Introduction
Esophageal squamous cell carcinoma (ESCC) is the
major histopathological subtype of esophageal cancer in China,
where esophageal cancer is the fourth most common cancer with a
total of 477,900 new cases and 375,000 deaths approximated to have
occurred in 2015 (1). ESCC is often
closely associated with extensive lymphatic and vascular invasion,
and early symptoms are usually absent (2,3). At
present, the clinical approach to ESCC is surgical treatment
combined with radiotherapy and chemotherapy (4,5). However,
ESCC still shows a poor prognosis due to high lymphatic metastatic
recurrence rates as well as chemotherapy resistance. Intrinsic and
acquired resistance of patients with ESCC to radio- and
chemotherapy is one of the major challenges in cancer treatment
(4,5).
Therefore, the 5 year survival rate for patients with ESCC with
resectable disease over the past 30 years is <20% due to
chemotherapy resistance (2,3). A regimen of cisplatin, docetaxel and
5-fluorouracil is recommended as the category 1 treatment for ESCC
with distant metastasis, according to the guidelines of The
National Comprehensive Cancer Network (5). Cisplatin, a DNA-damaging agent, is
widely used as a chemotherapeutic drug for the treatment of various
human malignancies, including esophageal cancer (6). However, several series of previous
studies have already demonstrated tumor cell resistance to
cisplatin both in vivo and in vitro (5,7,8). Therefore, it is very important to
identify new biological markers which may predict the response to
cisplatin resistance in ESCC.
Several previous studies have found that the
mechanisms involved in cisplatin resistance include reduced
intracellular drug accumulation, increased DNA damage repair, the
inhibition of apoptosis, as well as increased generation of
reactive oxygen species (ROS) (6,9). Elevated
ROS are found in all cisplatin-resistant cell lines, including
those derived from patients who did not respond to cisplatin
treatment (9). Mitochondria are the
organelles where the majority of cellular ROS are generated and
play a central role in ROS-mediated apoptosis (10). Super dismutase [Mn], mitochondrial
(SOD-2), an important primary antioxidant enzyme located in
mitochondria, plays a vital role in ROS metabolism in that it
converts highly toxic superoxide (O2•-) into less toxic
hydrogen peroxides (H2O2) in the
mitochondria, a site that is vulnerable to ROS attack and important
to cell apoptosis (11,12). As part of the nucleoid complex, SOD-2
may protect mitochondrial DNA (mtDNA) from ROS-mediated damage by
interacting with Polγ, glutathione peroxidase, and mtDNA itself
(13). Zhou and Du (14) reported that acquisition of resistance
in pancreatic cancer cells to 2-methoxyestradiol is associated with
the upregulation of SOD-2. Those previous studies suggested that
SOD-2 and ROS may play an important role in cisplatin resistance
during tumor chemotherapy (13,14).
However, whether SOD-2 contributes to cell proliferation and
cisplatin resistance in ESCC is unknown, and needs to be
investigated further.
Our recent study indicated that tumor necrosis
factor-α (TNF-α)-mediated SOD-2 upregulation through the NF-κB
pathway plays an important role in epithelial-mesenchymal
transition (EMT) and migration in A549 cells (15). Chung-man Ho et al (16) reported that TNF-α upregulated SOD-2 in
A549 cells, which suggested that inflammation in the lung tissues
may contribute to high levels of manganese SOD and decreased
catalase, hence creating an intracellular environment favorable to
DNA damage and the promotion of cancer. Kinugasa et al
(17) reported that the NF-κB pathway
contributes to SOD-2 upregulation in well-defined transformed oral
and esophageal human epithelial cell lines. These findings support
that the TNF-α-mediated NF-κB pathway may play a critical role in
regulating SOD-2 expression in tumor cells. Therefore, the present
study examined whether TNF-α mediated SOD-2 upregulation is
involved in cisplatin resistance in ESCC.
Materials and methods
Materials
Rabbit anti-nuclear factor (NF)-κB/p65 (cat. no.
1546-1) and rabbit anti-SOD-2 (cat. no. 2299-s) antibodies were
purchased from Epitomics; Abcam. Rabbit anti-phosphorylated
(p)-NF-κB/p-p65 (cat. no. 3033P) and Survivin (cat. no. 2808)
antibodies were purchased from Cell Signaling Technology, Inc.
CyclinD1 (cat. no. ab134175) and rabbit anti-Bax (cat. no. ab32503)
antibodies were purchased from Abcam. Histone H3 (cat. no. AF0863)
antibody was purchased from Affinity Biosciences. β-actin (cat. no.
AC026) antibody was purchased from ABclonal Biotech Co., Ltd.
Peroxidase-labeled anti-rabbit IgG secondary antibody (cat. no.
074-1506) and anti-mouse IgG secondary antibody (cat. no. 074-1806)
were purchased from KPL, Inc. Cytokine TNF-α was purchased from
Peprotech, Inc. Cisplatin was purchased from Qilu Pharmaceutical
Co., Ltd.
Human ESCC samples
Paraffin-embedded human ESCC cancer samples were
obtained from The Department of Pathology, Second Hospital of Hebei
Medical University between January 2010 and July 2014. A total of
60 cases of ESCC and 30 cases of paired adjacent non-cancerous
normal esophageal tissues (from >2 cm away from the tumor
margin) were obtained in the present study. All 60 patients (44
male and 16 female) were <75 years of age, and were diagnosed
before chemotherapy, radiotherapy or other treatments. The
patients, who agreed to the use of their samples in scientific
research, all provided written informed consent. The present study
was approved by the Ethics Committee of Hebei Medical University
(Hebei, China), and the handling of the information and specimens
collected was conducted in accordance with their ethical and legal
standards. Diagnosis of ESCC was performed by two specialized
pathologists based on the criteria stipulated by the World Health
Organization (18,19). The TNM stage of ESCC was based on the
8th Edition American Joint Committee on Cancer/Union for
International Cancer Control staging manuals (18). The clinical histopathological data of
the patients and the overall survival information of the patients
are available until death or the end of the investigation.
Immunohistochemical staining
Paraffin-embedded human ESCC samples were collected.
All samples were sectioned to 5 µm thickness and
immunohistochemically stained, as previously described (20). Briefly, after blocking, the sections
were incubated with antibodies against TNF-α, SOD-2, CyclinD1 and
Survivin (1:200) overnight at 4°C. Visualization was achieved with
peroxidase-labeled streptavidin-biotin (1 h at room temperature)
and diaminobenzidine (2–5 min at room temperature) staining.
Positive cells were observed under a light microscope
(magnification, ×200).
The degree of immunostaining was scored
independently by two specialized pathologists, according to a
previous study (21). Briefly, the
proportion of tumor cells was scored as: i) 0 (no positive tumor
cells); ii) 1 (<30% positive tumor cells), iii) 2 (30–60%
positive tumor cells); and iv) 3 (>60% positive tumor cells).
The intensity of staining was graded as: i) 0 (for no staining);
ii) 1 (weak staining, light yellow); iii) 2 (moderate staining,
yellow brown); and iv) 3 (strong staining, brown). The final
staining index was calculated as the staining intensity score times
the proportion of positive tumor cells, and scores ≤4 were defined
as low expression of SOD-2.
Cell culture and treatment
Human Eca109 cells and OE-21 cells were obtained
from The Resource Center of Peking Union Medical College Hospital
of China. Eca109 and OE-21 cells were maintained in RPMI 1640
medium containing 10% FBS (both Gibco; Thermo Fisher Scientific,
Inc.). and penicillin/streptomycin, at 37°C and in a 5%
CO2 humidified atmosphere. The expression of SOD-2 was
measured by western blotting. Cells (1×105 cells/well)
were seeded into 6-well plates. Transfections were carried out
using Lipofectamine® 2000 reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
The control small interfering RNA (siRNA), SOD-2 siRNAs and NF-κB
siRNAs were obtained from Shanghai GenePharma Co., Ltd. The siRNA
sequences are presented in Table I.
