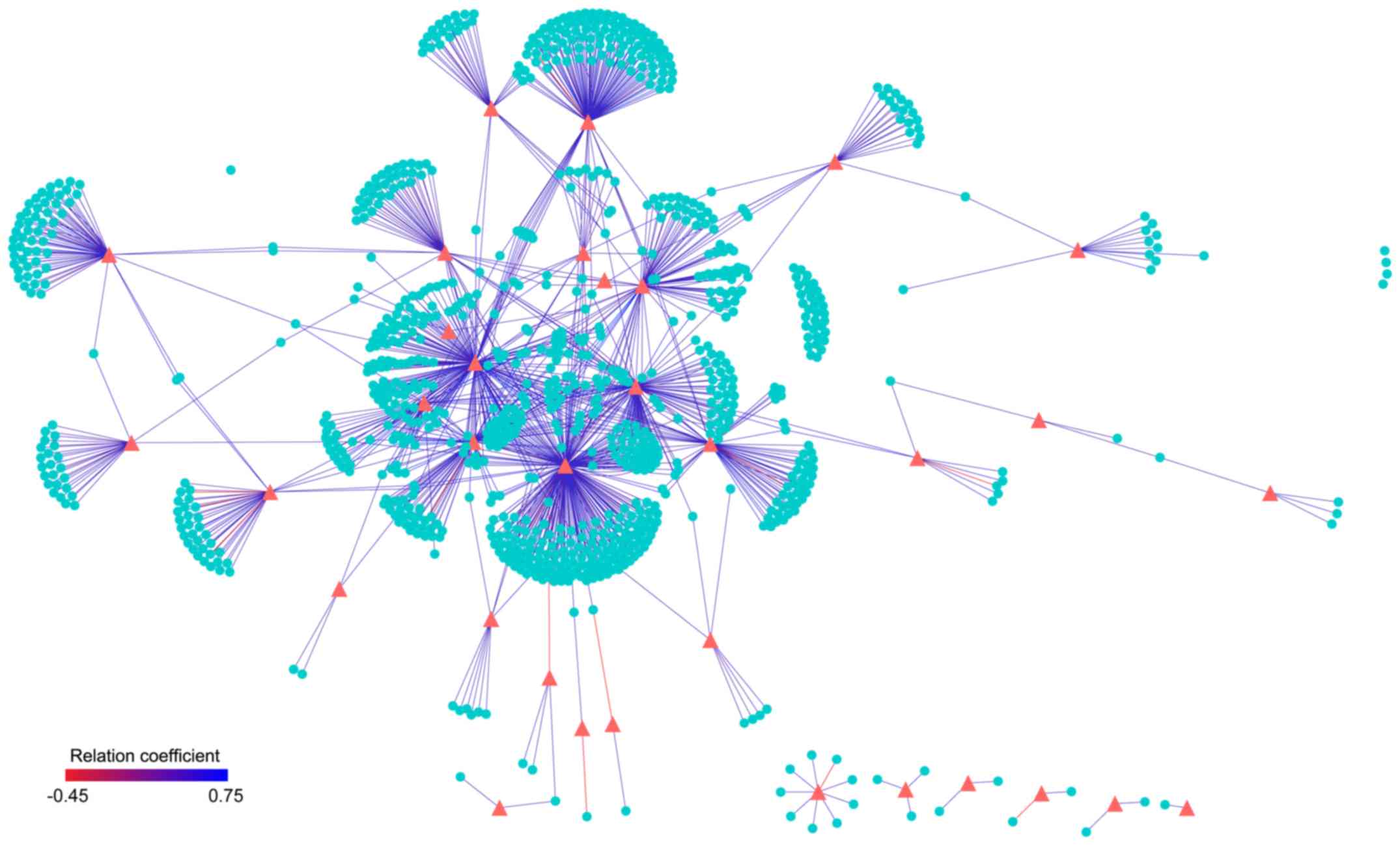
These mapped ceRNA pairs were also verified to
display high correlations in expression level. The Spearman
correlation coefficients between mRNA and lncRNA in these 13,419
ceRNAs pairs were calculated. Based on the results, 1,405 ceRNA
relationships were significantly correlated (FDR <0.01), of
which 1,378 ceRNAs showed a significant positive correlation
(Fig. 2). Cumulative binominal
distributions demonstrated that there was a significant positive
correlation (P<1.0e-16) for non-randomized ceRNAs in the
independent data set; this demonstrated the consistency of the
obtained ceRNAs in different datasets.