The cells were cultured for 24 h to allow for successful knockdown,
then the cells transfected with 0.05 µM control siRNA, SOD-2 siRNAs
or NF-κB siRNAs were treated with TNF-α (20 ng/ml) for 24 h,
according to the design of each experiment.
 | Table I.siRNA sequences. |
Table I.
siRNA sequences.
| siRNA | Forward, 5′-3′ | Reverse, 3′-5′ |
|---|
| Control siRNA |
UUCUCCGAACGUGUCACGUTT |
ACGUGACACGUUCGGAGAATT |
| NF-κB siRNA |
CGCCAUCUAUGACAGUAAATT |
UUUACUGUCAUAGAUGGCGTT |
| SOD-2 siRNA |
GGGUUGGCUUGGUUUCAAUTT |
AUUGAAACCAAGCCAACCCTT |
In some experiments, the cells were re-treated with
different concentrations of cisplatin for 24 h, and cell death and
apoptosis were measured.
RNA isolation and reverse
transcription-quantitative PCR
After treatment, the Eca109 cells were homogenized
with TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) and total RNA of cells was isolated using an
RNeasy-kit (Qiagen GmbH) according to the manufacturer's protocol
(15). The total RNA was reverse
transcribed to cDNA in a total volume of 20 µl using a reaction
mixture kit (Promega Corporation). The RT temperature protocol was
as follows: Start for 10 min at 25°C, 1 h at 42°C, 10 min at 80°C,
and then the reaction stopped at 4°C. Transcript levels of SOD-2
were evaluated by RT-qPCR using a SYBR Green kit (Promega
Corporation). The PCR thermocycling conditions included: 10 min at
95°C followed by 40 cycles of denaturation for 15 sec at 95°C, and
annealing for 1 min at 60°C. Gene expression was normalized against
the housekeeping gene β-actin. The following primers were used:
SOD-2 primer: Forward 5′-3′CACTGCAAGGAACAACAGGC, reverse
3′-5′ACCAGGCTTGATGCACATCTT; and β-actin primer: Forward
5′-3′AGCGAGCATCCCCCAAAGTT, reverse 3′-5′GGGCACGAAGGCTCATCATT.
Relative gene expression was calculated using the comparative
2−∆∆Cq method (22).
Western blotting
After treatment, the Eca109 cells were harvested and
homogenized in 100 µl lysis buffer (10 µl Tris HCl pH 7.5; 15 µl 1M
NaCl; 10 µl EDTA pH 8.0; 1 µl Tritanx-100; 64 µl ddH2O).
Total protein was extracted by centrifuging at 10,625 × g for 30
min at 4°C. To detect nuclear translocation of NF-κB p65, the
nuclear proteins were extracted using a Thermo Scientific Pierce
NE-PER kit (Thermo Fisher Scientific, Inc.). Protein concentrations
were determined using a standard bicinchoninic acid assay kit
(Thermo Fisher Scientific, Inc.). Then, 30–100 µg protein per
sample was separated by SDS-PAGE on 10% gels, and transferred to
PVDF nylon membranes. After blocking with 5% non-fat milk in TBS
and Tween-20 for 2 h at room temperature, the membranes were
incubated with NF-κB p65, CyclinD1, Survivin and SOD-2 antibodies
(1:1,000) or Bax (1:500) at 4°C overnight. The membranes were
incubated with the antibody against β-actin (ABclonal Biotech Co.,
Ltd.) at 4°C overnight, which was used as the control. The
membranes were incubated with p-NF-κB p65 and histone H3 (1:1,000)
at 4°C overnight. Histone H3 was used as the loading control for
nuclear fraction. The membranes were then washed and incubated with
anti-rabbit IgG secondary antibody (1:2,000) or anti-mouse IgG
secondary antibody labeled with peroxidase (1:5,000) for 1.5 h at
37°C. The protein signals were acquired using Pierce ECL Western
Blotting Substrate (Thermo Fisher Scientific, Inc.) and an ECL
detection system. The resulting images were quantified by
densitometric analysis using BIO-1D 11.0 software [Gold Sim
(Beijing) International Co.].
Cell Counting Kit-8 (CCK-8) assay
Eca109 cells (2×104/ml) were seeded into
96-well plates and treated with or without TNF-α for 24 h. The
cells transfected with negative control-siRNA or SOD-2 siRNA were
treated with different concentrations (1, 2, 4, 8, 16 and 32 µg/ml)
of cisplatin for 24 h, according to the design of experiments. Cell
death was measured by a CCK-8 assay (MedChemExpress), according to
the manufacturer's protocol. Experiments were carried out at least
three times.
Colony formation assay
After Eca109 cells were transfected with control
siRNA or SOD-2 siRNA, the cells were counted and seeded (2,000
cells/well) in 96-well plates. The cells were treated with TNF-α
and fresh culture medium containing TNF-α was replaced every 3
days. The number of colonies was counted at 9–12 days when the
colonies contained >50 cells. The cells were fixed with 10%
formalin for 1 h at room temperature and stained with crystal
violet for 8 min at room temperature. Colony formation ability was
determined by colony formation number. Cell numbers were counted
under a light microscope (magnification, ×50). Experiments were
repeated at least three times.
Transwell migration assay
Eca109 cells (1×105 cells/well) were
plated in medium without serum in the top chamber of a transwell
plate (Corning, Inc.). The bottom chamber contained standard medium
with 10% FBS plus TNF-α. After 48 h of incubation, the cells in the
top chamber had migrated to the lower surface of the membrane, were
then fixed with 10% formalin for 1 h at room temperature, stained
with crystal violet for 8 min at room temperature, and imaged under
a microscope. Cell numbers in the bottom chamber were counted under
a light microscope (magnification, ×400). Experiments were carried
out at least three times.
Flow cytometric analysis
After treatment with a low concentration of
cisplatin (2 µg/ml), Eca109 cells were harvested and stained with
PE Annexin V and propidium iodide (PI) kit (BD Biosciences),
according to the manufacturer's protocol. The cell apoptosis rates
were measured by fluorescence-activated cell sorting analysis using
a flow cytometer, according to our previous study (23). Briefly, Eca109 cells were stained with
5 µl Annexin V for 15 min and then 5 µl PI for 5 min at room
temperature in the dark. Both early apoptotic cells (Annexin
V-positive and PI-negative) and late apoptotic cells (Annexin
V-positive and PI-positive) were included in apoptosis rate
determinations. The data was analyzed using FlowJo software 7.6
(FlowJo, LLC). Experiments were carried out at least three
times.
Statistical analysis
Statistics were calculated using SPSS 21.0 software
(IMB, Corp.). Associations between SOD-2 expression and clinical
pathologic characteristics were assessed with χ2 test.
Spearman's correlation analysis was applied to determine the
relationship between SOD-2 and TNF-α expression as well as CyclinD1
and Survivin in ESCC samples. Overall survival was measured from
the date of surgical operation to the date of death, or the last
clinical follow-up time before November 2016, according to the
Kaplan-Meier method. The log-rank test was used to compare the
survival distribution. P<0.05 was considered to indicate a
statistically significant difference.
One-way ANOVA or two-way ANOVA was applied according
to one or two variables in the present study in vitro. When
there was a significant ANOVA result, post-hoc analysis was
performed using the Least Significant Difference test to assess
multiple comparisons. Data are presented as the mean ± SD of at
least three independent experiments. P<0.05 was considered to
indicate a statistically significant difference.