Topological analysis of the
network
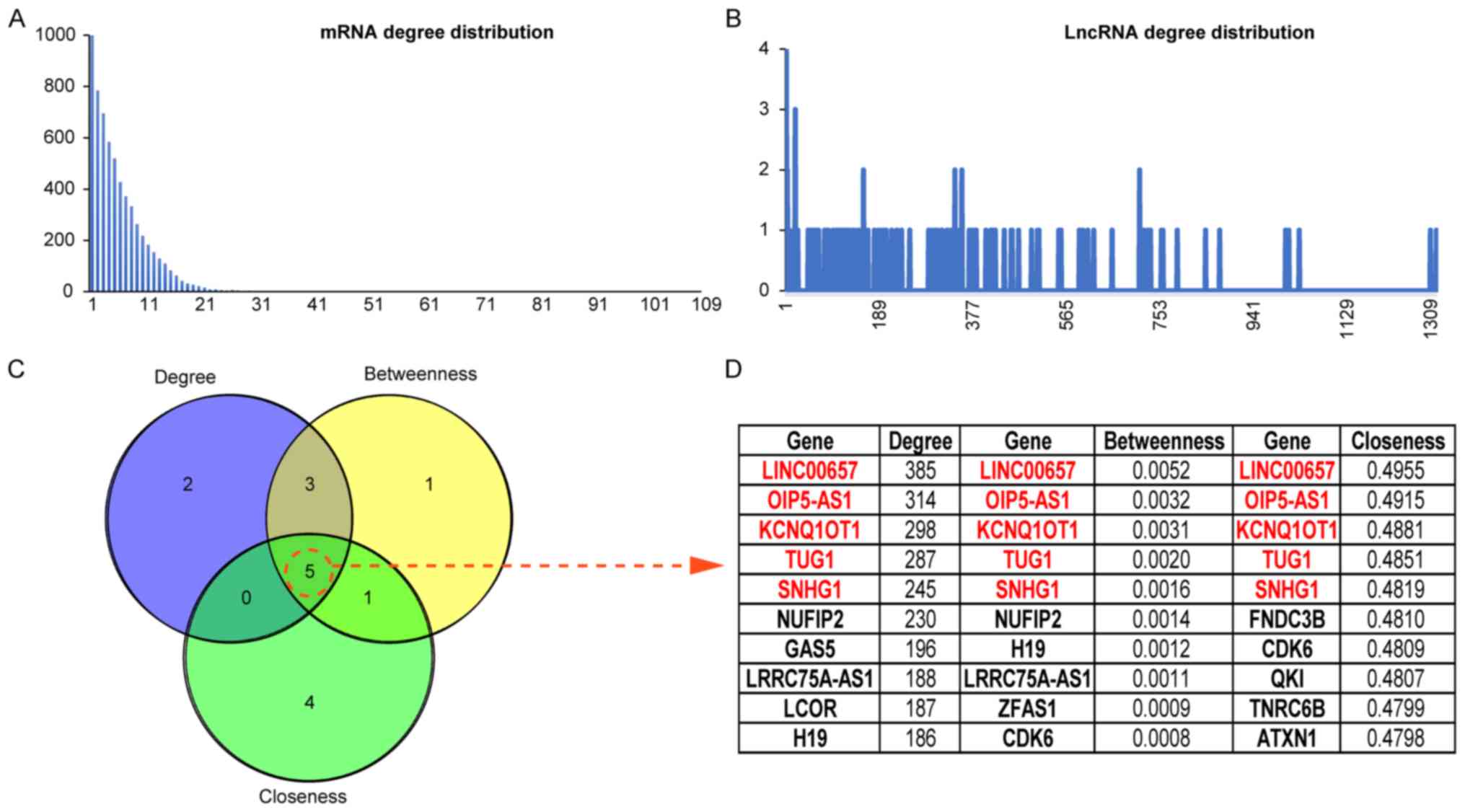
To further analyze the topological properties of the
ceRNA networks, the degree, closeness and betweenness of the
lncRNAs and mRNAs were calculated in the network. The distributions
of lncRNAs and mRNAs are shown in Fig. 4A
and B, and the average degree of a lncRNA node was
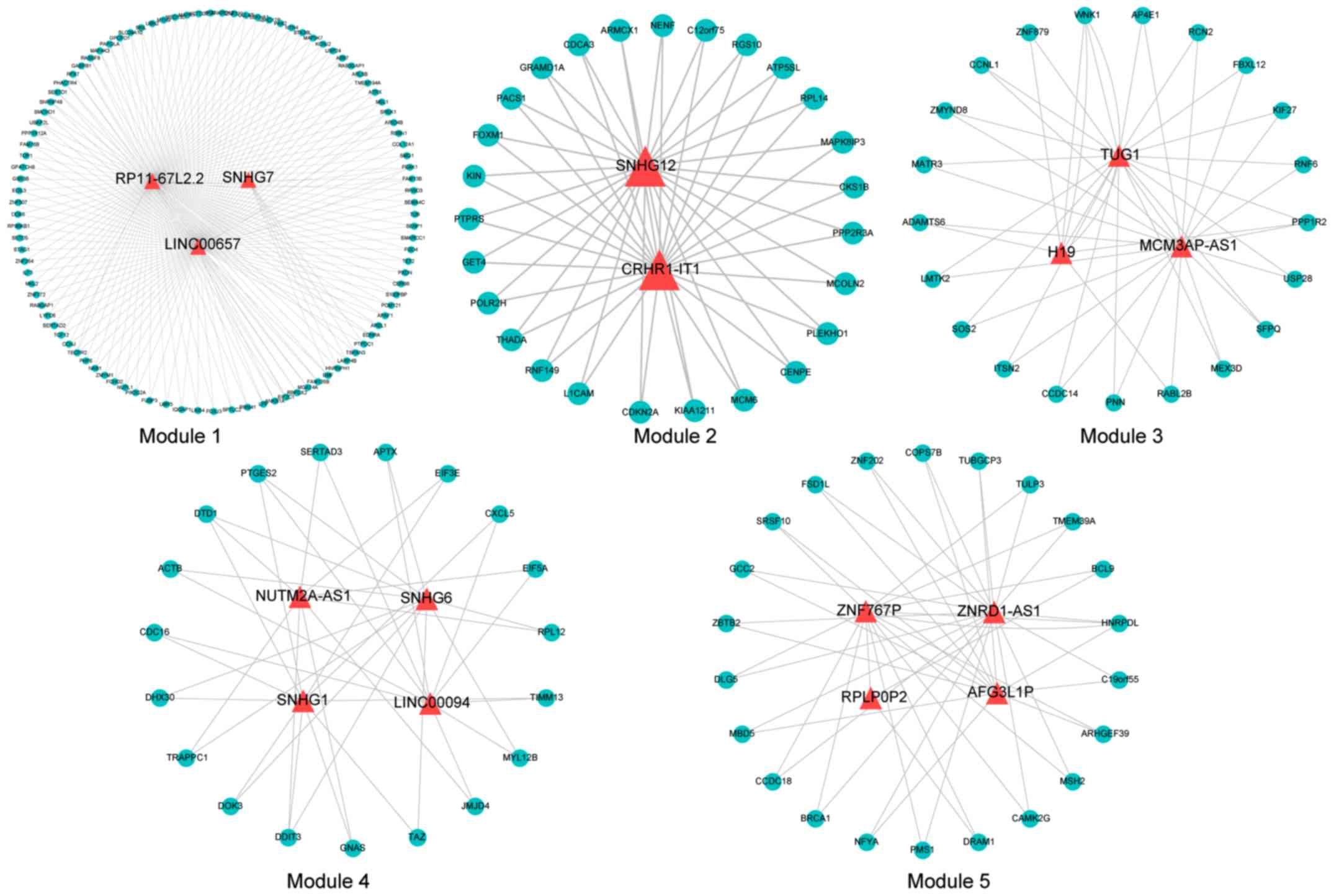
significantly higher than that of a mRNA node. Using the mcode
plugin in Cytoscape software, the ceRNA network was further mined
to obtain 5 modules (Fig. 3),
containing a total of 16 lncRNAs. The topological features of the
nodes are ranked from large to small, and the genes in TOP10 in 3
dimensions are listed (Fig. 4C and
D). Five lncRNAs were found in each dimension list and 3
lncRNAs were included in the mined ceRNA modules (LINC00657 in
module 1, TUG1 in module 3, and SNHG1 in module 2).
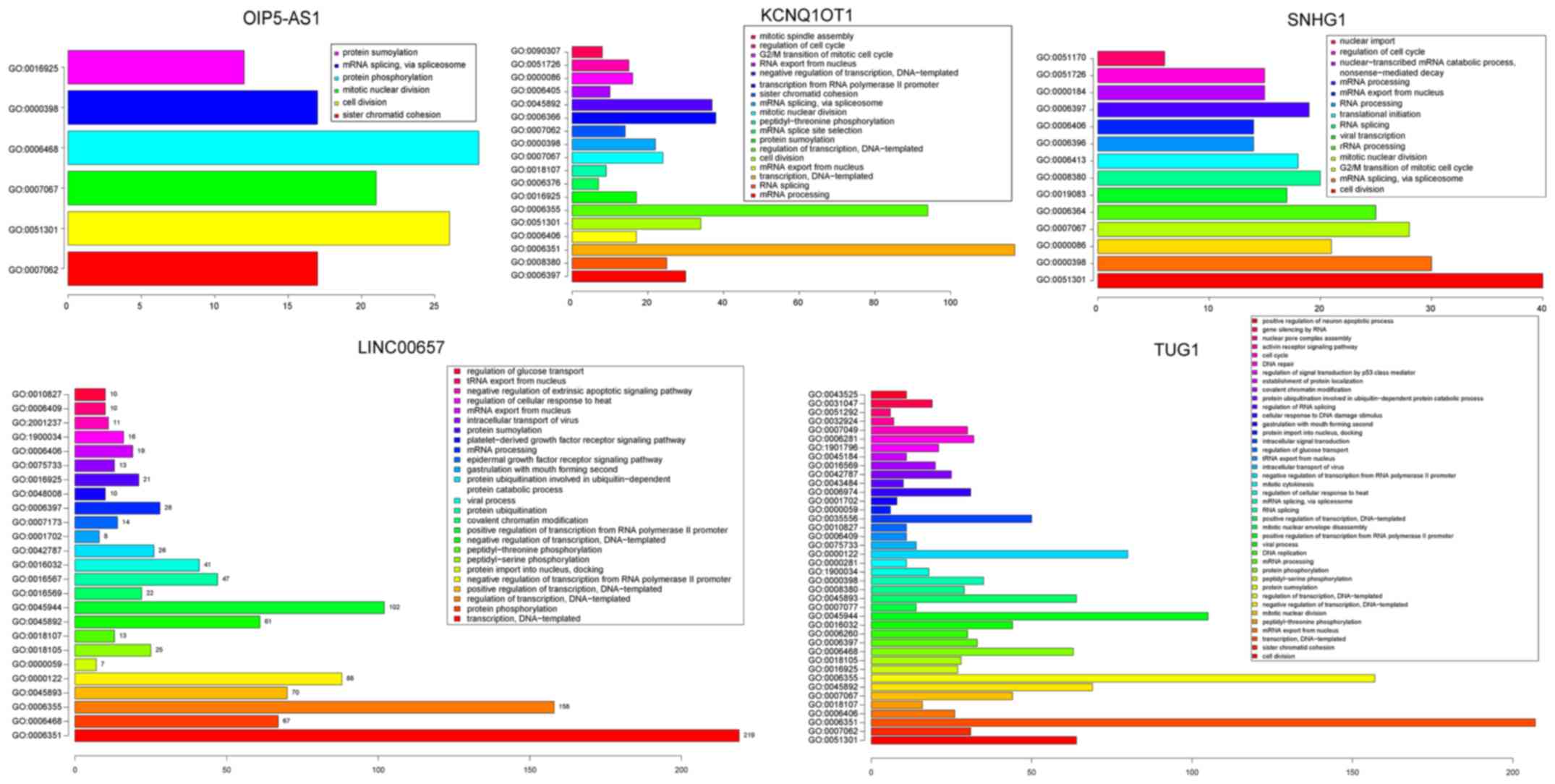
Moreover, GO enrichment analysis was conducted on
the mRNAs from the ceRNA network (David, FDR <0.05; Fig. 5; Table
SII). For lncRNA LINC00657, significant biological pathways
were found, including ‘protein phosphorylation’, ‘regulation of
transcription’ and ‘epidermal growth factor receptor signaling
pathway’. Significantly enriched biological pathways included ‘cell
division’, ‘G2/M transition of mitotic cell cycle’ and ‘mitotic
nuclear division’ for lncRNA TUG1 and SNHG1. These pathways have
been confirmed to be closely related to the occurrence and
progression of HCC (25–30). In addition, Zhang et al
reported that patients in the SNHG1 high-expression group had a
worse recurrence-free survival (P=0.0073, log-rank test) and
overall survival (P=0.0068, log-rank test) than those in the SNHG1
low-expression group, indicating that SNHG1 may serve as a
potential prognostic target (31).
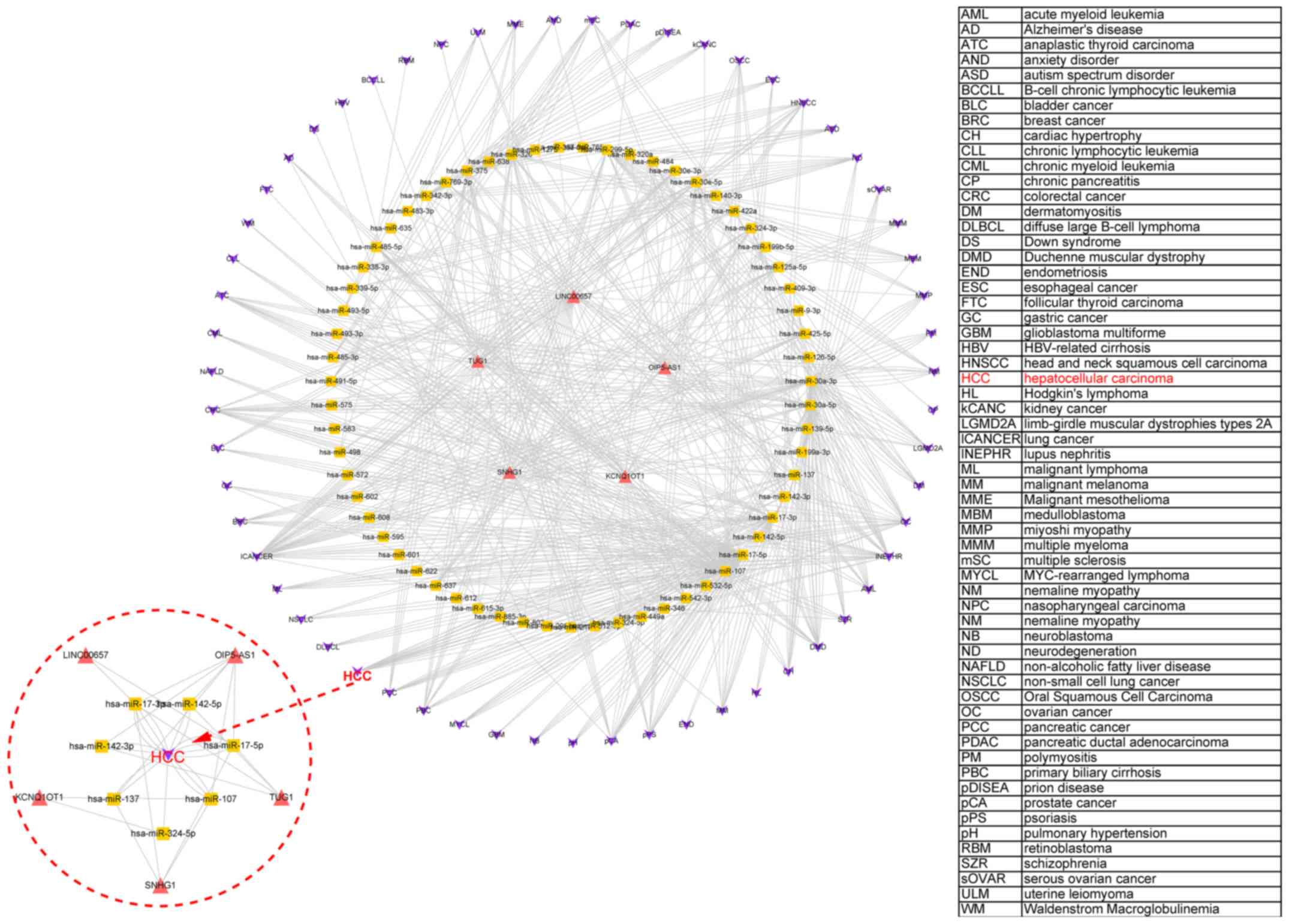
In addition, the miR2Disease database (www.mir2disease.org/) was used to elucidate the
relationship between miRNA expression and various diseases,
including HCC (Fig. 6). Through the
interaction between lncRNAs and miRNAs, an lncRNA-miRNA-disease
network was constructed for the abovementioned 5 high-ranked
lncRNAs. This showed that all 5 lncRNAs could regulate the
occurrence of various diseases including HCC by affecting the
expression of miRNAs.
Discussion
Competing endogenous RNAs (ceRNAs) may explain
certain biological phenomena (autophagy and apoptosis,
morphogenesis), and also can be involved in the inhibition of miRNA
activity (32). As early as 2007,
Ebert et al developed an miRNA sponge to inhibit the
activity of specific miRNAs (33).
Compared with the miRNA sponge, ceRNAs have the advantage of
inhibiting many miRNAs and can play a role in network regulation by
changing the type and number of miRNA binding sites (33). Tang et al developed a human
ceRNA, known as a short tandem target mimic (STTM) that effectively
inhibits many miRNAs (34,35). This suggests that ceRNA has good
application prospects in the treatment of diseases.
The regulatory mechanisms of lncRNAs in cancer
formation are diverse and complicated compared to miRNAs. Although
a large number of studies have shown that lncRNAs play an important
role in the development of tumors (36), the precise molecular mechanisms remain