Results
Increased expression of SOD-2 in human
esophageal squamous cell carcinoma
Human ESCC samples were collected and the expression
of SOD-2 in ESCC as well as its role in the overall survival of
patients with ESCC was analyzed. In total, 60 cases of ESCC were
collected and the expression of SOD-2 was detected by
immunohistochemical staining. After blinding the identity of the
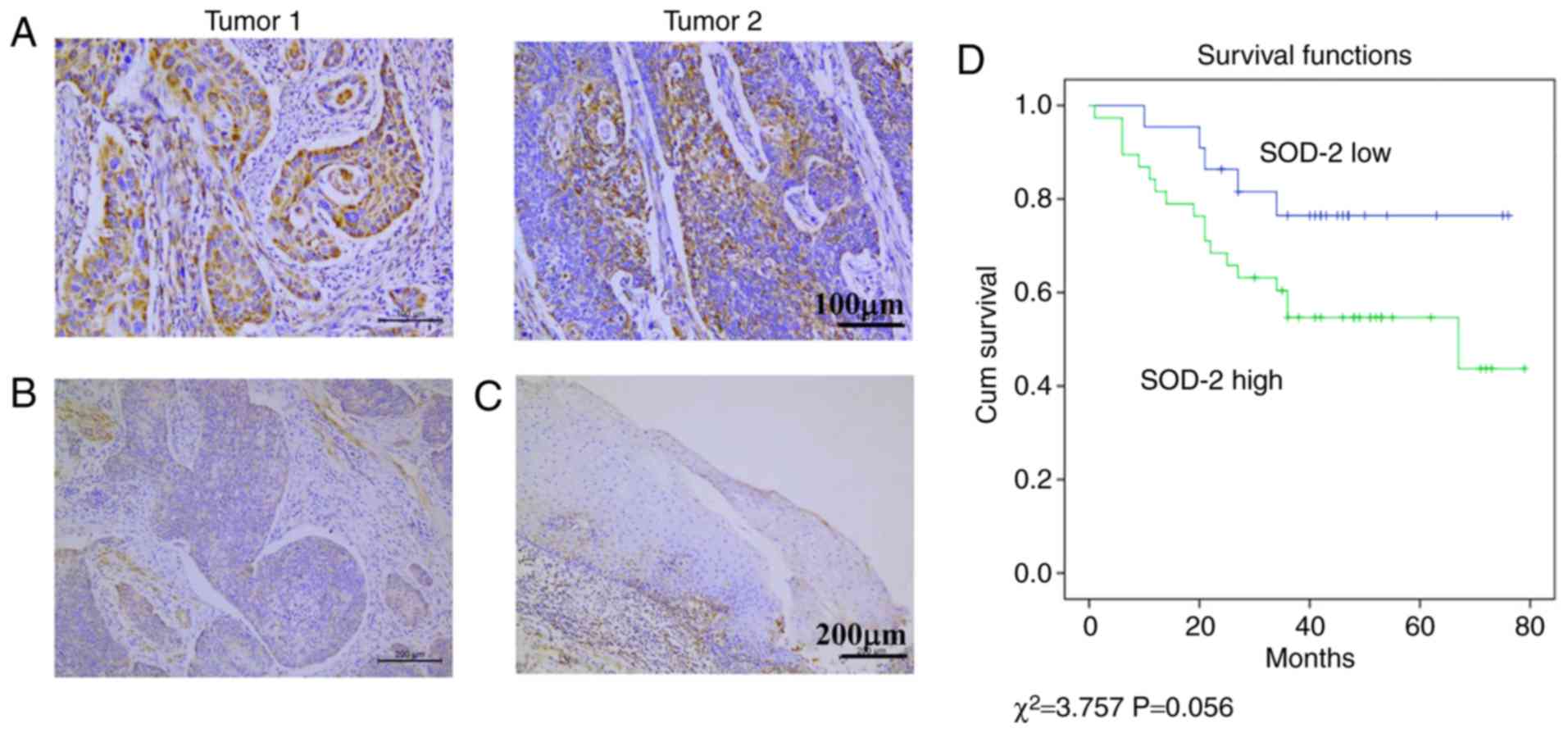
tissue sections, high expression of SOD-2 was observed in the
cytoplasm of some human ESCC cells (Fig.
1A). Low expression of SOD-2 was also observed in some human
ESCC cells (Fig. 1B) as well as
normal esophageal epithelial cells (Fig.
1C). Of the 60 ESCC cases, 38 (63.3%) stained positively for
SOD-2 with higher expression of SOD-2 compared with normal
esophageal epithelial cells (Table
II). Kaplan-Meier analysis indicated that higher expression of
SOD-2 was associated with a poor overall survival in patients with
ESCC (Fig. 1D; P=0.056). To further
explore the role of SOD-2 in human ESCC, the association between
SOD-2 and clinical parameters, including age, sex, differentiation
and TNM stage in 60 cases of human ESCC was assessed. It was
determined that high expression of SOD-2 was significantly
associated with lymph node metastasis and TNM stage in the
patients, but was not associated with other clinical features, such
as age, sex and tumor differentiation of patients (Table III).
| Table II.Positive rate of SOD-2
expression. |
Table II.
Positive rate of SOD-2
expression.
|
|
| SOD-2
expression |
|
|---|
|
|
|
|
|
|---|
| Groups | n | Low (%) | High (%) | P-value |
|---|
| Normal | 30 | 6 (20) | 24 (80) | <0.05 |
| Cancer | 60 | 38 (63.3) | 22 (36.7) |
|
| Table III.SOD-2 expression and
clinicopathological characteristics in 60 cases of esophageal
squamous cell carcinoma. |
Table III.
SOD-2 expression and
clinicopathological characteristics in 60 cases of esophageal
squamous cell carcinoma.
|
|
| SOD-2
expression |
|
|---|
|
|
|
|
|
|---|
| Clinicopathological
variables | n | Low | High | P-value |
|---|
| Age (years) |
|
|
| 0.492 |
|
≤60 | 16 | 7 | 9 |
|
|
>60 | 44 | 15 | 29 |
|
| Sex |
|
|
| 0.815 |
|
Female | 18 | 7 | 11 |
|
|
Male | 42 | 15 | 27 |
|
|
Differentiation |
|
|
| 0.184 |
|
Well/moderate | 43 | 18 | 25 |
|
|
Poor | 17 | 4 | 13 |
|
| Lymph node
metastasis |
|
|
| 0.019 |
|
Negative | 29 | 15 | 14 |
|
|
Positive | 31 | 7 | 24 |
|
| TNM stage |
|
|
| 0.02 |
| I | 11 | 8 | 3 |
|
| II | 25 | 8 | 17 |
|
|
III | 24 | 6 | 18 |
|
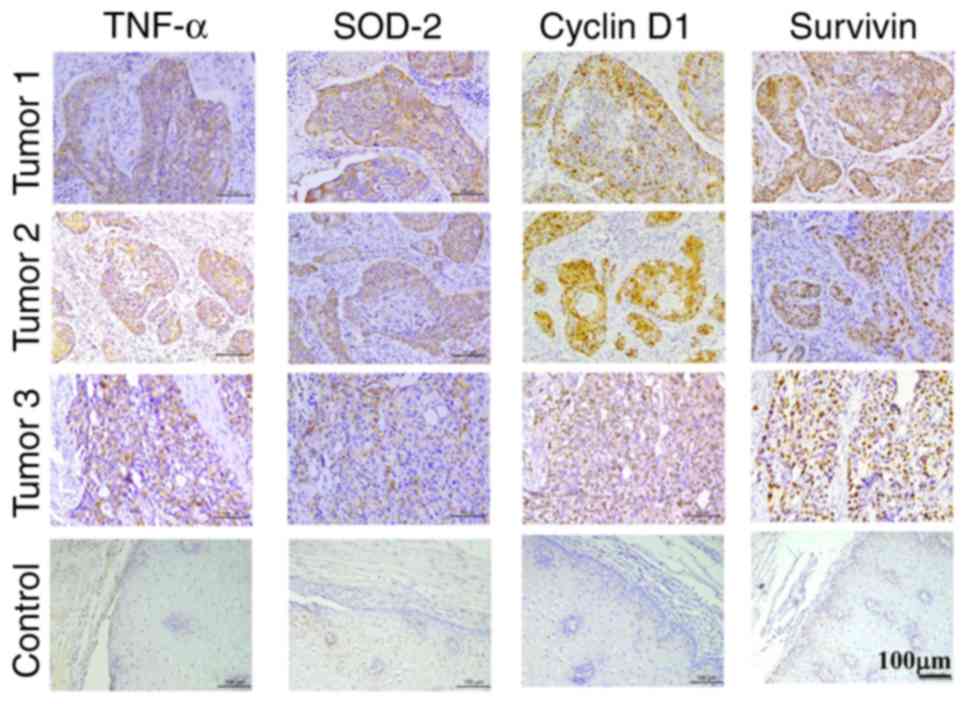
Furthermore, the correlation between TNF-α and
SOD-2, CyclinD1, and Survivin in ESCC samples was analyzed.