unclear. lncRNAs can regulate their downstream target genes through
signaling, decoy, guide and scaffold action mode (37). A previous study found that lncRNAs can
also serve as ceRNAs or miRNA sponge, through its microRNA response
elements (MREs) competitive binding miRNAs, and inhibit the
function and activity of miRNAs, thereby regulating the
transcriptional level of miRNA target gene mRNA expression
(38); and are involved in tumor
proliferation, invasion, metastasis and angiogenesis and other
biological behaviors (39). For
example, lncRNA-NEAT1 was found to be upregulated in esophageal
squamous cell carcinoma (ESCC), and was demonstrated to function as
an endogenous sponge to downregulate miR-129, leading to its target
mRNA CTBP2 depression. CTBP2 restoration overturned cellular
proliferation and suppression of invasion regulated by NEAT1
depletion or miR-129 overexpression. This suggested that the lncRNA
NEAT1/miR-129/CTBP2 axis regulates cell progression in ESCC
(40). lncRNA-SNHG1was found to be
upregulated in osteosarcoma (OS) tissues and cell lines, and
knockdown of SNHG1 was found to inhibit cell growth and metastasis
in vitro and in vivo, and also showed better overall
survival for OS patients. Additionally, SNHG1 increased the
oncogene NOB1 through sponging miR-326 as a ceRNA, finally
promoting cell growth, migration and invasion in OS. These findings
uncovered that the SNHG1/miR-326/NOB1 signaling axis plays a key
role in OS progression also suggesting the potential application of
SNHG1 and miR-326 as biomarkers in OS diagnosis and treatment
(41).
The ceRNA hypothesis presents a new concept for the
research of lncRNAs. With the development of genome-wide
sequencing, especially gene chip and second-generation sequencing,
more and more non-coding RNAs have been shown to have special and
important functions related to biological processes such as tumor
formation, invasion and metastasis. lncRNAs expand the regulatory
network through interactions with miRNAs to connect with other
transcriptome members. In this study, LINC00657, TUG1, SNHG1,
KCNQ1OT1 and OIP5-AS1 were ranked as important nodes in the ceRNA
network by a series of topological features (Fig. 4C). A recent study by Liu et al
found that knockdown of LINC00657 significantly inhibited the
growth and progression of tumor cells and LINC00657 was found to be
significantly upregulated in breast cancer, indicating that it
exerts an oncogenic function in breast cancer (42). In another study, LINC00657 was found
to be downregulated in HCC and a potential ceRNA regulatory network
involving LINC00657 and miR-106a-5p was found to display action in
the modulation of PTEN. These results may contribute to a better
understanding of the role of LINC00657 and provide a new
therapeutic target for HCC. TUG1 is a recently discovered
proto-oncogene (43). Huang et
al reported that TUG1 is upregulated in HCC and promotes cell
growth by silencing the KLF2 gene (44). Zhang et al found that
lncRNA-SNHG1 is a potential prognostic marker and therapeutic
target (31), which was consistent
with our findings. KCNQ1OT1 is an lncRNA gene located at the KCNQ1
locus and belongs to the ‘imprinted gene’ and only expresses the
paternal allele. Its transcripts regulate the 15.5 centromere at
p-terminal of chromosome 11 (45).