Immunohistochemical staining determined the expression of TNF-α,
SOD-2, CyclinD1 and Survivin in the same ESCC samples (Fig. 2). Positive TNF-α expression was highly
correlated with SOD-2, CyclinD1 and Survivin expressions (Table IV), while positive SOD-2 expression
was significantly correlated with CyclinD1 and Survivin expressions
in human ESCC samples (Table V). The
present results suggested that increased SOD-2 in ESCC may be
correlated with TNF-α expression, as well as proliferation-related
proteins CyclinD1 and Survivin, which suggested that TNF-α may
regulate SOD-2, contributing to cell proliferation in ESCC.
| Table IV.Correlation between TNF-α with
CyclinD1, Survivin as well as SOD-2. |
Table IV.
Correlation between TNF-α with
CyclinD1, Survivin as well as SOD-2.
|
| CyclinD1 | Survivin | SOD-2 |
|---|
|
|
|
|
|
|---|
| TNF-α | High | Low | r | P-value | High | Low | r | P-value | High | Low | r | P-value |
|---|
| 39 (high) | 31 | 8 | 0.499 | <0.001 | 34 | 5 | 0.293 | 0.023 | 33 | 6 | 0.602 | <0.05 |
| 21 (low) | 6 | 15 |
|
| 13 | 8 |
|
| 5 | 16 |
|
|
| Table V.Correlation between SOD-2 with
CyclinD1 as well as Survivin. |
Table V.
Correlation between SOD-2 with
CyclinD1 as well as Survivin.
|
| CyclinD1 | Survivin |
|---|
|
|
|
|
|---|
| SOD-2 | High | Low | r | P-value | High | Low | r | P-value |
|---|
| 38 (high) | 30 | 8 | 0.681 | <0.001 | 31 | 7 | 0.291 | 0.036 |
| 22 (low) | 7 | 15 |
|
| 16 | 6 |
|
|
TNF-α induces cell proliferation
associated with upregulation of SOD-2 expression in human ESCC
The human ESCC cell line Eca-109 was treated with
TNF-α to determine if TNF-α contributes to SOD-2 expression in ESCC
in vitro. It was determined that a low concentration of
TNF-α could significantly increase cell viability and colony
formation in Eca109 cells, which suggests that TNF-α could induce
cell proliferation in ESCC (Fig.
3A-C; P<0.05). It was additionally identified that TNF-α
could enhance cell migration in Eca-109 cells (Fig. 3D and E; P<0.05). The western
blotting results indicated that TNF-α upregulated SOD-2 as well as
CyclinD1 and Survivin expressions in Eca-109 cells (Fig. 3F and G). Additionally, it was observed
that Bax was significantly downregulated in cells treated with
TNF-α (Fig. 3F and G). These results
suggested that TNF-α induces cell proliferation associated with
upregulation of SOD-2 in human ESCC cells.
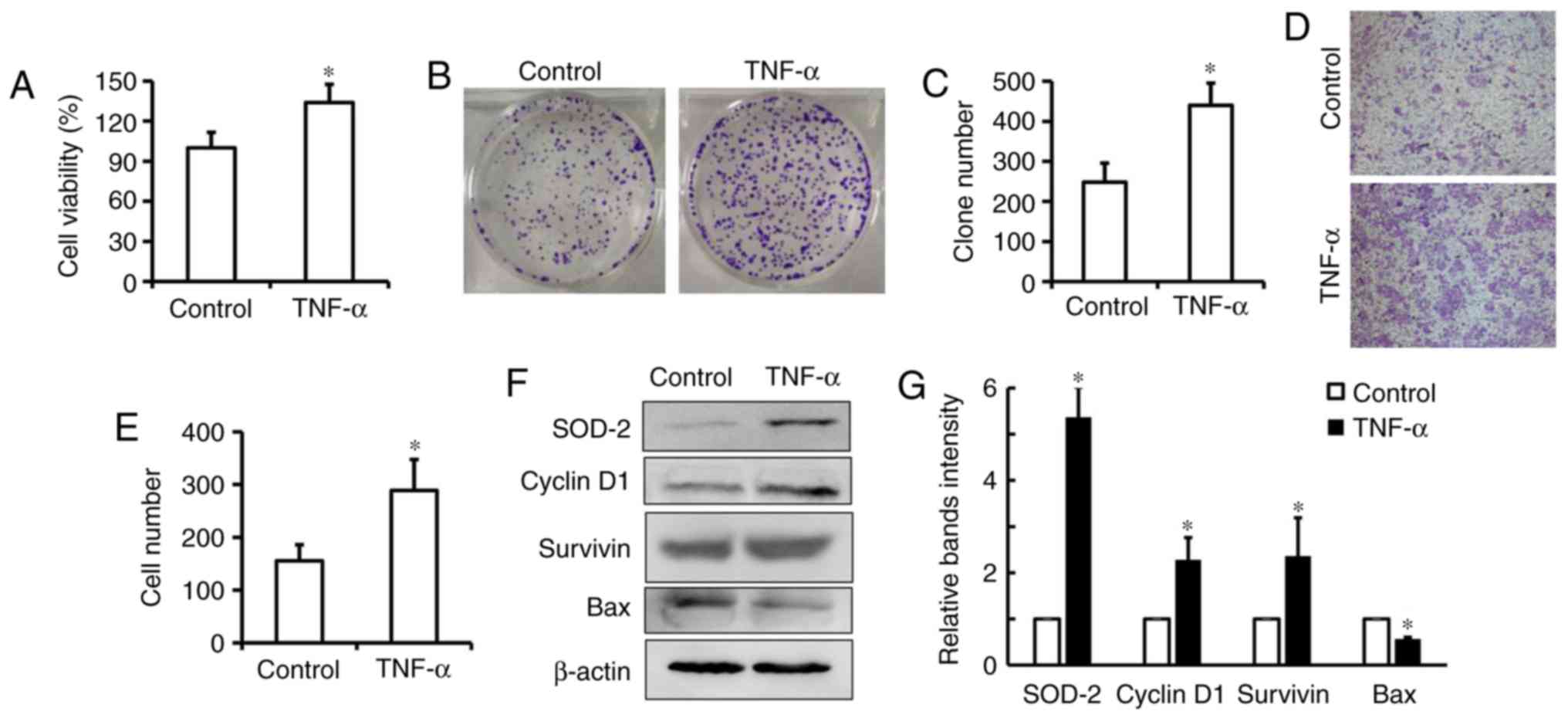 | Figure 3.TNF-α induces cell proliferation in
esophageal cancer cells. (A) Eca109 cells were treated with 20
ng/ml TNF-α for 24 h, and cell proliferation was measured by a Cell
Counting Kit-8 assay. (B and C) After 20 ng/ml TNF-α-treatment for
9–12 days, colony formation was counted and the number was
represented as percent of total cells at the beginning of the
assay. (D and E) After 20 ng/ml TNF-α-treatment for 48 h, the
migratory ability of Eca109 cells was measured by a transwell
migration assay and the cell number was determined by counting the
number of positive cells in the higher field. Magnification, ×400.