Aberrant expression of imprinted genes elicits a variety of human
diseases with complex mutations and phenotypic defects (46). The research concerning KCNQ1OT1 in
regards to tumors is still in its infancy, yet a previous studies
found that KCNQ1OT1 can promote the formation of HCC (47). OIP5-AS1 may have a ‘sponge’ function
or serve as a ceRNA for RNA-binding protein HuR, which enhances
cell proliferation. Competitive binding of HuR and miR-424 to
OIP5-AS1 affects HuR binding to target mRNAs in HeLa cells,
including those that encoded proliferative proteins (48). Another study showed that lncRNA
OIP5-AS1 was downregulated and had a negative regulation effect on
miR-410 expression in multiple myeloma MM tissues, thus promoting
KLF10-mediated PTEN/AKT signaling in MM cells. Additionally, the
OIP5-AS1-miR-410-KLF10/PTEN/AKT signaling axis was hypothesized to
exert key functions in cell proliferation, cell cycle progression
and apoptosis inhibition of MM and may represent a therapeutic
target for MM patients (49).
Currently, there is a limited number of cell function studies
concerning OIP5-AS1 in the ceRNA network and we will carry out the
above research.
The ceRNA hypothesis demonstrates a completely new
mode of post-transcriptional regulation. Research has shown that
complex organisms contain a high proportion of non-coding DNA.
These non-coding DNA transcripts can participate in large-scale
post-transcriptional regulation network, and become an integral
part of life activities. However, how lncRNAs function by competing
with ceRNAs has not been adequately investigated (47). Therefore, the establishment and
application of bioinformatic research methods play an important
role in the study of lncRNAs as a regulator of the ceRNA gene
expression network. In the present study, based on the methods of
bioinformatics, lncRNAs were obtained by different algorithms and
the 5 lncRNAs that were found to function as ceRNAs in HCC were
summarized, which provided an important basis of lncRNAs as the
gene expression regulatory network of ceRNAs. Future research will
investigate the biological function of these 5 lncRNAs, especially
OPI5-AS1 in HCC, with the aim to reveal its ceRNA mechanism in the
occurrence and development of HCC and provide a new class of
molecules for tumor prediction and diagnosis. Overall, the ceRNA
network was constructed and various significant lncRNAs in HCC were
identified. The greatest utility of the ceRNA hypothesis may be to
understand how ceRNA networks affect post-transcriptional
regulation, and how mbalances in ceRNA networks may contribute to
cancer. A large number of differentially expressed lncRNAs in HCC
were identified which may be potential biomarkers for early
diagnosis of HCC and targets for drug therapy, opening up new ideas
for the clinical diagnosis and treatment of HCC.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported in part by the
National Natural Science Foundation of China (NSFC 81702526).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
HH and DC designed the study. HH, DC, HP, SC and HT
collected and analyzed the data. HH, DC, XW, PY and SJ wrote the
manuscript. All authors discussed the results and contributed to
the final draft of the manuscript. All authors read and approved
the manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Shimizu M, Tanaka T and Moriwaki H:
Obesity and hepatocellular carcinoma: Targeting obesity-related
inflammation for chemoprevention of liver carcinogenesis. Semin
Immunopathol. 35:191–202. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sakai H, Shirakami Y and Shimizu M:
Chemoprevention of obesity-related liver carcinogenesis using
pharmaceutical and nutraceutical agents. World J Gastroenterol.
22:394–406. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dhanasekaran R, Limaye A and Cabrera R:
Hepatocellular carcinoma: Current trends in worldwide epidemiology,
risk factors, diagnosis, and therapeutics. Hepat Med. 4:19–37.
2012.PubMed/NCBI
|
|
4
|
Farazi TA, Spitzer JI, Morozov P and
Tuschl T: miRNAs in human cancer. J Pathol. 223:102–115. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Guttman M, Donaghey J, Carey BW, Garber M,
Grenier JK, Munson G, Young G, Lucas AB, Ach R, Bruhn L, et al:
lincRNAs act in the circuitry controlling pluripotency and
differentiation. Nature. 477:295–300. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ponting CP, Oliver PL and Reik W:
Evolution and functions of long noncoding RNAs. Cell. 136:629–641.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Calin GA, Liu CG, Ferracin M, Hyslop T,
Spizzo R, Sevignani C, Fabbri M, Cimmino A, Lee EJ, Wojcik SE, et
al: Ultraconserved regions encoding ncRNAs are altered in human
leukemias and carcinomas. Cancer Cell. 12:215–229. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wilusz JE, Sunwoo H and Spector DL: Long
noncoding RNAs: Functional surprises from the RNA world. Genes Dev.