After 20 ng/ml TNF-α-treatment for 24 h, the expression of
CyclinD1, Survivin, SOD-2 and Bax was measured by (F) western
blotting and (G) densitometric analysis. Data are presented as the
mean ± SD of three independent experiments. *P<0.05 vs.
respective control. TNF-α, tumor necrosis factor-α; SOD-2, super
dismutase [Mn], mitochondrial. |
TNF-α upregulates SOD-2 through the
NF-κB pathway to induce cell proliferation in human ESCC
To further explore whether upregulation of SOD-2
plays a critical role in TNF-α-induced cell proliferation in human
ESCC, SOD-2 expression was blocked by transfecting cells with SOD-2
siRNA (Fig. 4A). It was observed that
blocking SOD-2 expression significantly inhibited TNF-α-elevated
cell viability and colony formation in ESCC cells (Fig. 4B-D; P<0.05). As shown in Fig. 4B and D, the cell viability and colony
formation in ESCC cells were also inhibited by SOD-2 siRNA, which
suggested that SOD-2 may play a critical role in cell proliferation
in ESCC. Furthermore, blocking SOD-2 expression significantly
inhibited TNF-α-induced cell migration in ESCC cells (Fig. 4E and F; P<0.05). Increased CyclinD1
and Survivin levels induced by TNF-α were also inhibited in Eca109
cells transfected with siRNA (Fig. 4G and
H; P<0.05). These results suggested that upregulation of
SOD-2 by TNF-α contributes to cell proliferation in Eca109 cells.
To further explore the mechanism of upregulated-SOD-2 by TNF-α in
Eca-109 cells, the NF-κB pathway downstream of TNF-α was blocked by
siRNA. It was identified that blocking the NF-κB pathway by siRNA
significantly inhibited TNF-α-increased cell migration in Eca109
cells (Fig. 4I and J; P<0.05).
Increased SOD-2 as well as CyclinD1 and Survivin induced by TNF-α
was inhibited in Eca109 cells transfected with NF-κB siRNA
(Fig. 4K and L; P<0.05).
Therefore, the present data suggested that TNF-α could upregulate
SOD-2 through the NF-κB pathway to contribute to cell proliferation
in Eca109 cells.
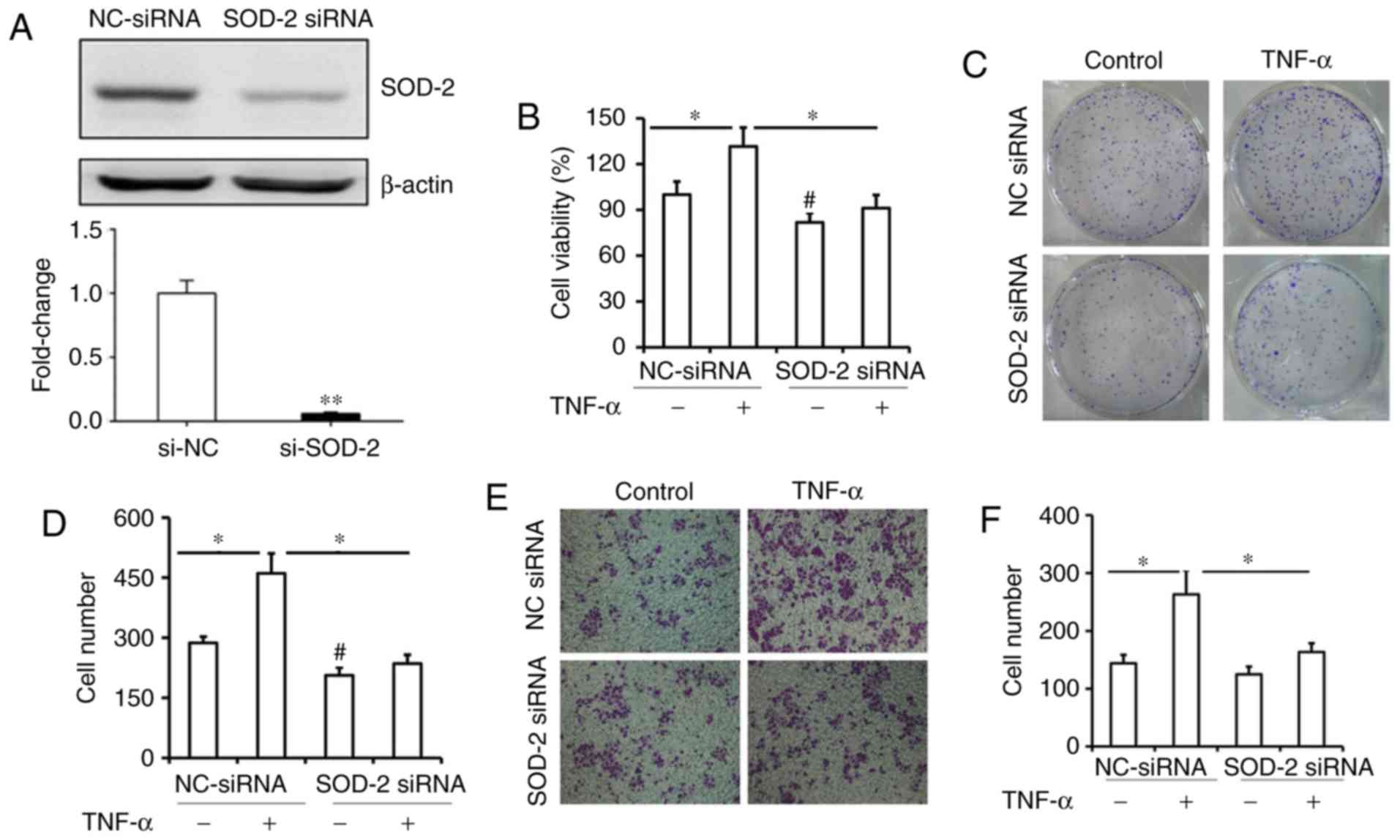 | Figure 4.SOD-2 contributes to TNF-α-induced
proliferation in esophageal cancer cells through the NF-κB pathway.
(A) Expression of SOD-2 mRNA and protein levels was significantly
inhibited after transfection with SOD-2 siRNA in Eca109 cells.
Eca109 cells were transfected with SOD-2 siRNA or control siRNA,
and then treated with 20 ng/ml TNF-α. **P<0.01 vs. si-NC. (B)
After TNF-α-treatment for 24 h, cell proliferation in both cell
samples were measured by a Cell Counting Kit-8 assay. *P<0.05;
#P<0.05 vs. NC-siRNA. (C and D) After treatment for
9–12 days, colony formation was also assessed, and the number is
represented as percent of total cells at the beginning of the
assay. *P<0.05; #P<0.05 vs. NC-siRNA. (E and F)
After TNF-α-treatment for 48 h, the migratory ability of Eca109
cells was measured by a transwell migration assay, and the number
of migrated cells in every field was counted. Magnification, ×400.
*P<0.05. (G and H) After TNF-α-treatment for 24 h, the
expression of CyclinD1, Survivin and SOD-2 was measured by western
blotting. *P<0.05 vs. respective NC-siRNA; #P<0.05
vs. respective TNF-α+NC siRNA. (I and J) Eca109 cells transfected
with control or NF-κB siRNA were treated with 20 ng/ml TNF-α. After
TNF-α-treatment for 48 h, the migratory ability of Eca109 cells was
measured by a transwell migration assay, and the number of migrated
cells in every field was counted. Magnification, ×400. *P<0.05.
(K and L) After TNF-α-treatment for 24 h, the expression of NF-κB,
SOD-2, CyclinD1 and Survivin was detected by western blotting. Data
are presented as the mean ± SD of three independent experiments.
*P<0.05 vs. respective NC-siRNA; #P<0.05 vs.
respective TNF-α+NC siRNA. SOD-2, super dismutase [Mn],
mitochondrial; TNF-α, tumor necrosis factor-α; NC, negative
control; siRNA, small interfering RNA; p, phosphorylated. |
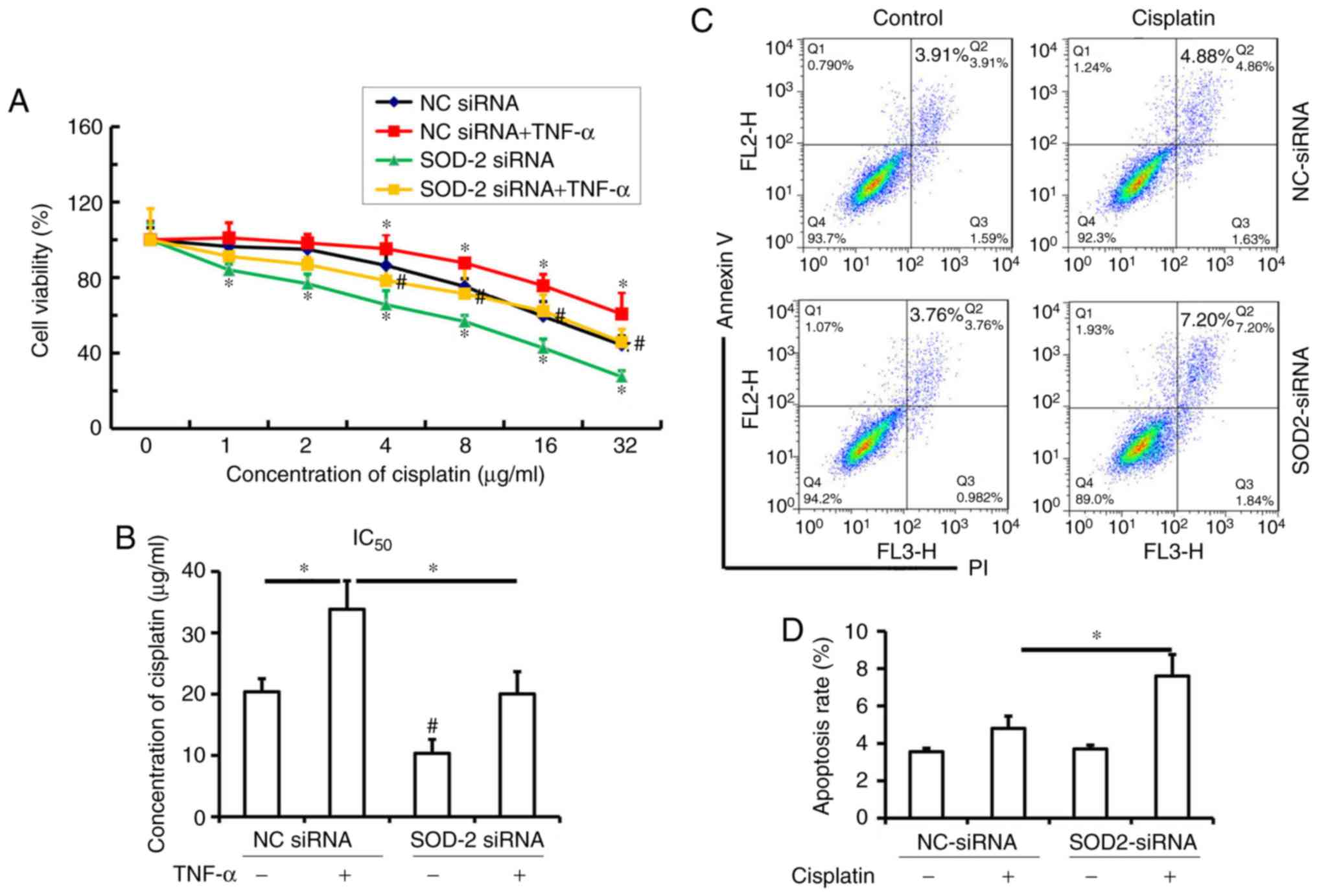
SOD-2 affects the chemosensitivity of
ESCC to cisplatin
Since upregulation of SOD-2 by TNF-α contributes to
cell proliferation and migration in Eca109 cells, the present study
investigated whether TNF-α induces cisplatin resistance, as well as
the role of SOD-2 in the chemosensitivity of ESCC to cisplatin. It
was observed that TNF-α increased cisplatin resistance over a range
of concentrations (4–32 µg/ml) in the NC siRNA group, while SOD-2
blocking enhanced cisplatin chemosensitivity over a range of
concentrations (1–32 µg/ml) in ESCC cells transfected with SOD-2
siRNA compared with the NC siRNA group (Fig. 5A; P<0.05). Transfection with SOD-2
siRNA significantly increased the chemosensitivity of ESCC to
cisplatin (4–32 µg/ml) in TNF-α-induced Eca-109 cells compared with
the TNF-α+NC siRNA group (Fig. 5A;
P<0.05). The IC50 of cisplatin in TNF-α-induced
Eca-109 cells was higher than that in the control cells (Fig. 5B; P<0.05). However, transfection
with SOD-2 siRNA significantly decreased the IC50 of
cisplatin both in TNF-α-induced Eca-109 cells and control cells
(Fig. 5B; P<0.05). Therefore, it
was demonstrated that SOD-2 contributes to TNF-α-induced cisplatin
resistance in ESCC.
Furthermore, in order to confirm that SOD-2
contributes to cisplatin resistance in Eca-109 cells, the effect of
a low concentration of cisplatin on the apoptosis rate of Eca-109
cells transfected with SOD-2 siRNA was determined. It was
identified that 2 µg/ml cisplatin did not induce apoptosis in
Eca-109 cells but could cause apoptosis in cells transfected with
SOD-2 siRNA (Fig. 5C and D;
P<0.05). Therefore, the present results suggested that
inhibition of SOD-2 significantly increases the chemosensitivity of
ESCC to cisplatin, which suggested that SOD-2 may play a critical
role in cisplatin resistance in ESCC.