23:1494–1504. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Arvey A, Larsson E, Sander C, Leslie CS
and Marks DS: Target mRNA abundance dilutes microRNA and siRNA
activity. Mol Syst Biol. 6:3632010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ebert MS and Sharp PA: Emerging roles for
natural microRNA sponges. Curr Biol. 20:R858–R861. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang P, Ning S, Zhang Y, Li R, Ye J, Zhao
Z, Zhi H, Wang T, Guo Z and Li X: Identification of
lncRNA-associated competing triplets reveals global patterns and
prognostic markers for cancer. Nucleic Acids Res. 43:3478–3489.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cao Y, Wang P, Ning S, Xiao W, Xiao B and
Li X: Identification of prognostic biomarkers in glioblastoma using
a long non-coding RNA-mediated, competitive endogenous RNA network.
Oncotarget. 7:41737–41747. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang J, Liu X, Wu H, Ni P, Gu Z, Qiao Y,
Chen N, Sun F and Fan Q: CREB up-regulates long non-coding RNA,
HULC expression through interaction with microRNA-372 in liver
cancer. Nucleic Acids Res. 38:5366–5383. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang P, Zhi H, Zhang Y, Liu Y, Zhang J,
Gao Y, Guo M, Ning S and Li X: miRSponge: A manually curated
database for experimentally supported miRNA sponges and ceRNAs.
Database. 2015(pii): bav0982015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu J, Li Y, Lu J, Pan T, Ding N, Wang Z,
Shao T, Zhang J, Wang L and Li X: The mRNA related ceRNA-ceRNA
landscape and significance across 20 major cancer types. Nucleic
Acids Res. 43:8169–8182. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Giza DE, Vasilescu C and Calin GA:
MicroRNAs and ceRNAs: Therapeutic implications of RNA networks.
Expert Opin Biol Ther. 14:1285–1293. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu K, Yan Z, Li Y and Sun Z: Linc2GO: A
human lincRNA function annotation resource based on ceRNA
hypothesis. Bioinformatics. 29:2221–2222. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Das S, Ghosal S, Sen R and Chakrabarti J:
lnCeDB: Database of human long noncoding RNA acting as competing
endogenous RNA. PLoS One. 9:e989652014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zheng T, Chou J, Zhang F, Liu Y, Ni H, Li
X, Zheng L, Tang T, Jin L and Xi T: CXCR4 3′UTR functions as a
ceRNA in promoting metastasis, proliferation and survival of MCF-7
cells by regulating miR-146a activity. Eur J Cell Biol. 94:458–469.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang K, Li Q, Kang X, Wang Y and Wang S:
Identification and functional characterization of lncRNAs acting as
ceRNA involved in the malignant progression of glioblastoma
multiforme. Oncol Rep. 36:2911–2925. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wei C, Luo T, Zou S, Zhou X, Shen W, Ji X,
Li Q and Wu A: Differentially expressed lncRNAs and miRNAs with
associated ceRNA networks in aged mice with postoperative cognitive
dysfunction. Oncotarget. 8:55901–55914. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kersey PJ, Lawson D, Birney E, Derwent PS,
Haimel M, Herrero J, Keenan S, Kerhornou A, Koscielny G, Kähäri A,
et al: Ensembl Genomes: Extending Ensembl across the taxonomic
space. Nucleic Acids Res. 38:D563–D569. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schulze K, Imbeaud S, Letouzé E,
Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C,
Shinde J, Soysouvanh F, et al: Exome sequencing of hepatocellular
carcinomas identifies new mutational signatures and potential
therapeutic targets. Nat Genet. 47:505–511. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hofmann E, Muller J and Schuknecht B:
Glomus tumor? Aberrant internal carotid artery. Radiologe.
30:555–556. 1990.(In German). PubMed/NCBI
|
|
26
|
Koshland DE Jr: Health care: More access
and more cures. Science. 262:14951993. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pang EY, Bai AH, To KF, Sy SM, Wong NL,
Lai PB, Squire JA and Wong N: Identification of PFTAIRE protein
kinase 1, a novel cell division cycle-2 related gene, in the motile
phenotype of hepatocellular carcinoma cells. Hepatology.
46:436–445. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xu X, Jiang C, Wang S, Tai Y, Wang T, Kang
L, Fan Z, Li S, Li L, Fu J, et al: HPIP is upregulated in liver
cancer and promotes hepatoma cell proliferation via activation of
G2/M transition. IUBMB Life. 65:873–882. 2013.PubMed/NCBI
|
|
29
|
Yan H, Li Z, Shen Q, Wang Q, Tian J, Jiang
Q and Gao L: Aberrant expression of cell cycle and material
metabolism related genes contributes to hepatocellular carcinoma
occurrence. Pathol Res Pract. 213:316–321. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yasuda E, Kumada T, Takai S, Ishisaki A,
Noda T, Matsushima-Nishiwaki R, Yoshimi N, Kato K, Toyoda H,
Kaneoka Y, et al: Attenuated phosphorylation of heat shock protein
27 correlates with tumor progression in patients with
hepatocellular carcinoma. Biochem Biophys Res Commun. 337:337–342.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang M, Wang W, Li T, Yu X, Zhu Y, Ding
F, Li D and Yang T: Long noncoding RNA SNHG1 predicts a poor
prognosis and promotes hepatocellular carcinoma tumorigenesis.
Biomed Pharmacother. 80:73–79. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tay Y, Rinn J and Pandolfi PP: The
multilayered complexity of ceRNA crosstalk and competition. Nature.
505:344–352. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ebert MS, Neilson JR and Sharp PA:
MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian
cells. Nat Methods. 4:721–726. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tang G and Tang X: Short tandem target
mimic: A long journey to the engineered molecular landmine for
selective destruction/blockage of microRNAs in plants and animals.
J Genet Genomics. 40:291–296. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tang G, Yan J, Gu Y, Qiao M, Fan R, Mao Y
and Tang X: Construction of short tandem target mimic (STTM) to
block the functions of plant and animal microRNAs. Methods.
58:118–125. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wei Y, Chang Z, Wu C, Zhu Y, Li K and Xu
Y: Identification of potential cancer-related pseudogenes in lung
adenocarcinoma based on ceRNA hypothesis. Oncotarget.
8:59036–59047. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wu H, Wu R, Chen M, Li D, Dai J, Zhang Y,
Gao K, Yu J, Hu G, Guo Y, et al: Comprehensive analysis of
differentially expressed profiles of lncRNAs and construction of
miR-133b mediated ceRNA network in colorectal cancer. Oncotarget.
8:21095–21105. 2017.PubMed/NCBI
|
|
38
|
Su X, Xing J, Wang Z, Chen L, Cui M and
Jiang B: microRNAs and ceRNAs: RNA networks in pathogenesis of
cancer. Chin J Cancer Res. 25:235–239. 2013.PubMed/NCBI
|
|
39
|
Shi X, Sun M, Liu H, Yao Y and Song Y:
Long non-coding RNAs: A new frontier in the study of human
diseases. Cancer Lett. 339:159–166. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li Y, Chen D, Gao X, Li X and Shi G:
LncRNA NEAT1 regulates cell viability and invasion in esophageal
squamous cell carcinoma through the miR-129/CTBP2 axis. Dis
Markers. 2017:53146492017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang J, Cao L, Wu J and Wang Q: Long
non-coding RNA SNHG1 regulates NOB1 expression by sponging miR-326
and promotes tumorigenesis in osteosarcoma. Int J Oncol. 52:77–88.
2018.PubMed/NCBI
|
|
42
|
Liu H, Li J, Koirala P, Ding X, Chen B,
Wang Y, Wang Z, Wang C, Zhang X and Mo YY: Long non-coding RNAs as
prognostic markers in human breast cancer. Oncotarget.
7:20584–20596. 2016.PubMed/NCBI
|
|
43
|
Li Z, Shen J, Chan MT and Wu WK: TUG1: A
pivotal oncogenic long non-coding RNA of human cancers. Cell
Prolif. 49:471–475. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Huang MD, Chen WM, Qi FZ, Sun M, Xu TP, Ma
P and Shu YQ: Long non-coding RNA TUG1 is up-regulated in
hepatocellular carcinoma and promotes cell growth and apoptosis by
epigenetically silencing of KLF2. Mol Cancer. 14:1652015.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sunamura N, Ohira T, Kataoka M, Inaoka D,
Tanabe H, Nakayama Y, Oshimura M and Kugoh H: Regulation of
functional KCNQ1OT1 lncRNA by β-catenin. Sci Rep.
6:206902016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhang Z, Weaver DL, Olsen D, deKay J, Peng
Z, Ashikaga T and Evans MF: Long non-coding RNA chromogenic in situ
hybridisation signal pattern correlation with breast tumour
pathology. J Clin Pathol. 69:76–81. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wan J, Huang M, Zhao H, Wang C, Zhao X,
Jiang X, Bian S, He Y and Gao Y: A novel tetranucleotide repeat
polymorphism within KCNQ1OT1 confers risk for hepatocellular
carcinoma. DNA Cell Biol. 32:628–634. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kim J, Abdelmohsen K, Yang X, De S,
Grammatikakis I, Noh JH and Gorospe M: LncRNA OIP5-AS1/cyrano
sponges RNA-binding protein HuR. Nucleic Acids Res. 44:2378–2392.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yang N, Chen J, Zhang H, Wang X, Yao H,
Peng Y and Zhang W: LncRNA OIP5-AS1 loss-induced microRNA-410
accumulation regulates cell proliferation and apoptosis by
targeting KLF10 via activating PTEN/PI3K/AKT pathway in multiple
myeloma. Cell Death Dis. 8:e29752017. View Article : Google Scholar : PubMed/NCBI
|