Discussion
SOD-2, located in the mitochondria, is the major
antioxidant enzyme in the regulation of oxidative stress, and it
functions by catalyzing the conversion of superoxide to hydrogen
peroxide in cells (12,23). Alterations in MnSOD enzymatic function
or protein expression can have serious repercussions on
mitochondrial activity, ultimately resulting in the development of
an assortment of illnesses or participating in the appearance of
malignant cellular phenotypes characterized by glycolytic
metabolism (24). SOD-2 expression is
upregulated in colorectal, lung, gastric/esophageal and cervical
cancer cells compared with normal tissues (25,26). In
the present study, human ESCC samples were collected, and the
expression of SOD-2 in ESCC as well as its role in overall survival
was analyzed in patients with ESCC. High expression of SOD-2 was
observed in human ESCC samples, which was associated with poor
overall survival in patients with ESCC. The present results
suggested that SOD-2 may function as an oncogene in ESCC. Several
previous studies have shown that SOD-2 was upregulated in tongue
squamous cell carcinoma, salivary adenoid cystic carcinoma, ovarian
clear cell carcinoma and lung cancer, which was associated with
increased metastasis (21,27,28). A
positive correlation between TNF-α and SOD-2 expression was
identified in human ESCC samples. Furthermore, a positive SOD-2
expression was significantly correlated with CyclinD1 and Survivin
expression in human ESCC samples. The presented findings suggested
that TNF-α/NF-κB might regulate SOD-2 expression associated with
cell proliferation in ESCC.
TNF-α, as a well-known transcriptional target of
NF-κB, has been reported to be an important regulator in tumor
metastasis and proliferation (29,30). Our
previous study reported that the TNF-α-mediated NF-κB pathway could
upregulateSOD-2 expression, and contribute to EMT and migration in
A549 cells (15). Therefore, it was
necessary to examine whether high expression of SOD-2 contributes
to cell proliferation using human ESCC cell lines Eca-109 and OE-21
cells in vitro. In the present study, it was demonstrated
that SOD-2 expression in OE-21 was very low compared with that in
Eca109 cells (data not shown). Therefore, one ESCC cell line,
Eca109, was used in the present experiments. After cells were
treated with a low concentration of TNF-α for 24 h, it was observed
that TNF-α-induced cell proliferation was associated with
upregulation of SOD-2 in Eca-109 cells. To further investigate if
upregulation of SOD-2 plays a critical role in TNF-α-induced cell
proliferation in ESCC, SOD-2 expression was blocked by transfecting
cells with SOD-2 siRNA. It was observed that blocking SOD-2
expression significantly inhibited TNF-α-elevated cell viability
and colony formation in ESCC cells, which indicated that
upregulation of SOD-2 by TNF-α plays an important role in cell
proliferation in ESCC. A previous study suggested that inflammatory
cytokines, such as TNF-α and IL-6, could result in the subsequent
expression of tumor-related genes, such as hypoxia-inducible factor
1-α and SOD-2 (31). In recent
studies, NF-κB si-RNA was applied to block the NF-κB pathway
(15,32). Blocking the NF-κB pathway
significantly inhibited TNF-α-induced SOD-2 upregulation and cell
migration in Eca109 cells. Therefore, the present results suggested
that SOD-2 could be upregulated by the TNF-α/NF-κB pathway in ESCC.
Although siRNA was applied to inhibit the NF-κB pathway, future
studies may aim to use specific inhibitors of the NF-κB pathway or
specific TNF-α inhibitors to further validate this conclusion.
It has been shown that cisplatin-resistant lung
cancer cells, regardless of the signaling pathway status, share a
common parameter of increase in ROS (33). The ROS-activated GCN2-eIF2α-ATF4-xCT
pathway contributes to mitochondrial dysfunction-enhanced cisplatin
resistance in human gastric cancer cells (33). SOD-2, located in mitochondria, is
essential for the removal of superoxide radicals and is critical
for maintenance of cellular ROS homeostasis and bioenergetic
balance (12). The present study
aimed to investigate if SOD-2 is involved in cisplatin resistance
to ESCC. It was observed that TNF-α induced cisplatin resistance in
Eca109 cells, while transfecting with SOD-2 siRNA significantly
increased the chemosensitivity of ESCC to cisplatin. Blocking SOD-2
could significantly enhance the ability of low concentrations of
cisplatin to cause cell death in ESCC. The present results
suggested that increased SOD-2 may play a critical role in
cisplatin resistance in ESCC. Several previous studies have shown
that overexpression of SOD-2 inhibits mtDNA oxidation from damage
induced by high glucose, UV or acute ethanol exposure (34–36). These
findings support that SOD-2 may play an important role in cisplatin
resistance in ESCC during tumor chemotherapy.
In conclusion, it was determined that a higher
expression of SOD-2 in human ESCC samples was associated with TNF-α
expression and cell proliferation, as well as poor overall survival
in patients with ESCC. SOD-2 upregulated by the TNF-α/NF-κB pathway
contributed to cell proliferation and played a critical role in
cisplatin resistance in ESCC in vitro. A tumor-associated
inflammation environment in the lung may induce high levels of
SOD-2, which contributes to cisplatin resistance in ESCC.
Therefore, SOD-2 may have a potent effect during cisplatin
treatment to enhance drug resistance in ESCC and targeting SOD-2
may become a promising substitute for chemotherapy in ESCC.
Acknowledgements
The authors would like to thank Miss Shelly M Xie,
an international student in School of International Education,
Hebei Medical University, for assistance with editing the
language.
Funding
The present study was supported by The National
Natural Science Foundation of China (grant nos. 31570894, 81670939
and 81672706), The Foundation of Hebei Educational Committee (key
program; grant no. ZD2015010), The Hebei Province Natural Science
Foundation (grant no. H2015206208), The Hebei Province Natural
Science Foundation for Youth (grant no. C2013206251), The
Foundation of Hebei Educational Committee (grant no. SLRC2017045)
and The Natural Science Foundation for Distinguished Young Scholars
of Hebei Province (grant no. H2018206120 to SH and grant no.
H2017206332 to ZJ).
Availability of data and materials
Not applicable.
Authors' contributions
JZ and MZ designed, performed and analyzed most of
the experiments. BL provided technical support for the flow
cytometer experiment and contributed to the flow cytometry
analysis. XH helped perform the immunohistochemical staining and
analysis. YL, QZ and WW helped collect the human samples and
performed the statistical analysis of the human samples. PL
provided intellectual support and helped design the
cisplatin-resistance experiments. LX provided intellectual support
and contributed to the design of the experiments. XZ and HS
supervised and designed the study and experiments, analyzed data
and co-wrote the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The present study has been approved by the Ethics
Committee of Hebei Medical University (Hebei, China), and the
handling of the information and specimens collected was conducted
in accordance with their ethical and legal standards. The patients,
who agreed to the use of their samples in scientific research, all
provided written informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Messager M, Warlaumont M, Renaud F, Marin
H, Branche J, Piessen G and Mariette C: Recent improvements in the
management of esophageal anastomotic leak after surgery for cancer.
Eur J Surg Oncol. 43:258–269. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zeng H, Zheng R, Guo Y, Zhang S, Zou X,
Wang N, Zhang L, Tang J, Chen J, Wei K, et al: Cancer survival in
China, 2003–2005: A population-based study. Int J Cancer.
136:1921–1930. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou P, Zhang R, Wang Y, Xu D, Zhang L,
Qin J, Su G, Feng Y, Chen H, You S, et al: Cepharanthine
hydrochloride reverses the mdr1 (P-glycoprotein)-mediated
esophageal squamous cell carcinoma cell cisplatin resistance
through JNK and p53 signals. Oncotarget. 8:111144–111160. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu T, Li R, Zhao H, Deng J, Long Y, Shuai
MT, Li Q, Gu H, Chen YQ and Leng AM: eIF4E promotes tumorigenesis
and modulates chemosensitivity to cisplatin in esophageal squamous
cell carcinoma. Oncotarget. 7:66851–66864. 2016.PubMed/NCBI
|
|
6
|
Siddik ZH: Cisplatin: Mode of cytotoxic
action and molecular basis of resistance. Oncogene. 22:7265–7279.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fang C, Chen YX, Wu NY, Yin JY, Li XP,
Huang HS, Zhang W, Zhou HH and Liu ZQ: MiR-488 inhibits
proliferation and cisplatin sensibility in non-small-cell lung
cancer (NSCLC) cells by activating the eIF3a-mediated NER signaling
pathway. Sci Rep. 7:403842017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li M, Chen W, Zhang H, Zhang Y, Ke F, Wu
X, Zhang Y, Weng M, Liu Y and Gong W: MiR-31 regulates the
cisplatin resistance by targeting Src in gallbladder cancer.
Oncotarget. 7:83060–83070. 2016.PubMed/NCBI
|
|
9
|
Wangpaichitr M, Sullivan EJ,
Theodoropoulos G, Wu C, You M, Feun LG, Lampidis TJ, Kuo MT and
Savaraj N: The relationship of thioredoxin-1 and cisplatin
resistance: Its impact on ROS and oxidative metabolism in lung
cancer cells. Mol Cancer Ther. 11:604–615. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Trachootham D, Lu W, Ogasawara MA, Nilsa
RD and Huang P: Redox regulation of cell survival. Antioxid Redox
Signal. 10:1343–1374. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Miriyala S, Holley AK and St Clair DK:
Mitochondrial superoxide dismutase-signals of distinction.
Anticancer Agents Med Chem. 11:181–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kienhöfer J, Häussler DJ, Ruckelshausen F,
Muessig E, Weber K, Pimentel D, Ullrich V, Bürkle A and Bachschmid
MM: Association of mitochondrial antioxidant enzymes with
mitochondrial DNA as integral nucleoid constituents. FASEB J.
23:2034–2044. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou J and Du Y: Acquisition of resistance
of pancreatic cancer cells to 2-methoxyestradiol is associated with
the upregulation of manganese superoxide dismutase. Mol Cancer Res.
10:768–777. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yi L, Shen H, Zhao M, Shao P, Liu C, Cui
J, Wang J, Wang C, Guo N, Kang L, et al: Inflammation-mediated
SOD-2 upregulation contributes to epithelial-mesenchymal transition
and migration of tumor cells in aflatoxin G1-induced lung
adenocarcinoma. Sci Rep. 7:79532017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chung-man Ho J, Zheng S, Comhair SA,
Farver C and Erzurum SC: Differential expression of manganese
superoxide dismutase and catalase in lung cancer. Cancer Res.
61:8578–8585. 2001.PubMed/NCBI
|
|
17
|
Kinugasa H, Whelan KA, Tanaka K,
Natsuizaka M, Long A, Guo A, Chang S, Kagawa S, Srinivasan S, Guha
M, et al: Mitochondrial SOD2 regulates epithelial-mesenchymal
transition and cell populations defined by differential CD44
expression. Oncogene. 34:5229–5239. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rice TW, Ishwaran H, Hofstetter WL, Kelsen
DP, Apperson-Hansen C and Blackstone EH; Worldwide Esophageal
Cancer Collaboration Investigators, : Recommendations for
pathologic staging (pTNM) of cancer of the esophagus and
esophagogastric junction for the 8th edition AJCC/UICC staging
manuals. Dis Esophagus. 29:897–905. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rice TW, Gress DM, Patil DT, Hofstetter
WL, Kelsen DP and Blackstone EH: Cancer of the esophagus and
esophagogastric junction-Major changes in the American Joint
Committee on Cancer eighth edition cancer staging manual. CA Cancer
J Clin. 67:304–317. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu C, Shen H, Yi L, Shao P, Soulika AM,
Meng X, Xing L, Yan X and Zhang X: Oral administration of aflatoxin
G1 induces chronic alveolar inflammation associated with
lung tumorigenesis. Toxicol Lett. 232:547–556. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chang B, Yang H, Jiao Y, Wang K, Liu Z, Wu
P, Li S and Wang A: SOD2 deregulation enhances migration, invasion
and has poor prognosis in salivary adenoid cystic carcinoma. Sci
Rep. 6:259182016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shen H, Liu J, Wang Y, Lian H, Wang J,
Xing L, Yan X, Wang J and Zhang X: Aflatoxin G1-induced oxidative
stress causes DNA damage and triggers apoptosis through MAPK
signaling pathway in A549 cells. Food Chem Toxicol. 62:661–669.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Miao L and St Clair DK: Regulation of
superoxide dismutase genes: Implications in disease. Free Radic
Biol Med. 47:344–356. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Johnson F and Giulivi C: Superoxide
dismutases and their impact upon human health. Mol Aspects Med.
26:340–352. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kinnula VL and Crapo JD: Superoxide
dismutases in malignant cells and human tumors. Free Radic Biol
Med. 36:718–744. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu Z, Li S, Cai Y, Wang A, He Q, Zheng C,
Zhao T, Ding X and Zhou X: Manganese superoxide dismutase induces
migration and invasion of tongue squamous cell carcinoma via
H2O2-dependent Snail signaling. Free Radic Biol Med. 53:44–50.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hemachandra LP, Shin DH, Dier U, Iuliano
JN, Engelberth SA, Uusitalo LM, Murphy SK and Hempel N:
Mitochondrial superoxide dismutase has a protumorigenic role in
ovarian clear cell carcinoma. Cancer Res. 75:4973–4984. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kitakata H, Nemoto-Sasaki Y, Takahashi Y,
Kondo T, Mai M and Mukaida N: Essential roles of tumor necrosis
factor receptor p55 in liver metastasis of intrasplenic
administration of colon 26 cells. Cancer Res. 62:6682–6687.
2002.PubMed/NCBI
|
|
30
|
Tomita Y, Yang X, Ishida Y, Nemoto-Sasaki
Y, Kondo T, Oda M, Watanabe G, Chaldakov GN, Fujii C and Mukaida N:
Spontaneous regression of lung metastasis in the absence of tumor
necrosis factor receptor p55. Int J Cancer. 112:927–933. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Atsumi T, Singh R, Sabharwal L, Bando H,
Meng J, Arima Y, Yamada M, Harada M, Jiang JJ, Kamimura D, et al:
Inflammation amplifier, a new paradigm in cancer biology. Cancer
Res. 74:8–14. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shao P, Guo N, Wang C, Zhao M, Yi L, Liu
C, Kang L, Cao L, Lv P, Xing L, et al: Aflatoxin G1
induced TNF-α-dependent lung inflammation to enhance DNA damage in
alveolar epithelial cells. J Cell Physiol. 234:9194–9206. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang SF, Chen MS, Chou YC, Ueng YF, Yin
PH, Yeh TS and Lee HC: Mitochondrial dysfunction enhances cisplatin
resistance in human gastric cancer cells via the ROS-activated
GCN2-eIF2α-ATF4-xCT pathway. Oncotarget. 7:74132–74151.
2016.PubMed/NCBI
|
|
34
|
Takai D, Park SH, Takada Y, Ichinose S,
Kitagawa M and Akashi M: UV-irradiation induces oxidative damage to
mitochondrial DNA primarily through hydrogen peroxide: Analysis of
8-oxodGuo by HPLC. Free Radic Res. 40:1138–1148. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Larosche I, Letteron P, Berson A, Fromenty
B, Huang TT, Moreau R, Pessayre D and Mansouri A: Hepatic
mitochondrial DNA depletion after an alcohol binge in mice:
Probable role of peroxynitrite and modulation by manganese
superoxide dismutase. J Pharmacol Exp Ther. 332:886–897. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Madsen-Bouterse SA, Zhong Q, Mohammad G,
Ho YS and Kowluru RA: Oxidative damage of mitochondrial DNA in
diabetes and its protection by manganese superoxide dismutase. Free
Radic Res. 44:313–321. 2010. View Article : Google Scholar : PubMed/NCBI
|